
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-selective nitrenoid insertions utilizing postfunctionalized bifunctional rhodium(II) catalysts†
Jan-Philipp
Berndt
a,
Yevhenii
Radchenko
a,
Jonathan
Becker
b,
Christian
Logemann
 b,
Dhaka R.
Bhandari
b,
Radim
Hrdina
*a and
Peter R.
Schreiner
*a
b,
Dhaka R.
Bhandari
b,
Radim
Hrdina
*a and
Peter R.
Schreiner
*a
aJustus Liebig University, Institute of Organic Chemistry, Heinrich-Buff-Ring 17, 35392 Giessen, Germany. E-mail: prs@uni-giessen.de; Radim.Hrdina@org.chemie.uni-giessen.de
bInstitute of Inorganic and Analytical Chemistry, Heinrich-Buff-Ring 17, 35392 Giessen, Germany
First published on 6th February 2019
Abstract
We report a new strategy for the preparation of dirhodium(II) complexes with the general formula Rh2(A)4 that allows the isolation of a dirhodium tetracarboxylate complex with a free amino group available for postfunctionalization. The postfunctionalization of this complex enables the incorporation of a variety of functional groups, including double and triple bonds as well as nucleophilic moieties, thus paving the way to new classes of polymeric as well as bifunctional catalysts, and polymetallic complexes. Furthermore, we demonstrate that a urea containing dirhodium(II) complex enables site-selective nitrenoid insertions by remote hydrogen bonding control.
Introduction
Dirhodium(II) complexes act as sensors,1 show antitumor activity,2 are capable of cross-linking DNA,3 and can be used to control peptide structures by binding carboxylate side chains to the Rh core,4 thus enabling site-specific modifications of polypeptides and proteins.5 They act as Lewis acids to activate alkynes,6 as well as enynes7 or serve as hydrogenation catalysts.8 The well-known Rh(II) carbenoid9 and nitrenoid10 transfer catalysts are capable of catalyzing X–H insertions,11 cyclopropanations,11a,12 aziridinations,13 ylide formation,11a,14 and allylic oxidations.15 One of the main challenges in Rh(II) mediated carbenoid and nitrenoid insertions is the control of site-selectivity.16 Davies and coworkers tackled this issue by applying highly site-selective Rh(II) catalysts for carbenoid C–H bond insertions.17 Bach's group designed a catalyst enabling hydrogen bonding of quinolones, to perform regio- and enantioselective C–H aminations and aziridinations of these substrates.18 Generally, Rh(II) carboxylates, carboxamidates, and phosphates are prepared in two steps.19 The first represents the synthesis of the ligand, which is then subjected to ligand exchange. Early procedures for the preparation of Rh(II) carboxylates made use of Rh(OH)3 (ref. 20) or Rh(Cl)3 (ref. 21) and large excess of the ligands. In 1992, a more atom economic protocol for the preparation of these complexes using Rh(II) carbonate,22 was reported.23 Ball's group used cis-[Rh2(tfa)2(OAc)2] as a precursor for the preparation of metalloenzymes by exchange of the trifluoroacetate ligands with peptide carboxylate side chains.4a The most widely used procedures subject ligands to an exchange with Rh(II) acetate, in which product formation is favored by removal of acetic acid.24 Functional groups such as unsaturated bonds or nucleophilic moieties are thereby not tolerated (for highly substituted ones see ref. 18a and 25) because of the Lewis acidity of the Rh complex.26Focusing on an alternative way to prepare functionalized Rh complexes, we decided to reverse the known approach by designing an appropriate spacer that is introduced first and allows efficient postfunctionalization, thereby enhancing the functional group diversity (Fig. 1).27
