
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-free dual labeling of DNA by triazines and cyclopropenes as minimal orthogonal and bioorthogonal functions†
Ulrike
Reisacher
a,
Damian
Ploschik
a,
Franziska
Rönicke
a,
Gergely B.
Cserép
b,
Péter
Kele
 b and
Hans-Achim
Wagenknecht
*a
b and
Hans-Achim
Wagenknecht
*a
aInstitute of Organic Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany. E-mail: Wagenknecht@kit.edu
bChemical Biology Research Group, Institute of Organic Chemistry, Research Centre for Natural Sciences, Hungarian Academy of Sciences, Magyar tudósok krt. 2, H-1117 Budapest, Hungary
First published on 4th March 2019
Abstract
Two different and small functions for inverse electron demand Diels–Alder reactions were applied for dual labeling of DNA: the 1,2,4-triazine was attached to the 5-position of 2′-deoxyuridine triphosphate, and the 1-methylcyclopropene to the 7-position of 7-deaza-2′-deoxyadenosine triphosphate. These two modified nucleotides were sequence-selectively incorporated into oligonucleotides by DNA polymerases. These products were labeled by two different fluorescent dyes using postsynthetic reactions that are not only bioorthogonal in general, but also mutually orthogonal.
Introduction
Bioorthogonal modification strategies are important ways to label biomolecules with fluorescent markers and probes.1–9 The broadly applied Cu(I)-catalyzed azide–alkyne cycloaddition has a broad substrate scope, gives high yields,10,11 and works even in live cells,12,13 but is not really bioorthogonal due to the cytotoxicity of Cu(I) salts.14 Among the variety of currently explored bioorthogonal reactions, the “photoclick” reaction between diaryltetrazoles as 1,3-dipole precursors and olefins as dipolarophiles,15,16 and the inverse electron demand Diels–Alder reaction (iEDDA) between 1,2,4,5-tetrazines as electron-poor dienes and strained olefins as dienophiles are the most potent ones,17,18 because they occur with second order rate constants that are comparable or even higher than those of the Cu(I)-catalyzed cycloadditions. These reactions were established for protein labeling, but cannot simply be transferred to nucleic acid chemistry.19–21 In particular 1,2,4,5-tetrazines as bioorthogonal reactive groups for iEDDA chemistry in DNA are very difficult to handle due to their fast reactivity that goes along with hydrolytic lability.17,18 We experienced this lability when we firstly incorporated the 1,2,4,5-tetrazine as bioorthogonal reactive group into DNA,19,22 and postsynthetic modifications did not exceed 40% yield. Prescher et al. identified 1,2,4-triazines as alternative because they are remarkably stable in aqueous buffers and even in the presence of biological nucleophiles inside cells.23 We developed a postsynthetic modification strategy for DNA based on 1,2,4-triazines and increased the labeling yields to 80% with highly reactive bicyclo[6.1.0]nonynes as counterparts.24 On the other side, reactive functions for iEDDA chemistry in DNA and RNA are strained alkenes as dienophiles, which were realized by norbornene,25trans-cyclooctene26–29 and bicyclo[6.1.0]nonyne.30 Expectedly, they react very fast in iEDDA-type reactions but their size may interfere with the biological function. In contrast, simple vinyl groups are the smallest possible dienophiles in DNA, but their rate constants are low due to the missing activation by ring strain.31,32 We established 1-methylcyclopropenes as dienophiles and modifications of two nucleotides (dU and dA) in DNA19,33 as the best possible compromise between small size and fast iEEDA reactivity.34 Kath-Schorr et al. published a Romesberg-type35 artificial base pair in RNA modified with 1-methylcyclopropenes for iEDDA reactions.36Dual bioorthogonality is particularly challenging but desired for dual molecular imaging using energy transfer systems. In principle, it may be achieved by combining an iEDDA reaction with a strain-promoted azide–alkyne cycloaddition. Cyclooctynes, however, are prone to react both by iEDDA and strain-promoted azide–alkyne cycloaddition, the latter being the slower one, which can be solved for proteins by firstly adding the azide and secondly the tetrazine label.37 The combination of methylcyclopropene as iEDDA dienophile and dibenzocyclooctyne as the dipolarophile for the azide–alkyne cycloaddition finally yielded a copper-free, mutually orthogonal strategy for proteins.38 Similar dual labeling methods were developed for proteins,39 also for carbohydrates,40 but only rarely for nucleic acids.41,42 So far, the applied combinations of reactions for nucleic acids include the Cu(I)-catalyzed cycloaddition, as described above, or sequentially by different ethynyl protecting groups,43 or, for instance, with thiol-ene44,45 or hydrazine46 bioconjugation chemistries.
Herein, we show that two bioorthogonally reactive groups, 1-methylcyclopropenes and 1,2,4-triazines can be combined for dual and copper-free labeling of DNA. The 1,2,4-triazine was attached to the 5-position of 2′-deoxyuridine triphosphate 1, and the 1-methylcyclopropene to the 7-position of 7-deaza-2′-deoxyadenosine triphosphate 2.33 This allows sequence selective incorporation of two differently modified nucleotides by DNA polymerases. The primer extension product can subsequently be labeled by two iEDDA reactions which are not only bioorthogonal in general, but also orthogonal to each other.
