DOI:
10.1039/C8SC05016C
(Edge Article)
Chem. Sci., 2019,
10, 3330-3339
Downsizing feature of microphase-separated structures via intramolecular crosslinking of block copolymers†
Received
11th November 2018
, Accepted 11th January 2019
First published on 14th January 2019
Abstract
A novel strategy for downsizing the feature of microphase-separated structures was developed via the intramolecular crosslinking reaction of block copolymers (BCPs) without changing the molecular weight. A series of BCPs consisting of poly[styrene-st-(p-3-butenyl styrene)] and poly(rac-lactide) (SBS–LA) was subjected to Ru-catalyzed olefin metathesis under highly diluted conditions to produce intramolecularly crosslinked BCPs (SBS(cl)–LAs). Small-angle X-ray scattering measurement and transmission electron microscopy observation of the SBS(cl)–LAs revealed feature size reduction in lamellar (LAM) and hexagonally close-packed cylinder (HEX) structures in the bulk state, which was surely due to the restricted chain dimensions of the intramolecularly crosslinked SBS block. Notably, the degree of size reduction was controllable by varying the crosslink density, with a maximum decrease of 22% in the LAM spacing. In addition, we successfully observed the downsizing of the HEX structure in the thin film state using atomic force microscopy, indicating the applicability of the present methodology to next-generation lithography technology.
Introduction
The self-assembly of block copolymers (BCPs) can produce various periodic nanostructures depending on the volume fraction of each block (f), such as lamellar, hexagonally close-packed cylinder, bicontinuous gyroid, and body-centered cubic structures. Therefore, it has attracted great interest in nanomanufacturing fields.1–8 In particular, “bottom-up” BCP lithography, in which line patterns can be fabricated on the underlying substrate using a microphase-separated thin film with ca. 10–100 nm features as a resist, has received substantial attention as a next-generation technology to replace the conventional “top-down” photolithography.9–20 To further advance BCP lithography, a very important requirement is to create nanostructures with shorter periodicity, i.e., smaller domain-spacing (d), and thus it is a current major focus in the field of BCP self-assembly. Since the feature size of the microphase-separated structures reflects the radius of gyration (Rg) of the BCP, the d value can be decreased by reducing the degree of polymerization (N). Yet, microphase separation requires the product of Flory–Huggins interaction parameter (χ) and N to exceed the critical value of χN = 10.5 for linear symmetric diblock copolymers (f = 0.5).21,22
A rational approach to achieve a smaller d value is decreasing the N value while increasing the χ value. Thus, a number of “high-χ BCPs”, such as silicon-containing,23–28 metal-doped,29–31 and oligosaccharide-based BCPs32–35 have been developed. However, only limited pairs of monomers are available for this approach. An alternative approach that is applicable to a wide range of polymers is reducing the Rg of BCP by varying the macromolecular architecture. In the pioneering work by Hawker et al., the macrocyclic BCP was found to exhibit a microphase-separated structure with a smaller d value than its linear counterpart.36 Furthermore, we and several other groups reported that the miktoarm star architecture is also effective for decreasing the d value, by increasing the branching number while retaining the total molecular weight and f.37–40 Thus, the preparation of BCPs with specific chain architectures is a promising way for shrinking the microphase-separated structures without decreasing the N value. Nevertheless, polymers with such architectures require an elaborate synthesis consisting of multiple reaction steps. Therefore, a synthetically more accessible method to reduce the d value through regulating the polymer chain dimensions is of significant interest.
Here, we highlight a crosslinking technique, which involves the intramolecular reaction of a crosslinkable precursor under highly diluted conditions, as a novel approach for reducing the d value of microphase-separated structures. Several reports by Meijer et al.,41–44 Pomposo et al.,45–48 and Sawamoto et al.49,50 clearly demonstrated the remarkable reduction in Rg as well as in the hydrodynamic radius of a single polymer molecule in certain solvents due to the crosslink formation, presenting the potential of this approach for chain compaction even in the bulk. Importantly, intramolecular crosslinking not only is a simple procedure, but also has the capability of controlling the chain dimensions by adjusting the crosslink density.51–54 Barner-Kowollik et al. have indeed demonstrated size regulation of intramolecularly crosslinked polymers by varying the content of crosslinkable functionality or the amount of the crosslinking agent.55–58 Thus, the intramolecular crosslinking of BCPs would allow fine tuning of the d value as well as modulating the phase structure. However, the microphase separation behavior of the intramolecularly crosslinked BCPs has not been investigated yet.
In the present study, we demonstrate shrinking the microphase-separated structures through restricting the BCP chain dimensions by the intramolecular crosslinking of linear BCPs. Polystyrene-b-poly(rac-lactide) (PS-b-PLA), which has been widely investigated by Hillmyer et al.,59–65 was employed as a model system for examining the feasibility of this approach. We designed poly{[styrene-stat-(p-3-butenyl styrene)]-block-(rac-lactide)} polymers (P(S-st-BS)-b-PLA or SBS–LA), in which the SBS block can be intramolecularly crosslinked upon the olefin metathesis reaction to produce the crosslinked-linear BCPs, i.e., SBS(cl)–LAs (Scheme 1). Indeed, the SBS(cl)–LAs exhibited a smaller d value in the microphase-separated structures as compared to the corresponding linear SBS–LA precursors. The minimum d value achieved in this study was 12.0 nm, corresponding to a sub-10 nm feature size. Furthermore, we succeeded in reducing the domain size up to 22% by varying the crosslink density of the SBS block.
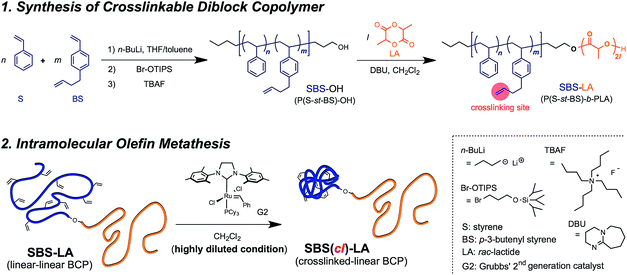 |
| | Scheme 1 Synthesis of the crosslinked-linear BCP via the intramolecular olefin metathesis reaction of the linear–linear BCP possessing pendant double bonds as the crosslinking sites. | |
Results and discussion
Intramolecular crosslinking of SBS–LAs
A series of polystyrene-b-poly(rac-lactide)s possessing pendant olefin groups on the polystyrene block (SBS–LA, Table 1) were successfully synthesized in two steps according to Scheme 1. First, the hydroxyl-terminated poly[styrene-st-(p-3-butenyl styrene)] (SBS–OH) was prepared via living anionic copolymerization of styrene (S) and p-3-butenyl styrene (BS) in a THF/toluene co-solvent (Fig. S1, Table S1†), under which the side chain double bond of BS remains intact due to the reduced polarity of the solvent.66 This is followed by the termination reaction with Br–OTIPS and the deprotection with tetrabutylammonium fluoride (TBAF). The subsequent ring-opening polymerization of rac-lactide (LA) using the obtained SBS–OH as the macroinitiator yielded the desired SBS–LAs. The total molecular weight (Mn,total) and the weight fraction of the SBS block (FSBS) were systematically varied to adjust the d values and morphologies of the microphase-separated structures. The mole fraction of BS units in the SBS block (fBS) was fixed at around 0.3 or 0.5 (SBS–LAs with fBS of ca. 0.5 have an asterisk in their sample names, i.e., SBS*–LAs.). Note that the SBS8k and  blocks were designed to be comparable in degree of polymerization (DP), in order to examine the effect of olefin contents on the chain compaction as well as on the microphase separation behavior.
blocks were designed to be comparable in degree of polymerization (DP), in order to examine the effect of olefin contents on the chain compaction as well as on the microphase separation behavior.
Table 1 Molecular characteristics of SBS–LAs
| Sample name |
M
n,SBS (DP, fBS)a |
M
n,LA (DP)a |
M
n,total
|
F
SBS
|
M
n,SEC
|
Đ
|
|
Determined by 1H NMR in CDCl3.
SBS weight fraction determined using Mn,SBS and Mn,LA.
Determined by SEC in THF using polystyrene standards.
|
| SBS5k–LA7k |
4910 (40, 0.28) |
6750 (47) |
11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 700 |
0.42 |
13700 |
1.03 |
| SBS8k–LA5k |
7940 (65, 0.31) |
4680 (32) |
12600 |
0.63 |
12500 |
1.03 |
| SBS8k–LA8k |
7940 (65, 0.31) |
8250 (57) |
16200 |
0.49 |
16500 |
1.03 |
|
9460 (71, 0.49) |
6420 (45) |
15900 |
0.60 |
16800 |
1.03 |
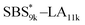
|
9460 (71, 0.49) |
11400 (79) |
20900 |
0.45 |
23300 |
1.03 |
| SBS27k–LA13k |
27000 (224, 0.29) |
13400 (93) |
40400 |
0.67 |
36600 |
1.03 |
| SBS27k–LA28k |
27000 (224, 0.29) |
27900 (193) |
54900 |
0.49 |
54400 |
1.03 |
Recently, our group reported the intramolecular olefin metathesis of linear precursors possessing pendant olefins using Grubbs' 2nd generation catalyst (G2), whose high reactivity and excellent functional group tolerance enabled the crosslinking of a variety of polymers under mild conditions.67,68
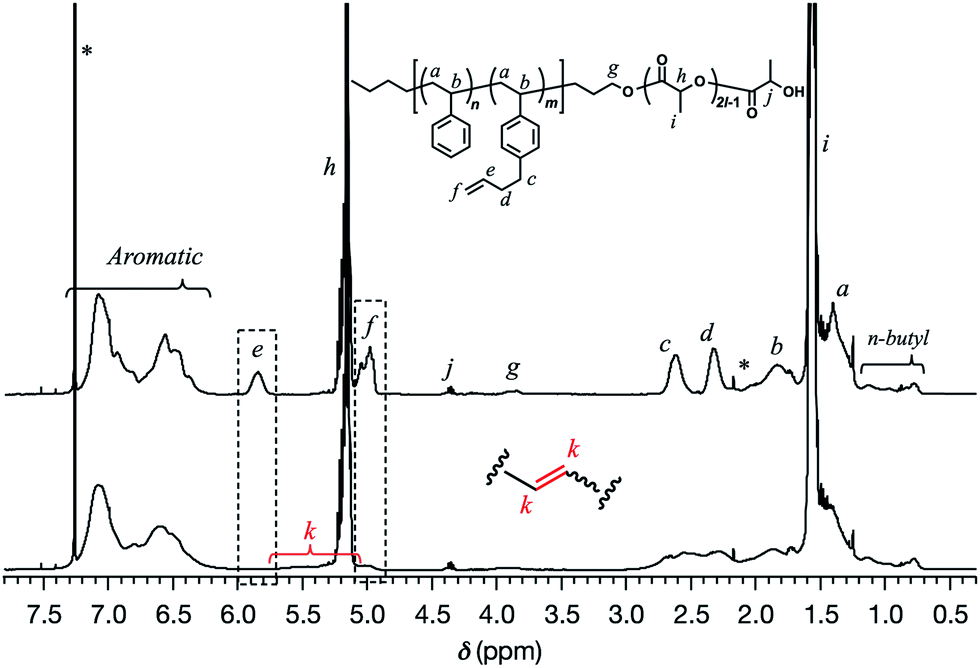
With a series of SBS–LAs in hand, the intramolecular olefin metathesis was conducted in the presence of G2 to produce the crosslinked-linear diblock copolymers (SBS(cl)–LAs). First, the intramolecular olefin metathesis of SBS5k–LA7k was carried out under highly diluted conditions ([SBS5k–LA7k]0 = 0.30 g L−1) in CH2Cl2 at 30 °C for 3 h in the presence of 1.0 mol% of G2 with respect to the pendant double bond. The reaction proceeded homogeneously without gelation, giving a soluble product after treatment with ethyl vinyl ether, followed by reprecipitation to remove the catalyst and the preparative SEC purification to isolate the main products from the minor byproducts with high molecular weights. In the 1H NMR spectrum of the product, the intensity of the signals due to the terminal olefin protons (e and f) decreased, and those due to the internal olefin protons (k) newly appeared (Fig. 1). The conversion of the terminal olefins (conv.olefin) was calculated to be 93% by comparing the intensities of e signals before and after the crosslinking, after normalization with the intensity of signals derived from the aromatic protons. Notably, all signals from the SBS block were broadened after the crosslinking reaction although those from the LA block remained unchanged, indicating that only the SBS block was intramolecularly crosslinked without an unwanted reaction on the LA block. The IR spectrum of the product also supported the high olefin conversion ratio. The characteristic absorption band at 1642 cm−1 due to the terminal olefin was no longer observed in the spectrum of the product (Fig. S2†). Thus, the 1H NMR and IR studies confirmed the crosslink formation via the olefin metathesis reaction.
