
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-selective functionalization of Si6R6 siliconoids†
Yannic
Heider‡
,
Nadine E.
Poitiers‡
 ,
Philipp
Willmes
,
Kinga I.
Leszczyńska
,
Volker
Huch
and
David
Scheschkewitz
*
,
Philipp
Willmes
,
Kinga I.
Leszczyńska
,
Volker
Huch
and
David
Scheschkewitz
*
Krupp-Chair of General and Inorganic Chemistry, Saarland University, 66123 Saarbrücken, Germany. E-mail: scheschkewitz@mx.uni-saarland.de
First published on 14th March 2019
Abstract
The recent progress in the synthesis of partially substituted neutral silicon clusters (siliconoids) revealed unique structures and electronic anisotropies that are reminiscent of bulk and nano surfaces of silicon. Here, we report the selective 2-lithiation of the global minimum Si6R6 siliconoid at a different vertex than in the previously reported isomeric 4-lithiated derivative (R = 2,4,6-iPr3C6H2). In order to enable an intuitive distinction of the vertices of the global minimum Si6R6 scaffold (which can be considered the silicon analogue of benzene in terms of thermodynamic stability), we introduce a novel nomenclature in analogy to the ortho–meta–para nomenclature of disubstituted benzenes. By treatment of the 2-lithiated Si6 cluster with Me3SiCl, SiCl4 H3B·SMe2, (Me2N)2PCl as well as with carboxylic acid chlorides RCOCl (R = tBu, Ph) various 2-functionalized Si6 clusters were obtained and characterized in solution and – in most cases – the solid state. The structural and spectroscopic effect of the position of the newly introduced functional group is discussed by comparison to the corresponding 4-functionalized derivatives.
Introduction
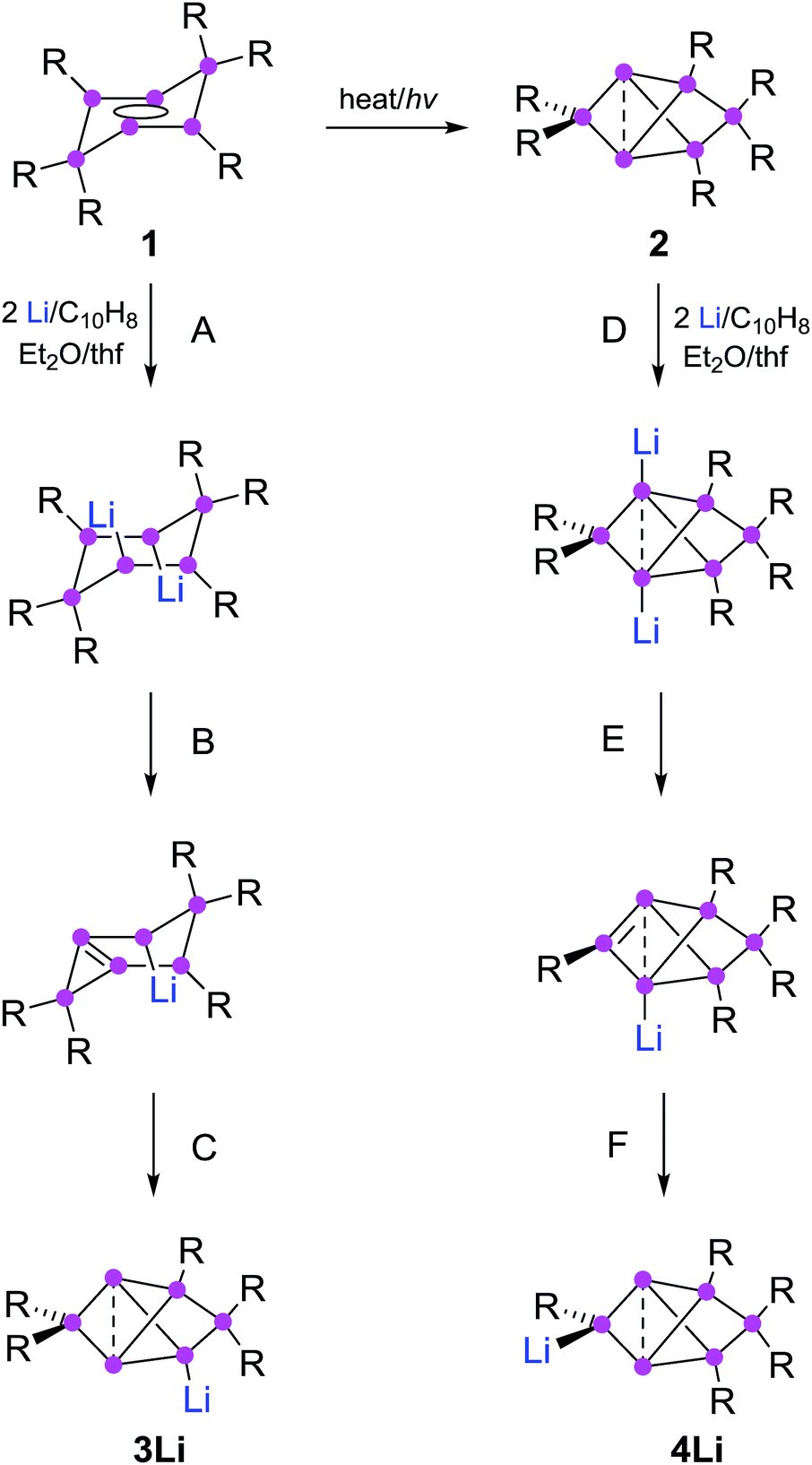
Partially substituted neutral silicon clusters (siliconoids)1–4 are fleeting intermediates during the production of silicon from molecular precursors and can typically only be detected in the gas phase.5–9 The synthesis of stable derivatives has attracted considerable interest as the unsubstituted vertices of siliconoids are reminiscent of the free valences at bulk and nano surfaces of silicon, the so-called “dangling bonds”.10–13 Since the report on the first stable siliconoid Si5R6 with one “naked” vertex in a hemispheroidal coordination environment by one of us,14 numerous examples have been prepared by the groups of Wiberg,15 Breher,16 Kyushin,17,18 Iwamoto,19 Fässler20 and ourselves.14,21–23 The Si6R6 isomers 1 (ref. 22) and 2 (ref. 23) are lower energy isomers of the hypothetical hexasilabenzene and as such prime examples of the often drastic differences between carbon and silicon (Scheme 1). While of the known C6H6 isomers, benzene is by far the lowest in energy, the tricyclic 2 corresponds to the global minimum isomer of Si6H6 and can therefore be considered as the silicon analogue of benzene on grounds of thermodynamic stability.23,24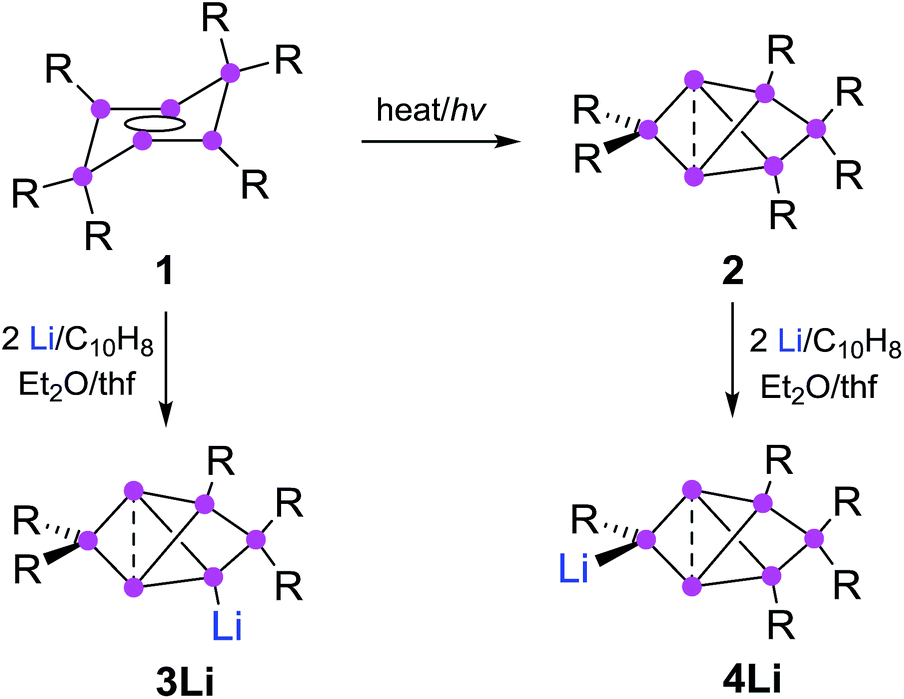 | ||
| Scheme 1 Synthesis of 3Li (ref. 32) and 4Li (this work) from dismutational hexasilabenzene isomer 1 and from the global minimum isomer 2 (Tip = 2,4,6-triisopropylphenyl). | ||
The functionalization of such clusters is a prerequisite for the further development of their chemistry and ultimately the application of their fascinating electronic properties in extended materials. Compared to the related Zintl anions of the heavier group 14 elements,25–31 which are (poly)anionic, completely unsubstituted deltahedral clusters, siliconoids are partially substituted yet exhibit a similarly wide dispersion of 29Si NMR shifts.14,16–18,22,24 Very recently, the protonation of silicon Zintl anions to partially H-substituted anionic siliconoids was reported independently by the groups of Fässler and Gschwind/Korber.30,31 We had previously described the reductive functionalization of the dismutational Si6R6 isomer 1 to the anionic siliconoid 3Li as well as its reactivity with several representative electrophiles of groups 13 to 15.32,33