Results and discussion
The synthesis of the new triphosphate 1 is described in the ESI (see Fig. S1,† descriptions on pages 4–9, and Fig. S6–S11†). The synthesis of 2 and its application in primer extension and single postsynthetic labeling are published.33 The acceptance of 1 for enzymatic incorporation was first tested by using the fluorescein-marked primer P1 and three different commercially available DNA polymerases (Vent (exo-), Deep Vent (exo-) and Hemo KlenTaq). T1 was used as template for standing start experiments, in which 1 is the first nucleotide to be incorporated in order to show its fidelity (Fig. 1 and S2†). The triazine-modified nucleotide 1 was recognized as an analog for TTP and incorporated by all tested DNA polymerases at 37 °C. When 1 was mixed with the other unmodified dNTPs (dCTP, dGTP, dATP, excluding TTP) P1 was extended to the full length product complementary to T1. Successful incorporation was also observed in running start experiments with template T2, in which 1 gets incorporated during the elongation process. In both cases (with T1 and T2) the full length products show a slightly slower gel mobility compared to the full length products that were yielded with exclusively unmodified dNTPs (dCTP, dGTP, dATP, and TTP) which is due to the additional triazine moiety. It is important to mention here, that we found a better acceptance of the modified triphosphate 1 by all tested DNA polymerases compared to our previously reported triazine-modified nucleotide,47 even at physiological temperature (37 °C) which is outside the respective temperature optima. Presumably, this could be due to the smaller size of the triazine label of 1.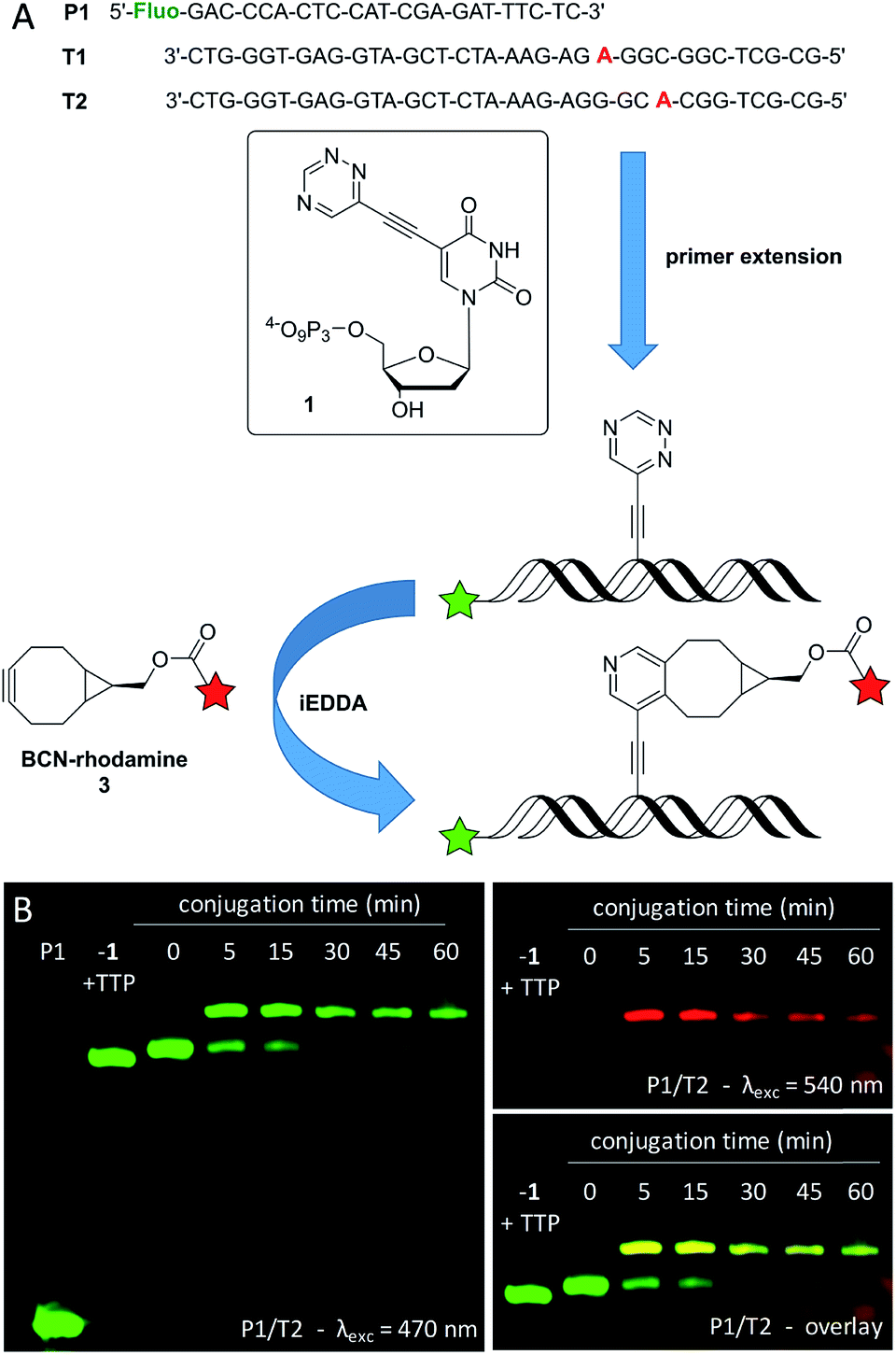 | ||
| Fig. 1 (A) Schematic illustration of the enzymatic incorporation of 1 into oligonucleotides by extension of primer P1 with templates T1 (standing start) and T2 (running start), and the postsynthetic iEDDA labeling with BCN-rhodamine 3. (B) PAGE analysis of P1/T2 elongated with 1, dATP, dCTP and dGTP using Vent (exo-) polymerase at 37 °C for 30 min and labeling of the extension product with 3. | ||
For further investigations of the subsequent postsynthetic labeling the Vent (exo-) polymerase was chosen for the elongation of P1/T2, whereby an elongation time of 30 min at 37 °C was sufficient in order to yield the full length product. The fully elongated oligonucleotide was desalinated, lyophilized, resolved and then labeled in the presence of excess (1000 equiv.) of the BCN-modified rhodamine dye 3. The presence of a new band with a significantly slower gel mobility indicates the successful conjugation between the triazine moiety at the oligonucleotide and dye 3. This conjugate is evidenced by the additional red fluorescence that is specific for the rhodamine dye. Within 5 min, a labeling yield of approximately 70% was obtained, which could be further increased to a maximum of about 80% after 45 min. The labeled primer extension product was additionally verified by the reaction of 1 with BCN-rhodamine 3 (for MALDI-TOF MS of this conjugate see Fig. S12†) and subsequent primer extension with this conjugate triphosphate (similar to literature48) yielding an identical gel shift (see Fig. S5†).