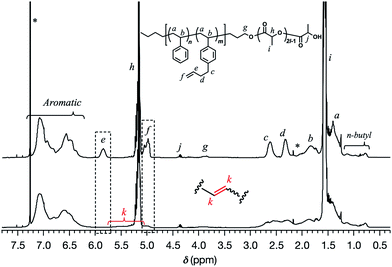 |
| | Fig. 1
1H NMR spectra of SBS5k–LA7k (upper) and SBS5k(cl)–LA7k (lower) in CDCl3 (400 MHz). | |
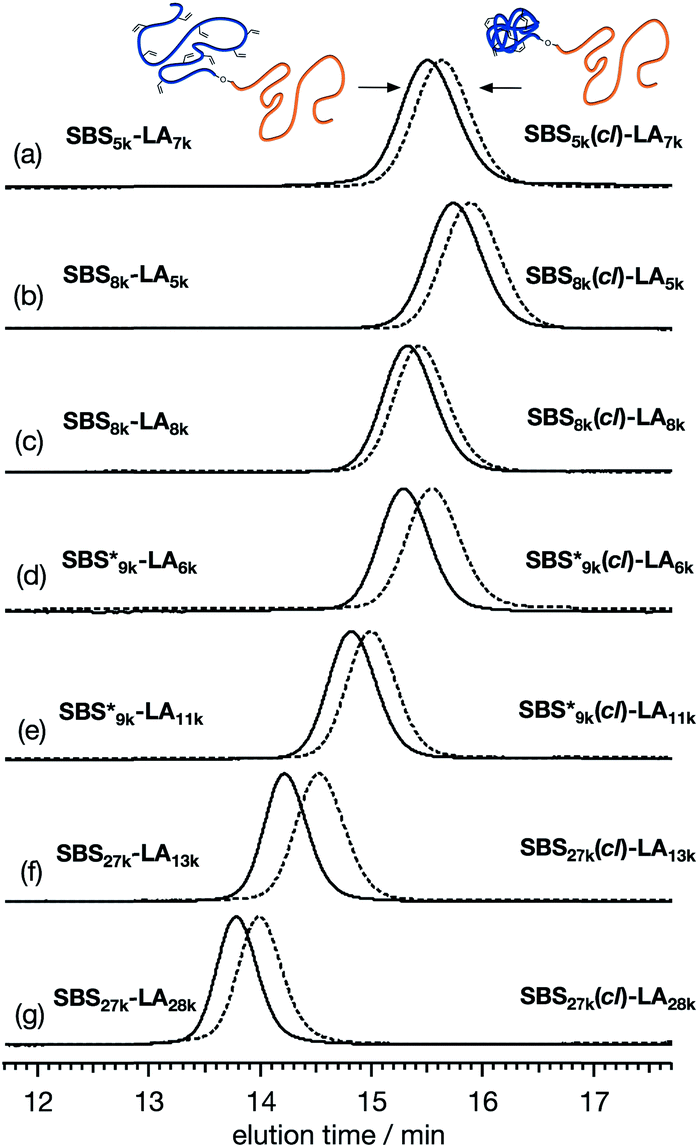
To further verify the intramolecular crosslink formation, SEC measurement was performed on SBS5k–LA7k and its reaction product. As can be seen in Fig. 2(a), the SEC trace of the crosslinked product exhibited a monomodal peak in the lower molecular weight region as compared to that of SBS5k–LA7k, implying a decrease in the hydrodynamic volume through the crosslinking reaction (the SEC trace of the product obtained before preparative SEC purification is shown in Fig. S3†). Under the assumption that the LA block maintains its linear structure after intramolecular crosslinking, we can define the shrinking factor of the SBS block, 〈G〉SBS, using the following equations:
| | 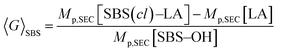 | (1) |
| | | Mp,SEC[LA] = Mp,SEC[SBS–LA] − Mp,SEC[SBS–OH] | (2) |
where
Mp,SEC[SBS(
cl)–LA],
Mp,SEC[SBS–OH], and
Mp,SEC[SBS–LA] stand for the molecular weights calculated from the peak top of the corresponding SEC traces. The 〈
G〉
SBS value in
Fig. 2(a) was 0.78, which was comparable to the 〈
G〉 value (0.79) obtained from the intramolecular crosslinking of SBS
5k–OH (Table S1
†). Based on the
1H NMR, IR, and SEC analyses, we concluded that the crosslinked-linear diblock copolymer,
i.e., SBS
5k(
cl)–LA
7k, was successfully obtained. In a similar manner, SBS–LAs with different compositions and molecular weights were also successfully converted to the corresponding SBS(
cl)–LAs with sufficient conv.
olefin values (
Table 2,
Fig. 2(b)–(g), and S3–S15
†). It is worth noting that the 〈
G〉
SBS values for
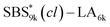
and
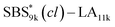
(0.67 and 0.66) were apparently lower than those of SBS
8k(
cl)–LA
5k and SBS
8k(
cl)–LA
8k (0.81 and 0.84) despite the comparable DP values of the SBS block, indicating that a higher crosslinking density led to further reduction in the chain dimensions.
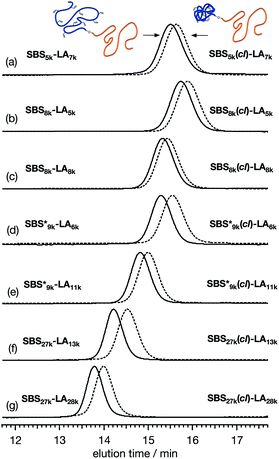 |
| | Fig. 2 SEC traces of (a) SBS5k–LA7k and SBS5k(cl)–LA7k, (b) SBS8k–LA5k and SBS8k(cl)–LA5k, (c) SBS8k–LA8k and SBS8k(cl)–LA8k, (d)  and and  , (e) , (e) 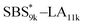 and and 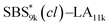 , (f) SBS27k–LA13k and SBS27k(cl)–LA13k, and (g) SBS27k–LA28k and SBS27k(cl)–LA28k (eluent, THF; flow rate, 1.0 mL min−1). SBS–LAs and the corresponding SBS(cl)–LAs are presented using solid and dashed lines, respectively. , (f) SBS27k–LA13k and SBS27k(cl)–LA13k, and (g) SBS27k–LA28k and SBS27k(cl)–LA28k (eluent, THF; flow rate, 1.0 mL min−1). SBS–LAs and the corresponding SBS(cl)–LAs are presented using solid and dashed lines, respectively. | |
Table 2 Characterization data for SBS–LAs and SBS(cl)–LAs
| Sample name |
Conv.olefina (%) |
M
p,SEC
|
Đ
|
〈G〉SBSc |
T
g,SBS
(°C) |
T
g,LA
(°C) |
Determined by 1H NMR in CDCl3.
Determined by SEC in THF using polystyrene standards.
Calculated using eqn (1) and (2). Mp,SEC[SBS5k–OH] = 5360, Mp,SEC[SBS8k–OH] = 7290, 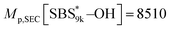 , Mp,SEC[SBS19k–OH] = 20100.
Determined by DSC at the heating rate of 10 °C min−1. , Mp,SEC[SBS19k–OH] = 20100.
Determined by DSC at the heating rate of 10 °C min−1.
|
| SBS5k–LA7k |
93 |
13400 |
1.03 |
0.78 |
59 |
51 |
| SBS5k(cl)–LA7k |
12200 |
1.03 |
91 |
54 |
| SBS8k–LA5k |
88 |
12500 |
1.03 |
0.81 |
60 |
49 |
| SBS8k(cl)–LA5k |
11100 |
1.03 |
109 |
54 |
| SBS8k–LA8k |
79 |
16500 |
1.03 |
0.84 |
63 |
52 |
| SBS8k(cl)–LA8k |
15300 |
1.03 |
110 |
55 |
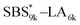
|
86 |
16900 |
1.03 |
0.67 |
— |
49 |
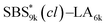
|
14100 |
1.03 |
138 |
52 |
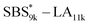
|
89 |
23600 |
1.03 |
0.66 |
— |
51 |
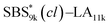
|
20700 |
1.03 |
143 |
55 |
| SBS27k–LA13k |
88 |
39300 |
1.03 |
0.45 |
76 |
52 |
| SBS27k(cl)–LA13k |
28200 |
1.03 |
140 |
53 |
| SBS27k–LA28k |
87 |
54400 |
1.03 |
0.54 |
80 |
55 |
| SBS27k(cl)–LA28k |
45200 |
1.04 |
137 |
53 |
Thermal properties
Since the intramolecularly crosslinked polymers obviously differ in molecular mobility from typical polymers with random coils, their bulk thermal property is an important consideration when evaluating the microphase separation behavior. Thus, the obtained SBS–LAs, SBS(cl)–LAs, and the corresponding homopolymers (SBS–OHs, SBS(cl)–OHs, and PLA) were characterized by differential scanning calorimetry (DSC) to determine their glass transition temperature (Tg) (Tables 2 and S1†). The DSC curves of SBS–LAs and SBS(cl)–LAs during the second heating process are shown in Fig. S16.† See also Fig. S17† for the DSC curves of SBS–OHs, SBS(cl)–OHs, and PLA.
