
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnisotropic strain release in a thermosalient crystal: correlation between the microscopic orientation of molecular rearrangements and the macroscopic mechanical motion†
Tomohiro
Seki
 *a,
Takaki
Mashimo
a and
Hajime
Ito
*ab
*a,
Takaki
Mashimo
a and
Hajime
Ito
*ab
aDivision of Applied Chemistry, Frontier Chemistry Center, Faculty of Engineering, Hokkaido University, Sapporo, Hokkaido 060-8628, Japan. E-mail: seki@eng.hokudai.ac.jp; hajito@eng.hokudai.ac.jp
bInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Sapporo, Hokkaido 060-8628, Japan
First published on 6th March 2019
Abstract
The salient effect, which refers to a jumping phenomenon of organic and organometallic molecular crystals typically triggered by phase transitions in response to external stimuli, has been investigated intensively in the last five years. A challenging topic in this research area is the question of how to characterize the release of microscopic strain accumulated during phase transitions, which generates macroscopic mechanical motion. Herein, we describe the thermosalient effect of the triphenylethenyl gold 4-chlorophenyl isocyanide complex 1, which jumps reversibly at approximately −100 °C upon cooling at 50 °C min−1 and heating at 30 °C min−1. Single-crystal X-ray diffraction measurements and differential scanning calorimetric analyses of 1 suggest the occurrence of a thermal phase transition at this temperature. Detailed structural analyses indicate that anisotropic changes to the molecular arrangement occur in 1, whereby the crystallographic a axis contracts upon cooling while the b axis expands. Simultaneously, macroscopic changes of the crystal dimensions occur. This is observed as bending, i.e., as an inclination of the crystal edges, and in the form of splitting, which occurs in a perpendicular direction to the major crystal axis. This study thus bridges the gap between macroscopic mechanical responses that are observed in high-speed photographic images and microscopic changes of the crystal structure, which are evaluated by X-ray diffraction measurements with face indexing.
Introduction
Studies on the mechanical motion of molecular crystals upon exposure to external stimuli have attracted much attention owing to potential applications in actuators and sensors based on organic compounds. For example, crystals of diarylethene derivatives show bending/twisting upon photoirradiation due to a ring-closing isomerization in the crystals.1 Another fascinating mechanical response is the thermally induced walking and rolling of crystals of an azobenzene derivative.2 One challenging theme in this field is the characterization of the relationship between microscopic molecular arrangements and macroscopic mechanical motion.The salient effect of molecular crystals, which refers to a jumping phenomenon of molecular crystals upon external stimulation (Fig. 1, bottom), is another mode of mechanical motion that has quickly developed into a hot topic in this research area during the last decade.3 Typically, temperature changes and photoirradiation initiate salient effects, which are accordingly called thermo-4 and photosalient effects.4c,5 Although crystal-jumping phenomena have been reported only sporadically for a long time,6 the group of Naumov and other groups have recently reported salient effects for various molecular crystals.4,5
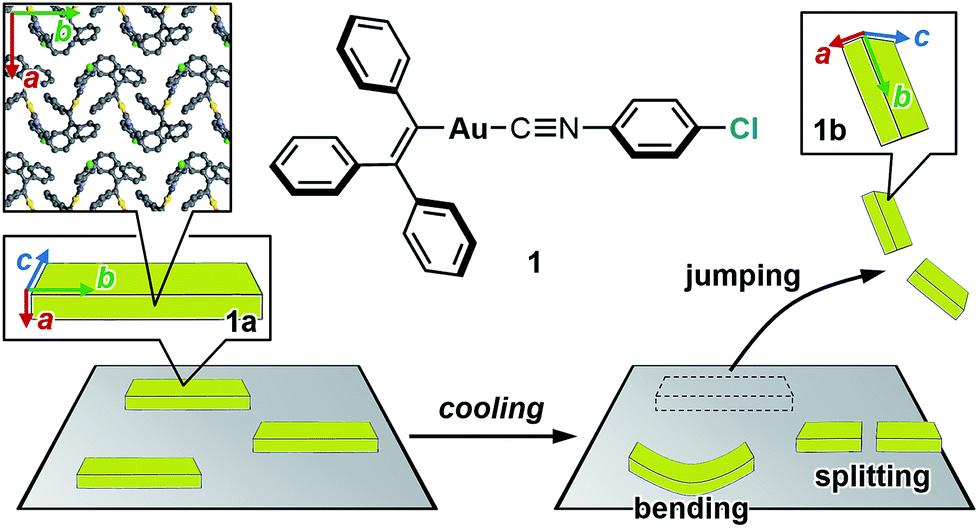 | ||
| Fig. 1 Chemical structure of 1 and schematic representation of the salient effect. Colored arrows indicate the crystallographic axes of crystals of 1. | ||
The mechanism of the salient effect has previously been discussed.4e,g,i,5a Certain external stimuli induce a phase transition in molecular crystals. Then, a molecular rearrangement generates strain within these crystals. The accumulation of this microscopic strain results in macroscopic mechanical power that manifests in rapid crystal motion (‘jumping’).
Several groups have intensively investigated how molecules in the crystal rearrange before and after the jumping. Upon phase transitions, most salient molecules show a slight and anisotropic crystal-structure change without alteration of the space group. For example, thermosalient crystals of 1,2,4,5-tetrabromobenzene show a thermal phase transition and jumping upon heating, whereby the crystallographic b and c axes slightly elongate (Δb = +0.475 Å; Δc = +0.052 Å; RT → 69.5 °C), while the a axis contracts (Δa = −0.323 Å).4a,b Similar trends have been reported for other salient crystals.4a,b,d,e,g,i,k,5e
Compared to crystal-structure analyses, which are conducted on the microscale, macroscopic morphological changes of salient crystals upon jumping have been investigated less extensively. This should be due to the fact that crystal jumping is a significantly more rapid motion, which would render detailed conventional observations on the microscale difficult. To observe such rapid motion, high-speed photographic images of jumping crystals have been recorded by several groups,4a,b,d,e,g,i,5a,b,d which revealed various mechanical responses of the salient crystals, including explosions, flipping, twisting, and simply jumping. Moreover, these macroscopic mechanical responses have been correlated with anisotropic changes of the crystal lattice. One of the limited examples has been reported by Skoko, in which a macroscopic crystal expansion along the major crystal axis of a salient crystal was correlated with an expansion of the crystallographic b axis (Δb > 0) upon jumping.4l
Herein, we report the thermosalient effect of the triphenylethenyl gold isocyanide complex 1 and the orientational relationship between microscopic molecular rearrangements and macroscopic motion (Fig. 1). The thermosalient effect of 1 occurs at approximately −100 °C upon cooling (50 °C min−1) or heating (30 °C min−1). In addition to the jumping, other macroscopic mechanical types of motion, such as bending and splitting, were also observed. Single-crystal structure analyses and DSC measurements of 1 indicate a thermal phase transition with a small molecular rearrangement in the crystal during the jump. Upon the phase transition of 1, the crystallographic a axis slightly contracts, while the b axis elongates. These directions correspond to the crystal thickness and length, respectively (insets in Fig. 1). Thus, the phase-transition-induced contraction and elongation of the macroscopic crystal agrees well with the observed responses of 1: bending along the major crystal axis and splitting perpendicular to the major crystal axis (Fig. 1). The present study thus represents a rare example of salient materials that exhibit a clear orientational correlation between microscopic molecular rearrangement patterns estimated by single-crystal X-ray diffraction analyses and macroscopic mechanical motion observed through high-speed photographic images. This confirmation represents a successful experimental observation of the generation/release of stress, which has so far been scarce in studies of the salient effect.