To demonstrate that both modified nucleotides 1 and 2 that carry the two different reactive groups can be incorporated into one DNA strand in a sequence-specific manner, we performed primer extension with T2 using a dNTP mixture of 1, 2 and the unmodified dCTP and dGTP (Fig. 2A). It is important to note here that the sequence of T2 was designed such that there is only a single incorporation site for each of the both modified nucleotides. Since the Vent (exo-) polymerase was efficient for the incorporation of the single modifications it was used for the dual incorporation, too. As it was already known from previous experiments that a desalination step between the enzymatic synthesis and the labeling reaction results in better yields, it was applied for the following experiments, too. The PAGE analysis (Fig. 2B, left column) after primer extension shows only the fully extended 35 nt products before the dye was added. The comparison with the unmodified primer extension product which was obtained with all four unmodified dNTPs shows again a slightly different gel mobility indicating the successful incorporation of the two reactive groups as oligonucleotide modifications. The successful single and dual postsynthetic labeling of the 1-methylcyclopropene and the 1,2,4-triazine functions as reactive groups in the primer extension products was evidenced by careful PAGE analysis. If the primer extension product was only subject to single labeling with either the BCN-rhodamine dye 3 or the tetrazine-TAMRA dye 4, new gel bands appeared with slower mobility in the green fluorescence channel of the 5′-fluorescein label and the additional rhodamine fluorescence in the red channel. The red-labeled primer extension product was additionally verified by the reaction of 2 with tetrazine-TAMRA 4 (for MALDI-TOF MS of this conjugate see Fig. S13†) and subsequent primer extension with this conjugate triphosphate yielding an identical gel shift (see Fig. S5†). An additional gel shift is observed when the primer extension product was labeled with both reagents, 3 and 4. It is important to note that the order in which the dyes were added to the modified oligonucleotide played an important role. When the conjugation of the cyclopropene moiety with the tetrazine-modified dye 4 was performed first, the BCN-modified dye 3 obviously formed an additional undesired side product according to PAGE analysis (see Fig. S3†) yielding three different labeling products in total. We propose a Diels–Alder reaction between the pyrazoline (product between 1-methylcyclopropene and tetrazine functions) and the BCN function of 3 to a “double-click” product (see Fig. S3†). This side product was additionally verified on the nucleotide triphosphate level by the reaction of 2 with both BCN-rhodamine 3 and tetrazine-TAMRA 4 (for MALDI-TOF MS of this conjugate see Fig. S14†). However, primer extension with this doubly labeled nucleotide triphosphate was not successfully probably due to high steric demand. To avoid this side reaction, the labeling reaction between the triazine-modified oligonucleotides and BCN-dye 3 was firstly carried out and the excess of the dye was removed by an additional desalting step prior to the second labeling by tetrazine-dye 4.