For SBS5k–LA7k and SBS5k(cl)–LA7k, two sets of baseline shifts due to the separate glass transitions of SBS and LA blocks were observed, indicating the formation of microphase-separated structures (Fig. 3). Importantly, the Tg of the SBS5k(cl) block was determined to be 91 °C, which was much higher than that of the SBS5k block, i.e., 59 °C, while there was no significant difference in Tg for the LA blocks between SBS5k–LA7k (51 °C) and SBS5k(cl)–LA7k (54 °C). The increased Tg of the crosslinked SBS block compared to its linear counterparts strongly reflected a decrease in the segmental chain mobility, which again confirmed the formation of the intramolecularly crosslinked structure. A similar thermal behavior was observed for all other SBS(cl)–LAs (Fig. S16†). It should also be noted that the determined Tg values of SBS–LAs and SBS(cl)–LAs were comparable with those of the corresponding homopolymers (Fig. S17 and Table S1†). We could not detect the baseline shift of the 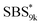 block in
block in 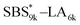 and
and 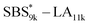 (Fig. S16(d) and (e)†). Considering that the Tg value of
(Fig. S16(d) and (e)†). Considering that the Tg value of 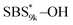 was 46 °C (Table S1†), the baseline shift due to the
was 46 °C (Table S1†), the baseline shift due to the 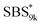 block seems to overlap with that of the LA blocks at around 50 °C. Surprisingly, the Tg values of the
block seems to overlap with that of the LA blocks at around 50 °C. Surprisingly, the Tg values of the 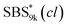 block (138 °C for
block (138 °C for 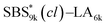 and 143 °C for
and 143 °C for 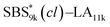 ) were considerably higher than that of the corresponding linear
) were considerably higher than that of the corresponding linear 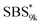 block (around 50 °C). To our knowledge, such a dramatic increase in the Tg values has not been reported for intramolecularly crosslinked polymers to date.68–73 This is probably due to the substantial incorporation of the crosslinkable units into the precursor (50 mol%) and the high conversion ratio to the crosslinkages (conv.olefin = ca. 90%). The Tg values of SBS27k(cl) blocks in SBS27k(cl)–LA10k (140 °C) and SBS27k(cl)–LA28k (137 °C) were also very high, which is surely due to their high molecular weights.
block (around 50 °C). To our knowledge, such a dramatic increase in the Tg values has not been reported for intramolecularly crosslinked polymers to date.68–73 This is probably due to the substantial incorporation of the crosslinkable units into the precursor (50 mol%) and the high conversion ratio to the crosslinkages (conv.olefin = ca. 90%). The Tg values of SBS27k(cl) blocks in SBS27k(cl)–LA10k (140 °C) and SBS27k(cl)–LA28k (137 °C) were also very high, which is surely due to their high molecular weights.
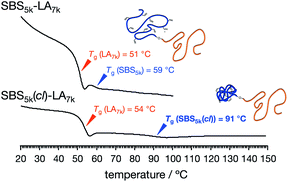 |
| | Fig. 3 DSC curves during the 2nd heating process of SBS5k–LA7k (upper) and SBS5k(cl)–LA7k (lower). | |
Microphase separation behavior in the bulk state
To investigate the microphase-separated structures of SBS–LAs and SBS(cl)–LAs, a small-angle X-ray scattering (SAXS) study was carried out on their bulk samples. Prior to the SAXS experiments, the samples were thermally annealed for 1 h under vacuum at temperatures higher than their measured Tg values. The resulting morphological characteristics of all samples are summarized in Table 3. The SAXS profile of SBS8k–LA8k (FSBS = 0.49) showed a primary scattering peak at q* = 0.413 nm−1 with higher-ordered scattering peaks at 2q*, 3q*, and 4q* positions, indicative of a well-ordered lamellar (LAM) morphology (Fig. 4(a), upper). The d value of the LAM structure was determined to be 15.2 nm based on the relationship of d = 2π/q*. The SAXS profile of SBS8k(cl)–LA8k also exhibited scattering peaks (q* = 0.469 nm−1, 2q*, and 3q*) corresponding to the LAM morphology with a d value of 13.4 nm (Fig. 4(a), lower). Importantly, these results clearly demonstrated a decrease in the d value by 12% after the intramolecular crosslinking process, indicating the compact chain dimensions of the resultant polymers in the bulk state. Because the chain dimensions of the LA block are presumed to be almost unchanged even after the intramolecular crosslinking process, the decrease in d should be attributed to a reduction in the chain volume of the SBS block. Transmission electron microscopy (TEM) observation of the microtomed samples was performed to further confirm the above hypothesis. TEM images of SBS8k–LA8k and SBS8k(cl)–LA8k exhibited the line patterns corresponding to the LAM morphology without any staining, in which the dark and bright parts were assigned to the SBS (SBS(cl)) block and the LA block, respectively (Fig. 4(b) and (c)). The large wrinkles in the horizontal direction are defects that occurred during ultra-microtome cutting. The estimated d value of 15.5–16.0 nm for SBS8k–LA8k is in good agreement with that determined from the SAXS profile (d = 15.2 nm). In the case of SBS8k(cl)–LA8k, the d value was estimated to be 14.0–14.5 nm from the TEM image, which is slightly different from the SAXS result (d = 13.4 nm). Nevertheless, the LAM period of SBS8k(cl)–LA8k was found to be smaller than that of SBS8k–LA8k, according to both the real space TEM image and the SAXS results in the reciprocal space.
Table 3 Morphological characteristics of SBS–LAs and SBS(cl)–LAs in the bulk
| Linear–linear |
Crosslinked-linear |
| Sample name |
Morphology |
d
(nm) |
Sample name |
Morphology |
d
(nm) |
% Decreasec |
|
Determined by SAXS.
d value was converted to the center-to-center distance between cylinders (dC–C).
Decreasing ratio of the d (dC–C) value.
|
| SBS5k–LA7k |
LAM |
13.4 |
SBS5k(cl)–LA7k |
LAM |
12.0 |
10 |
| SBS8k–LA5k |
HEX |
14.5b |
SBS8k(cl)–LA5k |
HEX |
12.9b |
11 |
| SBS8k–LA8k |
LAM |
15.2 |
SBS8k(cl)–LA8k |
LAM |
13.4 |
12 |
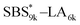
|
HEX |
18.1b |
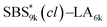
|
HEX |
15.0b |
17 |
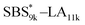
|
LAM |
19.8 |
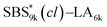
|
LAM |
15.5 |
22 |
| SBS27k–LA13k |
HEX |
29.4b |
SBS27k(cl)–LA13k |
HEX |
22.9b |
22 |
| SBS27k–LA28k |
LAM |
34.9 |
SBS27k(cl)–LA28k |
HEX |
31.9b |
(9) |
 |
| | Fig. 4 (a) SAXS profiles of SBS8k–LA8k (upper) and SBS8k(cl)–LA8k (lower). Cross-sectional TEM images of (b) SBS8k–LA8k and (c) SBS8k(cl)–LA8k. The scale bar is 100 nm. | |
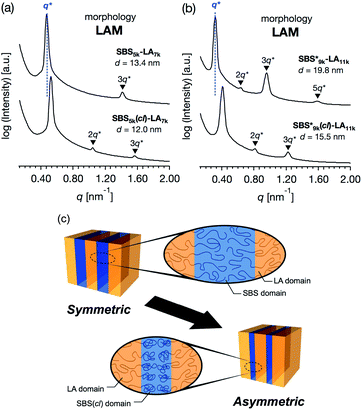
SBS5k–LA7k (FSBS = 0.42), 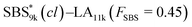 , and the corresponding SBS(cl)–LAs also exhibited the LAM morphology, as expected from their FSBS values (Fig. 5(a) and (b)). From the SAXS results of the polymers with the lowest molecular weights (SBS5k–LA7k and SBS5k(cl)–LA7k), the d value was reduced from 13.4 to 12.0 nm after intramolecular crosslinking. These d values equal to sub-10 nm half-pitch, demonstrating that the presented approach can be applied at such small size scales. Remarkably,
, and the corresponding SBS(cl)–LAs also exhibited the LAM morphology, as expected from their FSBS values (Fig. 5(a) and (b)). From the SAXS results of the polymers with the lowest molecular weights (SBS5k–LA7k and SBS5k(cl)–LA7k), the d value was reduced from 13.4 to 12.0 nm after intramolecular crosslinking. These d values equal to sub-10 nm half-pitch, demonstrating that the presented approach can be applied at such small size scales. Remarkably, 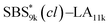 exhibited the maximum downsizing in d in this work, being 22% smaller compared to the corresponding linear
exhibited the maximum downsizing in d in this work, being 22% smaller compared to the corresponding linear 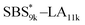 (d = 15.5 vs. 19.8 nm). As described above, the SBS8k and
(d = 15.5 vs. 19.8 nm). As described above, the SBS8k and 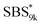 blocks were designed to be different in fBS but with the comparable DP values, which enabled us to examine the effect of olefin contents on the size of the microphase-separated structures. As expected, the decrease ratio in d is greater for
blocks were designed to be different in fBS but with the comparable DP values, which enabled us to examine the effect of olefin contents on the size of the microphase-separated structures. As expected, the decrease ratio in d is greater for 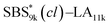 than for SBS8k(cl)–LA8k (22% vs. 12%), in spite of their comparable DPs in the crosslinkable SBS block. This result clearly demonstrates that increasing the crosslinking density of BCP is an effective approach to further reducing the feature sizes of the microphase-separated structures.
than for SBS8k(cl)–LA8k (22% vs. 12%), in spite of their comparable DPs in the crosslinkable SBS block. This result clearly demonstrates that increasing the crosslinking density of BCP is an effective approach to further reducing the feature sizes of the microphase-separated structures.
 |
| | Fig. 5 SAXS profiles for the bulk samples of (a) SBS5k–LA7k and SBS5k(cl)–LA7k annealed at 150 °C and (b) 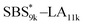 and and 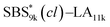 annealed at 180 °C. (c) Schematic illustration of the structural transition in LAM morphology via the intramolecular cross-linking. SBS domains and LA domains are shown in blue and yellow, respectively. annealed at 180 °C. (c) Schematic illustration of the structural transition in LAM morphology via the intramolecular cross-linking. SBS domains and LA domains are shown in blue and yellow, respectively. | |
Notably, the even-ordered peaks in the SAXS profiles of SBS5k–LA7k and 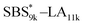 were absent (or suppressed) (Fig. 5(a) and (b), upper), indicating that the two phases of the LAM morphology were highly symmetric.74 On the other hand, the SAXS profiles of intramolecularly crosslinked products, i.e., SBS5k(cl)–LA7k and
were absent (or suppressed) (Fig. 5(a) and (b), upper), indicating that the two phases of the LAM morphology were highly symmetric.74 On the other hand, the SAXS profiles of intramolecularly crosslinked products, i.e., SBS5k(cl)–LA7k and 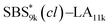 , clearly exhibited a scattering peak at the 2q* position with an intensity similar to those at the 3q* position (Fig. 5(a) and (b), lower), indicative of the transition from the symmetric LAM to an asymmetric one (Fig. 5(c)). These results support our assumption that the major reason for the decrease in d is reduction of feature sizes of the SBS phase.