Results and discussion
Thermosalient effect
Complex 1 was prepared according to a procedure similar to that used for the preparation of a series of arylgold(arylisocyanide) complexes reported by us.5e,7 Complex 1 was characterized by 1H and 13C NMR spectroscopy, HR-MS, and elemental analyses. Crystalline samples of 1 were prepared by recrystallization, i.e., solutions of 1 (50 mg) in CH2Cl2 (2 mL) were carefully layered with hexane (8 mL) to generate yellow crystalline platelets after a few days (Fig. S1†). These crystals, which are stable at room temperature, are henceforth referred to as 1a.8Crystals of 1 exhibit a prominent thermosalient effect upon exposure to temperature changes. Fig. 2 shows crystals of 1 on a heating/cooling plate under ambient light observed using a high-speed camera. Upon cooling at 50 °C min−1, crystals of 1 start to jump as high as ∼1 cm between −80 to −150 °C (Fig. 2 and S3 and ESI Movies S1–S3†), confirming the thermosalient behavior. Hereafter, the crystals obtained after mechanical movement upon cooling will be called 1b.9 The thermosalient effect of 1 can be induced by a wide range of cooling rates (2–80 °C min−1), and slower cooling rates tend to increase the temperature for crystal jumping.10 Although our previously reported gold isocyanide complex showed a photosalient effect, only a small proportion of the crystals (∼5%) jumped upon photoexcitation.5e Conversely, almost all crystals of 1 engage in jumping upon cooling (ESI Movie S3†). Upon heating from −150 °C at ∼30 °C min−1, crystals of 1 also exhibit a thermosalient effect (Fig. S4†). The resulting crystals jump again upon cooling, demonstrating the reversibility of this process. Several of the hitherto reported thermosalient crystals only jump upon heating or cooling, while reversible thermosalient crystals are less common.4b,e,l,o We also confirmed that semicrystalline powder samples of 1 do not show a thermosalient effect (Fig. S5†), which demonstrates the importance of crystallinity and crystal size for the mechanical response.4b
 | ||
| Fig. 2 Photographs of 1 upon cooling (50 °C min−1), derived from a movie using a high-speed camera (2000 fps). The approximate temperature is −100 °C. | ||
Detailed observations provided us with three representative types of behavior for the thermosalient effect of 1. Fig. 3 summarizes the typical mechanical behavior of 1. In addition to jumping (Fig. 3i), we observed bending of the crystals of 1 (Fig. 3ii). During bending, the faces of the two termini of the crystals bend upwards along the major crystal axis (Fig. 3ii and S6 and ESI Movies S4 and S5†), before typically relaxing to recover the original shape. Further decreasing the temperature induces a thermosalient effect in these crystals. The reproducible bending along the major crystal axis was confirmed by cooling a crystal of 1 on a goniometer head using a flow of nitrogen gas (Fig. S7†). The splitting of crystals is another typical mechanical response of 1 (Fig. 3iii). Careful observation of several splitting crystals indicated that the cross-section is always obtained along the direction orthogonal to the major crystal axis (Fig. 3iii and S8†).11 Further observation of the jumping behavior of 1 using high-speed recordings revealed that most of the crystals split when the crystals jumped (Fig. 2 and S8 as well as ESI Movies S6 and S7†).4a,b The following discussion of the molecular arrangements may help to clarify the origin of the observed trends in the mechanical response of 1.