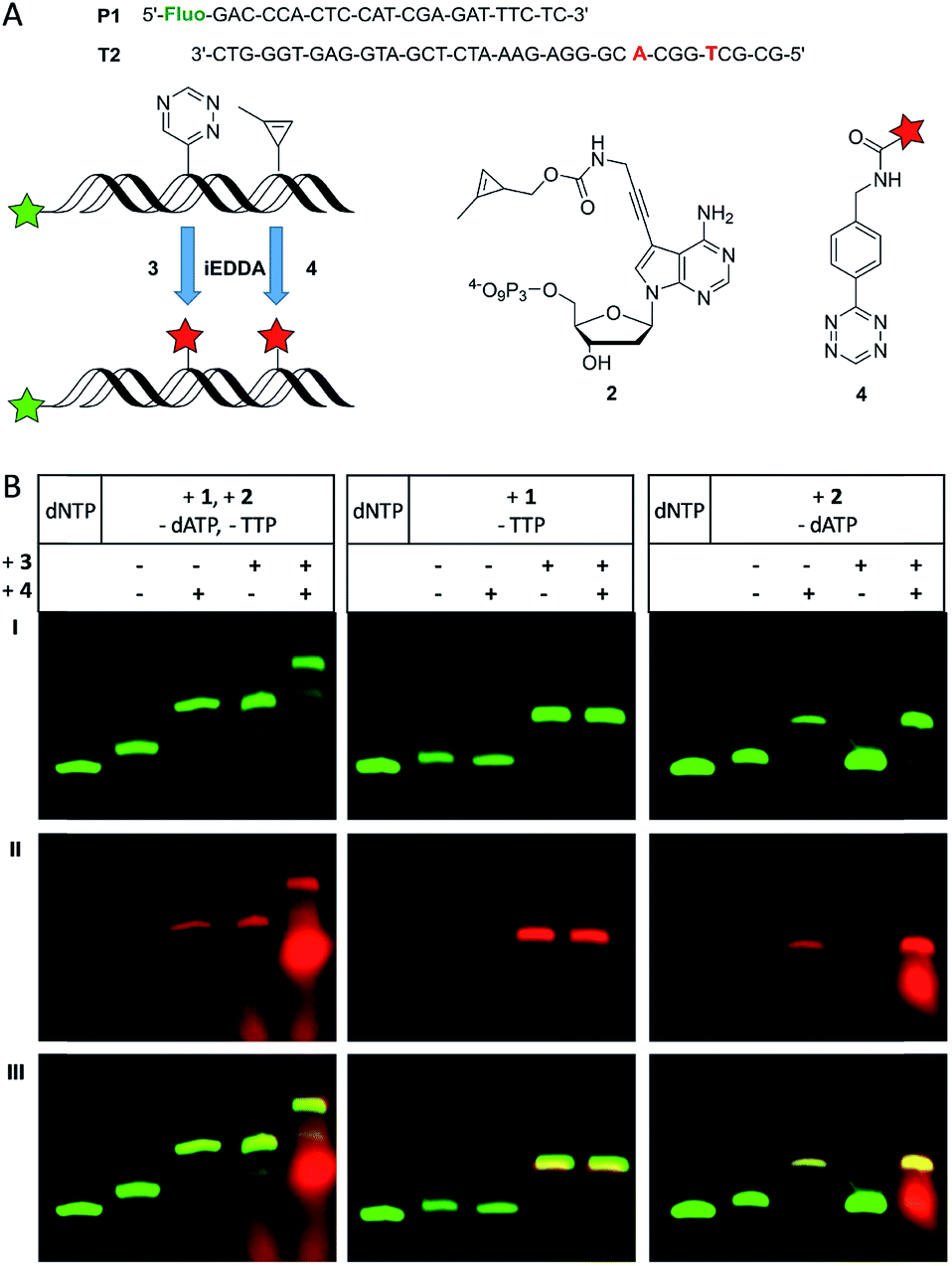 | ||
| Fig. 2 (A) Schematic overview over the dual labeling of DNA using P1/T2 and 1/2, and subsequent bioorthogonal conjugation with the respective dyes 3 and 4. (B) PAGE analysis of labeling reactions using elongated P1 (Vent (exo-), 37 °C, 30 min, yielded by different triphosphate mixtures including 1 and/or 2 in combination with the other unmodified dNTPs) and the fluorescent dyes 3 (30 min incubation) and 4 (15 min incubation). The lanes “dNTP” refer to the fully extended P1 using only unmodified triphosphates and serve as references. Row (I) fluorescein channel: λexc = 470 nm, λem = 535 nm; row (II) rhodamine/TAMRA channel: λexc = 540 nm, λem = 605 nm; row (III) overlay of (I) and (II). | ||
To show that the described handling does not affect the efficiency of each labeling step nor does it interfere with the specificity of the given bioorthogonal reactions control experiments were done using elongation products of P1/T2 with only one of the modified triphosphates. When only 1 was incorporated (Fig. 2B, middle column) an additional gel band compared to the full-length product is visible both in the green and red channel upon reaction with 3 indicating a successful bioconjugation as expected. However, no additional product was found after subsequent incubation with 4, since the combination of a tetrazine with a triazine moiety is not reactive. When both dyes, 3 and 4, were present, there is still only one labeling product visible on the gel. These results show that the additional tetrazine-dye 4 does not interfere with the bioconjugation between the triazine and the BCN-dye 3. A similar result was obtained in control experiments when only 2 was incorporated into oligonucleotides (Fig. 2B, right column), and efficient single labeling was obtained with tetrazine-dye 4. Addition of BCN-dye 3 did not alter this reactivity. These results support the mutual orthogonality of the 1-methylcyclopropene and triazine functions in bioorthogonal modification chemistry.