Herein, we show that reduction of the global minimum isomer 2 under similar conditions selectively affords the regiomeric anionic Si6 siliconoid 4Li instead of 3Li by cleavage of an aryl substituent in the 2-position of the bridged propellane scaffold (Scheme 1). In order to account for the rapidly increasing number of species with the thermodynamically favored bridged-propellane scaffold and unequivocally distinguish between the different vertices, we propose a novel terminology for this structural motif, inspired by the well-established ortho–meta–para nomenclature for disubstituted benzenes.34–37 The functionalization of 3Li and 4Li with selected electrophiles is shown to result in several sets of regiomeric derivatives allowing for the systematic comparison of the structural and spectroscopic consequences of the functional group's position.
Results and discussion
Nomenclature of Si6 siliconoids
Structures with [1.1.1.]propellane motif have intrigued experimentalists and theorists alike ever since the early 1970s,38,39 because of their non-classical structure containing bridgehead atoms in an umbrella-type hemispheroidal coordination environment. The bonding situation between the bridgehead atoms of [1.1.1]propellanes can be described by biradical or ionic contributions to the electronic ground state40–43 and was discussed by Shaik et al. as a “charge-shift-bond”.44,45 The Si6 siliconoids 2 and 3Li show a closely related structure having two propeller blades bridged by one SiTip2 moiety. Strongly deshielded 29Si NMR signals had been explained by a cluster-like delocalization of the two electrons in question.23 Electron density determinations of the Si6 siliconoid 2 confirmed the absence of direct bonding between the bridgehead silicon vertices.46 For nomenclature purposes,47,48 the Si6 scaffold is nonetheless formally considered as tetracyclic system with a direct connection between the hemispheroidally coordinated vertices that are depicted in the schemes as a dashed line.The high thermodynamic stability of Si6 siliconoid 2 as the alleged global minimum of the Si6H6 potential energy surface23,24 suggests a considerable prevalence of this structural motif. This received first corroboration by the successful synthesis of mixed group 14 systems49 and a Sn6 derivative recently.50 Saturated variations of the six-atom scaffold occur in numerous other species throughout main group chemistry.51–54 In the past, the tetracyclic core structure has been variously referred to as “edge-capped trigonal bipyramid”55 “doubly edge-bridged tetrahedron”56 or “bridged propellane”.23 As these terminologies do not seem to do justice to the ubiquity of the structural motif, we propose a novel term that echoes the relationship to the iconic benzene molecule and – at the same time – takes into account the extraordinary polarization of 2 and related species.39,57–64 We thus suggest the term “benzpolarene”– in analogy to benzvalene – for the tetracyclic arrangement of vertices in the cluster core of 2 and 3Li. In addition, we feel that the availability of the first Si6 siliconoid regiomers described herein requires a descriptive nomenclature not unlike the well-established ortho, meta and para prefixes used for disubstituted benzenes. The prefixes thus proposed in the following reflect the characteristic bonding situation of each vertex (Chart 1).
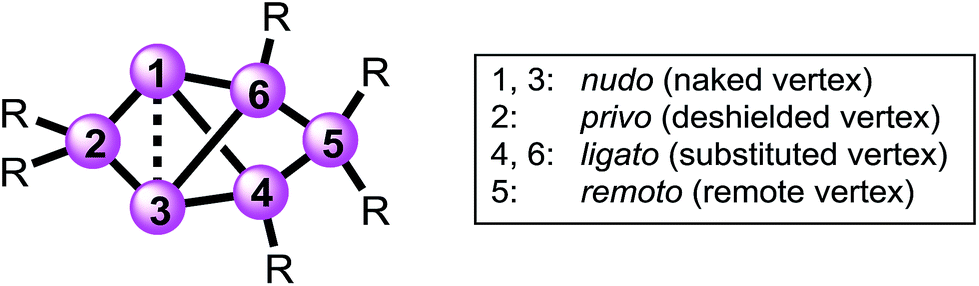 | ||
| Chart 1 Proposed prefixes for unique assignment of vertices in bridged propellane-type (“benzpolarene”) siliconoids. | ||
The latin words for “naked” (lat. nudus), “bonded” (lat. ligatus), “remote” (lat. remotus) and “deprived” (lat. privus) served as inspirations. The nudo prefix is assigned to the unsubstituted (“naked”) bridgehead silicon atoms in 1,3-position, the ligato prefix to the mono-substituted vertices (4,6-position) bonded to one substituent each, the remoto prefix to the remote bridge in 5-position and the privo prefix to the characteristically deshielded (“deprived” of electrons) atom in 2-position.