, clearly exhibited a scattering peak at the 2q* position with an intensity similar to those at the 3q* position (Fig. 5(a) and (b), lower), indicative of the transition from the symmetric LAM to an asymmetric one (Fig. 5(c)). These results support our assumption that the major reason for the decrease in d is reduction of feature sizes of the SBS phase.
The SAXS profile of SBS8k–LA5k (FSBS = 0.63) showed a primary scattering peak at q* = 0.500 nm−1 and higher-ordered ones at 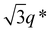 , 2q*,
, 2q*, 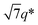 , and 3q* positions, which correspond to the hexagonally close-packed cylinder (HEX) morphology with d = 12.6 nm (Fig. 6(a), upper). For the HEX, the d value was converted to the center-to-center distance between cylinders
, and 3q* positions, which correspond to the hexagonally close-packed cylinder (HEX) morphology with d = 12.6 nm (Fig. 6(a), upper). For the HEX, the d value was converted to the center-to-center distance between cylinders 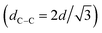 to be 14.5 nm. The SAXS profile of SBS8k(cl)–LA5k also suggested the HEX morphology with dC–C = 12.9 nm, as revealed by the scattering peaks observed at q* (0.563 nm−1), 2q*, and
to be 14.5 nm. The SAXS profile of SBS8k(cl)–LA5k also suggested the HEX morphology with dC–C = 12.9 nm, as revealed by the scattering peaks observed at q* (0.563 nm−1), 2q*, and 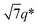 (Fig. 6(a), lower), representing an 11% decrease in the dC–C value compared with the corresponding linear SBS8k–LA5k. Since the HEX-forming SBS–LAs were designed to have longer SBS blocks (matrix) and shorter LA blocks (cylinder domain), the structure transition model in Fig. 6(b) is assumed. In this model, the volume of the SBS matrix is reduced after intramolecular crosslinking to decrease the dC–C while the LA cylinder diameter is maintained. Thus, we successfully reduced the feature size in both the LAM and HEX morphologies.
(Fig. 6(a), lower), representing an 11% decrease in the dC–C value compared with the corresponding linear SBS8k–LA5k. Since the HEX-forming SBS–LAs were designed to have longer SBS blocks (matrix) and shorter LA blocks (cylinder domain), the structure transition model in Fig. 6(b) is assumed. In this model, the volume of the SBS matrix is reduced after intramolecular crosslinking to decrease the dC–C while the LA cylinder diameter is maintained. Thus, we successfully reduced the feature size in both the LAM and HEX morphologies.
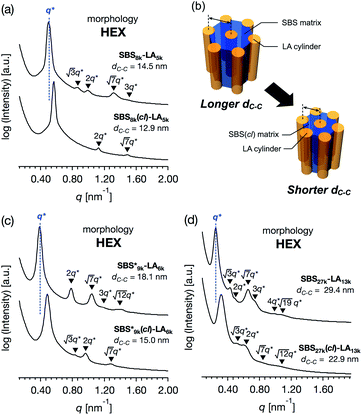 |
| | Fig. 6 SAXS profiles for the bulk samples of (a) SBS8k–LA5k and SBS8k(cl)–LA5k annealed at 150 °C, (c) 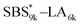 and and 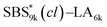 annealed at 180 °C and (d) SBS27k–LA13k and SBS27k(cl)–LA13k annealed at 180 °C. (b) Schematic illustration of the structural transition in HEX morphology via the intramolecular cross-linking. SBS domains and LA domains are shown in blue and yellow, respectively. annealed at 180 °C and (d) SBS27k–LA13k and SBS27k(cl)–LA13k annealed at 180 °C. (b) Schematic illustration of the structural transition in HEX morphology via the intramolecular cross-linking. SBS domains and LA domains are shown in blue and yellow, respectively. | |
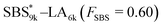 , SBS19k–LA10k (FSBS = 0.65), and the corresponding SBS(cl)–LAs also exhibited the HEX morphology as expected from their FSBS values. A similar result was obtained for the case of
, SBS19k–LA10k (FSBS = 0.65), and the corresponding SBS(cl)–LAs also exhibited the HEX morphology as expected from their FSBS values. A similar result was obtained for the case of 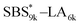 , and
, and 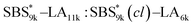 has a dC–C value 17% smaller (15.0 nm) compared to the linear
has a dC–C value 17% smaller (15.0 nm) compared to the linear 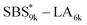 (18.1 nm, Fig. 6(c)), representing a larger decrease ratio than that for SBS8k(cl)–LA5k (11%, Fig. 6(a)) because of the higher crosslink density. In the case of the high-molecular-weight SBS27k–LA13k, a larger volume reduction of the SBS block is expected after the crosslinking reaction as indicated by the 〈G〉SBS values. Indeed, SBS27k(cl)–LA13k exhibited a 22% reduction in dC–C (22.9 nm) compared to that of the corresponding linear SBS27k–LA13k (29.4 nm, Fig. 6(d)).
(18.1 nm, Fig. 6(c)), representing a larger decrease ratio than that for SBS8k(cl)–LA5k (11%, Fig. 6(a)) because of the higher crosslink density. In the case of the high-molecular-weight SBS27k–LA13k, a larger volume reduction of the SBS block is expected after the crosslinking reaction as indicated by the 〈G〉SBS values. Indeed, SBS27k(cl)–LA13k exhibited a 22% reduction in dC–C (22.9 nm) compared to that of the corresponding linear SBS27k–LA13k (29.4 nm, Fig. 6(d)).
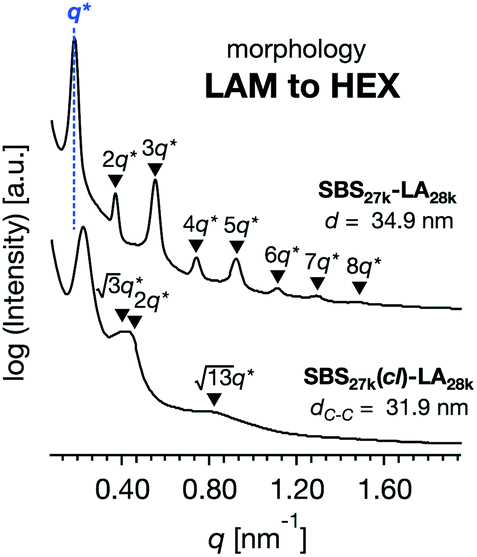
While SBS27k–LA28k (FSBS = 0.49) exhibited a well-ordered LAM morphology, the SAXS profile of SBS27k(cl)–LA28k showed scattering peaks (q*, 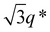 , 2q*, and
, 2q*, and 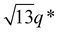 ) corresponding to the HEX morphology (Fig. 7). Such a change in phase was only observed in this BCP with the highest molecular weight, and it should also be attributed to a significant change in the effective volume fraction of the SBS block due to the intramolecular crosslinking. Therefore, the presented methodology can be applied for not only downsizing the feature but also varying the morphology in the microphase-separated structures.
) corresponding to the HEX morphology (Fig. 7). Such a change in phase was only observed in this BCP with the highest molecular weight, and it should also be attributed to a significant change in the effective volume fraction of the SBS block due to the intramolecular crosslinking. Therefore, the presented methodology can be applied for not only downsizing the feature but also varying the morphology in the microphase-separated structures.
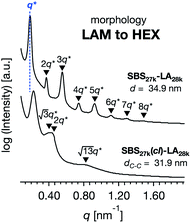 |
| | Fig. 7 SAXS profiles for the bulk samples of SBS27k–LA28k and SBS27k(cl)–LA28k annealed at 180 °C. | |
Microphase separation behavior in the thin film state
For the lithographic application, microphase separation in the thin film state is of particular importance. We therefore finally investigated the thin film morphologies of the HEX-forming SBS27k–LA13k and SBS27k(cl)–LA13kvia atomic force microscopy (AFM) observation. To obtain vertically oriented microphase-separated structures, thin films ca. 50 nm in thickness were prepared by spin-coating onto Si substrates, and THF vapor annealing was applied to the films.75,76 The AFM height images of SBS27k–LA28k and SBS27k(cl)–LA13k thin films are shown in Fig. 8(a) and (b), respectively, where the dot patterns corresponding to the HEX structure oriented perpendicular to the film surface were observed. The insets show the 2D fast Fourier transform (FFT) profiles obtained from each height image. As expected, the LA cylinders embedded within the SBS matrix were more tightly packed in the SBS27k(cl)–LA13k thin film than in the SBS27k–LA13k thin film (Fig. 8(c)), strongly supporting our assumption about the structural change in HEX morphology after crosslinking (Fig. 6(b)). The dC–C values extracted from the 2D FFT profiles indeed decreased from 27.9 nm (SBS27k–LA13k) to 24.5 nm (SBS27k(cl)–LA13k) due to the intramolecular crosslinking. Thus, we confirmed the downsizing of the microphase-separated structures in the thin film state as well as in the bulk state. It is worth noting that a uniformly packed structure was observed in the thin film even after the intramolecular crosslinking (Fig. 8(b)) despite the decreased molecular mobility, as indicated by the 1H NMR and DSC results. Overall, we successfully demonstrated feature size reduction as well as nanoscale organization of the crosslinked-linear BCPs into a well-ordered morphology in the thin film state, supporting the applicability of the presented approach to lithographic technology.