However, since rhodamine and TAMRA have very similar fluorescence properties it is not possible to differentiate between those two labels by the red fluorescence channel of PAGE analysis. To overcome this problem, the tetrazine-modified BODIPY 5 was used as reaction partner for the cyclopropene moiety. However, the BODIPY dye as bioorthogonal marker has again very similar fluorescence properties as the 5′-fluorescein label in the green fluorescence channel. Therefore, primer P2 carrying a Cy5 label was applied (Fig. 3A) that emits in the far red channel. With those three differently emitting dyes dual labeling experiments were performed, similar to those described above. The PAGE analysis (Fig. 3B) shows the fully extended P2 by only unmodified dNTPs in the far red fluorescent channel of the Cy5 label, imaged in blue color to differentiate from the red rhodamine channel. Again, a shift in the gel mobility of the full length product is visible when both modifications (1 and 2) were incorporated. When the primer extension product was incubated with either 3 or 5 separately, only one new gel band with slower mobility is visible showing the respective labeling products: the conjugate with BCN-rhodamine 3 is evidenced by the additional rhodamine fluorescence in the red channel, whereas the product with tetrazine-BODIPY 5 is visible by the additional BODIPY fluorescence in the green channel. The latter green-labeled primer extension product was additionally verified by the reaction of 2 with tetrazine-BODIPY 5 (for MALDI-TOF MS of this conjugate see Fig. S15†) and subsequent primer extension with this conjugate triphosphate yielding an identical gel shift (see Fig. S5†). Orthogonal labeling experiments with both dyes were carried out as described above. Thereby, two additional bands with slower gel mobility appeared in the far red channel (imaged in blue color), whereby the upper band shows a higher intensity. The lower band shows a fluorescence in the red and green channel, indicating a mixture of singly labeled primer extension products by either 3 or 5. The upper band shows a clear fluorescence in the red channel indicating the presence of the rhodamine dye, but no fluorescence in the green channel. With respect to the clear evidence from the previous primer extension experiments with the two different red-emitting dyes 3 and 4 by the same excitation wavelength (presented in Fig. 2) we assume an energy transfer between the dyes of 3 and 5. In fact, when the gel is excited at 470 nm within the selective absorption range of BODIPY and emission is recorded using an emission filter at 605 nm (outside the emission range of BODIPY but inside the emission range of rhodamine) a fluorescence signal matching the upper band is visible. As a result of this