DOI:
10.1039/C8SC05416A
(Edge Article)
Chem. Sci., 2019,
10, 4025-4031
Wave-shaped polycyclic hydrocarbons with controlled aromaticity†
Received
4th December 2018
, Accepted 27th February 2019
First published on 28th February 2019
Abstract
Controlling the aromaticity and electronic properties of curved π-conjugated systems has been increasingly attractive for the development of novel functional materials for organic electronics. Herein, we demonstrate an efficient synthesis of two novel wave-shaped polycyclic hydrocarbons (PHs) 1 and 2 with 64 π-electrons. Among them, the wave-shaped π-conjugated carbon skeleton of 2 is unambiguously revealed by single-crystal X-ray crystallography analysis. The wave-shaped geometry is induced by steric congestion in the cove and fjord regions. Remarkably, the aromaticity of these two structural isomers can be tailored by the annulated direction of cyclopenta[b]fluorene units. Isomer 1 (Eoptg = 1.13 eV) behaves as a closed-shell compound with weakly antiaromatic feature, whereas its structural isomer 2 displays a highly stable tetraradical character (y0 = 0.23; y1 = 0.22; t1/2 = 91 days) with a narrow optical energy gap of 0.96 eV. Moreover, the curved PH 2 exhibits remarkable ambipolar charge transport in solution-processed organic thin-film transistors. Our research provides a new insight into the design and synthesis of stable functional curved aromatics with multiradical characters.
Introduction
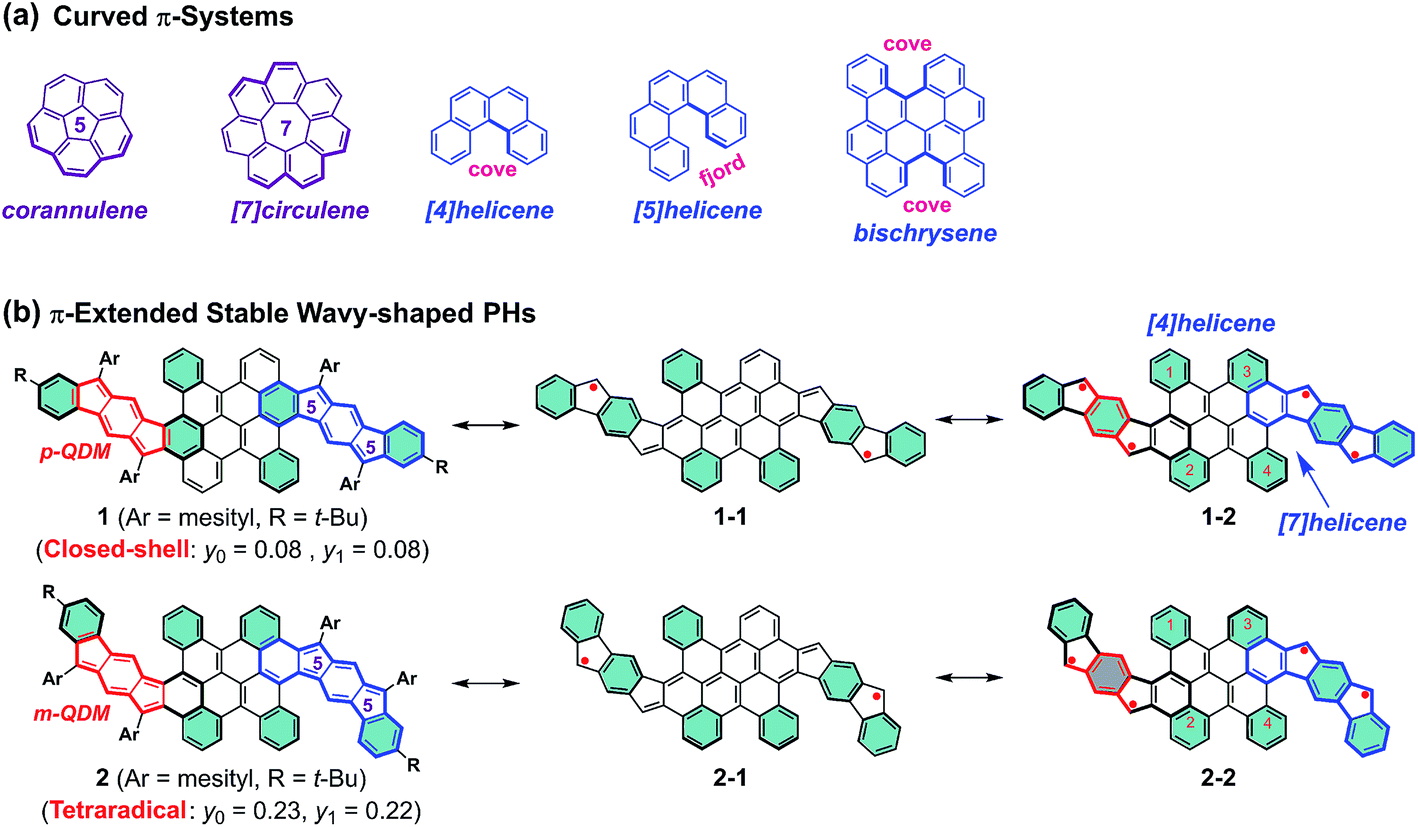
Curved π-conjugated molecules1,2 have grown to become an important field of research due to their fascinating intermolecular packing, extraordinary chiroptical properties and dynamic behavior resulting from their contorted conformation, which have potential application in chiroptical sensing and photovoltaics.3–5 To date, two distinct strategies have been established for the synthesis of curved polycyclic aromatic hydrocarbons (PAHs). The most prominent way is the incorporation of non-hexagonal rings which induce the curved nature of PAHs,6–10 such as bowl-shaped corannulene11 and saddle-shaped [7]circulene12 (Fig. 1a). Another attractive approach to influence the curvature of PAHs is the incorporation of steric strain in their skeleton periphery.13–15 For instance, the curved structures of [4]helicene and [5]helicene result from the steric congestion of the hydrogen atoms in the cove and fjord regions, respectively (Fig. 1a).16,17 Interestingly, the strain-induced curvature in a π-conjugated system often yields an unusual electronic structure and unprecedented physical properties, such as narrowed singlet–triplet energy gap,18 increased the two-photon absorption cross sections,19etc. Apart from the tunable molecular geometry of PAHs, aromaticity is also a fundamental concept to understand their physical properties and chemical reactivity. For example, several types of pro-aromatic and antiaromatic π-conjugated molecules have been exploited, which possess a singlet open-shell biradical nature or display small energy gaps.20–22 Note also that most of the reported biradicaloids adopt a planar structure and they are thermodynamically unstable.23–25 Therefore, the development of novel stable polycyclic hydrocarbon (PH)-based radicals for functional applications in organic electronics still remains challenging, mostly due to the lack of efficient synthetic methods and appropriate building blocks.26
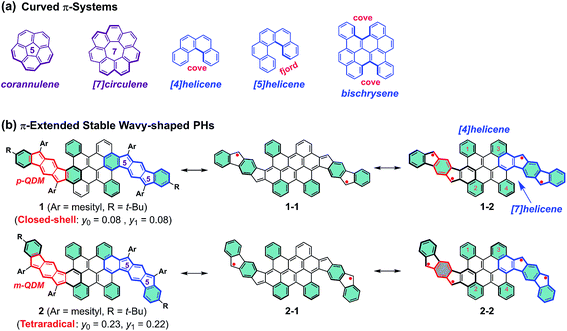 |