Functionalization in ligato position
In addition to the previously reported persilabenzpolarenes,32,33 we investigated two further reactions of siliconoid 3Li with electrophiles. The novel ligato functionalized siliconoids 5a,b were thus obtained by treatment of 3Li in benzene at room temperature with Me3SiCl and benzoyl chloride, respectively (Scheme 2). The reactions proceed quantitatively according to 29Si NMR spectroscopy. The siliconoids 5a,b were isolated as single crystals and fully characterized by X-ray analysis, NMR spectroscopy, UV/Vis (Table 1) and by IR spectroscopy in case of the CO containing species.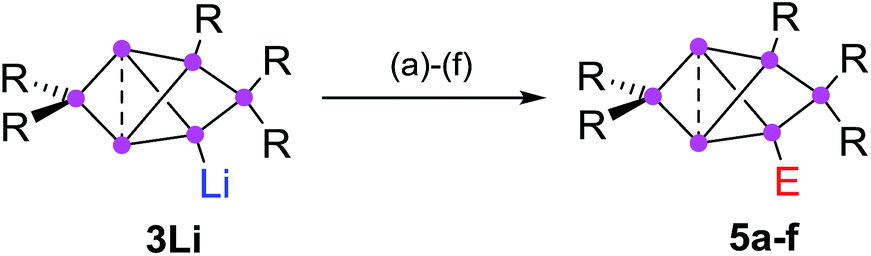 | ||
| Scheme 2 Synthesis of ligato-functionalized siliconoids 5a–f. Reagents: (a) Me3SiCl, (b) PhCOCl, (c) tBuCOCl, (d) ClP(NMe2)2, (e) SiCl4, (f) BH3·SMe2. 5a: E = Me3Si, 5b: E = COPh and the previously reported325c: E = COtBu, 5d: E = P(NMe2)2, 5e: E = SiCl3, 5f: E = BH3−. | ||
| Position of E | Comp. | Functional group (E) | Si1, Si3 [ppm] | Si2 [ppm] | Si4 [ppm] | λ max [nm] | ΔSi1–Si3 [Å] | ΔSi4–Si6 [Å] | σ m , 67 | Hemispheroidalitybϕ [Å] |
|---|---|---|---|---|---|---|---|---|---|---|
| a For substituents BH3− and Tip no Hammett parameters are available. The Hammett parameters of similar compounds were used for the correlation plots in Fig. 4: B(OH)3− for BH3− and (C6H5-4-CHMe2) for the Tip substituent (see ref. 68). b The hemispheroidality ϕ is the distance of a naked cluster vertex from the plane spanned by its three substituents. Its value is taken as a measure for the degree of hemispheroidality of the vertex. For a detailed explanation see ref. 4. | ||||||||||
| — | 2 | Tip23 | −274.2 | 174.6 | −7.5 | 473 | 2.7076(8) | 2.9037 | 0.08 | 1.3535 |
| ligato | 3Li | Li32 | −230.9, −232.6 | 152.2 | −66.8 | 364 | 2.5506(9) | 3.2243 | — | 1.2805 |
| privo | 4Li | Li | −222.2, −231.4 | 267.9 | −43.8 | 468 | 2.5562(10) | 2.9171(11) | — | 1.3078 |
| ligato | 5a | TMS | −257.8, −266.6 | 169.9 | −3.7 | 459 | 2.6176(5) | 2.9643(8) | −0.04 | 1.3283 |
| ligato | 5b | COPh | −263.0, −279.0 | 174.7 | −26.5 | 477 | 2.6598(9) | 2.8854(8) | 0.34 | 1.3333 |
| ligato | 5c | COtBu32 | −264.7, −271.1 | 171.8 | −27.4 | 475 | 2.6430(6) | 2.9095 | 0.27 | 1.3458 |
| ligato | 5d | P(NMe2)2 (ref. 32) | −256.0, −261.4 | 168.7 | −33.8 | 475 | 2.6231(5) | 2.9508 | 0.18 | 1.3498 |
| ligato | 5e | SiCl3 (ref. 32) | −252.3, −264.2 | 175.4 | 12.9 | 460 | 2.635(1) | 2.8920 | 0.48 | 1.3409 |
| ligato | 5f | BH3 (ref. 32) | −257.3, −265.0 | 161.2 | −4.8 | 475 | 2.620(1) | 2.9988 | −0.48 | 1.3153 |
| privo | 6a | TMS | −242.0, −253.3 | 193.6 | −15.9 | 469 | 2.6118(6) | 2.9482(6) | −0.04 | 1.3308 |
| privo | 6b | COPh | −268.5, −271.1 | 166.2 | −16.2 | — | — | — | 0.34 | — |
| privo | 6c | COtBu | −263.1, −265.8 | 173.1 | −14.5 | 473 | 2.6350(5) | 2.9641(7) | 0.27 | 1.3439 |
| privo | 6d | P(NMe2)2 | −246.0, −256.1 | 186.5 | −16.9 | — | — | — | 0.18 | — |
| privo | 6e | SiCl3 | −251.6, −258.9 | 161.7 | −6.4 | — | — | — | 0.48 | — |
| privo | 6f | BH3− | −243.3, −255.6 | 237.3 | −28.8 | 454 | 2.6024(8) | 2.9431(7) | −0.48 | 1.3155 |
The 29Si NMR spectra of 5a,b show the typical distribution of chemical shifts for ligato functionalized persilabenzpolarenes as recently reported by our group for 5c–f:32,33 the two unsubstituted bridgehead silicon atoms give rise to two 29Si signals in a range of −257 to −280 ppm (Table 1). The C2v symmetry of the benzpolarene scaffold of 2 is lowered to Cs in the substituted cases. As we had shown by VT NMR studies for some of the ligato-functionalized species,32 hindered rotation further reduces the symmetry so that the two seemingly identical nudo atoms become diastereotopic. The resonances of the SiTip2 groups in privo position are strongly deshielded with signals at 169.9 (5a) and 174.7 ppm (5b). The surprisingly downfield shifted signals (for tetracoordinate silicon atoms) had been rationalized by invoking magnetically induced cluster currents or – in a complementary manner – by the strong LUMO contribution at this atom.49,65,66 The 29Si NMR chemical shifts of the remaining cluster vertices are located in the typical range for saturated silicon atoms and vary only slightly with the introduced functionality. The longest wavelength absorption bands in the UV/Vis are observed at λmax = 459 nm (5a) and 477 nm (5b). The characteristic CO stretching mode in the IR of 5b at υ = 1605 cm−1 compares well with that of 5c.32,33 Single crystals were obtained in 45% (5a) and 66% (5b) yield and the structures confirmed by X-ray diffraction studies (Fig. 1).67 The distances between the unsubstituted bridgehead silicon atoms Si1 and Si3 are slightly shorter than in the global minimum isomer,23 in line with the observations for the previously reported ligato functionalized persilabenzpolarenes 3Li and 5c–f.32,33 The distance between the ligato positions Si4 and Si6 of 5a–f increases with decreasing distance between the nudo positions Si1 and Si3, presumably in order to minimize strain (Table 1). Apparently, the variation of the ligato functionality of the benzpolarene structures 5a–f directly influences the bonding between the unsubstituted silicon atoms Si1 and Si3.