control excitation, this band can be assigned to the doubly labeled primer extension product of P2. This explains also the absence of a signal in the green channel for this product by an obviously efficient energy transfer between BODIPY and rhodamine. Since both reactive groups, the triazine and the 1-methylcyclopropene, undergo bioorthogonal labeling with the respective dye in high yields, mostly the double labeled product and no unlabeled oligonucleotide was obtained. For control experiments with single incorporations of either 1 or 2 see Fig. S4.† The observed energy transfer between the two labels is an interesting aspect for applications in molecular imaging of living cells. Hence, we characterized this energy transfer additionally by steady-state in vitro fluorescence measurements (see Fig. S16 and S17†).
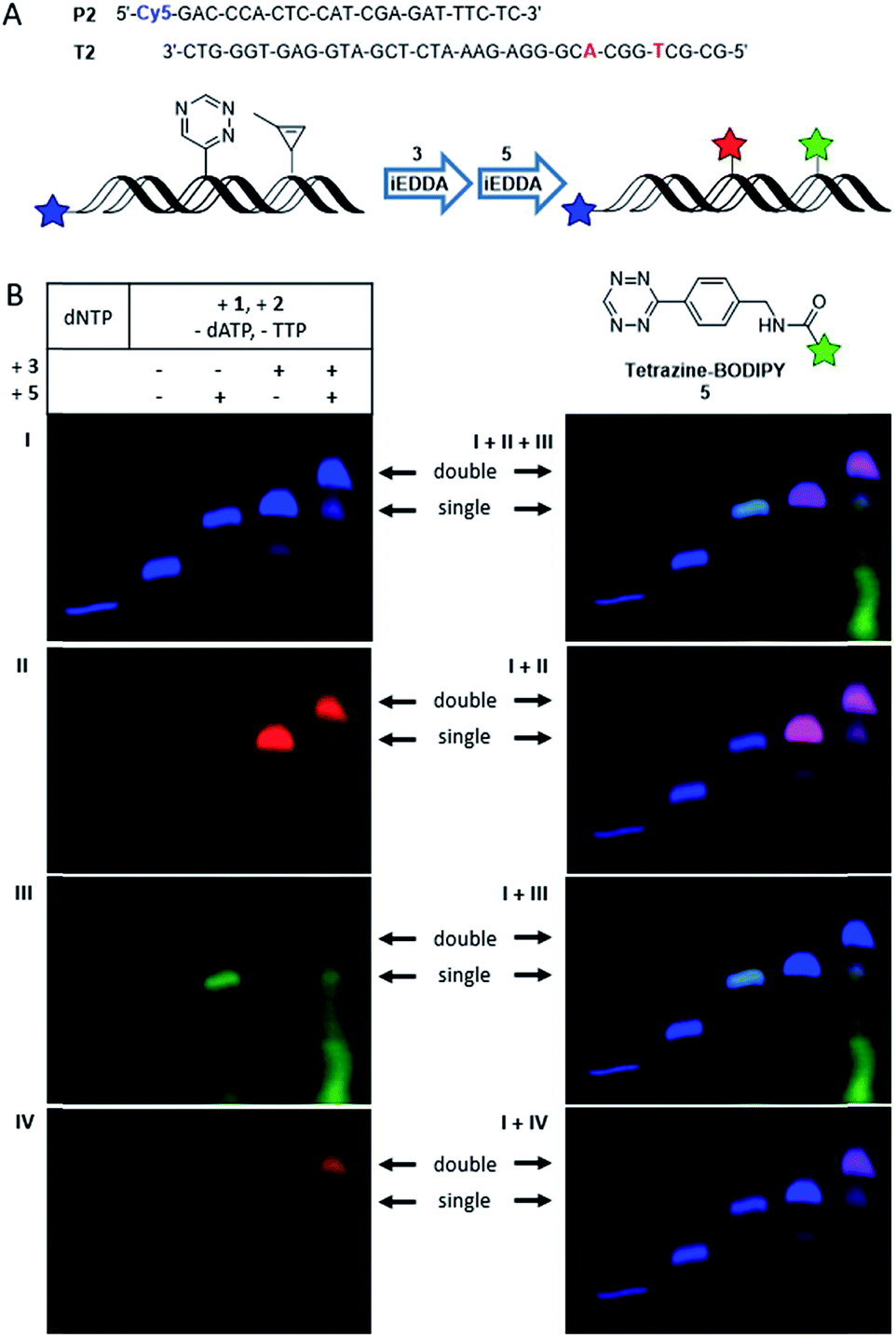 | ||
| Fig. 3 (A) Schematic illustration of a dual labeling approach using Cy5-labeled Primer P2, BCN-rhodamine 3 for conjugation with the triazine moiety and tetrazine-modified BODIPY 5 for orthogonal conjugation with the cyclopropene component. (B) PAGE analysis of labeling reactions using elongated P2 (Vent (exo-), 37 °C, 30 min with a triphosphate mixture containing 1, 2, dCTP and dGTP) and the fluorescent dyes 3 (30 min incubation) and 5 (15 min incubation). “dNTP” shows the full-length product when only natural triphosphates are used (I) Cy5 λexc = 630 nm, λem = 700 nm; (II) rhodamine λexc = 540 nm, λem = 605 nm; (III) BODIPY λexc = 470 nm, λem = 535 nm; (IV) BODIPY λexc = 470 nm, λem = 605 nm. Overlays of the individual images are shown on the right side, whereby a mixed color occurs whenever different signals are present: cyan for Cy5 (blue) + BODIPY (green); magenta for Cy5 (blue) + rhodamine (red). For control experiments with only one of the two modified triphosphates, see ESI.† | ||
The DNA primer extension product of P2/T2 labeled with both BCN-rhodamine 3 and tetrazine-BODIPY 5 were applied in cell biology. HeLa cells were transfected by this DNA construct using Screenfect® agent. The cell images 24 h after transfection (Fig. 4) showed not only the BODIPY fluorescence when excited at 488 nm in the green emission channel (502–522 nm) but also the rhodamine fluorescence in the red emission channel (560–580 nm) (lane I). Control experiments with DNA primer extension products that were either labeled with BCN-rhodamine 3 or tetrazine-BODIPY 5 revealed only the corresponding single fluorescence in the respective emission channels (lanes II and III). This clearly shows that the energy transfer which was indicated by the gels (Fig. 3) is also observable inside cells. This supports the application of our double iEDDA modification chemistry for nucleic acid probes in molecular imaging.