 | ||
| Fig. 3 Photographs that show jumping, bending, and splitting as typical mechanical responses of crystals of 1 upon cooling. | ||
Crystal structure analyses
In order to investigate the mechanism of the thermosalient effect of 1, single-crystal X-ray diffraction (XRD) analyses were performed. The XRD measurements of crystals of 1a at 25 °C revealed that 1a crystallizes in the P21/c space group (Fig. 4 and S9 as well as Table S1†). Only one crystallographically independent molecule is contained within the crystalline lattice (Fig. 4a). Hereafter, we denote the three benzene rings of the triphenylethenyl moiety as A, B, and C, while the isocyanide benzene ring is labelled D (Fig. 4a). Molecules of 1 in crystals of 1a form dimers (Fig. 4b) that are characterized by two sets of CH–π interactions between the H atom of ring D and the π-plane of ring C (2.648 Å; dashed blue lines in Fig. 4b). In these dimers, an Au⋯Au distance of 5.2462(5) Å was observed, which suggests the absence of aurophilic interactions.12 Between these dimers, intermolecular π–π stacking interactions manifest at rings C and D (3.523 Å; dashed red lines in Fig. 4c), leading to the formation of infinite chains extending along the [011] direction, i.e., the diagonal direction relative to the b- and c-axis. Similarly, the same molecular chains also extend along the [01−1] direction (Fig. 4c), and thus form a molecular sheet along the bc-plane (Fig. S10†).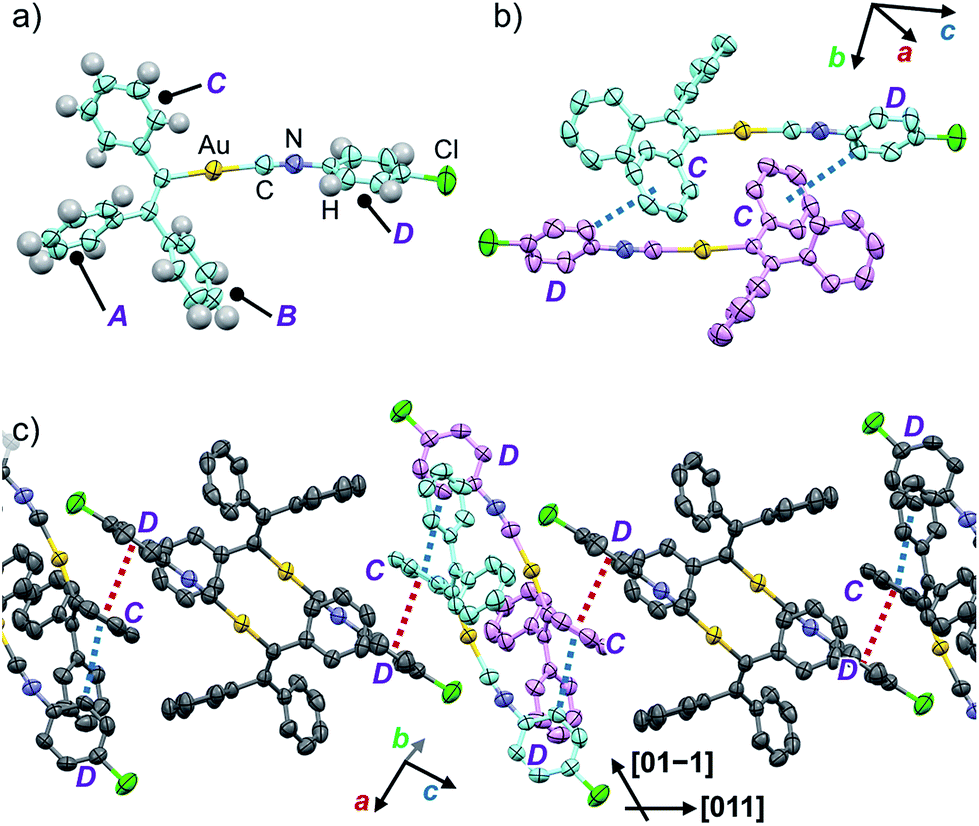 | ||
| Fig. 4 Single-crystal structure of 1a measured at 25 °C with thermal ellipsoids at 50% probability. (a) A monomer unit, (b) a dimer unit, and (c) an extended molecular chain segment. H atoms are omitted for clarity in (b) and (c). | ||
The single-crystal structure of 1b was investigated at −150 °C, which revealed that the molecular rearrangements were only minimal, manifesting in an anisotropic lattice change from 1a at 25 °C. The XRD analyses of 1b at −150 °C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13 indicated that 1b also crystallizes in the P21/c space group similar to 1a (Fig. 5 and S11 as well as Table S1†). The overall molecular arrangements of 1a and 1b are similar (Fig. 5 and S9 and S11†). However, a careful observation revealed a twisting of the isocyanide moiety (ring D) upon cooling. In other words, a juxtaposition of the structures of monomers 1a and 1b shows a twisting of 24.99° (Fig. 6a). Such subtle molecular rearrangements14 upon cooling lead to a slight rearrangement of the overall molecular packing in an anisotropic manner. For example, the crystallographic a and b axes contract (Δa = −0.96 Å; −7.8%) and expand (Δb = +0.44 Å; +4.0%), respectively, upon cooling (25 → −150 °C), while the c axis remains almost unaffected (Δc = −0.18 Å; −1.1%). Previously reported salient organic crystals show similar anisotropic lattice changes in the unit cell dimensions upon phase transitions between similar phases.4a,b,d,e,g,i,k,5e The crystal packing overlay of 1a and 1b, viewed along the c axis (Fig. 6b) provides a visual understanding of the anisotropic crystal lattice change upon cooling. In Fig. 6b, molecules of 1a at 25 °C are colored red and those of 1b at −150 °C are colored light blue within the fixed unit cells, containing four molecules each (Z = 4). Focusing on molecules of 1a and 1b outside the unit cell, we found that upon cooling (25 → −150 °C), the molecules move away from each other along the direction of the b axis and approach each other along the a axis (dotted arrows in Fig. 6b). These results are consistent with the aforementioned anisotropic crystallographic changes upon cooling (Δa < 0; Δb > 0).
13 indicated that 1b also crystallizes in the P21/c space group similar to 1a (Fig. 5 and S11 as well as Table S1†). The overall molecular arrangements of 1a and 1b are similar (Fig. 5 and S9 and S11†). However, a careful observation revealed a twisting of the isocyanide moiety (ring D) upon cooling. In other words, a juxtaposition of the structures of monomers 1a and 1b shows a twisting of 24.99° (Fig. 6a). Such subtle molecular rearrangements14 upon cooling lead to a slight rearrangement of the overall molecular packing in an anisotropic manner. For example, the crystallographic a and b axes contract (Δa = −0.96 Å; −7.8%) and expand (Δb = +0.44 Å; +4.0%), respectively, upon cooling (25 → −150 °C), while the c axis remains almost unaffected (Δc = −0.18 Å; −1.1%). Previously reported salient organic crystals show similar anisotropic lattice changes in the unit cell dimensions upon phase transitions between similar phases.4a,b,d,e,g,i,k,5e The crystal packing overlay of 1a and 1b, viewed along the c axis (Fig. 6b) provides a visual understanding of the anisotropic crystal lattice change upon cooling. In Fig. 6b, molecules of 1a at 25 °C are colored red and those of 1b at −150 °C are colored light blue within the fixed unit cells, containing four molecules each (Z = 4). Focusing on molecules of 1a and 1b outside the unit cell, we found that upon cooling (25 → −150 °C), the molecules move away from each other along the direction of the b axis and approach each other along the a axis (dotted arrows in Fig. 6b). These results are consistent with the aforementioned anisotropic crystallographic changes upon cooling (Δa < 0; Δb > 0).