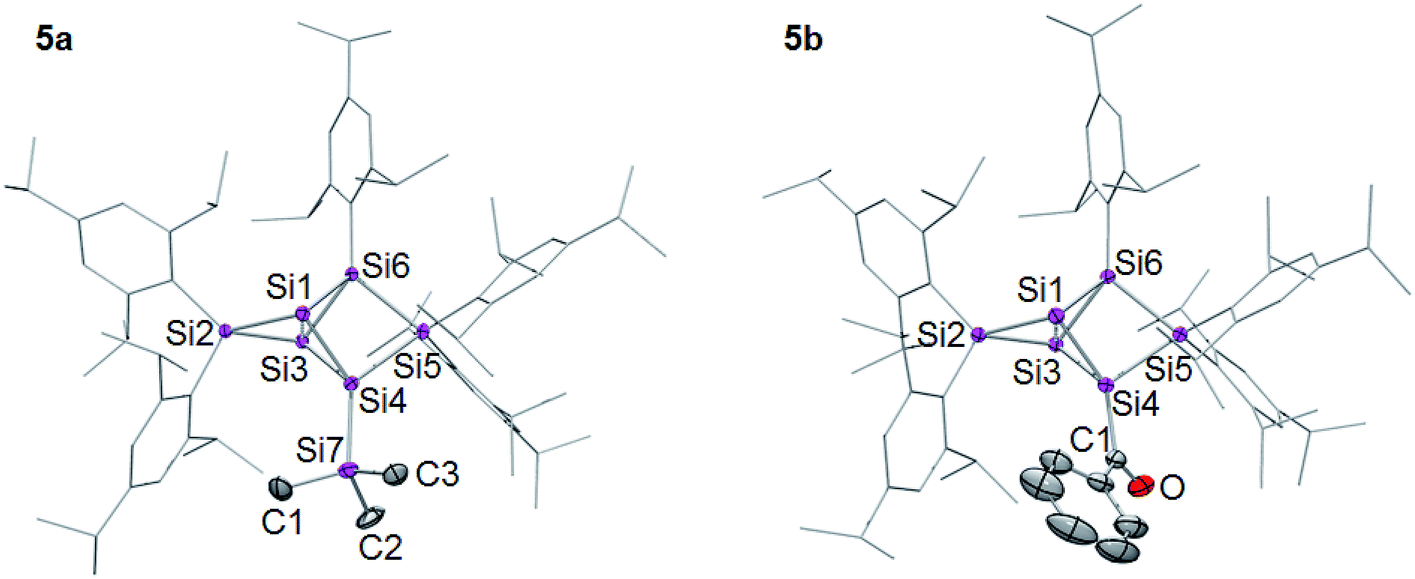 | ||
| Fig. 1 Structures of 5a,b in the solid state (thermal ellipsoids at 50% probability). Hydrogen atoms and co-crystallized pentane omitted. a: E = SiMe3, b: E = COPh. | ||
Synthesis of privo lithiated siliconoid 4Li
The reduction of the dismutational isomer of hexasilabenzene 1 had yielded the lithiated siliconoid with a benzpolarene scaffold 3Li and thus a functionalized derivative of the Si6H6 global minimum isomer 2.32,33 In order to probe the possible intermediacy of 2, its reduction with lithium/naphthalene was investigated (Scheme 3).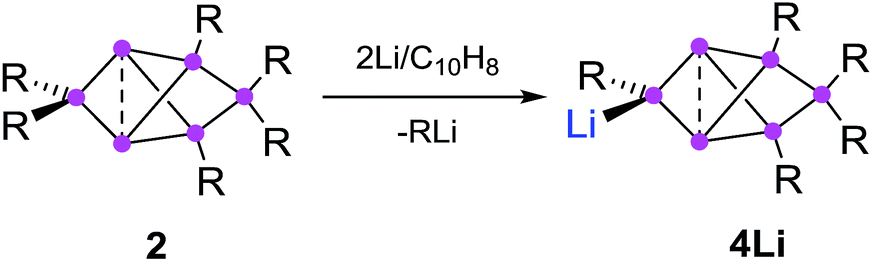 | ||
| Scheme 3 Synthesis of privo-lithiohexasilabenzpolarene 4Li by reduction with 2 equivalents of Li/C10H8. | ||
Treatment of 2 with 2.2 equivalents of Li/C10H8 in Et2O and thf indeed results in the complete and uniform conversion into a novel anionic Si6 species as confirmed by 29Si NMR spectroscopy. The six resonances show a similar chemical shift distribution as in 3Li, but with distinctly different values suggesting the functionalization had taken place in another position of the benzpolarene scaffold (Table 1). The reduction product 4Li was fully characterized by X-ray diffraction on single crystals, NMR spectroscopy and UV/Vis spectroscopy.