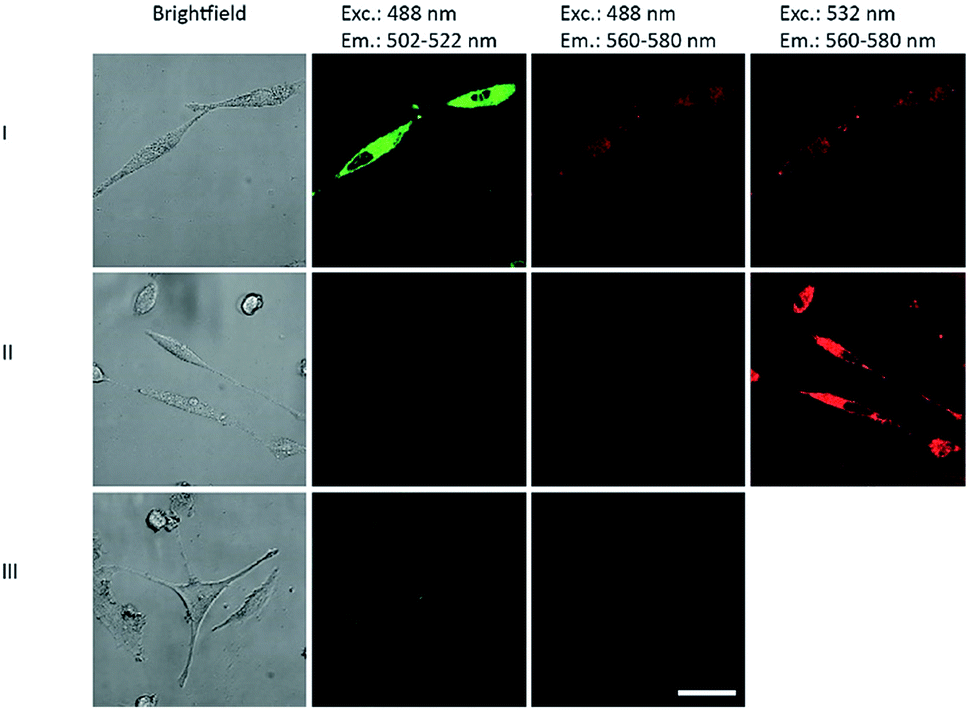 | ||
| Fig. 4 Images of HeLa cells 24 h after transfection with DNA primer extension products of P2/T2 (see ESI† page 25) labeled with both tetrazine-BODIPY 5 and BCN-rhodamine 3 (lane I), labeled only with 3 (lane II), and labeled only with 5 (lane III). Excitation and emission wavelengths are given as headings. | ||
Conclusions
In conclusion, we applied two different functions for inverse electron demand Diels–Alder reactions and dual labeling of DNA. The 1,2,4-triazine function was attached to the 5-position of 2′-deoxyuridine triphosphate 1, and the 1-methylcyclopropene function to the 7-position of 7-deaza-2′-deoxyadenosine triphosphate 2. Sequence selective incorporation of these two functionalized nucleotides was achieved by primer extension with standard DNA polymerases at 37 °C. The primer extension products were subsequently labeled by two different fluorescent dyes using two Diels–Alder reactions for postsynthetic modifications that are not only bioorthogonal in general, but also mutually orthogonal. This was successfully shown for BODIPY and rhodamine dyes and yield oligonucleotides with two different emission colors. Remarkably, both dyes as postsynthetically attached labels form an energy transfer pair which shows the high potential for this type of dual labeling in molecular imaging of living cells. Due to (i) the principle bioorthogonality of these type of iEDDA reactions, and (ii) the small steric size of the reactive groups, this chemistry should also be applicable to labelings inside living cells. Our preliminary cell experiments show the potential of the doubly labeled probes for molecular imaging and other applications in chemical biology of nucleic acids. Moreover, the chemistry may also be transferred to other biomolecules of interest, like carbohydrates and proteins.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (grant Wa 1386/15-2) and KIT is gratefully acknowledged.Notes and references
- R. D. Row and J. A. Prescher, Acc. Chem. Res., 2018, 51, 1073–1081 CrossRef CAS PubMed.
- F. Liu, Y. Liang and K. N. Houk, Acc. Chem. Res., 2017, 50, 2297–2308 CrossRef CAS PubMed.
- G. B. Csérep, A. Herner and P. Kele, Methods Appl. Fluoresc., 2015, 3, 042001 CrossRef PubMed.
- P. Shieh and C. R. Bertozzi, Org. Biomol. Chem., 2014, 12, 9307–9320 RSC.
- K. Lang and J. W. Chin, ACS Chem. Biol., 2014, 9, 16–20 CrossRef CAS PubMed.
- M. King and A. Wagner, Bioconjugate Chem., 2014, 25, 825–839 CrossRef CAS PubMed.
- C. P. Ramil and Q. Lin, Chem. Commun., 2013, 49, 11007–11022 RSC.
- M. F. Debets, J. C. M. v. Hest and F. P. J. T. Rutjes, Org. Biomol. Chem., 2013, 11, 6439–6455 RSC.
- N. K. Devaraj, S. Hilderbrand, R. Upadhyay, R. Mazitschek and R. Weissleder, Angew. Chem., Int. Ed., 2010, 49, 2869–2872 CrossRef CAS PubMed.
- V. V. Rostovstev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- V. Hong, N. F. Steinmetz, M. Manchester and M. G. Finn, Bioconjugate Chem., 2010, 21, 1912–1916 CrossRef CAS PubMed.
- M. Yang, J. Li and P. R. Chen, Chem. Soc. Rev., 2014, 43, 6511–6526 RSC.
- D. C. Kennedy, C. S. McKay, M. C. B. Legault, D. C. Danielson, J. A. Blake, A. F. Pegoraro, A. Stolow, Z. Mester and J. P. Pezacki, J. Am. Chem. Soc., 2011, 133, 17993–18001 CrossRef CAS PubMed.
- R. Huisgen, Angew. Chem., Int. Ed., 1962, 2, 565–598 CrossRef.
- C. P. Ramil and Q. Lin, Curr. Opin. Chem. Biol., 2014, 21, 89–95 CrossRef CAS PubMed.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2011, 22, 2263–2270 CrossRef CAS PubMed.
- S. Mayer and K. Lang, Synthesis, 2017, 49, 830–848 CAS.
- M. Merkel, S. Arndt, D. Ploschik, G. B. Cserép, U. Wenge, P. Kele and H.-A. Wagenknecht, J. Org. Chem., 2016, 81, 7527–7538 CrossRef CAS PubMed.