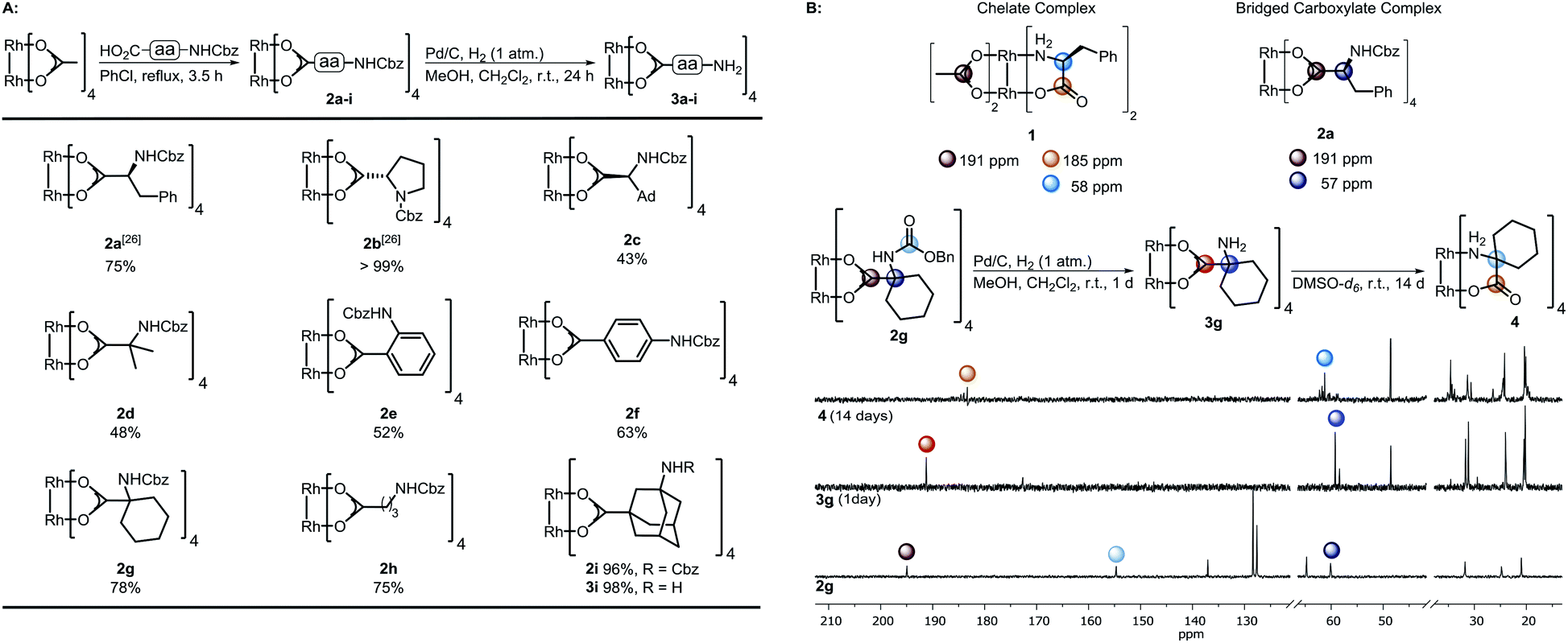 | ||
| Scheme 1 (A) Preparation of the complexes 2a–i and subsequent hydrogenation; (B) top: characteristic 13C-NMR shifts of complex 1 (ref. 28a) and dirhodium(II) bridged carboxylate 4a. Bottom: Hydrogenation of 2g to 3g and study of its time-dependent stability; NMR solvent: DMSO-d6. | ||
Results and discussion
The spacer has to bear an acid moiety for attachment to the complex and an additional functional group enabling postfunctionalization. We envisioned that amino acids may be excellent precursors, as the amino group allows efficient functionalization through amide bond formation. However, subjecting unprotected α-amino acids to ligand exchange with Rh2(OAc)4 is not possible as it results in complexes such as 1 involving the binding of the carboxylic acid and the amine to the Rh center (Scheme 1B).28 Amines irreversibly disrupt the carboxylate bridging structure, also intermolecularly, unless they are sterically crowded or the rhodium core is shielded.20,25,29 Hence, only a few examples of dirhodium complexes containing an amine have been reported.4a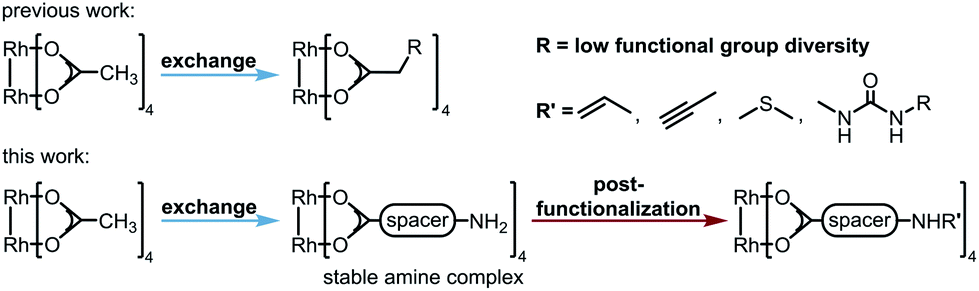 | ||
| Fig. 1 New approach for the preparation of dirhodium(II) complexes. | ||
We decided to investigate the stability of a variety of Rh2(A)4 complexes containing amines. For this purpose, we envisioned to apply Cbz-protected (benzyloxycarbonyl) amino acids to a ligand exchange with Rh2(OAc)4 and to cleave the Cbz group by hydrogenolysis subsequently (Scheme 1A). A variety of Cbz-protected amino acids were synthesized and subjected to ligand exchange using reported conditions.24a The novel dirhodium(II) carboxylates 2a–i were obtained in yields up to 99%. To our delight Cbz deprotection proceeded quantitatively. As anticipated, it was not possible to isolate complexes 3a–f, because of the presence of the free amino group leading to disruption of the bridged carboxylate structures. The stability of the deprotected complexes was studied with cyclohexyl derivative 3g (Scheme 1B) via time-dependent NMR. While hydrogenolysis of 2g was quantitative, 13C-NMR analysis of the reaction mixture revealed the disappearance of the Cbz group and appearance of the characteristic bridged carboxylate peak at 191 ppm and an additional carbonyl peak at 173 ppm (Scheme 1B). Thus, the spectrum indicates the presence of the desired complex 3g and another compound. NMR analysis after 14 days showed the disappearance of both carbonyl peaks and rise of a new peak at 183 ppm, typical for complexes such as 1. We concluded that a sterically crowded ligand should suppress the decomposition of the complex and decided to use γ-aminoadamantane carboxylic acid S2.30 The bulky cage should shield the Rh core and prevent coordination to the metal by the sterically demanding amine. Indeed, deprotection of 2i resulted in formation of complex 3i containing a free amine. The use of γ-aminobutyric acid further supported the notion that steric bulk is the predominant factor for the design of suitable ligands.