A strongly deshielded 29Si NMR signal at 267.9 ppm is significantly broadened (presumably due to coupling to the quadrupolar 7Li-nucleus) and only shows a cross-peak to the aromatic H atoms of a single Tip ligand in the 2D 1H/29Si correlation spectrum. These observations led us to conclude that the anionic functionality of 4Li is located at the tetracoordinate silicon atom in the privo position Si2. The 29Si chemical shift of 267.9 ppm is particularly remarkable as saturated silyl anions typically show resonances at much higher field often deep in the negative ppm region.69 According to our previous calculations,66 the magnetically induced cluster currents circumvent the privo position and thus cause its pronounced deshielding even in case of the peraryl-substituted benzpolarene 2. On the basis of a complementary explanation referring to the topology of the LUMO,49 this phenomenon is probably due to the pronounced silylene character of the privo atom. The presence of a directly attached electron-releasing substituent could lead to an increased localization of the vacant p orbital in the privo position and thus to the observed even more pronounced deshielding. The signals for the remoto SiTip2 and the two ligato SiTip units appear at δ = 15.3 and 100.2/−43.8 ppm, respectively. The reason for the large difference between the chemical shifts is unclear although the electronic environments of the ligato atoms are certainly dominated by their relative position to the anionic functionality and the lithium counter cation in privo position. The unsubstituted bridgehead silicon atoms in the nudo positions are apparently not compromised by the reduction and give rise to two signals at the usual high field at δ = −222.2 and −231.4 ppm, comparable to the corresponding signals of the ligato lithiated 3Li. The constitution of the reduction product of benzpolarene 2 was finally proven as the privo functionalized 4Li by X-ray diffraction on single crystals (Fig. 2).67
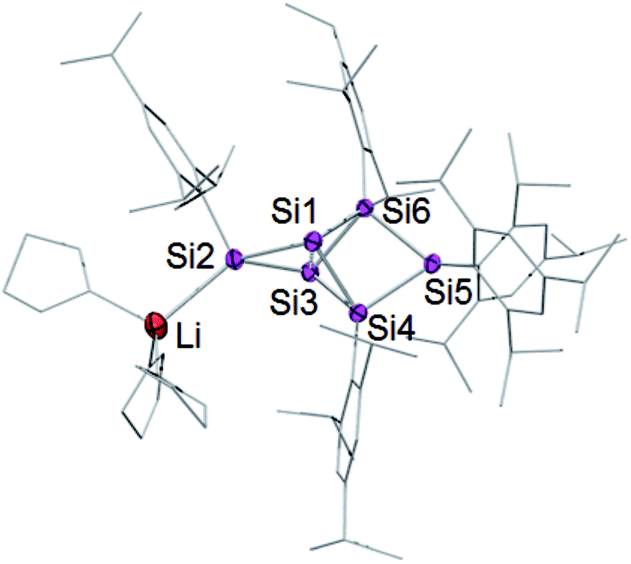 | ||
| Fig. 2 Structure of 4Li in the solid state (thermal ellipsoids at 50% probability). Hydrogen atoms and co-crystallized solvent molecules omitted. | ||
The distance between the bridgehead silicon atoms (Si1–Si3 2.5562(10) Å) is similar to that in 3Li, but shorter than in the fully Tip-substituted siliconoid 2 and the ligato functionalized siliconoids 5a–b. This shortening is tentatively attributed to delocalization of the lone-pair of the anionic silicon vertex into cluster bonding orbitals. The formation of the two regioisomeric derivatives is predominantly a consequence of the different topologies of the LUMOs of both the dismutational Si6R6 isomer 1 and the benzpolarene isomer 2. The initial reduction plausibly occurs at the unsubstituted vertices of the starting materials (A, D), which provide dominant contributions to the respective LUMOs.22,23 Other important LUMO contributions are located precisely at the silicon vertices to which the preferentially eliminated aryl groups are bonded. The subsequent isomerizations are likely driven by the very low energy of the benzpolarene scaffold (Scheme 4). The lithiated regioisomer 3Li is formed due to a syn TipLi elimination (B) followed by a cyclobutene–bicyclobutane rearrangement (C). In case of the reduction of the benzpolarene isomer 2, we suggest an orbital- and strain-controlled TipLi elimination followed by a 1,2-migration of the lithium counteraction (F) to yield 4Li.
 | ||
| Scheme 4 Mechanistic explanation of the regioselective formation of 3Li and 4Li. | ||
Functionalization in privo position
In order to evaluate the suitability of privo lithiated siliconoid 4Li as nucleophilic transfer reagent for the intact unsaturated Si6 scaffold, we treated it with several electrophiles (Me3SiCl, PhCOCl, tBuCOCl, ClP(NMe2)2, SiCl4, BH3·SMe2). Indeed, the corresponding privo substituted siliconoids 6a–f are obtained by straightforward combination of the reagents in toluene at room temperature (Scheme 5).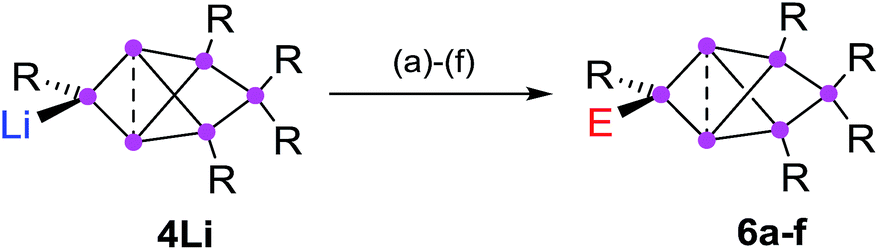 | ||
| Scheme 5 Synthesis of privo-functionalized siliconoids 6a–d. Reagents: (a) Me3SiCl, (b) PhCOCl, (c) tBuCOCl, (d) ClP(NMe2)2, (e) SiCl4, (f) BH3·SMe2. 6a: E = Me3Si, 6b: E = COPh, 6c: E = COtBu, 6d: E = P(NMe2)2, 6e: E = SiCl3, 6f: E = BH3−. | ||
According to 29Si NMR spectra, the reactions lead to full conversion of 4Li to the privo functionalized siliconoids 6a–f. Crystallization of 6a,c,f from concentrated hexane solutions affords single crystals in moderate to good yields (6a: 66%; 6c: 27%; 6f: 78%), which were fully characterized by multinuclear NMR spectroscopy, UV/Vis spectroscopy and X-ray diffraction (Fig. 3).67 In case of 6b,d,e, the reactions were only performed on the NMR scale and characterized by multinuclear NMR spectroscopy. The 29Si NMR data of the privo substituted species display a similarly wide dispersion in chemical shifts as the corresponding ligato isomers. The 29Si NMR spectra of 6a–f thus show two signals in the high-field region for the nudo positions and a strongly deshielded signal for the privo silicon atom, which carries one Tip substituent and the functional group E in this case.
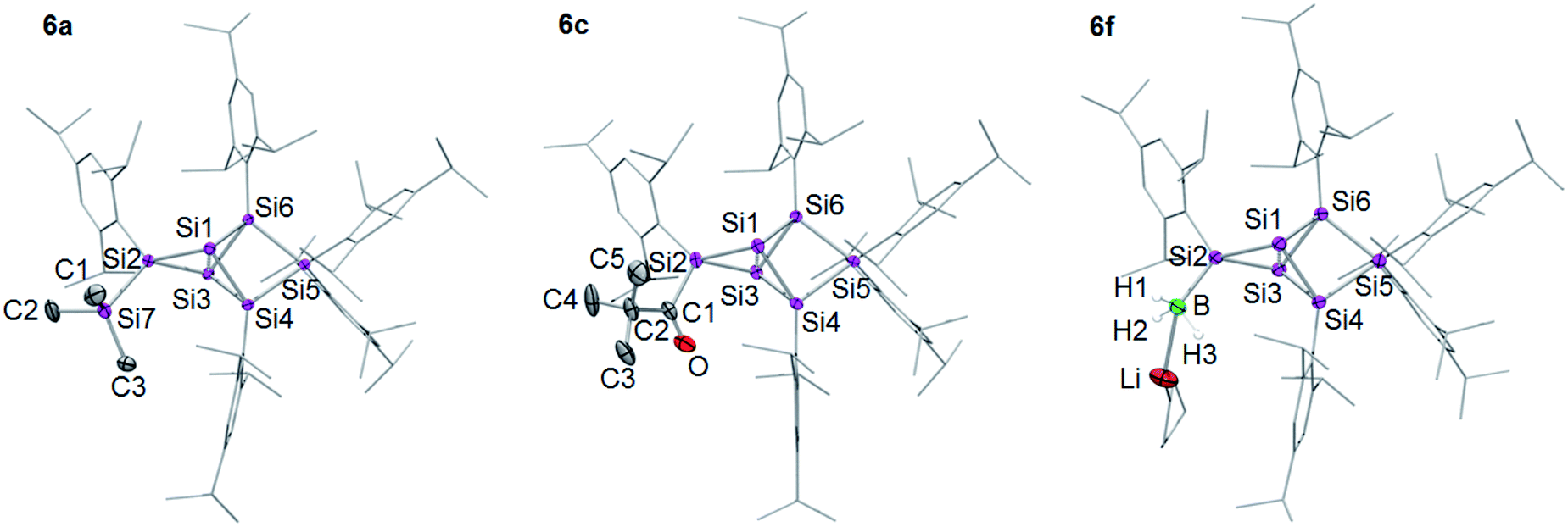 | ||
| Fig. 3 Structures of 6a, 6c and 6f in the solid state (thermal ellipsoids at 50% probability). Hydrogen atoms and co-crystallized solvent molecules omitted. a: E = TMS, c: E = COtBu, f: E = BH3−. | ||
The UV/Vis spectra of the isolated products 6a,c,f and 4Li show the position of the longest wavelength absorption maximum to strongly depend on the substituents of the Si6 scaffold (λmax; 6a 469 nm; 6c 473 nm; 6f 454 nm; 4Li 468 nm). As in case of the ligato functionalized species, it can presumably be assigned to the vertical HOMO–LUMO singlet excitation.