| | Fig. 1 (a) Representative curved π-systems. (b) The resonance structures of π-extended stable wave-shaped PHs 1 and 2 reported in this work (p-QDM and m-QDM units are highlighted in red color; the flanked indeno[1,2-b]fluorene (IF1) and indeno[2,1-b]fluorene (IF2) in 1 and 2 are highlighted in blue color). The substituents are omitted in the resonance structures for clarity and the benzene rings in cyan color denote Clar's aromatic sextet rings. | |
We herein demonstrate two novel curved PHs by extension of a bischrysene27,28 (Fig. 1a) system with two cyclopenta[b]fluorene units, which show unprecedented wave-shaped structures. As shown in Fig. 1b, isomer 1 was obtained firstly by annulation of two cyclopenta[b]fluorene units into the bischrysene backbone; as a result, two para-quinodimethane (p-QDM) units are embedded in its carbon skeleton. Compound 1 presented a closed-shell electronic structure with weak antiaromaticity from the experimental and theoretical results (vide infra). Its structural isomer 2, which contains two meta-quinodimethane (m-QDM) units, was subsequently synthesized by tuning the annulated direction of cyclopenta[b]fluorene moieties into bischrysene (Fig. 1b). Compound 2 adopts a remarkable wave-shaped geometry based on the single crystal structure analysis, in which the curvature induced by the steric strain from the cove and fjord regions yields a torsion angle of 22.1° and 31.8°, respectively. Interestingly, compared with the antiaromatic PH 1 (Eoptg = 1.13 eV), compound 2 possesses a moderate singlet tetraradical character (y0 = 0.23; y1 = 0.22) associated with a narrow optical energy gap of 0.96 eV, which results in the first case of a curved tetraradicaloid reported to date.25,29,30 Moreover, tetraradicaloid 2 exhibits high stability with a half-life (t1/2) as long as 91 days under ambient conditions and unique ambipolar charge-transport behavior in solution-processed organic field-effect transistors.
Results and discussion
Synthesis and structural characterization
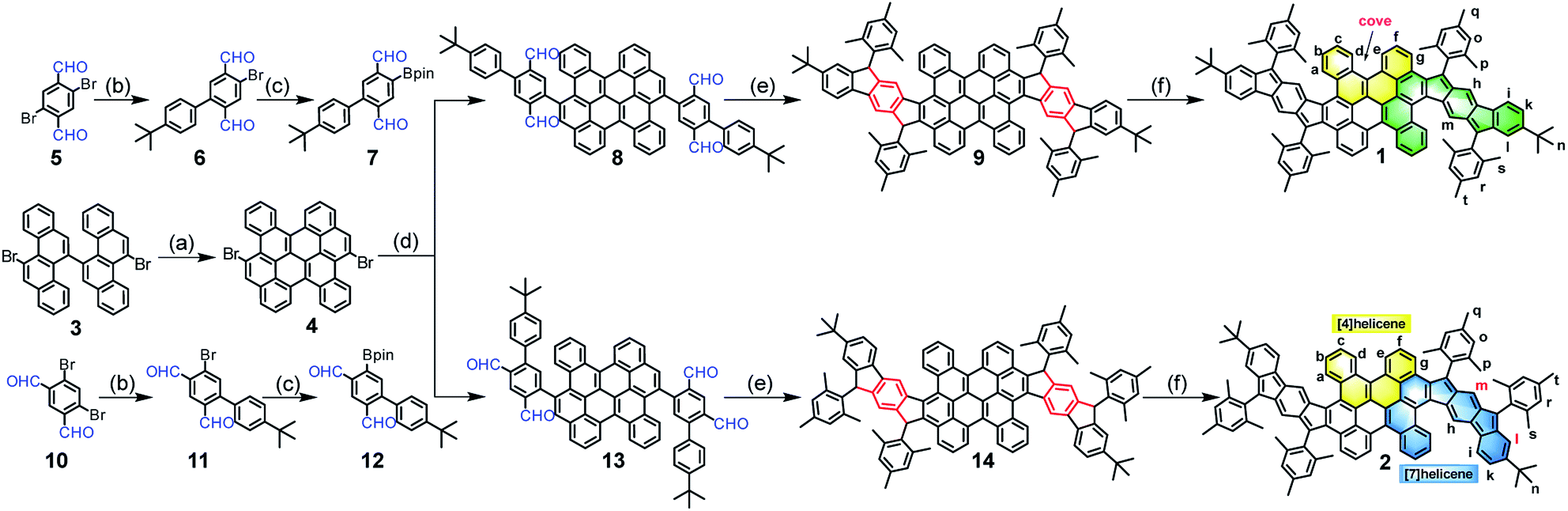
The synthetic routes are shown in Scheme 1. Firstly, the fused bischrysene 4 was obtained through the oxidative cyclodehydrogenation of 11,11′-dibromo-5,5′-bichrysene (3) in the presence of DDQ/TfOH in 72% yield. The monoboronic ester 7 was meanwhile obtained in 59% yield in two steps by Suzuki cross-coupling from 2,5-dibromoterephthalaldehyde (5) and followed by the palladium-catalyzed Miyaura-borylation with B2pin2. Subsequently, a Suzuki coupling reaction between 4 and 7 afforded the key intermediate tetraaldehyde 8 in 61% yield. Treatment of 8 with mesitylmagnesium bromide gave the corresponding tetraol compound, which was further subjected to an intramolecular Friedel–Crafts alkylation mediated by BF3·OEt2 to provide the precursor 9. Finally, the dehydrogenation of 9 by treatment with DDQ in dry toluene at 80 °C afforded PH 1 as a dark-brown solid in 66% yield with three steps from 8. Following a similar synthetic strategy, compound 2 was achieved as a deep-green solid starting from 4,6-dibromoisophthalaldehyde (10). Both PHs 1 and 2 are highly stable and can be purified by chromatography using silica columns without argon protection. Compounds 1 and 2 were firstly characterized by high-resolution MALDI-TOF mass spectroscopy (Fig. S1†). They have good solubility in common organic solvents, such as DCM, chloroform, THF, toluene, etc.