Bench-stable complex 3i was used for postfunctionalization and optimization of the amide bond formation showed that N-succinimidyl (OSu) acetate performed best, resulting in quantitative yield of tetra-acetylated S11 (Table S1†). The succinimidyl esters of 4-pentenoic acid, 4-pentynoic acid, and Boc-L-methionine (tert-butoxycarbonyl) were prepared by EDC-coupling. Postfunctionalization of 3i with the ester of 4-pentenoic acid afforded 77% of 5a (Scheme 2). The alkynyl containing complex 5b was isolated in comparable yield. A control experiment was performed by subjecting 1-alkynyl-3-adamantane carboxylic acid and Rh2(OAc)4 to a thermal ligand exchange. The formation of the desired complex was not observed, but decomposition of the starting material occurred. These new complexes are particular valuable for functionalizations via Huisgen cyclization,31 thiol–ene chemistry32 or olefin metathesis.33
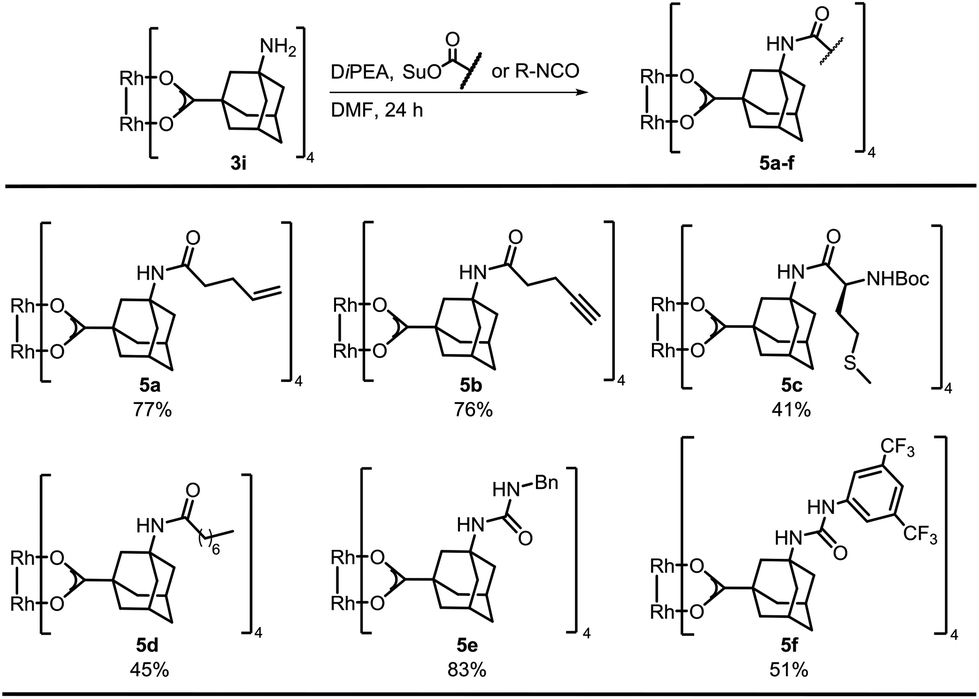 | ||
| Scheme 2 Postfunctionalization of complex 3i. | ||
Furthermore, we functionalized 3i with methionine. Carboxylic acid anhydrides can be employed instead of succinimidyl esters, as demonstrated with the synthesis of 5d.34 Isocyanates were used for the preparation of ureas, affording bifunctional Rh complexes 5e, f (Scheme 2). The structures of the urea complexes 5e and 5f were determined by crystal X-ray analysis. The ligands around the Rh–Rh bond align in local C2-symmetry (Scheme 3).35
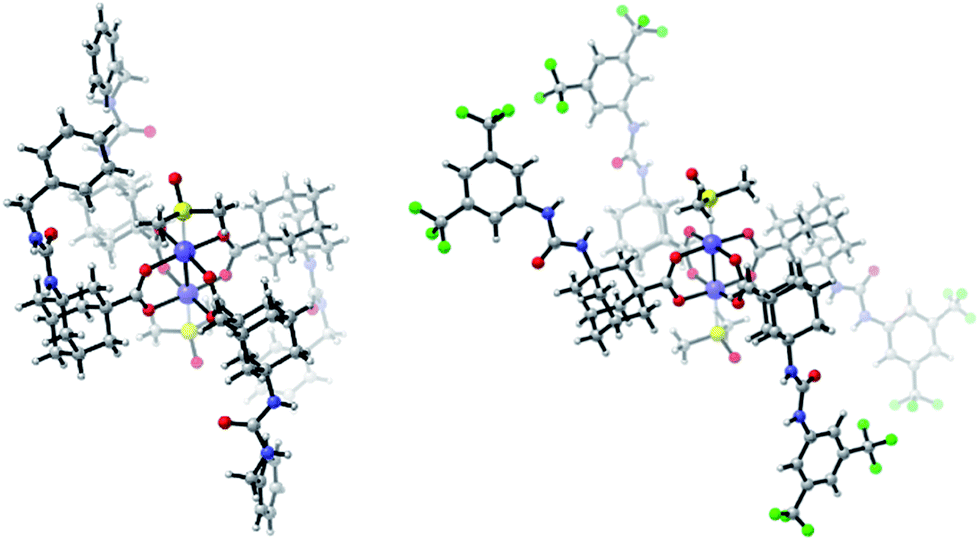 | ||
| Scheme 3 X-Ray crystal structures 5e (left) and 5f (right), DMSO molecules coordinate the Rh atoms. | ||
One of the main challenges in Rh(II) mediated aziridinations, C–H insertions, and cyclopropanations is the control of site-selectivity. Generally, aziridinations are faster than C–H insertions if sulfamates or sulfonamides are used.9b,36 However, this trend can change, especially when C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds are sterically crowded or when sulfonimideamides are used as nitrene precursors.37 Rh(II) catalyzed nitrenoid and carbenoid C–H insertions favor sites that stabilize positive charge. Thus, the reactivity scale for alkanes can be drawn as 3° > benzylic ∼ α-heteroatom > 2° ≫ 1.9b,36a However, catalyst design can alter this trend, e.g., sterically demanding catalysts favor insertions at sterically more accessible C–H bonds.17b,c,36a As the selective functionalization of, e.g., polyenes, would “greatly streamline the synthesis of complex target molecules”,16b we envisioned to apply bifunctional catalyst 5f in remote site-selective nitrenoid insertion directed by H-bonding.16a,18a,38 The non-covalent interactions between 5f, containing the key structural moiety 3,5-bis(trifluoromethyl)phenyl for H-bonding,39 and an acceptor, should create well-defined spatial relationships. We envisioned farnesol to be a worthwhile target for site-selective aziridination as it has three π-bonds possessing the reactivity trend A > B > C and nine allylic bonds, which may undergo C–H insertion (Table 1).40 First we installed a hydrogen-bonding acceptor on farnesol by converting the alcohol to carbamate 6. We also performed a conformational analysis in the gas phase using U-GFN2-xTB on nitrenoid complex 5fN with 6.41 Conformers entailing a reasonable alignment of 6 and 5fN were further optimized in toluene using GBSA as solvent model.42 