The distances between the bridgehead silicon atoms Si1–Si3 in the crystal structure of 6a,c,f (6a 2.6118(6), 6c 2.6350(5), 6f 2.6024(6) Å) are longer than in the ligato lithiated siliconoid 3Li32,33 and privo lithiated siliconoid 4Li, but slightly shorter than in the ligato functionalized siliconoids 5a,c,f. This is in line with a more effective σ donation in the privo position. While for siliconoids 5a–f, a reciprocal interdependency between the distances of Si1–Si3 and Si4–Si6 is observed, no such relationship is present in case of the privo functionalized siliconoids 6a–f.
The 29Si NMR resonances of the privo silicon atom are strongly influenced by the nature of the pending functionality. The signal is shifted to higher field with increasing electron-withdrawing power of the substituent: Li > BH3 > TMS > P(NMe2)2 > Tip > COtBu > COPh > SiCl3. This sequence correlates nicely with the Hammett parameter σm,70–73 which is based on the relative reaction kinetics of a second substitution in the meta position of benzene relative to the functionality in question.
Correlation with Hammett parameters σ
Fig. 4 shows the two correlations between the 29Si NMR chemical shift at the privo position of compounds 2, 5a–f and 6a–f and the Hammett parameter σm drawn separately for the two synthetically accessible positions of the functional group. The correlations with the σp Hammett parameter are similar, but slightly less satisfactory (see ESI†).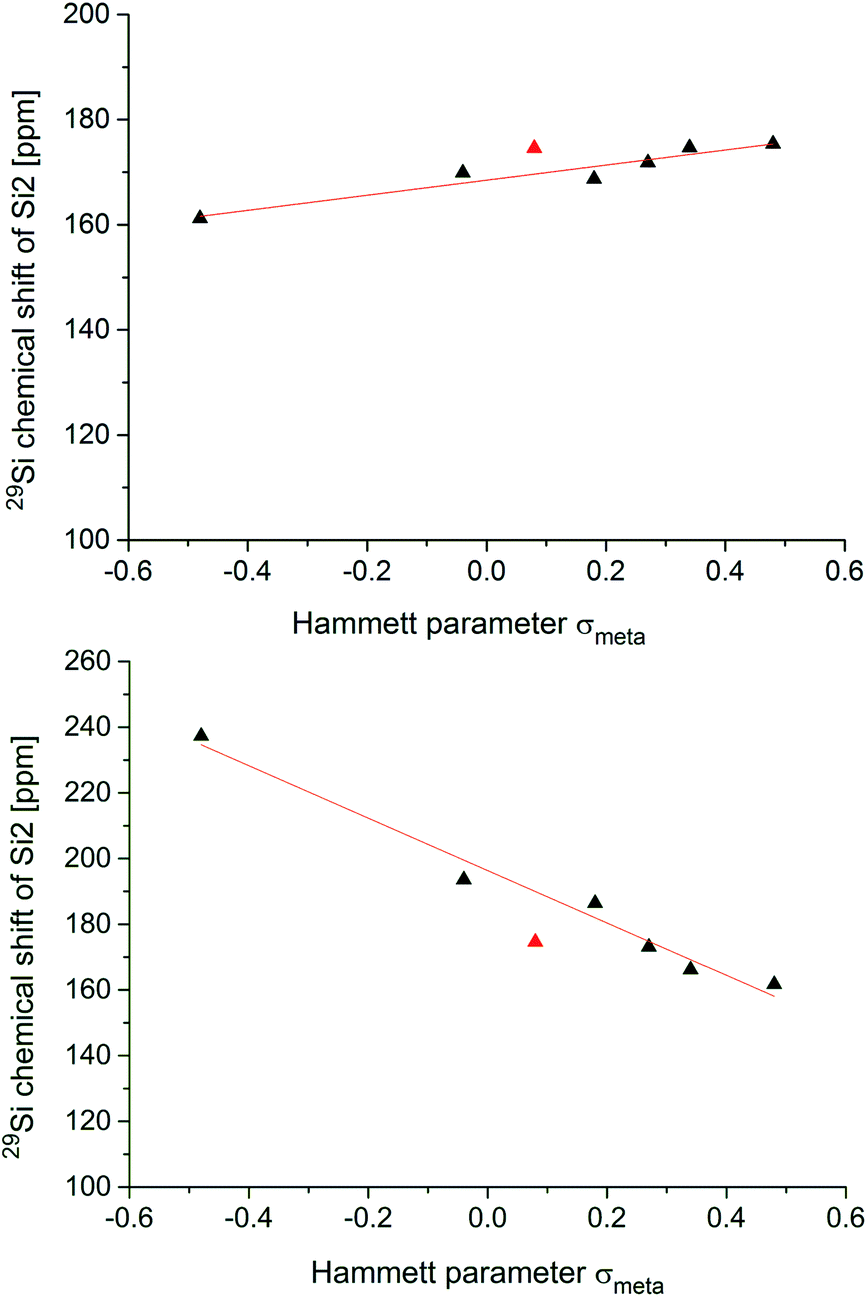 | ||
| Fig. 4 Plots of the Hammett parameters σmvs.29Si chemical shifts of the privo silicon vertices of ligato (5a–f; top) and privo functionalized hexasilabenzpolarenes (6a–f; bottom). The data pair marked with red triangles corresponds to the unfunctionalized hexaaryl derivative 2. | ||
The plot for the ligato functionalized compounds 5a–f (Fig. 3, top) shows a linear relationship (R2 = 0.912). The response of the 29Si chemical shift, however, is moderate as indicated in its range (160 to 180 ppm) and the resulting slope (m = 14.346 ppm). Electron-withdrawing substituents in ligato position result in a stronger deshielding of the privo atom in the 29Si NMR (5b,c,d,e) while electron-donating groups exert the opposite effect (5a,f). The σm value for the Tip substituent (red triangle in Fig. 4) had to be approximated by that of C6H5-4-CHMe2 (ref. 68) and was therefore disregarded for the linear fit. Surprisingly, there is no apparent correlation of the Hammett parameters with the 29Si chemical shifts of the nudo silicon atoms Si1 and Si3 (Table 1).