- S. Arndt and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2014, 53, 14580–14582 CrossRef CAS PubMed.
- B. Lehmann and H.-A. Wagenknecht, Org. Biomol. Chem., 2018, 16, 7579–7582 RSC.
- G. B. Cserép, O. Demeter, E. Bätzner, M. Kállay, H.-A. Wagenknecht and P. Kele, Synthesis, 2015, 47, 2738–2744 CrossRef.
- D. N. Kamber, Y. Liang, R. J. Blizzard, F. Liu, R. A. Mehl, K. N. Houk and J. A. Prescher, J. Am. Chem. Soc., 2015, 137, 8388–8391 CrossRef CAS PubMed.
- K. Peewasan and H.-A. Wagenknecht, ChemBioChem, 2017, 18, 1473–1476 CrossRef CAS PubMed.
- J. Schoch, S. Ameta and A. Jäschke, Chem. Commun., 2011, 47, 12536–12537 RSC.
- P. N. Asare-Okai, E. Augustin, D. Fabris and M. Royzen, Chem. Commun., 2014, 50, 7844–7847 RSC.
- A. M. Pyka, C. Domnick, F. Braun and S. Kath-Schorr, Bioconjugate Chem., 2014, 25, 1438–1443 CrossRef CAS PubMed.
- X. Ren, A. El-Sagheer and T. Brown, Analyst, 2015, 140, 2671–2678 RSC.
- K. Wang, D. Wang, K. Ji, W. Chen, Y. Zheng, C. Dai and B. Wang, Org. Biomol. Chem., 2015, 13, 909–915 RSC.
- X. Ren, M. Gerowska, A. H. El-Sagheer and T. Brown, Bioorg. Med. Chem., 2014, 22, 4384–4390 CrossRef CAS PubMed.
- H. Bußkamp, E. Batroff, A. Niederweiser, O. S. Abdel-Rahman, R. F. Winter, V. Wittmann and A. Marx, Chem. Commun., 2014, 50, 10827–10829 RSC.
- U. Rieder and N. W. Luedtke, Angew. Chem., Int. Ed., 2014, 53, 9168–9172 CrossRef CAS PubMed.
- D. Ploschik, F. Rönicke, H. Beike, R. Strasser and H.-A. Wagenknecht, ChemBioChem, 2018, 19, 1949–1953 CrossRef CAS PubMed.
- D. N. Kamber, L. A. Nazarova, Y. Liang, S. A. Lopez, D. M. Patterson, H.-W. Shih, K. N. Houk and J. A. Prescher, J. Am. Chem. Soc., 2013, 135, 13680–13683 CrossRef CAS PubMed.
- D. A. Malyshev and F. E. Romesberg, Angew. Chem., Int. Ed., 2015, 54, 11930–11944 CrossRef CAS PubMed.
- F. Eggert and S. Kath-Schorr, Chem. Commun., 2016, 52, 7284–7287 RSC.
- T. Plass, S. Milles, C. Koehler, J. Szymanski, R. Mueller, M. Wießler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2012, 51, 4166–4170 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova, B. Xie, D. N. Kamber and J. A. Prescher, J. Am. Chem. Soc., 2012, 134, 18638–18643 CrossRef CAS PubMed.
- I. Nikić, T. Plass, O. Schraidt, J. Szmański, J. A. G. Briggs, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2014, 53, 2245–2249 CrossRef PubMed.
- A. Niederwieser, A.-K. Späte, L. D. Nguyen, C. Jüngst, W. Reutter and V. Wittmann, Angew. Chem., Int. Ed., 2013, 52, 4265–4268 CrossRef CAS PubMed.
- J. Schoch, M. Staudt, A. Samanta, M. Wiessler and A. Jäschke, Bioconjugate Chem., 2012, 23, 1382–1386 CrossRef CAS PubMed.
- M.-L. Winz, E. C. Linder, J. Becker and A. Jäschke, Chem. Commun., 2018, 54, 11781–11784 RSC.
- P. M. E. Gramlich, S. Warncke, J. Gierlich and T. Carell, Angew. Chem., Int. Ed., 2008, 47, 3442–3444 CrossRef CAS PubMed.
- J. Matyasovsky, R. Pohl and M. Hocek, Chem.–Eur. J., 2018, 24, 14938–14941 CrossRef CAS PubMed.
- J. Matyasovský, P. Perlíková, V. Malnuit, R. Pohl and M. Hocek, Angew. Chem., Int. Ed., 2016, 55, 15856–15859 CrossRef PubMed.
- M. Krömer, K. Bártová, V. Raindlová and M. Hocek, Chem.–Eur. J., 2018, 24, 11890–11894 CrossRef PubMed.
- K. Peewasan and H.-A. Wagenknecht, ChemBioChem, 2017, 18, 1473–1476 CrossRef CAS PubMed.
- M. Slavíckova, M. Janousková, A. Simonová, H. Cahová, M. Kambová, H. Sanderová, L. Krásný and M. Hocek, Chem.–Eur. J., 2018, 24, 8311–8314 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of 1, images of NMR and mass spectra, images of PAGE analyses after primer extension. See DOI: 10.1039/c8sc05588b |
| This journal is © The Royal Society of Chemistry 2019 |