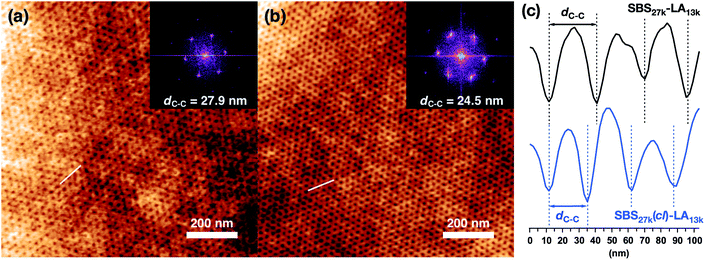 |
| | Fig. 8 AFM height images of (a) SBS27k–LA13k and (b) SBS27k(cl)–LA13k thin films with thicknesses of 56 and 42 nm, respectively. The insets show the 2D FFTs obtained from the height images. Scale bars are 200 nm. (c) AFM height cross sectional images along the white lines in (a) (upper) and (b) (lower). | |
Conclusions
We have successfully synthesized crosslinked-linear BCPs (SBS(cl)–LAs) from the linear–linear BCPs consisting of the crosslinkable SBS segment possessing pendant double bonds and the LA segment (SBS–LA), via the intramolecular olefin metathesis under highly diluted conditions. Morphological analyses of the obtained BCPs in the bulk state by SAXS measurement clearly revealed that the SBS(cl)–LAs had smaller d values in their LAM and HEX morphologies as compared to the corresponding SBS–LAs, due to the restricted chain dimensions of the SBS(cl) segments. Importantly, the amount of d decrease was controllable by varying the contents of the crosslinking sites, where the largest decrease (22%) was achieved in LAM morphology with ca. 50% of the crosslinkable BS unit incorporated into the SBS block. Although the macrocyclic or miktoarm architectures can effectively reduce the size of microphase-separated structures to some extent, further reduction in the d value requires increasing the number of the cyclic units or the branching, which presents a synthetic challenge and practical limitations. In contrast, the currently reported intramolecular crosslinking approach could easily reduce the d value further by simply controlling the crosslink density. We also confirmed the downsizing of the HEX morphology in the thin film state by AFM observation, and the uniformly packed structures were present even after the intramolecular crosslinking that reduced chain mobility of the crosslinked segment. To the best of our knowledge, this is the first experimental demonstration on how the intramolecular crosslinking of BCPs affects the microphase separation behaviors. Considering the fact that the sub-10 nm scale nanostructure was successfully downsized here and the crosslinked-linear BCP could form a well-ordered structure in the thin film, the current approach is highly promising for the development of next-generation lithography. Moreover, the ruthenium-catalyzed olefin metathesis has excellent functional group tolerance, so this approach is expandable to a wide variety of BCPs. To produce even smaller microphase-separated structures, we are currently working on the synthesis and morphological analysis of crosslinked–crosslinked BCPs in which each block is separately crosslinked. Our methodology using this novel-shaped BCP should eventually contribute to remarkable advancements in not only lithography but also other nanomanufacturing applications, such as mesoporous filtering membranes and high-performance memory devices.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the MEXT Grant-in-Aid for Research Activity Start-up (26888001), Grant-in-Aid for Young Scientists (B) (15K17862), Grant-in-Aid for Scientific Research (B) (16H04152), and Grant-in-Aid for Scientific Research on Innovative Areas “Hybrid Catalysis” (18H04639). K. W. was funded by a JSPS Fellowship for Young Scientists. T. I gratefully acknowledges the Nanotech CUPAL NRP program. The authors thank Prof. H. Ito (Hokkaido University, Japan) for his assistance with the AFM experiments.
Notes and references
- C. Park, J. Yoon and E. L. Thomas, Polymer, 2003, 44, 6725–6760 CrossRef CAS.
- H.-C. Kim, S.-M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146–177 CrossRef CAS PubMed.
- J. K. Kim, S. Y. Yang, Y. Lee and Y. Kim, Prog. Polym. Sci., 2010, 35, 1325–1349 CrossRef CAS.
- A. Horechyy, B. Nandan, N. E. Zafeiropoulos, P. Formanek, U. Oertel, N. C. Bigall, A. Eychmüller and M. Stamm, Adv. Funct. Mater., 2013, 23, 483–490 CrossRef CAS.
- H.-Y. Hsueh, C.-T. Yao and R.-M. Ho, Chem. Soc. Rev., 2015, 44, 1974–2018 RSC.
- M. Stefik, S. Guldin, S. Vignolini, U. Wiesner and U. Steiner, Chem. Soc. Rev., 2015, 44, 5076–5091 RSC.
- C. M. Bates and F. S. Bates, Macromolecules, 2017, 50, 3–22 CrossRef CAS.
- C.-C. Hung, Y.-C. Chiu, H.-C. Wu, C. Lu, C. Bouilhac, I. Otsuka, S. Halila, R. Borsali, S.-H. Tung and W.-C. Chen, Adv. Funct. Mater., 2017, 27, 1–10 CrossRef.
- M. P. Stoykovich, M. Müller, S. O. Kim, H. H. Solak, E. W. Edwards, J. J. de Pablo and P. F. Nealey, Science, 2005, 308, 1442–1446 CrossRef CAS PubMed.
- M. P. Stoykovich and P. F. Nealey, Mater. Today, 2006, 9, 20–29 CrossRef CAS.
- M. Li and C. K. Ober, Mater. Today, 2006, 9, 30–39 CrossRef CAS.
- R. Ruiz, H. Kang, F. A. Detcheverry, E. Dobisz, D. S. Kercher, T. R. Albrecht, J. J. de Pablo and P. F. Nealey, Science, 2008, 321, 936–939 CrossRef CAS PubMed.
- J. Bang, U. Jeong, D. Y. Ryu, T. P. Russell and C. J. Hawker, Adv. Mater., 2009, 21, 4769–4792 CrossRef CAS PubMed.
- C. M. Bates, M. J. Maher, D. W. Janes, C. J. Ellison and C. G. Willson, Macromolecules, 2014, 47, 2–12 CrossRef CAS.
- A. Nunns, J. Gwyther and I. Manners, Polymer, 2013, 54, 1269–1284 CrossRef CAS.
- S.-J. Jeong, J. Y. Kim, B. H. Kim, H.-S. Moon and S. O. Kim, Mater. Today, 2013, 16, 468–476 CrossRef CAS.
- C.-C. Liu, E. Han, M. S. Onses, C. J. Thode, S. Ji, P. Gopalan and P. F. Nealey, Macromolecules, 2011, 44, 1876–1885 CrossRef CAS.
- J. Kwak, A. K. Mishra, J. Lee, K. S. Lee, C. Choi, S. Maiti, M. Kim and J. K. Kim, Macromolecules, 2017, 50, 6813–6818 CrossRef CAS.
- R. Nakatani, H. Takano, A. Chandra, Y. Yoshimura, L. Wang, Y. Suzuki, Y. Tanaka, R. Maeda, N. Kihara, S. Minegishi, K. Miyagi, Y. Kasahara, H. Sato, Y. Seino, T. Azuma, H. Yokoyama, C. K. Ober and T. Hayakawa, ACS Appl. Mater. Interfaces, 2017, 9, 31266–31278 CrossRef CAS PubMed.
- A. Legrain, G. Fleury, M. Mumtaz, C. Navarro, J. Arias-Zapata, X. Chevalier, I. Cayrefourcq and M. Zelsmann, ACS Appl. Mater. Interfaces, 2017, 9, 43043–43050 CrossRef CAS PubMed.
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS PubMed.
- T.-Y. Lo, M. R. Krishnan, K.-Y. Lu and R.-M. Ho, Prog. Polym. Sci., 2018, 77, 19–68 CrossRef CAS.
- M. D. Rodwogin, C. S. Spanjers, C. Leighton and M. A. Hillmyer, ACS Nano, 2010, 4, 725–732 CrossRef CAS PubMed.
- J. D. Cushen, C. M. Bates, E. L. Rausch, L. M. Dean, S. X. Zhou, C. G. Willson and C. J. Ellison, Macromolecules, 2012, 45, 8722–8728 CrossRef CAS.
- M. J. Maher, C. T. Rettner, C. M. Bates, G. Blachut, M. C. Carlson, W. J. Durand, C. J. Ellison, D. P. Sanders, J. Y. Cheng and C. G. Willson, ACS Appl. Mater. Interfaces, 2015, 7, 3323–3328 CrossRef CAS PubMed.
- K. Aissou, M. Mumtaz, G. Fleury, G. Portale, C. Navarro, E. Cloutet, C. Brochon, C. A. Ross and G. Hadziioannou, Adv. Mater., 2015, 27, 261–265 CrossRef CAS PubMed.
- Y. Luo, D. Montarnal, S. Kim, W. Shi, K. P. Barteau, C. W. Pester, P. D. Hustad, M. D. Christianson, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Macromolecules, 2015, 48, 3422–3430 CrossRef CAS.
- W.-S. Young and T. H. Epps, Macromolecules, 2009, 68, 2672–2678 CrossRef.
- S. Park, D. H. Lee, J. Xu, B. Kim, S. W. Hong, U. Jeong and T. P. Russell, Science, 2009, 323, 1030 CrossRef CAS PubMed.
- Z. Sun, Z. Chen, W. Zhang, J. Choi, C. Huang, G. Jeong, E. B. Coughlin, Y. Hsu, X. Yang, K. Y. Lee, D. S. Kuo, S. Xiao and T. P. Russell, Adv. Mater., 2015, 27, 4364–4370 CrossRef CAS PubMed.
- J. D. Cushen, I. Otsuka, C. M. Bates, S. Halila, S. Fort, C. Rochas, J. A. Easley, E. L. Rausch, A. Thio, R. Borsali, C. G. Willson and C. J. Ellison, ACS Nano, 2012, 6, 3424–3433 CrossRef CAS PubMed.
- I. Otsuka, S. Tallegas, Y. Sakai, C. Rochas, S. Halila, S. Fort, A. Bsiesy, T. Baron and R. Borsali, Nanoscale, 2013, 5, 2637–2641 RSC.
- Y. Sakai-Otsuka, S. Zaioncz, I. Otsuka, S. Halila, P. Rannou and R. Borsali, Macromolecules, 2017, 50, 3365–3376 CrossRef CAS.
- T. Isono, B. J. Ree, K. Tajima, R. Borsali and T. Satoh, Macromolecules, 2018, 51, 428–437 CrossRef CAS.
- J. E. Poelma, K. Ono, D. Miyajima, T. Aida, K. Satoh and C. J. Hawker, ACS Nano, 2012, 6, 10845–10854 CrossRef CAS PubMed.
- T. Isono, I. Otsuka, Y. Kondo, S. Halila, S. Fort, C. Rochas, T. Satoh, R. Borsali and T. Kakuchi, Macromolecules, 2013, 46, 1461–1469 CrossRef CAS.
- W. Shi, Y. Tateishi, W. Li, C. J. Hawker, G. H. Fredrickson and E. J. Kramer, ACS Macro Lett., 2015, 4, 1287–1292 CrossRef CAS.
- H. Minehara, L. M. Pitet, S. Kim, R. H. Zha, E. W. Meijer and C. J. Hawker, Macromolecules, 2016, 49, 2318–2326 CrossRef CAS.
- K. Yue, C. Liu, M. Huang, J. Huang, Z. Zhou, K. Wu, H. Liu, Z. Lin, A.-C. Shi, W.-B. Zhang and S. Z. D. Cheng, Macromolecules, 2017, 50, 303–314 CrossRef CAS.
- N. Hosono, M. A. J. Gillissen, Y. Li, S. S. Sheiko, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2013, 135, 501–510 CrossRef CAS PubMed.
- G. M. ter Huurne, M. A. J. Gillissen, A. R. A. Palmans, I. K. Voets and E. W. Meijer, cJ. Am. Chem. Soc., 2015, 48, 3949–3956 CAS.
- Y. Liu, T. Pauloehrl, S. I. Presolski, L. Albertazzi, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2015, 137, 13096–13105 CrossRef CAS PubMed.
- O. Altintas, M. Artar, G. ter Huurne, I. K. Voets, A. R. A. Palmans, C. Barner-Kowollik and E. W. Meijer, Macromolecules, 2015, 48, 8921–8932 CrossRef CAS.
- A. Sanchez-Sanchez, S. Akbari, A. J. Moreno, F. L. Verso, A. Arbe, J. Colmenero and J. A. Pomposo, Macromol. Rapid Commun., 2013, 34, 1681–1686 CrossRef CAS PubMed.
- I. Perez-Baena, I. Asenjo-Sanz, A. Arbe, A. J. Moreno, F. L. Verso, J. Colmenero and J. A. Pomposo, Macromolecules, 2014, 47, 8270–8280 CrossRef CAS.
- A. Sanchez-Sanchez, D. A. Fulton and J. A. Pomposo, Chem. Commun., 2014, 50, 1871–1874 RSC.
- S. Basasoro, M. Gonzalez-burgos, A. J. Moreno, F. L. Verso, A. Arbe, J. Colmenero and J. A. Pomposo, Macromol. Rapid Commun., 2016, 37, 1060–1065 CrossRef CAS PubMed.
- T. Terashima, T. Sugita, K. Fukae and M. Sawamoto, Macromolecules, 2014, 47, 589–600 CrossRef CAS.
- K. Matsumoto, T. Terashima, T. Sugita, M. Takenaka and M. Sawamoto, Macromolecules, 2016, 49, 7917–7927 CrossRef CAS.
- E. Harth, B. Van Horn, V. Y. Lee, D. S. Germack, C. P. Gonzales, R. D. Miller and C. J. Hawker, J. Am. Chem. Soc., 2002, 124, 8653–8660 CrossRef CAS PubMed.
- C. F. Hansell, A. Lu, J. P. Patterson and R. K. O'Reilly, Nanoscale, 2014, 6, 4102–4107 RSC.
- J. B. Beck, K. L. Killops, T. Kang, K. Sivanandan, A. Bayles, M. E. Mackay, K. L. Wooley and C. J. Hawker, Macromolecules, 2009, 42, 5629–5635 CrossRef CAS PubMed.
- C. Song, L. Li, L. Dai and S. Thayumanavan, Polym. Chem., 2015, 6, 4828–4834 RSC.
- O. Altintas, J. Willenbacher, K. N. R. Wuest, K. K. Oehlenschlaeger, P. Krolla-Sidenstein, H. Gliemann and C. Barner-Kowollik, Macromolecules, 2013, 46, 8092–8101 CrossRef CAS.
- J. Willenbacher, K. N. R. Wuest, J. O. Mueller, M. Kaupp, H.-A. Wagenknecht and C. Barner-Kowollik, ACS Macro Lett., 2014, 3, 574–579 CrossRef CAS.
- K. N. R. Wuest, H. Lu, D. S. Thomas, A. S. Goldmann, M. H. Stenzel and C. Barner-Kowollik, ACS Macro Lett., 2017, 6, 1168–1174 CrossRef CAS.
- T. S. Fischer, S. Spann, Q. An, B. Luy, M. Tsotsalas, J. P. Blinco, H. Mutlu and C. Barner-Kowollik, Chem. Sci., 2018, 9, 4696–4702 RSC.
- A. S. Zalusky, R. Olayo-Valles, C. J. Taylor and M. A. Hillmyer, J. Am. Chem. Soc., 2001, 123, 1519–1520 CrossRef CAS.
- A. S. Zalusky, R. Olayo-Valles, J. H. Wolf and M. A. Hillmyer, J. Am. Chem. Soc., 2002, 124, 12761–12773 CrossRef CAS PubMed.
- R. Olayo-Valles, M. S. Lund, C. Leighton and M. A. Hillmyer, J. Mater. Chem., 2004, 14, 2729–2731 RSC.
- R. Olayo-Valles, S. Guo, M. S. Lund, C. Leighton and M. A. Hillmyer, Macromolecules, 2005, 38, 10101–10108 CrossRef CAS.
- A. Baruth, M. Rodwogin, A. Shankar, M. A. Torija, M. J. Erickson, M. A. Hillmyer and C. Leighton, ACS Appl. Mater. Interfaces, 2011, 3, 3472–3481 CrossRef CAS PubMed.
- A. Baruth, M. Seo, C. H. Lin, K. Walster, A. Shankar, M. A. Hillmyer and C. Leighton, ACS Appl. Mater. Interfaces, 2014, 6, 13770–13781 CrossRef CAS PubMed.
- M. E. Vanderlaan and M. A. Hillmyer, Macromolecules, 2016, 49, 8031–8040 CrossRef CAS.
- H. Zhang and E. Ruckenstein, Macromolecules, 1999, 32, 5495–5500 CrossRef CAS.
- K. Watanabe, R. Tanaka, K. Takada, M.-J. Kim, J.-S. Lee, K. Tajima, T. Isono and T. Satoh, Polym. Chem., 2016, 7, 4782–4792 RSC.
- R. Tanaka, K. Watanabe, T. Yamamoto, K. Tajima, T. Isono and T. Satoh, Polym. Chem., 2017, 8, 3647–3656 RSC.
- D. Mecerreyes, V. Lee, C. J. Hawker, J. L. Hedrick, A. Wursch, W. Volksen, T. Magbitang, E. Huang and R. D. Miller, Adv. Mater., 2001, 13, 204–208 CrossRef CAS.
- J. He, L. Tremblay, S. Lacelle and Y. Zhao, Soft Matter, 2011, 7, 2380 RSC.
- J. Rubio-Cervilla, F. Barroso-Bujans and J. A. Pomposo, Macromolecules, 2016, 49, 90–97 CrossRef CAS.
- F. Wang, H. Pu, M. Jin and D. Wan, Macromol. Rapid Commun., 2016, 37, 330–336 CrossRef CAS PubMed.
- J. Zhang, G. Gody, M. Hartlieb, S. Catrouillet, J. Moffat and S. Perrier, Macromolecules, 2016, 49, 8933–8942 CrossRef CAS.
- T. Hashimoto, H. Tanaka and H. Hasegawa, Macromolecules, 1985, 18, 1864–1868 CrossRef CAS.
- M. Vayer, M. A. Hillmyer, M. Dirany, G. Thevenin, R. Erre and C. Sinturel, Thin Solid Films, 2010, 518, 3710–3715 CrossRef CAS.
- M. Dirany, P. Lacroix-Desmazes, M. Vayer, R. Erre, B. Boutevin and C. Sinturel, J. Appl. Polym. Sci., 2011, 122, 2944–2951 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and additional data (1H NMR, SEC, IR, and DSC). See |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Satoshi
Katsuhara
a,
Hiroaki
Mamiya
b,
Takuya
Yamamoto
a,
Satoshi
Katsuhara
a,
Hiroaki
Mamiya
b,
Takuya
Yamamoto
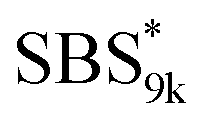 blocks were designed to be comparable in degree of polymerization (DP), in order to examine the effect of olefin contents on the chain compaction as well as on the microphase separation behavior.
blocks were designed to be comparable in degree of polymerization (DP), in order to examine the effect of olefin contents on the chain compaction as well as on the microphase separation behavior.
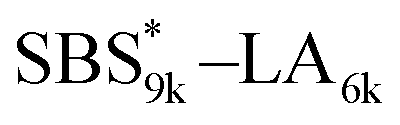
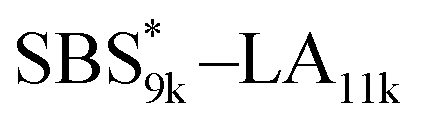
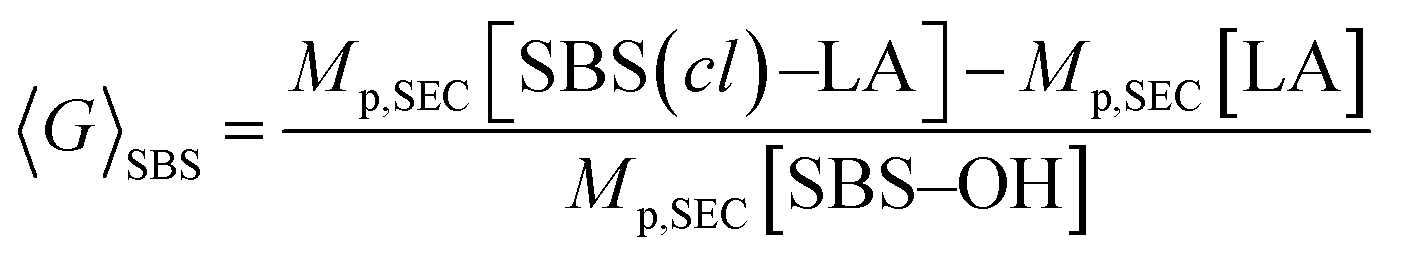
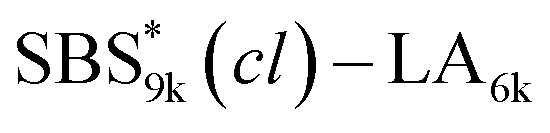 and
and 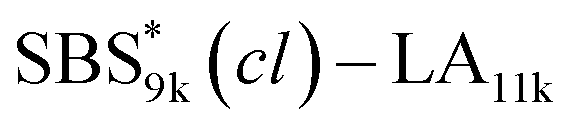 (0.67 and 0.66) were apparently lower than those of SBS8k(cl)–LA5k and SBS8k(cl)–LA8k (0.81 and 0.84) despite the comparable DP values of the SBS block, indicating that a higher crosslinking density led to further reduction in the chain dimensions.
(0.67 and 0.66) were apparently lower than those of SBS8k(cl)–LA5k and SBS8k(cl)–LA8k (0.81 and 0.84) despite the comparable DP values of the SBS block, indicating that a higher crosslinking density led to further reduction in the chain dimensions.
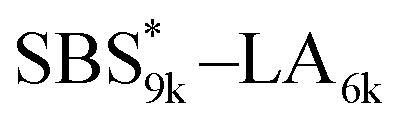 and
and 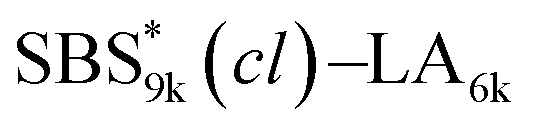 , (e)
, (e) 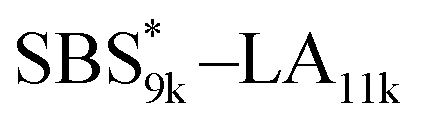 and
and 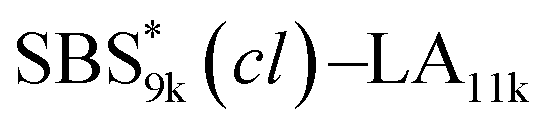 , (f) SBS27k–LA13k and SBS27k(cl)–LA13k, and (g) SBS27k–LA28k and SBS27k(cl)–LA28k (eluent, THF; flow rate, 1.0 mL min−1). SBS–LAs and the corresponding SBS(cl)–LAs are presented using solid and dashed lines, respectively.