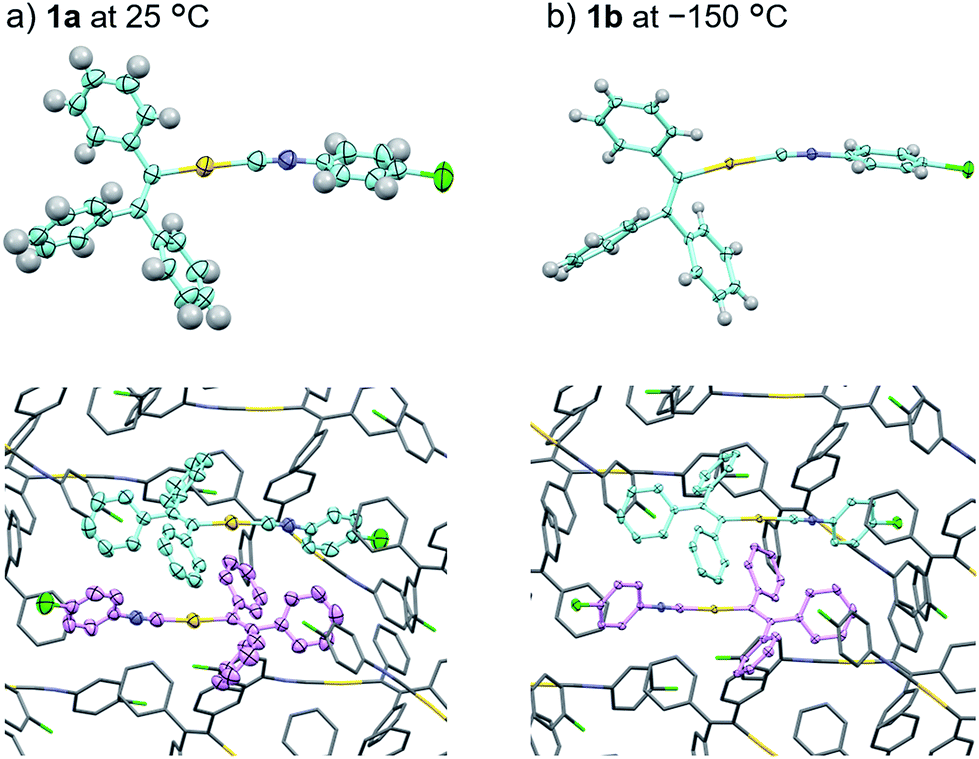 | ||
| Fig. 5 Comparison of the crystal structures of 1a at 25 °C (a) and 1b at −150 °C (b) with thermal ellipsoids at 50% probability. In the bottom figures, H atoms are omitted for clarity. | ||
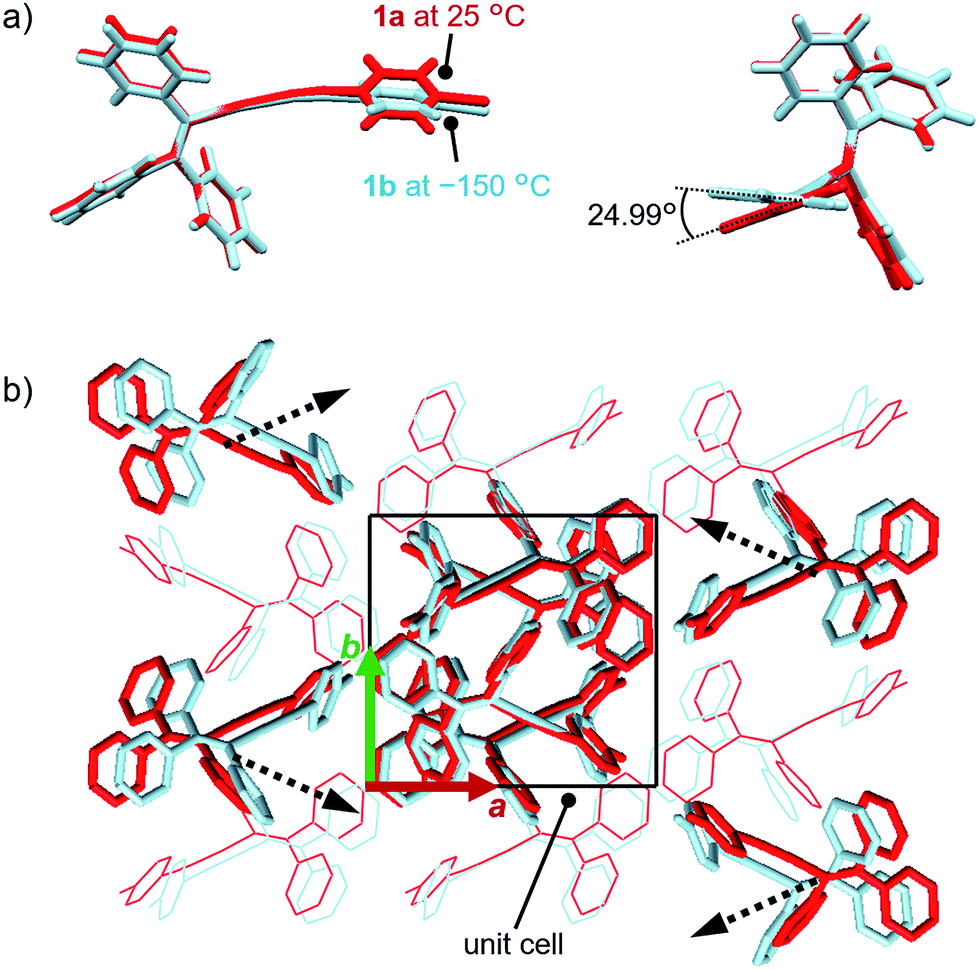 | ||
| Fig. 6 Juxtaposition of the crystal structures of 1a (red; 25 °C) and 1b (light blue; −150 °C). (a) Comparison of the monomer conformations. (b) Comparison of the molecular packing viewed along the c axis. Black dotted arrows indicate how molecules rearrange upon cooling (25 °C → −150 °C). | ||
Temperature-dependent single-crystal XRD measurements of 1 indicate an abrupt change in unit cell dimensions at approximately −50 °C, implying a thermal phase transition. Single-crystal XRD measurements of 1 were carried out between 25 and −150 °C in 25 °C increments. All measurements indicate that the crystal qualities of 1 are satisfactory and the results are summarized in Table S1.† The resulting single-crystal structures of 1 obtained at various temperatures are essentially identical (Fig. S13 and S14†). When the lengths of a and b of the unit cells are plotted as a function of the temperature, drastic changes are observed between −25 and −50 °C (Fig. 7). Thermal expansion coefficients (α) along these directions are estimated to be αa = +579.1 MK−1 and αb = −285.2 MK−1, respectively (Fig. S15†).15 These are one of the highest positive/negative thermal expansion coefficients for organic-based molecular crystals.4e,g,l,16 The degree of the change regarding the length of the c axis remains rather limited in this temperature region (Fig. S16†), which confirms the anisotropic nature of the crystal-lattice change. Simulated powder XRD patterns derived from the single-crystal XRD structures of 1 also show a clear change in this temperature region (Fig. S17†). All these results indicate the occurrence of a thermal phase transition from 1a to 1b.