Conformers at which the olefinic chain of 6 was oriented towards the outside of the cavity of 5fN were not considered as they do not lead to aziridination. The complex depicted in Scheme 4 is the energetically lowest-lying conformer. The computations place the shortest distance between nitrenoid and double bond B at d(N⋯πB) = 3.43 Å, followed by the least reactive double bond C at d(N⋯πC) = 4.24 Å, and d(N⋯πA) = 6.20 Å. Thus, based on steric arguments, catalyst 5f should favor double bond B, although the intrinsic reactivity of 6 should lead to the aziridination of double bond A as the major product. We commenced our study by applying Du Bois conditions43 with commercially available sulfonamide TcesNH2 (2,2,2-trichloroethyl sulfamate). The aziridination of 6 with bis[rhodium(α,α,α′,α′-tetramethyl-1,3-benzenedipropionic acid)] [Rh2(esp)2] afforded a 2.0
C bonds are sterically crowded or when sulfonimideamides are used as nitrene precursors.37 Rh(II) catalyzed nitrenoid and carbenoid C–H insertions favor sites that stabilize positive charge. Thus, the reactivity scale for alkanes can be drawn as 3° > benzylic ∼ α-heteroatom > 2° ≫ 1.9b,36a However, catalyst design can alter this trend, e.g., sterically demanding catalysts favor insertions at sterically more accessible C–H bonds.17b,c,36a As the selective functionalization of, e.g., polyenes, would “greatly streamline the synthesis of complex target molecules”,16b we envisioned to apply bifunctional catalyst 5f in remote site-selective nitrenoid insertion directed by H-bonding.16a,18a,38 The non-covalent interactions between 5f, containing the key structural moiety 3,5-bis(trifluoromethyl)phenyl for H-bonding,39 and an acceptor, should create well-defined spatial relationships. We envisioned farnesol to be a worthwhile target for site-selective aziridination as it has three π-bonds possessing the reactivity trend A > B > C and nine allylic bonds, which may undergo C–H insertion (Table 1).40 First we installed a hydrogen-bonding acceptor on farnesol by converting the alcohol to carbamate 6. We also performed a conformational analysis in the gas phase using U-GFN2-xTB on nitrenoid complex 5fN with 6.41 Conformers entailing a reasonable alignment of 6 and 5fN were further optimized in toluene using GBSA as solvent model.42 Conformers at which the olefinic chain of 6 was oriented towards the outside of the cavity of 5fN were not considered as they do not lead to aziridination. The complex depicted in Scheme 4 is the energetically lowest-lying conformer. The computations place the shortest distance between nitrenoid and double bond B at d(N⋯πB) = 3.43 Å, followed by the least reactive double bond C at d(N⋯πC) = 4.24 Å, and d(N⋯πA) = 6.20 Å. Thus, based on steric arguments, catalyst 5f should favor double bond B, although the intrinsic reactivity of 6 should lead to the aziridination of double bond A as the major product. We commenced our study by applying Du Bois conditions43 with commercially available sulfonamide TcesNH2 (2,2,2-trichloroethyl sulfamate). The aziridination of 6 with bis[rhodium(α,α,α′,α′-tetramethyl-1,3-benzenedipropionic acid)] [Rh2(esp)2] afforded a 2.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0 (7A:7B) ratio in favor of double bond A. Likewise, Rh2(OAc)4 afforded a 1.2:1.0 ratio (Table 1). The aziridination of double bond C was not observed.