, (f) SBS27k–LA13k and SBS27k(cl)–LA13k, and (g) SBS27k–LA28k and SBS27k(cl)–LA28k (eluent, THF; flow rate, 1.0 mL min−1). SBS–LAs and the corresponding SBS(cl)–LAs are presented using solid and dashed lines, respectively.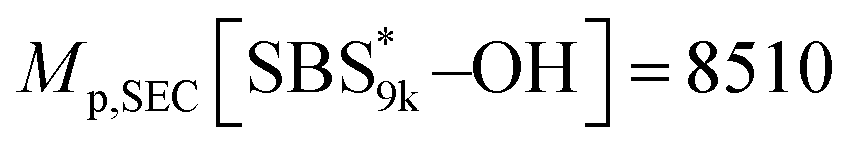 , Mp,SEC[SBS19k–OH] = 20
, Mp,SEC[SBS19k–OH] = 20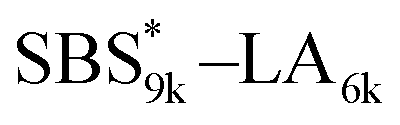
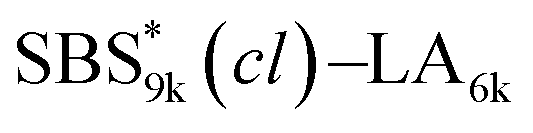
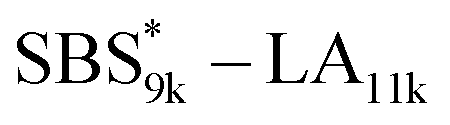
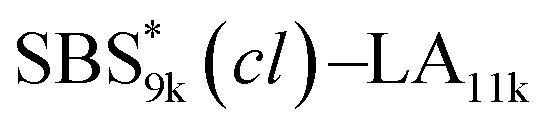
 block in
block in 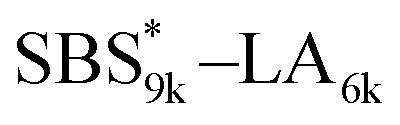 and
and 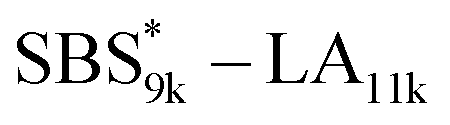 (Fig. S16(d) and (e)†). Considering that the Tg value of
(Fig. S16(d) and (e)†). Considering that the Tg value of 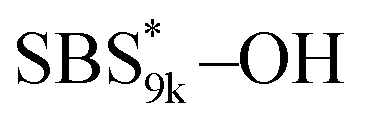 was 46 °C (Table S1†), the baseline shift due to the
was 46 °C (Table S1†), the baseline shift due to the 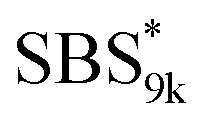 block seems to overlap with that of the LA blocks at around 50 °C. Surprisingly, the Tg values of the
block seems to overlap with that of the LA blocks at around 50 °C. Surprisingly, the Tg values of the 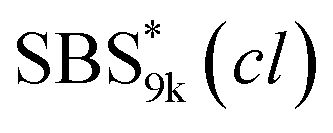 block (138 °C for
block (138 °C for 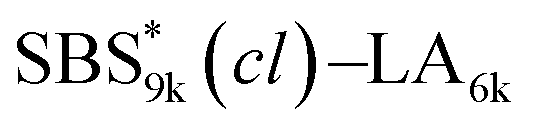 and 143 °C for
and 143 °C for 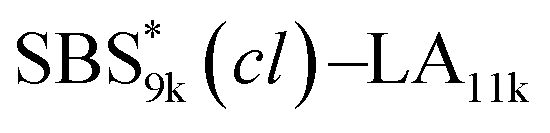 ) were considerably higher than that of the corresponding linear
) were considerably higher than that of the corresponding linear 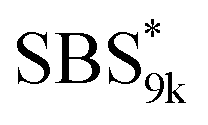 block (around 50 °C). To our knowledge, such a dramatic increase in the Tg values has not been reported for intramolecularly crosslinked polymers to date.68–73 This is probably due to the substantial incorporation of the crosslinkable units into the precursor (50 mol%) and the high conversion ratio to the crosslinkages (conv.olefin = ca. 90%). The Tg values of SBS27k(cl) blocks in SBS27k(cl)–LA10k (140 °C) and SBS27k(cl)–LA28k (137 °C) were also very high, which is surely due to their high molecular weights.
block (around 50 °C). To our knowledge, such a dramatic increase in the Tg values has not been reported for intramolecularly crosslinked polymers to date.68–73 This is probably due to the substantial incorporation of the crosslinkable units into the precursor (50 mol%) and the high conversion ratio to the crosslinkages (conv.olefin = ca. 90%). The Tg values of SBS27k(cl) blocks in SBS27k(cl)–LA10k (140 °C) and SBS27k(cl)–LA28k (137 °C) were also very high, which is surely due to their high molecular weights.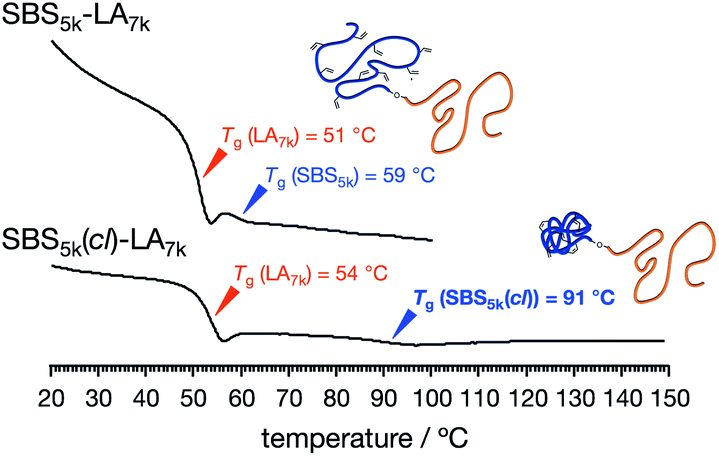
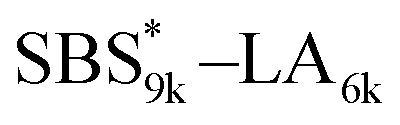
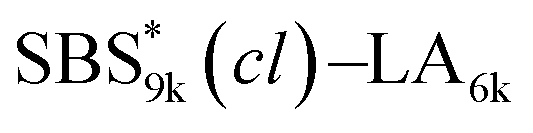
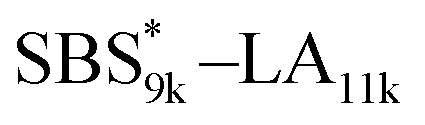
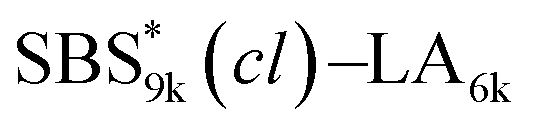

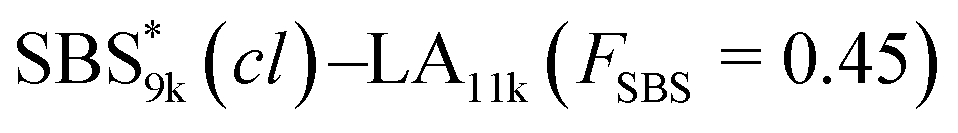 , and the corresponding SBS(cl)–LAs also exhibited the LAM morphology, as expected from their FSBS values (Fig. 5(a) and (b)). From the SAXS results of the polymers with the lowest molecular weights (SBS5k–LA7k and SBS5k(cl)–LA7k), the d value was reduced from 13.4 to 12.0 nm after intramolecular crosslinking. These d values equal to sub-10 nm half-pitch, demonstrating that the presented approach can be applied at such small size scales. Remarkably,
, and the corresponding SBS(cl)–LAs also exhibited the LAM morphology, as expected from their FSBS values (Fig. 5(a) and (b)). From the SAXS results of the polymers with the lowest molecular weights (SBS5k–LA7k and SBS5k(cl)–LA7k), the d value was reduced from 13.4 to 12.0 nm after intramolecular crosslinking. These d values equal to sub-10 nm half-pitch, demonstrating that the presented approach can be applied at such small size scales. Remarkably, 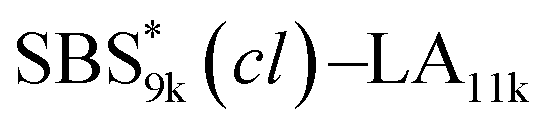 exhibited the maximum downsizing in d in this work, being 22% smaller compared to the corresponding linear
exhibited the maximum downsizing in d in this work, being 22% smaller compared to the corresponding linear 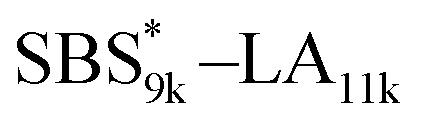 (d = 15.5 vs. 19.8 nm). As described above, the SBS8k and
(d = 15.5 vs. 19.8 nm). As described above, the SBS8k and 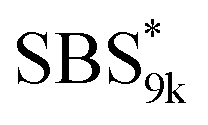 blocks were designed to be different in fBS but with the comparable DP values, which enabled us to examine the effect of olefin contents on the size of the microphase-separated structures. As expected, the decrease ratio in d is greater for
blocks were designed to be different in fBS but with the comparable DP values, which enabled us to examine the effect of olefin contents on the size of the microphase-separated structures. As expected, the decrease ratio in d is greater for 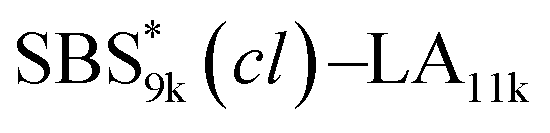 than for SBS8k(cl)–LA8k (22% vs. 12%), in spite of their comparable DPs in the crosslinkable SBS block. This result clearly demonstrates that increasing the crosslinking density of BCP is an effective approach to further reducing the feature sizes of the microphase-separated structures.