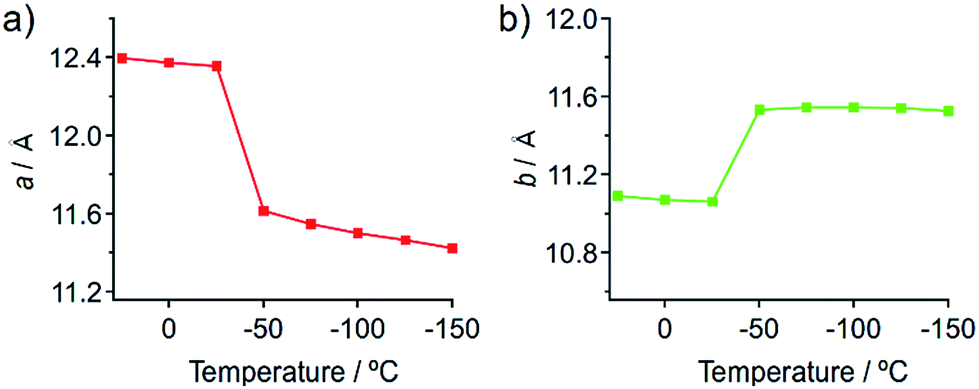 | ||
| Fig. 7 Changes of the length of the crystallographic a (a) and b (b) axes of the single crystals of 1 upon temperature changes. | ||
Differential scanning calorimetric (DSC) analyses of 1 clearly indicate the presence of reversible phase transitions upon temperature changes. Upon cooling 1a at 5 °C min−1, the DSC traces show sawtooth exothermic peaks around −40 to −60 °C with an onset temperature of −40 °C (Fig. 8). Sawtooth profiles are a common sign of phase transitions of molecular crystals with thermosalient effects.4b,i,k,n,o Upon heating 1b from −80 °C at 5 °C min−1, an endothermic peak is again observed at approximately −50 to −30 °C (Fig. 8), confirming the reversibility of the thermal phase transition of 1. The absence of the sawtooth profile upon heating was attributed to the lower probability of the jumping of 1 upon heating compared to that upon cooling. The broad DSC peak upon heating with a temperature range similar to that of the cooling scan indicates that the phase transition of 1 is enantiotropic. The phase-transition temperature estimated by the DSC studies (−30 to −60 °C) are relatively high compared to the temperature where the thermosalient effect occurs (−80 to −150 °C). This discrepancy should reflect the different heating rates employed: −50 °C min−1 for the observation of the thermosalient effect and −5 °C min−1 for the DSC measurements. Moreover, another possible reason is that the jumping motion, which requires a certain time in order to accumulate sufficient strain after the phase transition takes place. Thus, the thermal phase transition of 1 triggers mechanical behavior such as jumping. Powder samples of 1 (its crystal structure basically corresponds to that of crystals of 1a) do not show any peaks in the DSC profiles, indicating the absence of a clear phase transition (Fig. S18†).17–19 This result supports the notion that a phase transition is required for the jumping of 1, given that the powder samples do not show a thermosalient effect (Fig. S5†).4b
Mechanistic investigation
Face index experiments of 1 provided information on the orientational relationship between the microscopic molecular arrangements and the macroscopic crystal morphologies. A face index experiment of crystals of 1a at 25 °C indicates that the largest face of the crystal corresponds to the (100) plane, and that its sides correspond to the (010) and (001) planes (Fig. 9 and S20†). This result suggests that the largest face of the macroscopic crystal corresponds to the crystallographic bc plane. Microscopically, a molecular sheet is formed by π–π stacking interactions within the bc plane (vide supra; Fig. 4c and S10†). This result indicates that the macroscopic crystal growth occurs primarily along this molecular sheet. A face index experiment of 1b at −150 °C afforded results similar to those of 1a (Fig. S21†), supporting the notion that only small molecular movements occur upon the phase transition, which should maintain crystal integrity.4b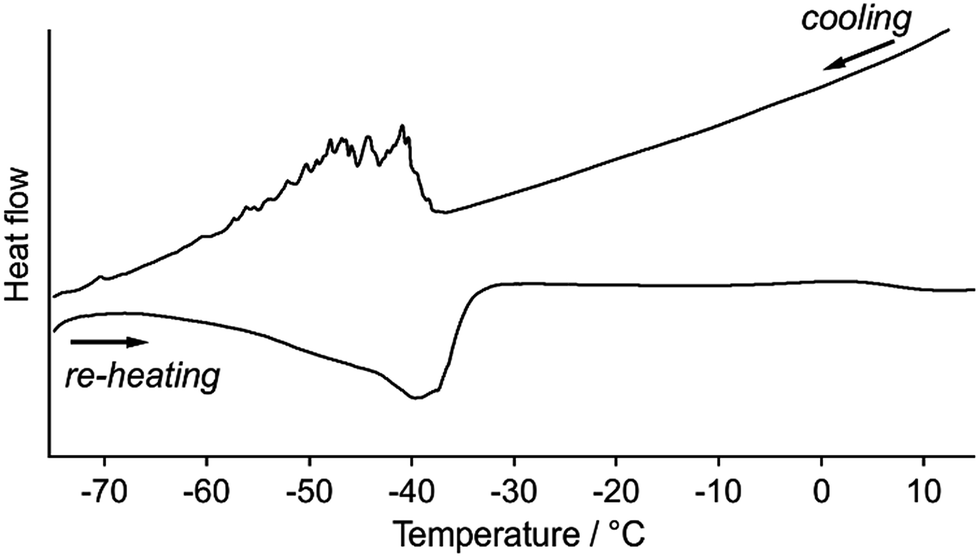 | ||
| Fig. 8 DSC traces for crystals of 1 at cooling/heating rates of 5 °C min−1. | ||
The pattern of microscopic changes of the molecular arrangements of 1 matches well with the trends of the mechanical motion of the macroscopic crystals. Based on the observed lattice changes upon cooling (Δa < 0; Δb > 0) and the face index results (Fig. 9), the macroscopic major crystal axis of 1 should elongate, while the crystal thickness should decrease upon cooling (green and red arrows, respectively, in Fig. 10i). This anisotropic change of the crystal dimensions can generate strain within the crystals to realize specific mechanical motion. Among the three typical types of mechanical motion (Fig. 3), the splitting and bending seem to exhibit orientational correlation with the dimensional changes of the crystals. For example, regarding the bending motion, it is important that the extension of the crystals of 1 upon cooling starts from the bottom domains given that the crystals are located on the cooling plate (Fig. 10ii). Thus, the bottom domains and the top domains of the crystals extend differently, resulting in the observed bending motion that preferentially occurs along the major crystal axis (Fig. 3ii and 10iii). Similarly, the crystal splitting of 1 upon cooling occurs predominantly perpendicular to the major crystal axis (vide supra; Fig. 3iii). This may be rationalized in terms of an extension of the crystals of 1 that generates strain to split the crystals preferentially along the c axis (Fig. 10iv). Such satisfactory orientational correlations between anisotropic crystal structure changes (Δa < 0; Δb > 0) on the microscale and reproducible mechanical motion (splitting and bending) on the macroscale have rarely been reported.20 Similar to the splitting and bending motion, rapid crystal jumping, which is difficult to evaluate visually, should take place based on the same mechanism, i.e., the crystal length and thickness increase and decrease upon cooling, respectively (Fig. 10i) to induce strain in the crystal lattice. Thus, in this study, we have successfully observed the macroscopic motion for the release of the accumulated strain that is generated by an anisotropic change of the lattice dimension on the microscopic scale.