:1.0 (7A:7B) ratio in favor of double bond A. Likewise, Rh2(OAc)4 afforded a 1.2:1.0 ratio (Table 1). The aziridination of double bond C was not observed.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst |
7A:7Bb |
Yield (7A + 7B)/% | Conv. (6)c/% |
|
a Conditions: 2 mol% [Rh], c = 1.0 M, 25 °C, Ph-H, ratio of 6:H2NTces:PhI(O2CtBu)2 (1:1:2).
b NMR ratio.
c Based on re-isolated starting material.
d
c = 0.05 M.
e
c = 0.01 M.
f 2 equiv. 6.
g 1.5 equiv. 6.
h PhI(O2CC(Me)2Ph)2 used.
i 1.2 equiv. PhI(O2CtBu)2.
j 2.3 equiv. MgO.
k 8 mol% 5f.
l 3.0 equiv. 6.
m 10.0 equiv. ethyl-N-ethyl carbamate.
|
||||
| 1 | Rh2(esp)2 | 2.0:1.0 |
29 | 81 |
| 2 | Rh2(O2CAd)4 | 1.4:1.0 |
30 | 55 |
| 3 | Rh2(OAc)4 | 1.2:1.0 |
14 | <20 |
| 4 | 5f | 1.0:1.9 |
31 | 80 |
| 5d | 5f | 1.0:2.5 |
22 | 85 |
| 6e | 5f | 1.0:5.0 |
23 | 73 |
| 7d,f | 5f | 1.0:3.4 |
38 | 100c |
| 8d,g | 5f | 1.0:3.8 |
31 | 100c |
| 9d,h | 5f | 1.0:2.1 |
28 | 82 |
| 10d,i | 5f | 1.0:3.6 |
21 | 70 |
| 11d,i,j | 5f | 1.0:4.9 |
20 | 74 |
| 12d,i,j,k | 5f | 1.0:3.6 |
26 | 67 |
| 13e,i,j,l | 5f | 1.0:4.0 |
40 | 100c |
| 14l | Rh2(esp)2 | 1.1:1.0 |
42 | 100c |
| 15e,m | 5f | 1.0:1.3 |
11 | 57 |
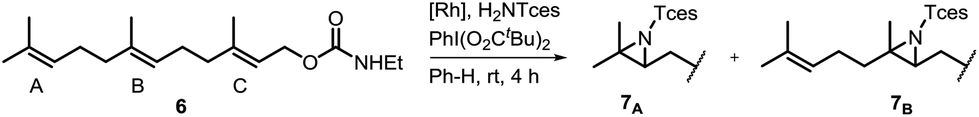
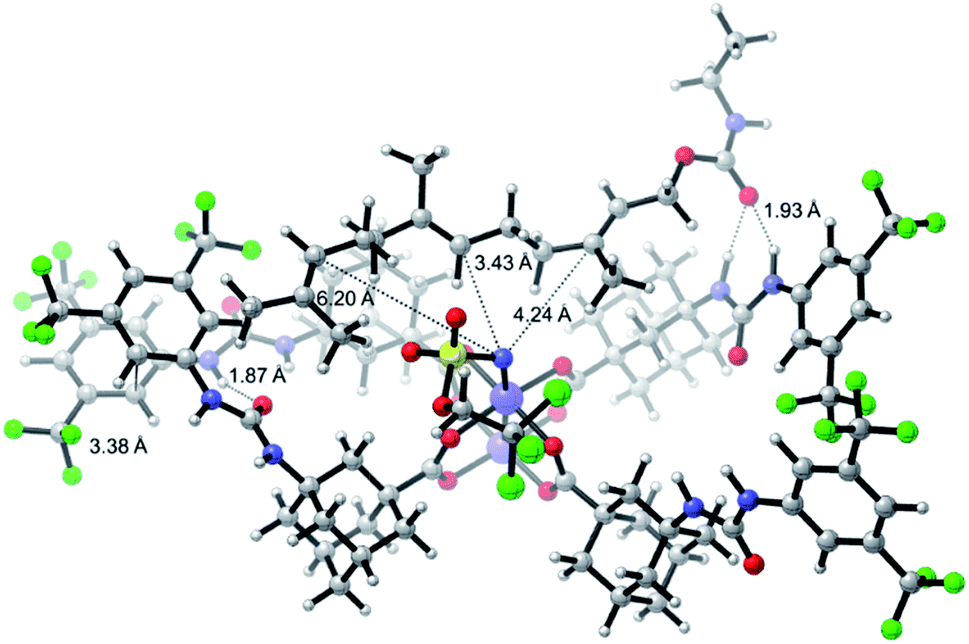 | ||
| Scheme 4 U-GFN2-xTB optimized structure of the nitrenoid complex 5fN with carbamate 6; solvent model GBSA (toluene). | ||
Bifunctional catalyst 5f favors double bond B with a 1.0:1.9 ratio, thereby overcoming the intrinsic reactivity of 6. Note that 5f exhibits the same reactivity as Rh2(esp)2, which was designed to circumvent the lack of reactivity in intermolecular reactions.44 With tetrakis[1-adamantanecarboxylate] dirhodium(II) [Rh2(O2CAd)4], a sterically similar bulky catalyst as compared to 5f, but not capable of hydrogen bonding, we observed a 1.4:1.0 ratio in favor of the intrinsically preferred product 7A. Higher dilution should suppress the aziridination of substrate not bound to catalyst 5f, thereby enhancing the ratio. Indeed, lower concentrations improved the ratio to up to 1.0:5.0 (entry 6). Furthermore, MgO increased the ratio by scavenging the released pivalic acid (entries 10 and 11), which disturbs hydrogen bonding between catalyst 5f and 6. We underscored our hypothesis of hydrogen bonding between 5f and 6 by using 10 equiv. of ethyl-N-ethylcarbamate as additive. The additive interacts with the urea moiety of 5f and thus competes for the hydrogen bonding with the substrate. As a consequence, the ratio of 7A and 7B decreased from 1.0:5.0 to 1.0:1.3 (entries 6 and 15). The optimized conditions catalysed by bifunctional complex 5f afforded 40% yield of 7A and 7B in a 1.0:4.0 ratio (entry 13). The catalysed aziridination utilizing benchmark catalyst Rh2(esp)2, exhibiting an exceptionally high activity,44 afforded a similar yield, but in a ratio of 1.1:1.0 (7A:7B). The comparable yields of benchmark catalyst Rh2(esp)2 and 5f confirm the high activity of 5f. Note, catalyst 5f can be used to achieve unique selectivity in the aziridination of polyenes. This proof-of-concept expands the limited number of examples utilizing non-covalent interactions for control of site-selectivity16a and shows that the novel bifunctional catalysts can be used to overcome intrinsic substrate reactivities by remote hydrogen bonding.
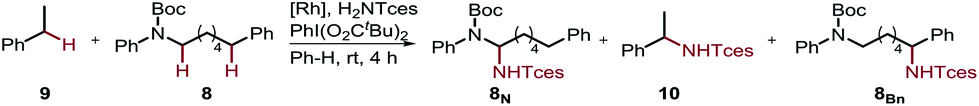
Furthermore, we performed a competition experiment for the nitrenoid C–H insertion of the benzylic position of Boc-protected amine 8vs. ethylbenzene 9, to demonstrate substrate recognition of 5fvia hydrogen bonding (Table 2). Benchmark catalyst Rh2(esp)2 afforded about a 1:1 ratio for the two benzylic positions 8Bn and 10, whereas bifunctional catalyst 5f is capable of discriminating between these two benzylic positions, thereby favoring 8Bn in a 1.6:8.1 ratio; Rh2(O2CAd)4, gave a 1:1 ratio. In accord with the reactivity trend, 8N was observed as the minor product.
Conclusions
We accomplished the isolation of the first stable Rh2(A)4 complex bearing a free amine. The acylation of this complex enables the incorporation of various functional groups. The bifunctional dirhodium complex was designed and tested in nitrenoid insertions. This catalyst is capable to overwrite the intrinsic reactivity of molecules by remote hydrogen bonding. Future work focuses on the application of other polyfunctional complexes prepared via this new procedure.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by LOEWE “SynChemBio” and by DFG (sp314/13-1). The authors thank S. Bernhardt for HPLC separations, S. Grimme for access to the GFN2-xTB code, and B. Spengler for access to MALDI measurements.Notes and references
- S. A. Hilderbrand, M. H. Lim and S. J. Lippard, J. Am. Chem. Soc., 2004, 126, 4972–4978 CrossRef CAS PubMed.
- (a) J. D. Aguirre, A. M. Angeles-Boza, A. Chouai, J.-P. Pellois, C. Turro and K. R. Dunbar, J. Am. Chem. Soc., 2009, 131, 11353–11360 CrossRef CAS PubMed; (b) H. T. Chifotides and K. R. Dunbar, Acc. Chem. Res., 2005, 38, 146–156 CrossRef CAS PubMed; (c) A. Erck, E. Sherwood, J. L. Bear and A. P. Kimball, Cancer Res., 1976, 36, 2204–2209 CAS.
- (a) S. U. Dunham, H. T. Chifotides, S. Mikulski, A. E. Burr and K. R. Dunbar, Biochemistry, 2005, 44, 996–1003 CrossRef CAS PubMed; (b) L. E. Joyce, J. D. Aguirre, A. M. Angeles-Boza, A. Chouai, P. K. L. Fu, K. R. Dunbar and C. Turro, Inorg. Chem., 2010, 49, 5371–5376 CrossRef CAS PubMed.
- (a) A. N. Zaykov, K. R. MacKenzie and Z. T. Ball, Chem.–Eur. J., 2009, 15, 8961–8965 CrossRef CAS PubMed; (b) A. N. Zaykov, B. V. Popp and Z. T. Ball, Chem.–Eur. J., 2010, 16, 6651–6659 CrossRef CAS PubMed.
- Z. T. Ball, Acc. Chem. Res., 2013, 46, 560–570 CrossRef CAS PubMed.
- M. Yang, S. J. Odelberg, Z. Tong, D. Y. Li and R. E. Looper, Tetrahedron, 2013, 69, 5744–5750 CrossRef CAS PubMed.
- (a) K. Ota and N. Chatani, Chem. Commun., 2008, 2906–2907 RSC; (b) K. Ota, S. I. Lee, J.-M. Tang, M. Takachi, H. Nakai, T. Morimoto, H. Sakurai, K. Kataoka and N. Chatani, J. Am. Chem. Soc., 2009, 131, 15203–15211 CrossRef CAS PubMed.
- (a) P. Legzdins, G. L. Rempel and G. Wilkinson, J. Chem. Soc., Chem. Commun., 1969, 825 RSC; (b) B. C. Y. Hui, W. K. Teo and G. L. Rempel, Inorg. Chem., 1973, 12, 757–762 CrossRef CAS.
- (a) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed; (b) H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857–1869 RSC; (c) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2904 CrossRef CAS PubMed.
- (a) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed; (b) F. Collet, C. Lescot and P. Dauban, Chem. Soc. Rev., 2011, 40, 1926–1936 RSC; (c) G. Dequirez, V. Pons and P. Dauban, Angew. Chem., Int. Ed., 2012, 51, 7384–7395 CrossRef CAS PubMed.
- (a) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS PubMed; (b) H. Lebel, C. Trudel and C. Spitz, Chem. Commun., 2012, 48, 7799–7801 RSC; (c) H. Lebel, L. Mamani Laparra, M. Khalifa, C. Trudel, C. Audubert, M. Szponarski, C. Dicaire Leduc, E. Azek and M. Ernzerhof, Org. Biomol. Chem., 2017, 15, 4144–4158 RSC.
- H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977–1050 CrossRef CAS PubMed.
- (a) P. Müller and C. Fruit, Chem. Rev., 2003, 103, 2905–2920 CrossRef PubMed; (b) H. Lebel, K. Huard and S. Lectard, J. Am. Chem. Soc., 2005, 127, 14198–14199 CrossRef CAS PubMed; (c) H. Lebel, C. Spitz, O. Leogane, C. Trudel and M. Parmentier, Org. Lett., 2011, 13, 5460–5463 CrossRef CAS PubMed.
- T. Ye and M. A. McKervey, Chem. Rev., 1994, 94, 1091–1160 CrossRef CAS.
- A. J. Catino, R. E. Forslund and M. P. Doyle, J. Am. Chem. Soc., 2004, 126, 13622–13623 CrossRef CAS PubMed.
- (a) H. J. Davis and R. J. Phipps, Chem. Sci., 2017, 8, 864–877 RSC; (b) Z. Huang and G. Dong, Acc. Chem. Res., 2017, 50, 465–471 CrossRef CAS PubMed.
- (a) K. Liao, T. C. Pickel, V. Boyarskikh, J. Bacsa, D. G. Musaev and H. M. L. Davies, Nature, 2017, 551, 609 CAS; (b) K. Liao, S. Negretti, D. G. Musaev, J. Bacsa and H. M. L. Davies, Nature, 2016, 533, 230 CrossRef CAS PubMed; (c) C. Qin and H. M. L. Davies, J. Am. Chem. Soc., 2014, 136, 9792–9796 CrossRef CAS PubMed; (d) K. Liao, W. Liu, Z. L. Niemeyer, Z. Ren, J. Bacsa, D. G. Musaev, M. S. Sigman and H. M. L. Davies, ACS Catal., 2018, 8, 678–682 CrossRef CAS.
- (a) T. Hoke, E. Herdtweck and T. Bach, Chem. Commun., 2013, 49, 8009–8011 RSC; (b) F. Zhong and T. Bach, Chem.–Eur. J., 2014, 20, 13522–13526 CrossRef CAS PubMed.
- (a) M. C. Pirrung and J. Zhang, Tetrahedron Lett., 1992, 33, 5987–5990 CrossRef CAS; (b) R. Hrdina, L. Guénée, D. Moraleda and J. Lacour, Organometallics, 2013, 32, 473–479 CrossRef CAS; (c) D. Poggiali, A. Homberg, T. Lathion, C. Piguet and J. Lacour, ACS Catal., 2016, 6, 4877–4881 CrossRef CAS; (d) N. McCarthy, M. A. McKervey, T. Ye, M. McCann, E. Murphy and M. P. Doyle, Tetrahedron Lett., 1992, 33, 5983–5986 CrossRef CAS; (e) D. M. Hodgson, D. A. Selden and A. G. Dossetter, Tetrahedron: Asymmetry, 2003, 14, 3841–3849 CrossRef CAS; (f) S. Kitagaki, M. Anada, O. Kataoka, K. Matsuno, C. Umeda, N. Watanabe and S.-i. Hashimoto, J. Am. Chem. Soc., 1999, 121, 1417–1418 CrossRef CAS.
- S. A. Johnson, H. R. Hunt and H. M. Neumann, Inorg. Chem., 1963, 2, 960–962 CrossRef CAS.
- P. A. Agaskar, F. A. Cotton, L. R. Falvello and S. Han, J. Am. Chem. Soc., 1986, 108, 1214–1223 CrossRef CAS.
- C. R. Wilson and H. Taube, Inorg. Chem., 1975, 14, 405–409 CrossRef CAS.
- G. H. P. Roos and M. A. McKervey, Synth. Commun., 1992, 22, 1751–1756 CrossRef CAS.
- (a) H. J. Callot and F. Metz, Tetrahedron, 1985, 41, 4495–4501 CrossRef CAS; (b) M. P. Doyle, W. R. Winchester, J. A. A. Hoorn, V. Lynch, S. H. Simonsen and R. Ghosh, J. Am. Chem. Soc., 1993, 115, 9968–9978 CrossRef CAS.
- T. Itoh, M. Kondo, H. Sakamoto, K. Wakabayashi, M. Kanaike, K. Itami and S. Masaoka, Dalton Trans., 2015, 44, 15334–15342 RSC.
- R. T. Buck, D. M. Coe, M. J. Drysdale, L. Ferris, D. Haigh, C. J. Moody, N. D. Pearson and J. B. Sanghera, Tetrahedron: Asymmetry, 2003, 14, 791–816 CrossRef CAS.
- H. Yang, P. Srivastava, C. Zhang and J. C. Lewis, ChemBioChem, 2014, 15, 223–227 CrossRef CAS PubMed.
- (a) N. R. Candeias, C. Carias, L. F. R. Gomes, V. André, M. T. Duarte, P. M. P. Gois and C. A. M. Afonso, Adv. Synth. Catal., 2012, 354, 2921–2927 CrossRef CAS; (b) R. F. M. Frade, N. R. Candeias, C. M. M. Duarte, V. André, M. Teresa Duarte, P. M. P. Gois and C. A. M. Afonso, Bioorg. Med. Chem. Lett., 2010, 20, 3413–3415 CrossRef CAS PubMed.
- J. Jaźwiński, J. Mol. Struct., 2005, 750, 7–17 CrossRef.
- L. Wanka, C. Cabrele, M. Vanejews and P. R. Schreiner, Eur. J. Org. Chem., 2007, 1474–1490 CrossRef CAS.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- Y. Dong, J. B. Matson and K. J. Edgar, Biomacromolecules, 2017, 18, 1661–1676 CrossRef CAS PubMed.
- Please note that Rh2(A)4 complexes containing amide moiety can be prepared by thermal ligand exchange (see ref. 26).
- (a) H. M. L. Davies, P. R. Bruzinski, D. H. Lake, N. Kong and M. J. Fall, J. Am. Chem. Soc., 1996, 118, 6897–6907 CrossRef CAS; (b) J. Hansen and H. M. L. Davies, Coord. Chem. Rev., 2008, 252, 545–555 CrossRef CAS PubMed.
- (a) K. W. Fiori, C. G. Espino, B. H. Brodsky and J. Du Bois, Tetrahedron, 2009, 65, 3042–3051 CrossRef CAS; (b) K. Guthikonda, P. M. Wehn, B. J. Caliando and J. Du Bois, Tetrahedron, 2006, 62, 11331–11342 CrossRef CAS.
- (a) C. Liang, F. Robert-Peillard, C. Fruit, P. Müller, R. H. Dodd and P. Dauban, Angew. Chem., Int. Ed., 2006, 45, 4641–4644 CrossRef CAS PubMed; (b) C. Liang, F. Collet, F. Robert-Peillard, P. Müller, R. H. Dodd and P. Dauban, J. Am. Chem. Soc., 2008, 130, 343–350 CrossRef CAS PubMed.
- Selected reviews on bifunctional ligands in metal catalysis; (a) S. Carboni, C. Gennari, L. Pignataro and U. Piarulli, Dalton Trans., 2011, 40, 4355–4373 RSC; (b) P. Dydio and J. N. H. Reek, Chem. Sci., 2014, 5, 2135–2145 RSC; (c) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC; (d) Y. Kuninobu, Synlett, 2018, 29, 2093–2107 CrossRef CAS; selected examples on urea directed transistion metal catalysis: (e) Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712 CrossRef CAS PubMed; (f) J. Wen, X. Fan, R. Tan, H.-C. Chien, Q. Zhou, L. W. Chung and X. Zhang, Org. Lett., 2018, 20, 2143–2147 CrossRef CAS PubMed.
- (a) K. M. Lippert, K. Hof, D. Gerbig, D. Ley, H. Hausmann, S. Guenther and P. R. Schreiner, Eur. J. Org. Chem., 2012, 2012, 5919–5927 CrossRef CAS; (b) G. Jakab, C. Tancon, Z. Zhang, K. M. Lippert and P. R. Schreiner, Org. Lett., 2012, 14, 1724–1727 CrossRef CAS PubMed.
- P. A. Lichtor and S. J. Miller, Nat. Chem., 2012, 4, 990 CrossRef CAS PubMed.
- For GFN-xTB see: S. Grimme, C. Bannwarth and P. Shushkov, J. Chem. Theory Comput., 2017, 13, 1989–2009 CrossRef CAS PubMed . GFN2-xTB will be published soon by Grimme and coworkers.
- Benzene is not included in the GFN2-xTB code, thus toluene was used.
- K. W. Fiori and J. Du Bois, J. Am. Chem. Soc., 2007, 129, 562–568 CrossRef CAS PubMed.
- C. G. Espino, K. W. Fiori, M. Kim and J. Du Bois, J. Am. Chem. Soc., 2004, 126, 15378–15379 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization data and copies of 1H and 13C NMR spectra. CCDC 1852225–1852227. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05733h |
| This journal is © The Royal Society of Chemistry 2019 |