than for SBS8k(cl)–LA8k (22% vs. 12%), in spite of their comparable DPs in the crosslinkable SBS block. This result clearly demonstrates that increasing the crosslinking density of BCP is an effective approach to further reducing the feature sizes of the microphase-separated structures.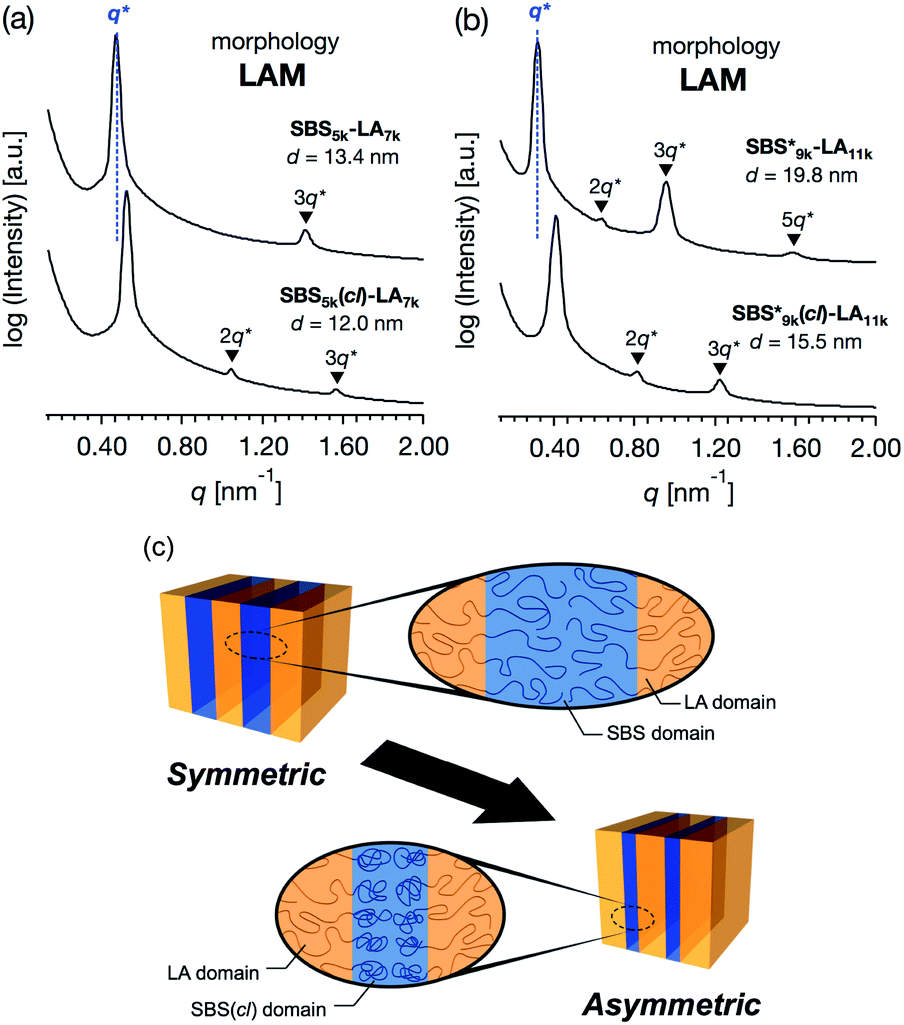
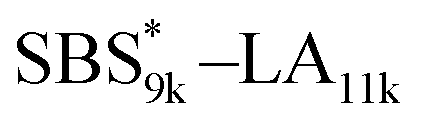 and
and 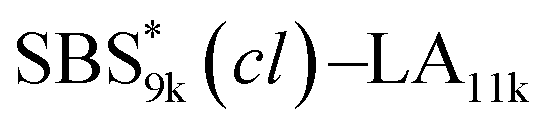 annealed at 180 °C. (c) Schematic illustration of the structural transition in LAM morphology via the intramolecular cross-linking. SBS domains and LA domains are shown in blue and yellow, respectively.
annealed at 180 °C. (c) Schematic illustration of the structural transition in LAM morphology via the intramolecular cross-linking. SBS domains and LA domains are shown in blue and yellow, respectively.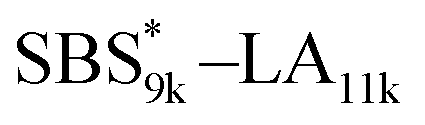 were absent (or suppressed) (Fig. 5(a) and (b), upper), indicating that the two phases of the LAM morphology were highly symmetric.74 On the other hand, the SAXS profiles of intramolecularly crosslinked products, i.e., SBS5k(cl)–LA7k and
were absent (or suppressed) (Fig. 5(a) and (b), upper), indicating that the two phases of the LAM morphology were highly symmetric.74 On the other hand, the SAXS profiles of intramolecularly crosslinked products, i.e., SBS5k(cl)–LA7k and 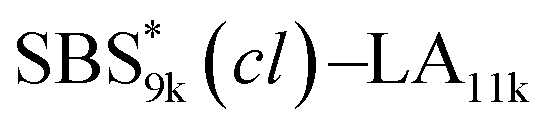 , clearly exhibited a scattering peak at the 2q* position with an intensity similar to those at the 3q* position (Fig. 5(a) and (b), lower), indicative of the transition from the symmetric LAM to an asymmetric one (Fig. 5(c)). These results support our assumption that the major reason for the decrease in d is reduction of feature sizes of the SBS phase.
, clearly exhibited a scattering peak at the 2q* position with an intensity similar to those at the 3q* position (Fig. 5(a) and (b), lower), indicative of the transition from the symmetric LAM to an asymmetric one (Fig. 5(c)). These results support our assumption that the major reason for the decrease in d is reduction of feature sizes of the SBS phase.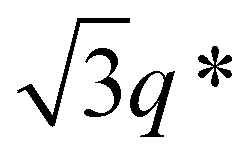 , 2q*,
, 2q*, 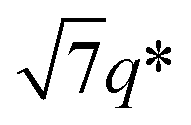 , and 3q* positions, which correspond to the hexagonally close-packed cylinder (HEX) morphology with d = 12.6 nm (Fig. 6(a), upper). For the HEX, the d value was converted to the center-to-center distance between cylinders
, and 3q* positions, which correspond to the hexagonally close-packed cylinder (HEX) morphology with d = 12.6 nm (Fig. 6(a), upper). For the HEX, the d value was converted to the center-to-center distance between cylinders 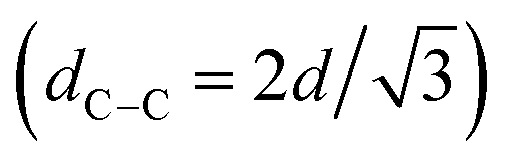 to be 14.5 nm. The SAXS profile of SBS8k(cl)–LA5k also suggested the HEX morphology with dC–C = 12.9 nm, as revealed by the scattering peaks observed at q* (0.563 nm−1), 2q*, and
to be 14.5 nm. The SAXS profile of SBS8k(cl)–LA5k also suggested the HEX morphology with dC–C = 12.9 nm, as revealed by the scattering peaks observed at q* (0.563 nm−1), 2q*, and 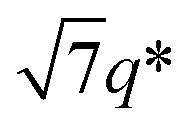 (Fig. 6(a), lower), representing an 11% decrease in the dC–C value compared with the corresponding linear SBS8k–LA5k. Since the HEX-forming SBS–LAs were designed to have longer SBS blocks (matrix) and shorter LA blocks (cylinder domain), the structure transition model in Fig. 6(b) is assumed. In this model, the volume of the SBS matrix is reduced after intramolecular crosslinking to decrease the dC–C while the LA cylinder diameter is maintained. Thus, we successfully reduced the feature size in both the LAM and HEX morphologies.
(Fig. 6(a), lower), representing an 11% decrease in the dC–C value compared with the corresponding linear SBS8k–LA5k. Since the HEX-forming SBS–LAs were designed to have longer SBS blocks (matrix) and shorter LA blocks (cylinder domain), the structure transition model in Fig. 6(b) is assumed. In this model, the volume of the SBS matrix is reduced after intramolecular crosslinking to decrease the dC–C while the LA cylinder diameter is maintained. Thus, we successfully reduced the feature size in both the LAM and HEX morphologies.
 and
and  annealed at 180 °C and (d) SBS27k–LA13k and SBS27k(cl)–LA13k annealed at 180 °C. (b) Schematic illustration of the structural transition in HEX morphology via the intramolecular cross-linking. SBS domains and LA domains are shown in blue and yellow, respectively.
annealed at 180 °C and (d) SBS27k–LA13k and SBS27k(cl)–LA13k annealed at 180 °C. (b) Schematic illustration of the structural transition in HEX morphology via the intramolecular cross-linking. SBS domains and LA domains are shown in blue and yellow, respectively. , SBS19k–LA10k (FSBS = 0.65), and the corresponding SBS(cl)–LAs also exhibited the HEX morphology as expected from their FSBS values. A similar result was obtained for the case of
, SBS19k–LA10k (FSBS = 0.65), and the corresponding SBS(cl)–LAs also exhibited the HEX morphology as expected from their FSBS values. A similar result was obtained for the case of  , and
, and  has a dC–C value 17% smaller (15.0 nm) compared to the linear
has a dC–C value 17% smaller (15.0 nm) compared to the linear  (18.1 nm, Fig. 6(c)), representing a larger decrease ratio than that for SBS8k(cl)–LA5k (11%, Fig. 6(a)) because of the higher crosslink density. In the case of the high-molecular-weight SBS27k–LA13k, a larger volume reduction of the SBS block is expected after the crosslinking reaction as indicated by the 〈G〉SBS values. Indeed, SBS27k(cl)–LA13k exhibited a 22% reduction in dC–C (22.9 nm) compared to that of the corresponding linear SBS27k–LA13k (29.4 nm, Fig. 6(d)).
(18.1 nm, Fig. 6(c)), representing a larger decrease ratio than that for SBS8k(cl)–LA5k (11%, Fig. 6(a)) because of the higher crosslink density. In the case of the high-molecular-weight SBS27k–LA13k, a larger volume reduction of the SBS block is expected after the crosslinking reaction as indicated by the 〈G〉SBS values. Indeed, SBS27k(cl)–LA13k exhibited a 22% reduction in dC–C (22.9 nm) compared to that of the corresponding linear SBS27k–LA13k (29.4 nm, Fig. 6(d)).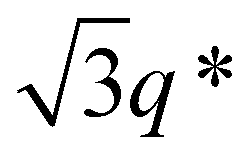 , 2q*, and
, 2q*, and 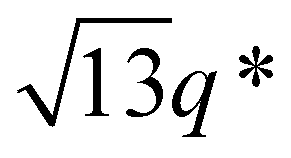 ) corresponding to the HEX morphology (Fig. 7). Such a change in phase was only observed in this BCP with the highest molecular weight, and it should also be attributed to a significant change in the effective volume fraction of the SBS block due to the intramolecular crosslinking. Therefore, the presented methodology can be applied for not only downsizing the feature but also varying the morphology in the microphase-separated structures.
) corresponding to the HEX morphology (Fig. 7). Such a change in phase was only observed in this BCP with the highest molecular weight, and it should also be attributed to a significant change in the effective volume fraction of the SBS block due to the intramolecular crosslinking. Therefore, the presented methodology can be applied for not only downsizing the feature but also varying the morphology in the microphase-separated structures.