In case of the privo functionalized benzpolarenes (6a–f), the correlation of the Hammett parameters σm is even better with a very good linear dependency (R2 = 0.978). This is due to a markedly stronger response than in case of ligato functionalization with a slope of m = −79.76 ppm and a consequently larger chemical shift range (160 to 240 ppm). The stronger influence of the functional group is readily explained by its direct attachment to the silicon atom in question (Si2) vs. an additional distance of two Si–Si bonds in case of ligato functionalization. Remarkably, the slope of the linear fit is negative proving a reciprocal relationship between the electron-withdrawing strength of the substituent and its deshielding effect in the privo position. We had shown previously that the formal substitution of the nudo silicon atoms by germanium or tin results in a pronounced deshielding of the privo positions as well, which we tentatively rationalized by the strong influence of the LUMO shape on the paramagnetic contribution to the chemical shift.49
In contrast to our findings, in the case of mono-substituted carbon-based benzenes, the correlation of the Hammett parameter σp with the chemical shift of the para-carbon atom is known,74i.e. the ring atom opposite to the one carrying the functional group.
Conclusion
Si6 siliconoids 1 and 2 can be selectively reduced to yield derivatives of the global minimum isomer of the Si6H6 potential energy surface with an anionic functionality at distinct vertices. In order to distinguish between the different positions of the tricyclic Si6 scaffold, we propose a nomenclature that refers to the characteristic environment of the four conceivable symmetry-independent positions: nudo, privo, ligato and remoto. The anisotropic electronic structure of the global minimum Si6H6 scaffold is accounted for by the introduction of “benzpolarene” as unique name for this ever more frequently occurring structural motif. The privo lithiated hexasilabenzpolarene is accessible by reductive cleavage of one of the Tip groups of the perarylated derivative, while the ligato lithiated isomer had been obtained from the dismutational isomer previously. The privo derivative is shown to be an equally suitable nucleophilic reagent for the transfer of the uncompromised benzpolarene framework. The electronic influence of the functional groups in two distinct positions is rationalized on the basis of linear correlations with the Hammett parameter σm. With the possibility of functionalization in different positions of the Si6 scaffold the construction of larger systems comprising Si6 siliconoid motifs has become a viable option, which is currently being investigated in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding by the Deutsche Forschungsgemeinschaft (DFG SCHE906/4-1 and 4-2) and COST Action CM1302 (Smart Inorganic Polymers) is gratefully acknowledged.Notes and references
- F. Breher, Coord. Chem. Rev., 2007, 251, 1007 CrossRef CAS.
- T. Iwamoto and S. Ishida, Chem. Lett., 2014, 43, 164 CrossRef CAS.
- S. Kyushin in Organosilicon Compounds: Theory and Experiment (Synthesis), ed. V. Y. Lee, Academic Press, 2017, vol. 1, ch. 3 Search PubMed.
- Y. Heider and D. Scheschkewitz, Dalton Trans., 2018, 47, 7104 RSC.
- T. M. I. Davidson, J. Organomet. Chem., 1970, 24, 97 CrossRef.
- G. A. Rechtsteiner, O. Hampe and M. F. Jarrold, J. Phys. Chem. B, 2001, 105, 4188 CrossRef CAS.
- H. Murakami and T. Kanayama, Appl. Phys. Lett., 1995, 67, 2341 CrossRef CAS.
- W. M. M. Kessels, M. C. M. Van De Sanden and D. C. Schram, Appl. Phys. Lett., 1998, 72, 2397 CrossRef CAS.
- M. Watanabe, H. Murakami, T. Miyazaki and T. Kanayama, Appl. Phys. Lett., 1997, 71, 1207 CrossRef CAS.
- Y. Ge and J. D. Head, Int. J. Quantum Chem., 2003, 95, 617 CrossRef CAS.
- G. Hadjisavvas, G. Kopidakis and P. Kelires, Phys. Rev. B, 2001, 64, 125413 CrossRef.
- M. F. Jarrold, Science, 1991, 252, 1085 CrossRef CAS.
- H. Neergaard Waltenburg and J. T. Yates Jr, Chem. Rev., 1995, 95, 1589 CrossRef.
- D. Scheschkewitz, Angew. Chem., Int. Ed., 2005, 44, 2954 CrossRef CAS PubMed.
- G. Fischer, V. Huch, P. Payer, S. K. Vasisht, M. Veith and N. Wiberg, Angew. Chem., Int. Ed., 2005, 44, 7884 CrossRef CAS PubMed.
- D. Nied, R. Köppe, W. Klopper, H. Schnöckel and F. J. Breher, Am. Chem. Soc., 2010, 132, 10264 CrossRef CAS PubMed.
- A. Tsurusaki, C. Iizuka, K. Otsuka and S. Kyushin, J. Am. Chem. Soc., 2013, 135, 16340 CrossRef CAS PubMed.
- S. Ishida, K. Otsuka, Y. Toma and S. Kyushin, Angew. Chem., Int. Ed., 2013, 52, 2507 CrossRef CAS PubMed.
- T. Iwamoto, N. Akasaka and S. Ishida, Nat. Commun., 2014, 5, 5353 CrossRef CAS PubMed.
- L. J. Schiegerl, A. J. Karttunen, W. Klein and T. F. Fässler, Chem. Eur. J., 2018, 24, 19171 CrossRef CAS PubMed.
- K. Abersfelder, S. Russell, H. S. Rzepa, A. J. P White, P. R. Haycock and D. Scheschkewitz, J. Am. Chem. Soc., 2012, 134, 16008 CrossRef CAS PubMed.
- K. Abersfelder, A. J. P. White, H. S. Rzepa and D. Scheschkewitz, Science, 2010, 327, 564 CrossRef CAS PubMed.
- K. Abersfelder, A. J. P. White, R. J. F. Berger, H. S. Rzepa and D. Scheschkewitz, Angew. Chem., Int. Ed., 2011, 50, 7936 CrossRef CAS PubMed.
- M. Moteki, S. Maeda and K. Ohno, Organometallics, 2009, 28, 2218 CrossRef CAS.
- E. Zintl and A. Z. Harder, Z. Phys. Chem. Abt. A, 1931, 154, 47 CAS.
- F. T. Fässler, Struct. Bond., 2011, 140, 91 CrossRef.