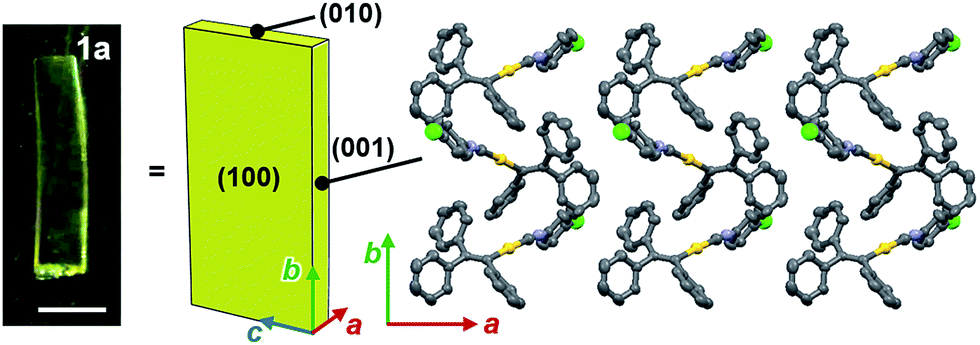 | ||
| Fig. 9 The results of the face index experiment for 1a. Scale bar = 0.5 mm. | ||
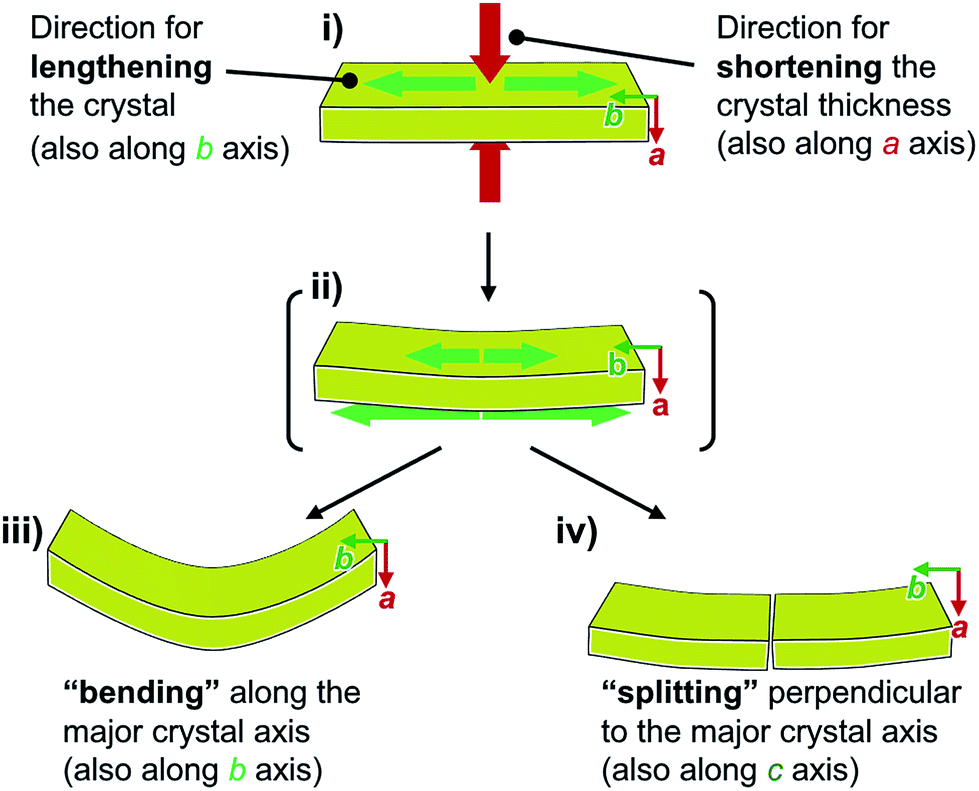 | ||
| Fig. 10 Schematic representation of the macroscopic behavior of crystals of 1 upon cooling, revealing the mechanism for splitting and bending. | ||
Summary
In this report, we investigated the thermosalient effect of triphenylethenyl gold isocyanide complex 1. X-ray diffraction (XRD) and differential scanning calorimetry analyses indicate that crystals and single crystals of 1 are subject to a phase transition between 1a and 1b at approximately −40 °C at 5 °C min−1. Using faster cooling rates of 50 °C min−1, the crystal jump is typically observed at approximately −100 °C. Single-crystal XRD studies of 1 also indicate that the crystal dimensions change anisotropically (Δa < 0; Δb > 0) upon the phase transition while the space group (P21/c) is retained. Face index experiments of 1a and 1b indicate that the crystallographic b axis corresponds to the major crystal axis. This suggests that the observed splitting and bending of crystals of 1 occurs due to an elongation of the major crystal axis during the thermal phase transition (Fig. 10). This clear orientational correlation between microscopic changes of the crystal dimensions and macroscopic mechanical responses should also be the key structural factor for the observed jumping motion of crystals of 1. This study thus provides rare experimental evidence that bridges the gap between microscopic crystal-structure information and macroscopic mechanical responses of crystals of 1. The information offered here should thus facilitate the understanding of the mechanistic details of the salient effect further.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by JSPS KAKENHI grants JP16H06034, JP17H05134, JP17H05344, and JP17H06370. This work was also supported by the Institute for Chemical Reaction Design and Discovery (ICReDD), established by the World Premier International Research Initiative (WPI) of MEXT, Japan. The authors would like to thank Prof. Dr Jian Ping Gong and Dr Muhammad Ilyas (Hokkaido University) for carrying out DSC analyses, and the Photron company for their assistance with the use of high-speed cameras.Notes and references
- (a) D. Kitagawa, H. Nishi and S. Kobatake, Angew. Chem., Int. Ed., 2013, 52, 9320–9322 CrossRef CAS PubMed; (b) D. Kitagawa, K. Kawasaki, R. Tanaka and S. Kobatake, Chem. Mater., 2017, 29, 7524–7532 CrossRef CAS; (c) D. Kitagawa, H. Tsujioka, F. Tong, X. Dong, C. J. Bardeen and S. Kobatake, J. Am. Chem. Soc., 2018, 140, 4208–4212 CrossRef CAS PubMed.
- T. Taniguchi, H. Sugiyama, H. Uekusa, M. Shiro, T. Asahi and H. Koshima, Nat. Commun., 2018, 9, 538 CrossRef PubMed.
- (a) N. K. Nath, M. K. Panda, S. C. Sahoo and P. Naumov, CrystEngComm, 2014, 16, 1850–1858 RSC; (b) P. Naumov, S. Chizhik, M. K. Panda, N. K. Nath and E. Boldyreva, Chem. Rev., 2015, 115, 12440–12490 CrossRef CAS PubMed; (c) P. Commins, I. T. Desta, D. P. Karothu, M. K. Panda and P. Naumov, Chem. Commun., 2016, 52, 13941–13954 RSC.
- (a) S. C. Sahoo, S. B. Sinha, M. S. Kiran, U. Ramamurty, A. F. Dericioglu, C. M. Reddy and P. Naumov, J. Am. Chem. Soc., 2013, 135, 13843–13850 CrossRef CAS PubMed; (b) S. C. Sahoo, M. K. Panda, N. K. Nath and P. Naumov, J. Am. Chem. Soc., 2013, 135, 12241–12251 CrossRef CAS PubMed; (c) S. Chandra Sahoo, N. K. Nath, L. Zhang, M. H. Semreen, T. H. Al-Tel and P. Naumov, RSC Adv., 2014, 4, 7640–7647 RSC; (d) M. K. Panda, T. Runcevski, S. Chandra Sahoo, A. A. Belik, N. K. Nath, R. E. Dinnebier and P. Naumov, Nat. Commun., 2014, 5, 4811 CrossRef CAS PubMed; (e) M. K. Panda, T. Runcevski, A. Husain, R. E. Dinnebier and P. Naumov, J. Am. Chem. Soc., 2015, 137, 1895–1902 CrossRef CAS PubMed; (f) E. Nauha, P. Naumov and M. Lusi, CrystEngComm, 2016, 18, 4699–4703 RSC; (g) M. K. Panda, R. Centore, M. Causa, A. Tuzi, F. Borbone and P. Naumov, Sci. Rep., 2016, 6, 29610 CrossRef CAS PubMed; (h) T. Takeda and T. Akutagawa, Chem.–Eur. J., 2016, 22, 7763–7770 CrossRef CAS PubMed; (i) D. P. Karothu, J. Weston, I. T. Desta and P. Naumov, J. Am. Chem. Soc., 2016, 138, 13298–13306 CrossRef CAS PubMed; (j) A. Khalil, E. Ahmed and P. Naumov, Chem. Commun., 2017, 53, 8470–8473 RSC; (k) S. Mittapalli, D. Sravanakumar Perumalla and A. Nangia, IUCrJ, 2017, 4, 243–250 CrossRef CAS PubMed; (l) I. Lončarić, J. Popović, V. Despoja, S. Burazer, I. Grgičević, D. Popović and Ž. Skoko, Cryst. Growth Des., 2017, 17, 4445–4453 CrossRef; (m) Y. Shibuya, Y. Itoh and T. Aida, Chem.–Asian J., 2017, 12, 811–815 CrossRef CAS PubMed; (n) H. Rawat, R. Samanta, B. Bhattacharya, S. Deolka, A. Dutta, S. Dey, K. B. Raju and C. M. Reddy, Cryst. Growth Des., 2018, 18, 2918–2923 CrossRef CAS; (o) L. O. Alimi, D. P. van Heerden, P. Lama, V. J. Smith and L. J. Barbour, Chem. Commun., 2018, 54, 6208–6211 RSC.
- (a) P. Naumov, S. C. Sahoo, B. A. Zakharov and E. V. Boldyreva, Angew. Chem., Int. Ed., 2013, 52, 9990–9995 CrossRef CAS PubMed; (b) R. Medishetty, A. Husain, Z. Bai, T. Runcevski, R. E. Dinnebier, P. Naumov and J. J. Vittal, Angew. Chem., Int. Ed., 2014, 53, 5907–5911 CrossRef CAS PubMed; (c) R. Medishetty, S. C. Sahoo, C. E. Mulijanto, P. Naumov and J. J. Vittal, Chem. Mater., 2015, 27, 1821–1829 CrossRef CAS; (d) P. Commins, A. Natarajan, C.-K. Tsai, S. I. Khan, N. K. Nath, P. Naumov and M. A. Garcia-Garibay, Cryst. Growth Des., 2015, 15, 1983–1990 CrossRef CAS; (e) T. Seki, K. Sakurada, M. Muromoto and H. Ito, Chem. Sci., 2015, 6, 1491–1497 RSC; (f) M. K. Mishra, A. Mukherjee, U. Ramamurty and G. R. Desiraju, IUCrJ, 2015, 2, 653–660 CrossRef CAS PubMed; (g) E. Hatano, M. Morimoto, K. Hyodo, N. Yasuda, S. Yokojima, S. Nakamura and K. Uchida, Chem.–Eur. J., 2016, 22, 12680–12683 CrossRef CAS PubMed; (h) C. E. Mulijanto, H. S. Quah, G. K. Tan, B. Donnadieu and J. J. Vittal, IUCrJ, 2017, 4, 65–71 CrossRef CAS PubMed; (i) E. Hatano, M. Morimoto, T. Imai, K. Hyodo, A. Fujimoto, R. Nishimura, A. Sekine, N. Yasuda, S. Yokojima, S. Nakamura and K. Uchida, Angew. Chem., Int. Ed., 2017, 56, 12576–12580 CrossRef CAS PubMed.
- (a) M. C. Etter and A. R. Siedle, J. Am. Chem. Soc., 1983, 105, 641–643 CrossRef CAS; (b) J. Ding, R. Herbst, K. Praefcke, B. Kohne and W. Saenger, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 739–742 CrossRef; (c) T. Steiner, W. Hinrichs, W. Saenger and R. Gigg, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 708–718 CrossRef; (d) I. Colombier, S. Spagnoli, A. Corval, P. Baldeck, M. Giraud, A. Léaustic and P. Yu, Mol. Cryst. Liq. Cryst., 2005, 431, 495–499 CrossRef CAS; (e) R. J. Davey, S. J. Maginn, S. J. Andrews, S. N. Black, A. M. Buckley, D. Cottier, P. Dempsey, R. Plowman, J. E. Rout, D. R. Stanley and A. Taylor, Mol. Cryst. Liq. Cryst., 2006, 242, 79–90 CrossRef; (f) T. Siegrist, C. Besnard, S. Haas, M. Schiltz, P. Pattison, D. Chernyshov, B. Batlogg and C. Kloc, Adv. Mater., 2007, 19, 2079–2082 CrossRef CAS; (g) A. Natarajan, C. Tsai, S. I. Khan, P. McCarren, K. Houk and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2007, 129, 9846–9847 CrossRef CAS PubMed; (h) R. Centore, M. Jazbinsek, A. Tuzi, A. Roviello, A. Capobianco and A. Peluso, CrystEngComm, 2012, 14, 2645–2653 RSC.
- (a) H. Ito, T. Saito, N. Oshima, N. Kitamura, S. Ishizaka, Y. Hinatsu, M. Wakeshima, M. Kato, K. Tsuge and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 10044–10045 CrossRef CAS PubMed; (b) H. Ito, M. Muromoto, S. Kurenuma, S. Ishizaka, N. Kitamura, H. Sato and T. Seki, Nat. Commun., 2013, 4, 2009 CrossRef PubMed; (c) T. Seki, K. Sakurada and H. Ito, Angew. Chem., Int. Ed., 2013, 52, 12828–12832 CrossRef CAS PubMed; (d) S. Yagai, S. Okamura, Y. Nakano, M. Yamauchi, K. Kishikawa, T. Karatsu, A. Kitamura, A. Ueno, D. Kuzuhara, H. Yamada, T. Seki and H. Ito, Nat. Commun., 2014, 5, 4013 CrossRef CAS PubMed; (e) T. Seki, T. Ozaki, T. Okura, K. Asakura, A. Sakon, H. Uekusa and H. Ito, Chem. Sci., 2015, 6, 2187–2195 RSC; (f) T. Seki, Y. Takamatsu and H. Ito, J. Am. Chem. Soc., 2016, 138, 6252–6260 CrossRef CAS PubMed; (g) S. Yagai, T. Seki, H. Aonuma, K. Kawaguchi, T. Karatsu, T. Okura, A. Sakon, H. Uekusa and H. Ito, Chem. Mater., 2016, 28, 234–241 CrossRef CAS; (h) T. Seki, N. Tokodai, S. Omagari, T. Nakanishi, Y. Hasegawa, T. Iwasa, T. Taketsugu and H. Ito, J. Am. Chem. Soc., 2017, 139, 6514–6517 CrossRef CAS PubMed; (i) M. Jin, T. Seki and H. Ito, J. Am. Chem. Soc., 2017, 139, 7452–7455 CrossRef CAS PubMed; (j) T. Seki, K. Kobayashi and H. Ito, Chem. Commun., 2017, 53, 6700–6703 RSC; (k) M. Jin, T. Sumitani, H. Sato, T. Seki and H. Ito, J. Am. Chem. Soc., 2018, 140, 2875–2879 CrossRef CAS PubMed.
- Under UV illumination (λ = 365 nm), 1a only exhibits weak red emission with a low absolute emission quantum yield (Φem ≤ 1%; Fig. S2†).
- The emission of 1b is also very weak (Φem ≤ 1%; Fig. S2†).
- Upon cooling at around 10 °C min−1, the crystals of 1 start to jump around at −50 °C.
- This mechanical behavior is not observed in all thermosalient crystals that show splitting. Other thermosalient crystals show splitting along the major crystal axis; for details, see ref. 3a and 4a.
- (a) P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412–4456 CrossRef PubMed; (b) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC.
- In order to investigate the structural origins of the thermosalient effect of 1, single crystals of 1a (typical size: 0.3 × 0.2 × 0.05 mm3) were slowly cooled (1 °C min−1) on a goniometer head using a flow of nitrogen gas to furnish 1b. During this procedure, crystals of 1 sometimes broke. Using lots of Paraton oil to fix the crystals decreases the probability of the crystals breaking.
- We also confirmed the slipping motion between two molecules, which is reflected in changes of the distance and angle of the Cl⋯Cl interaction (Fig. S12†).
- M. J. Cliffe and A. L. Goodwin, J. Appl. Crystallogr., 2012, 45, 1321–1329 CrossRef CAS.
- For the calculation of α values of 1, we use the data at temperature in which the lattice expansion occurs non-linearly. Thus, as suggested by Skoko (ref. 4l), the α values of 1 with those of other molecular crystals, whose α values are estimated for the linear expansion region, would not be directly compared.
- Temperature-dependent powder XRD measurements of powder samples of 1 do not show significant changes (Fig. S19†) different from the results of the single-crystal XRD measurements.
- Disappearance of the DSC peak upon powdering was previously reported, see: V. S. Minkov, V. A. Drebushchak, A. G. Ogienkoac and E. V. Boldyreva, CrystEngComm, 2011, 13, 4417–4426 RSC . See also ref. 4b.
- The powder XRD pattern of the powder form of 1 exhibits rather low peak intensity (Fig. S19†), indicating the low crystallinity. Thus, the powder samples also contain a certain amount of amorphous components as impurities, which would interfere with the phase transition from 1a to 1b upon cooling.
- Limited examples have been reported by Naumov and Skoko: see ref. 4g and l.
Footnote |
| † Electronic supplementary information (ESI) available: Additional spectroscopic, crystallographic, and thermal analysis data. CCDC 1884306–1884313. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05563g |
| This journal is © The Royal Society of Chemistry 2019 |