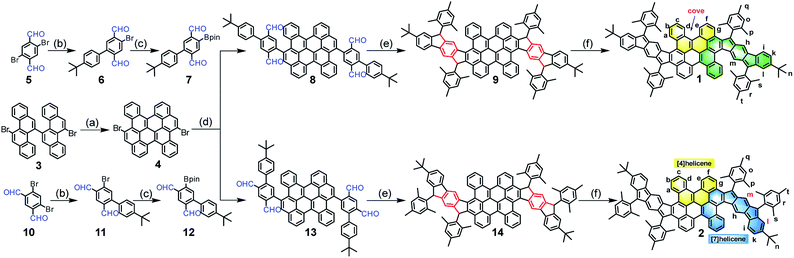 |
| | Scheme 1 Synthetic route of the wave-shaped PHs 1 and 2. Reagents and conditions: (a) DDQ/TfOH, DCM, −78 °C, 1 h, 72%; (b) (4-(tert-butyl)phenyl)boronic acid, Pd(PPh3)4, 2 M K2CO3, toluene/ethanol, 100 °C, 12 h, 6: 65%, 11: 68%; (c) Pd(PPh3)2Cl2, B2pin2, KOAc, 1,4-dioxane, 90 °C, 20 h, 7: 91%, 12: 88%; (d) Pd(PPh3)4, 2 M K2CO3, toluene/ethanol, 100 °C, 36 h, 8: 61%, 13: 66%; (e) (i) Mesitylmagnesium bromide, dry THF, rt, 4 h;(ii) BF3·OEt2, DCM, rt, 1 h, 9: 87%, and 14: 88%; (f) DDQ, toluene, 80 oC, 8 h, 1: 76%, 2: 67%. DDQ = 2,3-dichloro-5,6-dicyanobenzoquinone; TfOH = trifluoromethanesulfonic acid; DCM = dichloromethane; B2pin2 = bis(pinacolato) diboron; THF = tetrahydrofuran. | |
VT NMR spectra and magnetic properties of 2
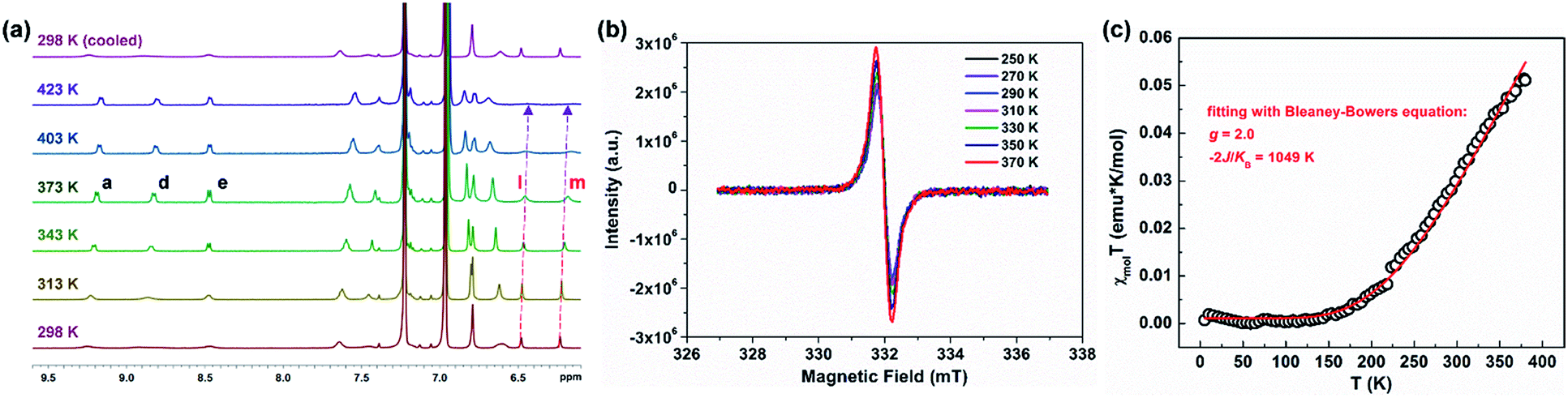
PH 1 shows a well-resolved 1H NMR spectrum at 303 K (Fig. S2†), which can be well assigned with the help of 2D NMR spectra such as COSY, ROESY and HSQC (Fig. S4–S6†). Its solid powder does not display any electron paramagnetic resonance (EPR) signal, thus supporting its closed-shell nature. In addition, variable-temperature (VT) 1H NMR experiments of 1 and 2 were performed to investigate the dynamic behavior in view of their helical structure (Fig. S11–S14†). For 1, the peaks d, e, b and g in the cove region became broad as the temperature increased (in degassed C2D2Cl4), and the rest of the signals slightly changed (Fig. S11†). This dynamic behavior points to a racemization in the cove regions in 1.28 Compared with 1, its structural isomer 2 displays broadened resonances in the aromatic region between 7.5 and 9.5 ppm at 298 K (Fig. 2a). Moreover, these signals became sharper with increasing temperature, and this allowed the assignment of the 1H resonances of 2 at 363 K with the help of 2D NMR spectra (Fig. S7–S10†). As the temperature was raised further to 423 K in o-C6D4Cl2 (Fig. 2a), the aromatic resonances became broadened again; especially the signals of protons l and m eventually disappeared. When the sample was cooled back to 298 K, all the proton NMR signals were recovered. These results indicate that a thermally accessible triplet state is populated at high temperatures of 2. Initiated by the dynamic process observed in the VT 1H NMR measurements, the isomerization process in the quadruple helicenes 1 and 2, which incorporates the [4]helicene and pentagon-embedded [7]helicene substructures (Fig. 1b), was also evaluated and the detailed analyses are illustrated in the ESI (Fig. S28–S30†). Moreover, the EPR measurement of 2 displays an unresolved signal (Fig. 2b), and the intensity increases by elevating the temperature (Fig. S15†), indicating the existence of thermally populated paramagnetic species. Superconducting quantum interfering device (SQUID) measurement was also carried out with a powdered sample of 2 in the range of 5–400 K (Fig. 2c). The result shows increasing susceptibility above 150 K, and careful fitting of the data by using the Bleaney–Bowers equation gives a 2J/kB of −1049 K (−2.08 kcal mol−1). All these experimental results confirm that PH 2 displays a singlet open-shell ground state.
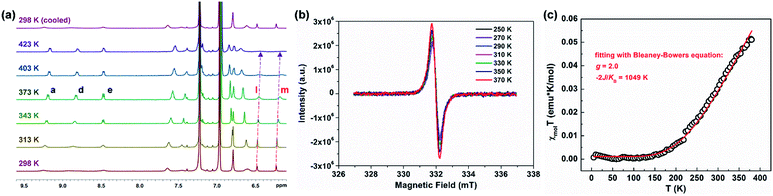 |
| | Fig. 2 (a) VT 1H NMR experiment of 2 in o-C6D4Cl2. (b) EPR spectrum of a powdered sample of 2 recorded at 250–370 K. (c) SQUID measurement of a powdered sample of 2 from 5 to 400 K, experimental data plotted as black “○” and fitted curve according to the Bleaney–Bowers equation with ge = 2.00 as a red line. | |
Ground-state geometry, bond length analysis and aromaticity evaluation
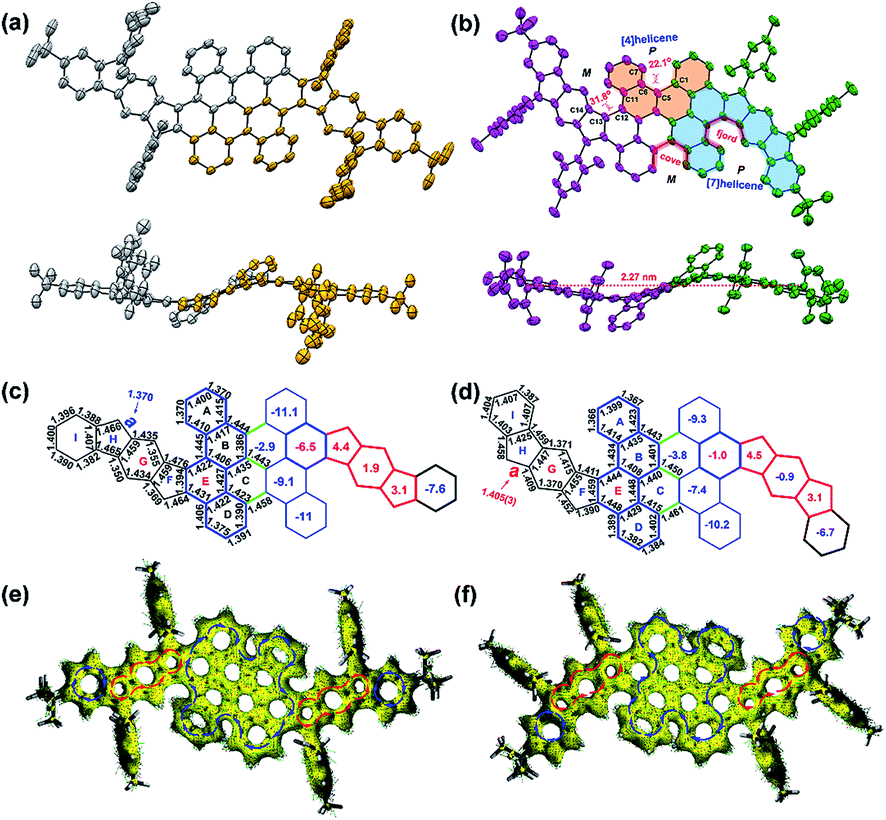
The single crystals of 2 were grown by slow evaporation from solutions in ethyl acetate/n-hexane, which are suitable for X-ray crystallographic analysis. PH 2 crystalizes in the space group P21/n. As shown in Fig. 3b, the twisted conjugated carbon skeleton of crystal 2 adopts a novel wave-shaped conformation with a length of 2.27 nm, which is induced by the combination of quadruple helical substructures. The [4]helicene substructures and [7]helicene analogues in 2 are induced by the steric strain from the cove and fjord regions with torsion angles of 22.1(3)° for C1–C5–C6–C7 and 31.8(3)° for C11–C12–C13–C14, which are larger than that of the cove region in [4]helicene (19.9°) and that of the fjord region in pristine [5]helicene (27.9°) respectively.16,17 According to the distribution of the bond lengths, the C–C bonds (1.44–1.46 Å) in the cove and fjord regions (red one in Fig. S20†) are significantly longer than the normal range for the bond order observed in aromatic systems (1.40 ± 0.02 Å) owing to the repulsive forces of congested hydrogen atoms. The crystal of 2 displays unique intermolecular π–π interactions in the packing pattern,15 and the enantiomers are alternately assembled in a layer-by-layer manner along the b-axis and tightly arrange themselves in an offset stacking array (Fig. S18b†); the enantiomers in each molecular layer are lying on the ac plane in an orderly way with slipped π–π stacking along the a-axis (Fig. S18c†). Consequently, the crystal of 2 has unconventional intermolecular π–π interactions in the packing pattern. Notably, the chrysene units (highlighted in blue color in Fig. 3d) in 2 connect with each other by three single C–C bonds (1.45–1.46 Å, green one in Fig. 3d). This suggests that the curved geometry results in less conjugation of the whole molecule compared with the reported planar PHs.19,20 Consequently, the communication of the radicals which are distributed along the wave-shaped structure can be minimized.25,29,30 In the cyclopenta[b]fluorene-fused systems, the length of the most critical bond a (Fig. S21†) is an indication of the degree of biradical character.31,32 For 1, the calculated length of bond a (1.370 Å) (Fig. 3c, UCAM-B3LYP/6-311G**) is significantly shorter than that of the parent IF1 (1.380 Å) (Fig. S21†),33,34 implying that the closed-shell resonance form contributes much more to the electronic ground state. Comparably, the intermediate value (1.405(3) Å) of bond a in 2 indicates the large contribution of the open-shell resonance structure in its ground state (Fig. 3d). However, the bond length of a in 2 is shorter than that of the parent IF2 (1.437 Å) (Fig. S21†),35,36 which is consistent with its relatively smaller open-shell character (y0 = 0.23; y1 = 0.22). Numerous attempts to crystallize PH 1 unfortunately failed under different conditions and only resulted in extremely thin crystal films. DFT calculations (UB3LYP/6-31G*) clearly predict that compound 1 also adopts a wave-shaped conformation in the ground state (Fig. S23†). Alternatively, a single crystal of its tetrahydro-precursor 9 was obtained by slow evaporation of its DCM/methanol solution. Interestingly, compound 9 contains cis and trans conformers in the crystal structure (Fig. S22†), in which the trans isomer adopts the wave-shaped conformation as shown in Fig. 3a.
 |
| | Fig. 3 Top view and side view of the X-ray crystallographic structure of (a) compound 9 (precursor for PH 1) and (b) tetraradicaloid 2; the hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and calculated NICS(1)av values in the rings A-I of (c) 1 and (d) 2. Calculated ACID plots of (e) 1 and (f) 2. Isovalue is 0.05, and the blue and red arrows indicate diamagnetic and paramagnetic ring current flows, respectively. | |
To better understand the electronic structure and evaluate the aromaticity, nucleus independent chemical shift (NICS) calculations and anisotropy of the induced current density (ACID) plot were performed. The calculated NICS(1)av37 values of 1 (Fig. 3c) indicate the antiaromatic character for rings F (4.4), G (1.9) and H (3.1). As shown in Fig. 1b, the indeno[1,2-b]fluorene (IF1) subunits in the open-shell resonance form 1–2 and closed-shell resonance form 1 possess the same aromatic sextet rings (Fig. S25†). This means that there is no driving force to form an open-shell resonance structure in 1. Combined with the above experimental results (resolved NMR results and EPR silence), PH 1 can be regarded as a closed-shell PH with weakly antiaromatic feature. In addition, the value for ring E in 1 (−6.5) is significantly smaller than that of ring E in 2 (−1.0), indicating that the former possesses higher aromaticity/conjugation due to the contribution from the inner bischrysene core. In the resonance form of 2–2, only the terminal sextets are due to benzene rings and the rest of the sextets are contributed by the core of the bischrysene backbone (Fig. S25†), which is different from the parent indeno[2,1-b]fluorene (IF2) where both sextets consist of benzene rings in their biradical form (IF2-1) (Fig. S25†). In addition, the ring E in 2 (Fig. 3d) exhibits smaller aromatic character (NICS(1)av = −1.0) compared with the terminal benzene rings (NICS(1)av = −1.24) in the biradical form IF2-1 (Fig. S25†). This result manifests that the driving force to form the tetraradical resonance 2–2 is smaller than that in parent IF2 to form its biradical resonance IF2-1 (y0 = 0.68), which is also consistent with the relatively smaller biradical and moderate tetraradical characters in 2 (y0 = 0.23: y1 = 0.22). Moreover, the NICS(1)av value of ring G (−0.9) in 2 (Fig. 3d) becomes negative compared with that of the central benzene ring (NICS(1) av = 7.95) in IF2 (Fig. S25†)36 or the ring G (1.9) in 1 (Fig. 3c), further supporting the large contribution of the tetraradical resonance form (2–2) to the ground-state structure of 2 (Fig. 1b). In addition, compounds 1 and 2 exhibit a similar pattern of ring current circuits (Fig. 3e and f, see more images in Fig. S26 and S27†), that is, the flanked indacene units show clear paramagnetic ring current and the inner bischrysene core displays obvious diamagnetic ring current, which is consistent with the NICS calculation. Therefore, the different behavior of the structural isomers 1 and 2 suggests that the aromaticity can be controlled by the molecular arrangement of the quinodimethane units.
Optical and electrochemical properties
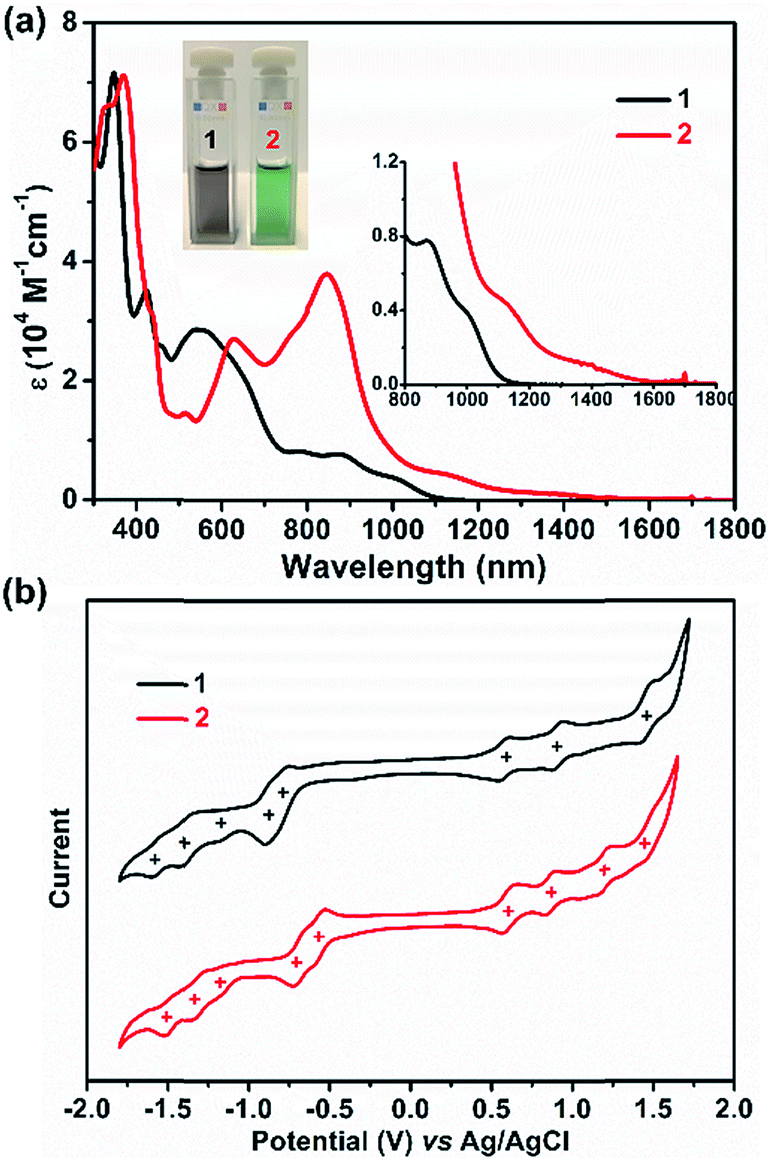
UV/Vis absorption spectra were recorded for 1 and 2, as illustrated in Fig. 4a. The grey solution of 1 in DCM exhibits an intense broad absorption peak between 500 and 728 nm, and two near-infrared (NIR) peaks at 873 and 1006 nm are also observed. In contrast, compound 2 displays a green color in DCM solution and presents intriguing absorption in the NIR region. For 2, an intense absorption peak in the far-red region with a maximum at 842 nm was found along with two smaller shoulder peaks at 775 and 623 nm, and two NIR peaks centered at 1142 and 1394 nm were also detected. Thus, the optical energy gaps (Eoptg) of 1 and 2 were determined from the onset of their UV/Vis absorptions, which are 1.13 and 0.96 eV, respectively. Not only the UV/Vis absorption but also the electrochemical behavior of 1 and 2 exhibit apparent differences (Fig. 4b). Compound 1 shows three reversible oxidation waves with half-wave potentials Eox1/2 at 0.58, 0.89 and 1.43 V and five quasi-reversible reduction waves with half-wave potentials Ered1/2 at −0.80, −0.88, −1.20, −1.40, and −1.56 V (vs. Ag/AgCl). In contrast, 2 manifests four quasi-reversible oxidation waves with half-wave potentials (Eox1/2) at 0.59, 0.88, 1.19, and 1.47 V and five reduction waves (Ered1/2) at −0.57, −0.71, −1.16, −1.32, and −1.50 V (vs. Ag/AgCl). Thus, the HOMO/LUMO energy levels are estimated to be −4.86/−3.68 and −4.97/−3.95 eV for 1 and 2, respectively, based on the onset potentials of the first oxidation/reduction waves. The corresponding electrochemical energy gaps (EECg) are estimated to be 1.18 and 1.02 eV for 1 and 2, respectively. Furthermore, time-dependent UV/Vis measurement was performed under ambient conditions to investigate the stability of 1 and 2. There are no significant changes of the absorption curves for both 1 and 2 for 10 days, suggesting that both molecules are highly stable under ambient conditions, and the half-life (t1/2) of tetraradicaloid 2 was found to be as long as 91 days (Fig. S16 and S17†).
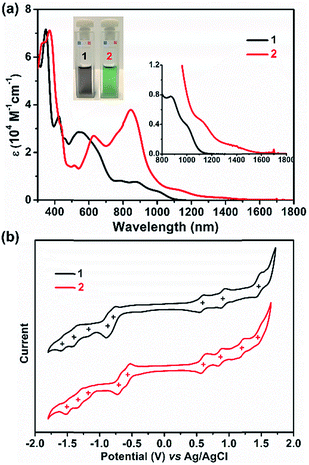 |
| | Fig. 4 (a) UV/Vis absorption of 1 and 2 recorded in dry DCM. Insets give a magnified view and the photos of their DCM solutions. (b) Cyclic voltammograms of 1 and 2 in a solution of Bu4NPF6 (0.1 M) in DCM with a scan rate of 50 mV s−1 at room temperature. | |
Organic field-effect transistor (OFET)
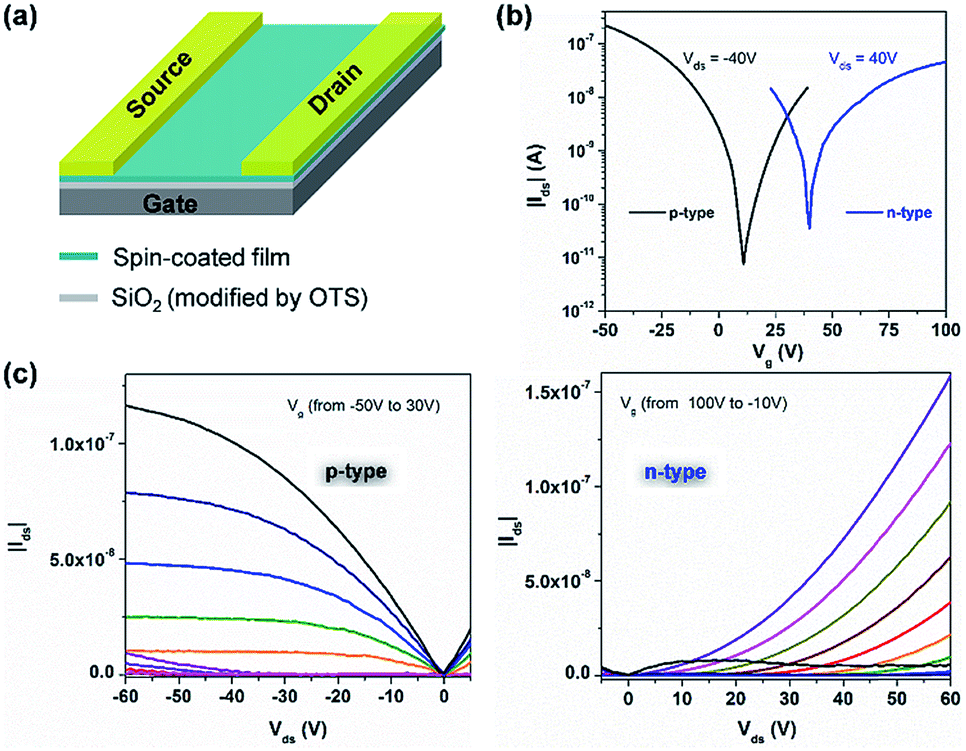
In view of the amphoteric redox character and high stability of 1 and 2, their charge transport behavior was evaluated using bottom-gate and top-contact field-effect transistors (Fig. 5a). Heavily doped silicon substrates with a 300 nm SiO2 dielectric (with a capacitance of 11 nF cm−2) modified with octadecyl trichlorosilane (OTS) through thermal vapor at 150 °C were used for the device fabrication. A homogeneous layer of 1 or 2 was spin-coated from 5 mg mL−1 CHCl3 and annealed at 200 °C for 2 hours under glovebox conditions to remove the residual solvent and to improve the film quality. Source and drain gold electrodes were thermal vapor deposited at a thickness of 50 nm. The channel length and width were 50 μm and 1000 μm, respectively. A Keithley 4200-SCS was used for all standard electrical measurements in a glovebox under a nitrogen atmosphere. The transfer and output curves shown in Fig. S31† indicate the unipolar charge transport behavior of transistors based on the spin-coated film of 1, with an average hole mobility (μh) of around 3.0 × 10−5 cm2 V−1 s−1. However, the transistors fabricated from 2 exhibit an interesting ambipolar charge transport behaviour (Fig. 5b and c). The average hole mobility (μh) is around 6.2 × 10−4 cm2 V−1 s−1 with a crossover point at around 10 V and the average electron mobility (μe) is about 1.3 × 10−4 cm2 V−1 s−1 with a crossover point at 40 V. The ambipolar performance of the thin film transistor of 2 can be attributed to the low energy gap of 2 (0.96 eV) and small injection barriers from gold electrodes (4.9 eV) to both HOMO and LUMO levels.38 And the hole and electron mobilities of 2 are comparable to those of previously reported organic biradical-based OFETs.32,39 In contrast, the electron transport of the thin film transistor of 1 is limited due to its relatively higher LUMO of −3.68 eV.40 Thus, our result presents the first case of a solution-processed ambipolar tetraradical-based transistor with balanced hole and electron mobilities, and the transition from unipolar to ambipolar behaviour induced by controlled aromaticity provides a new design strategy towards solution-processed ambipolar PH-based transistors.
 |
| | Fig. 5 Solution-processed thin-film transistors of 2. (a) Schematic illustration of the transistor. (b) Transfer characteristics for an ambipolar transistor based on the spin-coated film of 2. (c) Transistor output curves of the spin-coated film of 2. | |
Calculated electronic structure and tetraradical character
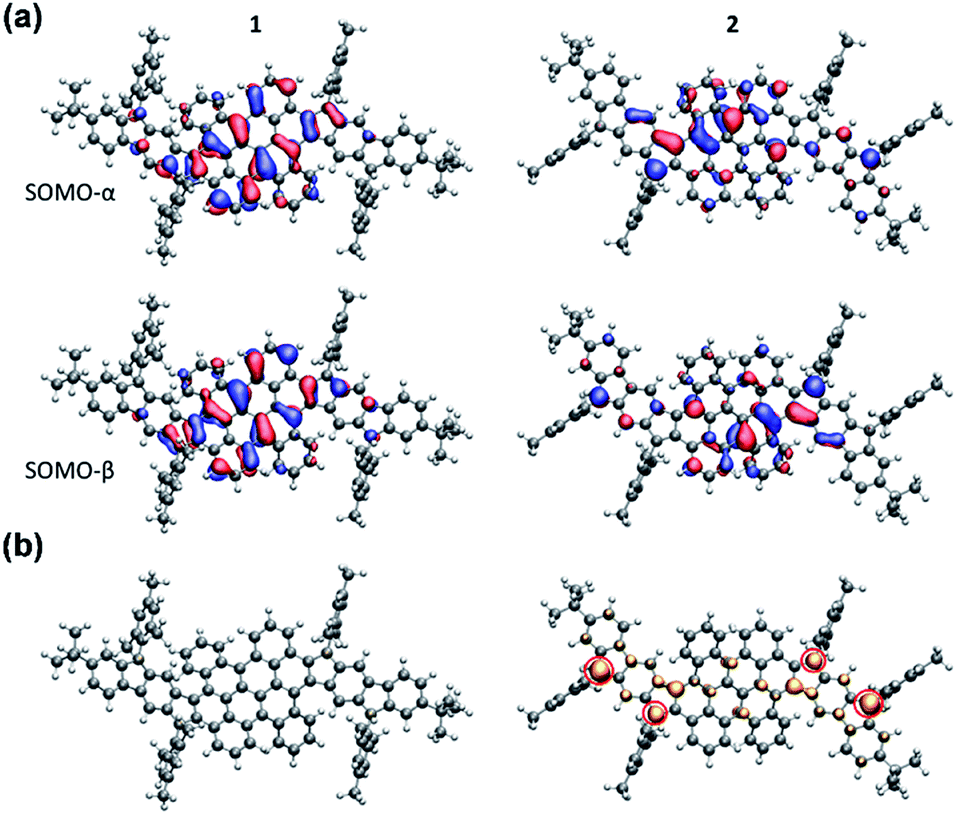
The electronic structure calculations were conducted using the Gaussian 09 suite to further elucidate the electronic properties and to understand the aromaticity of 1 and 2. The biradical character (y0) and tetraradical character (y1) were evaluated from the occupation number of the lowest unoccupied natural orbital (LUNO) and LUNO+1, respectively, obtained from CASSCF(4,4)/6-31G** calculations. PH 1 exhibits essentially a lower degree of biradical or tetraradical features (1: y0 = 0.08; y1 = 0.08) in the ground state and has a large singlet–triplet energy gap (ΔES–T = −9.55 kcal mol−1), suggesting its closed-shell electronic structure. In contrast, PH 2 was found to possess remarkable biradical and tetraradical characters (2: y0 = 0.23; y1 = 0.22). A smaller singlet–triplet energy gap (ΔES–T = −4.2 kcal mol−1) was also predicted from the calculation, which is consistent with the experimental data of 2. As shown in Fig. 6a, the calculated singly occupied molecular orbital (SOMO) profiles of α and β spins of 1 are both homogeneously distributed throughout the fused-bischrysene core, and that of 2 shows a typical disjoint feature for the singlet open-shell molecules.25 The odd electron density of the singlet ground states32 is depicted in Fig. 6b. In agreement with the lower radical character of 1, it shows only minor features in the molecular backbone which are almost not visible when plotting it at the same isovalue as that of 2. In contrast, the odd electron density of tetraradicaloid 2 shows pronounced features with the highest densities located at the four carbon atoms on the cyclopenta rings (marked with a red circle in Fig. 6b), which further implies the resonance structure 2–2 shown in Fig. 1b. Moreover, the delocalization of the unpaired electrons by resonance through the highly twisted π-system endows 2 with thermodynamic stability.41
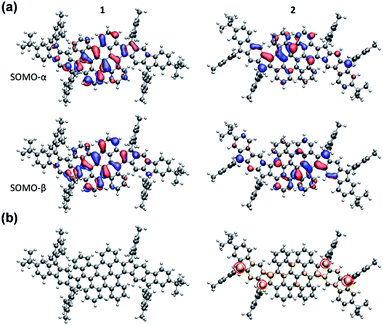 |
| | Fig. 6 (a) SOMOs of 1 and 2 in the singlet state. SOMOs are affected by spin contamination. (b) Odd electron densities of 1 and 2 in the singlet state. Corresponding closed-shell HOMO orbitals are given in the ESI.† | |
Conclusions
In summary, we have demonstrated an efficient synthesis towards the first wave-shaped π-conjugated PH 2 with a highly stable singlet tetraradical feature (y0 = 0.23; y1 = 0.22), which exhibits a narrow optical energy gap (Eoptg = 0.96 eV) and a half-life as long as 91 days under ambient conditions. Meanwhile, wave-shaped closed-shell PH 1 with weakly antiaromatic feature was also achieved by tuning the annulated direction of cyclopenta[b]fluorene moieties. From the crystal structure of 2 as well as supported by the DFT calculations, the curved geometry indeed exerts a significant role in increasing its thermodynamic stability, enabling its application in OFETs with unique ambipolar charge-carrier transport properties. Our studies underline the important effect of the curved geometry on the aromaticity and intrinsic stability of π-extended multiradicaloids, which offer the foundation for future explorations of these functional molecules in organic electronics.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the European Union's Horizon 2020 research and innovation program under grant agreement No. 696656 (Graphene Flagship Core2), the German Research Foundation (DFG) within the Cluster of Excellence “Center for Advancing Electronics Dresden (cfaed)” and EnhanceNano (No. 391979941) as well as the European Social Fund and the Federal State of Saxony (ESF-Project “GRAPHD”, TU Dresden). We thank Prof. Dr Martin Baumgarten (Max Planck Institute for Polymer Research, Germany) for the EPR measurement, and Dr Manfred Wagner (Max Planck Institute for Polymer Research, Germany) for the high temperature NMR measurement. J. J. W. thanks the DFG for funding a Rigaku Oxford Diffraction SuperNova system with a dual source (INST 269/618-1). K. Z. thanks the China Scholarship Council for financial support. The authors acknowledge the use of computational facilities at the Center for information services and high performance computing (ZIH) at TU Dresden.
Notes and references
- M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Acc. Chem. Res., 2015, 48, 267 CrossRef CAS PubMed.
-
H. Dodziuk, in Strained Hydrocarbons: Beyond the van't Hoff and Le Bel Hypothesis, Wiley-VCH, Weinheim, 2009, DOI:10.1002/9783527627134.fmatter.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 1051 RSC.
- R. Rieger and K. Müllen, J. Phys. Org. Chem., 2010, 23, 315 CAS.
- Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843 CrossRef CAS PubMed.
- K. Y. Cheung, X. Xu and Q. Miao, J. Am. Chem. Soc., 2015, 137, 3910 CrossRef CAS PubMed.
- J. Liu, S. Osella, J. Ma, R. Berger, D. Beljonne, D. Schollmeyer, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 8364 CrossRef CAS PubMed.
- C.-N. Feng, M.-Y. Kuo and Y.-T. Wu, Angew. Chem., Int. Ed., 2013, 52, 7791 CrossRef CAS PubMed.
- E. A. Jackson, B. D. Steinberg, M. Bancu, A. Wakamiya and L. T. Scott, J. Am. Chem. Soc., 2007, 129, 484 CrossRef CAS PubMed.
- B. D. Steinberg, E. A. Jackson, A. S. Filatov, A. Wakamiya, M. A. Petrukhina and L. T. Scott, J. Am. Chem. Soc., 2009, 131, 10537 CrossRef CAS PubMed.
- W. E. Barth and R. G. Lawton, J. Am. Chem. Soc., 1966, 88, 380 CrossRef CAS.
- K. Yamamoto, T. Harada, Y. Okamoto, H. Chikamatsu, M. Nakazaki, Y. Kai, T. Nakao, M. Tanaka, S. Harada and N. Kasai, J. Am. Chem. Soc., 1988, 110, 3578 CrossRef CAS.
- M. Rickhaus, M. Mayor and M. Juricek, Chem. Soc. Rev., 2016, 45, 1542 RSC.
- S. Xiao, S. J. Kang, Y. Wu, S. Ahn, J. B. Kim, Y.-L. Loo, T. Siegrist, M. L. Steigerwald, H. Li and C. Nuckolls, Chem. Sci., 2013, 4, 2018 RSC.
- T. Fujikawa, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 7763 CrossRef CAS PubMed.
- M. Gingras, G. Felix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007 RSC.
- R. Kuroda, J. Chem. Soc., Perkin Trans. 2, 1982, 789 RSC.
- P. Ravat, T. Šolomek, M. Rickhaus, D. Häussinger, M. Neuburger, M. Baumgarten and M. Juríček, Angew. Chem., Int. Ed., 2016, 55, 1183 CrossRef CAS PubMed.
- Y.-C. Hsieh, H.-Y. Fang, Y.-T. Chen, R. Yang, C.-I. Yang, P.-T. Chou, M.-Y. Kuo and Y.-T. Wu, Angew. Chem., Int. Ed., 2015, 54, 3069 CrossRef CAS PubMed.
- Z. Zeng, X. Shi, C. Chi, J. T. L. Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578 RSC.
- J. J. Dressler, M. Teraoka, G. L. Espejo, R. Kishi, S. Takamuku, C. J. Gómez-García, L. N. Zakharov, M. Nakano, J. Casado and M. M. Haley, Nat. Chem., 2018, 10, 1134 CrossRef CAS PubMed.
- C. Liu, M. E. Sandoval-Salinas, Y. Hong, T. Y. Gopalakrishna, H. Phan, N. Aratani, T. S. Herng, J. Ding, H. Yamada, D. Kim, D. Casanova and J. Wu, Chem, 2018, 4, 1586 CAS.
- A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata, R. Kishi, Y. Shigeta, M. Nakano, K. Tokunaga, K. Kamada and T. Kubo, J. Am. Chem. Soc., 2013, 135, 1430 CrossRef CAS PubMed.
- Z. Sun, Z. Zeng and J. Wu, Acc. Chem. Res., 2014, 47, 2582 CrossRef CAS PubMed.
- P. Hu, S. Lee, T. S. Herng, N. Aratani, T. P. Gonçalves, Q. Qi, X. Shi, H. Yamada, K.-W. Huang, J. Ding, D. Kim and J. Wu, J. Am. Chem. Soc., 2016, 138, 1065 CrossRef CAS PubMed.
- J. Ma, J. Liu, M. Baumgarten, Y. Fu, Y. Z. Tan, K. S. Schellhammer, F. Ortmann, G. Cuniberti, H. Komber, R. Berger, K. Mullen and X. Feng, Angew. Chem., Int. Ed., 2017, 56, 3280 CrossRef CAS PubMed.
- J. Liu, B. Li, Y. Tan, A. Giannakopoulos, C. Sanchez-Sanchez, D. Beljonne, P. Ruffieux, R. Fasel, X. Feng and K. Müllen, J. Am. Chem. Soc., 2015, 137, 6097 CrossRef CAS PubMed.
- T. A. Chen and R. S. Liu, Chem.–Eur. J., 2011, 17, 8023 CrossRef CAS PubMed.
- S. Nobusue, H. Miyoshi, A. Shimizu, I. Hisaki, K. Fukuda, M. Nakano and Y. Tobe, Angew. Chem., Int. Ed., 2015, 54, 2090 CrossRef CAS PubMed.
- X. Lu, T. Y. Gopalakrishna, H. Phan, T. S. Herng, Q. Jiang, C. Liu, G. Li, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 13052 CrossRef CAS.
- C. K. Frederickson, B. D. Rose and M. M. Haley, Acc. Chem. Res., 2017, 50, 977 CrossRef CAS PubMed.
- G. E. Rudebusch, J. L. Zafra, K. Jorner, K. Fukuda, J. L. Marshall, I. Arrechea-Marcos, G. L. Espejo, R. P. Ortiz, C. J. Gomez-Garcia, L. N. Zakharov, M. Nakano, H. Ottosson, J. Casado and M. M. Haley, Nat. Chem., 2016, 8, 753 CrossRef CAS.
- D. T. Chase, B. D. Rose, S. P. McClintock, L. N. Zakharov and M. M. Haley, Angew. Chem., Int. Ed., 2011, 50, 1127 CrossRef CAS PubMed.
- D. T. Chase, A. G. Fix, S. J. Kang, B. D. Rose, C. D. Weber, Y. Zhong, L. N. Zakharov, M. C. Lonergan, C. Nuckolls and M. M. Haley, J. Am. Chem. Soc., 2012, 134, 10349 CrossRef CAS.
- A. Shimizu, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Miyata and Y. Tobe, Angew. Chem., Int. Ed., 2013, 52, 6076 CrossRef CAS.
- H. Miyoshi, M. Miki, S. Hirano, A. Shimizu, R. Kishi, K. Fukuda, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Nakano and Y. Tobe, J. Org. Chem., 2017, 82, 1380 CrossRef CAS PubMed.
- J. C. Dobrowolski and P. F. J. Lipinski, RSC Adv., 2016, 6, 23900 RSC.
- K. Zhang, P. Wucher, T. Marszalek, M. Babics, A. Ringk, P. W. M. Blom, P. M. Beaujuge and W. Pisula, Chem. Mater., 2018, 30, 5032 CrossRef CAS.
- M. Chikamatsu, T. Mikami, J. Chisaka, Y. Yoshida, R. Azumi, K. Yase, A. Shimizu, T. Kubo, Y. Morita and K. Nakasuji, Appl. Phys. Lett., 2007, 91, 043506 CrossRef.
- K. Zhou, H. Dong, H.-l. Zhang and W. Hu, Phys. Chem. Chem. Phys., 2014, 16, 22448 RSC.
- H. Rath, S. Tokuji, N. Aratani, K. Furukawa, J. M. Lim, D. Kim, H. Shinokubo and A. Osuka, Angew. Chem., Int. Ed., 2010, 49, 1489 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra, 2D NMR spectra, single crystal X-ray diffraction, ACID plots, orbital plots, and cartesian coordinates of B3LYP/6-31G* optimized structures. CCDC 1829452 and 1870938. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05416a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Ke
Zhang
b,
Karl Sebastian
Schellhammer
c,
Yubin
Fu
a,
Ke
Zhang
b,
Karl Sebastian
Schellhammer
c,
Yubin
Fu