- S. Scharfe, F. Kraus, S. Stegmaier, S. Schier and T. F. Fässler, Angew. Chem., Int. Ed., 2011, 50, 3630 CrossRef CAS PubMed.
- J. M. Goicoechea and S. C. Sevov, J. Am. Chem. Soc., 2004, 126, 6860 CrossRef CAS PubMed.
- M. Waibel and T. F. Fässler, Inorg. Chem., 2013, 52, 5861 CrossRef CAS PubMed.
- T. Henneberger, W. Klein and T. F. Fässler, Z. Anorg. Allg. Chem., 2018, 644, 1018 CrossRef CAS.
- C. Lorenz, F. Hastreiter, K. Hioe, N. Lokesh, S. Gärtner, N. Korber and R. M. Gschwind, Angew. Chem., Int. Ed., 2018, 57, 12956 CrossRef CAS PubMed.
- P. Willmes, K. Leszczyńska, Y. Heider, K. Abersfelder, M. Zimmer, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2016, 55, 2907 CrossRef CAS PubMed.
- K. I. Leszczyńska, V. Huch, C. Präsang, J. Schwabedissen, R. J. F. Berger and D. Scheschkewitz, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201811331.
- K. Lonsdale, Nature, 1928, 122, 810 CrossRef CAS.
- E. D. Glending, R. Faust, A. Streitwieser, K. Peter, C. Vollhardt and F. Weinhold, J. Am. Chem. Soc., 1993, 115(23), 10952 CrossRef.
- E. G. Cox, Rev. Mod. Phys., 1958, 30(1), 159–162 CrossRef CAS.
- T. C. Dinadayalane, U. D. Prixakumar and G. N. J. Sastry, J. Phys. Chem. A, 2004, 108, 11433 CrossRef CAS.
- M. D. Newton and J. M. Schulman, J. Am. Chem. Soc., 1972, 93, 773 CrossRef.
- A. M. Dilmac, E. Spuling, A. de Meijere and S. Bräse, Angew. Chem., Int. Ed., 2017, 56, 5684 CrossRef CAS PubMed.
- F. Breher, Coord. Chem. Rev., 2007, 251, 1007 CrossRef CAS.
- P. P. Power, Chem. Rev., 2003, 103, 789 CrossRef CAS PubMed.
- H. Grützmacher and F. Breher, Angew. Chem., Int. Ed., 2002, 41, 4006 CrossRef.
- D. Nied and F. Breher, Chem. Soc. Rev., 2011, 40, 3455 RSC.
- S. Shaik, P. Maitre, G. Sini and C. P. Hiberty, J. Am. Chem. Soc., 1992, 114, 7861 CrossRef CAS.
- W. Wu, J. Gu, J. Song, S. Shaik and P. C. Hiberty, Angew. Chem., Int. Ed., 2009, 48, 1407 CrossRef CAS PubMed.
- D. Kratzert, D. Leusser, J. J. Holstein, B. Dittrich, K. Abersfelder and D. S. D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 4478 CrossRef CAS PubMed.
- IUPAC Nomenclature of Organic Chemistry 1957, J. Am. Chem. Soc., 1960, 82, 5545 CrossRef.
- International Union of Pure and Applied Chemistry, Nomenclature of Organic Chemistry, sections A, B, C, D, E, F, H, Pergamon Press, 1979 edn, 1979 Search PubMed.
- A. Jana, V. Huch, M. Repisky, R. J. F. Berger and D. Scheschkewitz, Angew. Chem., Int. Ed., 2014, 53, 3514 CrossRef CAS PubMed.
- C. P. Sindlinger and L. Wesemann, Chem. Sci., 2014, 5, 2739 RSC.
- M. Veith, Angew. Chem., Int. Ed., 1987, 26, 1 CrossRef.
- Y. Xiong, S. Yao, M. Brym and M. Driess, Angew. Chem., 2017, 119, 4595 CrossRef.
- Y. Peng, H. Fan, H. Zhu, H. W. Roesky, J. Magull and C. E. Hughes, Angew. Chem., Int. Ed., 2004, 43, 3443 CrossRef CAS PubMed.
- T. Iwamoto, K. Uchiyama, K. Chizuko and M. Kira, Chem. Lett., 2007, 36, 368 CrossRef CAS.
- K. Raghavachari and V. Logovinsky, Phys. Rev. Lett., 1985, 55, 2853 CrossRef CAS PubMed.
- D. Nieder, C. B. Yildiz, A. Jana, M. Zimmer, V. Huch and D. Scheschkewitz, Chem. Commun., 2016, 52, 2799 RSC.
- L. R. Sita and R. D. Bickerstaff, J. Am. Chem. Soc., 1989, 11, 6454 CrossRef.
- D. Nied, W. Klopper and F. Breher, Angew. Chem., 2009, 121, 1439 CrossRef.
- L. R. Sita and I. Kinoshita, J. Am. Chem. Soc., 1990, 112, 8839 CrossRef CAS.
- L. R. Sita and I. Kinoshita, J. Am. Chem. Soc., 1991, 113, 5070 CrossRef CAS.
- L. R. Sita and I. Kinoshita, J. Am. Chem. Soc., 1992, 114, 7024 CrossRef CAS.
- A. F. Richards, M. Brynda and P. P. Power, Organometallics, 2004, 23, 4009 CrossRef CAS.
- A. F. Richards, M. Brynda, M. M. Olmstead and P. P. Power, Organometallics, 2004, 23, 2841 CrossRef CAS.
- C. Drost, M. Hildebrand and P. Lönnecke, Main Group Met. Chem., 2002, 25, 93 CAS.
- J. Juselius, D. Sundholm and J. Gauss, Chem. Phys., 2004, 121, 3952 CAS.
- R. J. F. Berger, H. S. Rzepa and D. Scheschkewitz, Angew. Chem., Int. Ed., 2010, 49, 10006 CrossRef CAS PubMed.
- Details of crystal structure analyses are available in the ESI. CCDC 1877380 (5a), 1877381 (5b), 1877378 (4Li), 1877379 (6a), 1877382 (6c) and 1877383 (6f) contain the supplementary crystallographic data for this paper.†.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- H. W. Lerner, Coord. Chem. Rev., 2005, 249, 781 CrossRef CAS.
- L. P. Hammett, Trans. Faraday Soc., 1938, 34, 156 RSC.
- L. P. Hammett, Physical Organic Chemistry, McGraw Hill, 1940, p. 184 Search PubMed.
- H. H. Jaffe, Chem. Rev., 1953, 53, 191 CrossRef CAS.
- L. P. Hammett, Physical Organic Chemistry, 2nd edn, McGraw-Hill, New York, 1970 Search PubMed.
- H. Spiesecke and W. G. Schneider, J. Chem. Phys., 1961, 35, 731 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: CCDC 1877380 (5a), 1877381 (5b), 1877378 (4Li), 1877379 (6a), 1877382 (6c), 1877383 (6f). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05591b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |