
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thorium- and uranium-azide reductions: a transient dithorium-nitride versus isolable diuranium-nitrides†
Jingzhen
Du‡
 a,
David M.
King‡
b,
Lucile
Chatelain
a,
Erli
Lu
a,
Floriana
Tuna
a,
Eric J. L.
McInnes
a,
Ashley J.
Wooles
a,
Laurent
Maron
*c and
Stephen T.
Liddle
*a
a,
David M.
King‡
b,
Lucile
Chatelain
a,
Erli
Lu
a,
Floriana
Tuna
a,
Eric J. L.
McInnes
a,
Ashley J.
Wooles
a,
Laurent
Maron
*c and
Stephen T.
Liddle
*a
aSchool of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: steve.liddle@manchester.ac.uk
bSchool of Chemistry, The University of Nottingham, University Park, Nottingham, NG7 2RD, UK
cLPCNO, CNRS, INSA, Université Paul Sabatier, 135 Avenue de Rangueil, Toulouse 31077, France. E-mail: laurent.maron@irsamc.ups-tlse.fr
First published on 23rd February 2019
Abstract
Molecular uranium-nitrides are now well known, but there are no isolable molecular thorium-nitrides outside of cryogenic matrix isolation experiments. We report that treatment of [M(TrenDMBS)(I)] (M = U, 1; Th, 2; TrenDMBS = {N(CH2CH2NSiMe2But)3}3−) with NaN3 or KN3, respectively, affords very rare examples of actinide molecular square and triangle complexes [{M(TrenDMBS)(μ-N3)}n] (M = U, n = 4, 3; Th, n = 3, 4). Chemical reduction of 3 produces [{U(TrenDMBS)}2(μ-N)][K(THF)6] (5) and [{U(TrenDMBS)}2(μ-N)] (6), whereas photolysis produces exclusively 6. Complexes 5 and 6 can be reversibly inter-converted by oxidation and reduction, respectively, showing that these UNU cores are robust with no evidence for any C–H bond activations being observed. In contrast, reductions of 4 in arene or ethereal solvents gives [{Th(TrenDMBS)}2(μ-NH)] (7) or [{Th(TrenDMBS)}{Th(N[CH2CH2NSiMe2But]2CH2CH2NSi[μ-CH2]MeBut)}(μ-NH)][K(DME)4] (8), respectively, providing evidence unprecedented outside of matrix isolation for a transient dithorium-nitride. This suggests that thorium-nitrides are intrinsically much more reactive than uranium-nitrides, since they consistently activate C–H bonds to form rare examples of Th–N(H)–Th linkages with alkyl by-products. The conversion here of a bridging thorium(IV)-nitride to imido-alkyl combination by 1,2-addition parallels the reactivity of transient terminal uranium(IV)-nitrides, but contrasts to terminal uranium(VI)-nitrides that produce alkyl-amides by 1,1-insertion, suggesting a systematic general pattern of C–H activation chemistry for metal(IV)- vs. metal(VI)-nitrides. Surprisingly, computational studies reveal a σ > π energy ordering for all these bridging nitride bonds, a phenomenon for actinides only observed before in terminal uranium nitrides and uranyl with very short U–N or U–O distances.
Introduction
In recent years there have been major developments in uranium-ligand multiple bonding,1 and arguably none more so than U–N multiple bonds.2 For example, significant advances include bis-, tris-, and tetrakis-imido complexes,3–5 two parent imido complexes,6 reductive cleavage and functionalisation of N2,7 terminal N2 and NO complexes,8 and the emergence of isolable uranium-nitrides.9–11 Such studies are motivated by a desire to prepare actinide congeners of linkages known for decades in the d-block in order to better understand covalency in actinide chemical bonding and to map out intrinsic reactivity trends;12 however, equivalent metal–ligand bonds need to be compared with different actinide elements to elucidate periodic trends. In that regard, thorium analogues demand attention, but there are few mono-imido and bis-imido derivatives, and no parent imidos.13 There are no isolable molecular thorium-nitrides to date, but under cryogenic matrix isolation conditions elegant species such as ThN, F3ThN, NThN, Th(N)2Th, and NThO have been reported.14 A transient zero-valent thorium synthon produced a Th–NH2 linkage from N2,15 but it is not known whether this transformation involves a transient thorium-nitride or if a conventional biomimetic-type (H+/e−) pathway is followed.Building on our prior work on terminal uranium-nitrides,11 we have reported complexes containing parent terminal U = EH (E = N, P, As) and bridging UP(H)U, UPU, and UAsK2 linkages.6a,16a,b,e For thorium, we have prepared complexes exhibiting parent terminal Th![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PH, and bridging ThP(H)Th, ThAs(H)Th, ThAs(H)K, ThPTh and ThAsTh units.16c,d We previously observed that bridging ThPTh and ThAsTh linkages are stable and isolable in triamidoamine derivatives, but conversely UPU and UAsK2 linkages are highly unstable and readily decompose. In contrast, most bridging UNU units seem to be relatively stable,10b,f,g,j,k though none are known with triamidoamine ancillary ligands. Thus, how ThNTh units would fit into these stability patterns is unknown and cannot be predicted with any certainty. So, we have initially sought to prepare dithorium-nitrides, as well as diuranium analogues for comparison, supported by triamidoamines.
PH, and bridging ThP(H)Th, ThAs(H)Th, ThAs(H)K, ThPTh and ThAsTh units.16c,d We previously observed that bridging ThPTh and ThAsTh linkages are stable and isolable in triamidoamine derivatives, but conversely UPU and UAsK2 linkages are highly unstable and readily decompose. In contrast, most bridging UNU units seem to be relatively stable,10b,f,g,j,k though none are known with triamidoamine ancillary ligands. Thus, how ThNTh units would fit into these stability patterns is unknown and cannot be predicted with any certainty. So, we have initially sought to prepare dithorium-nitrides, as well as diuranium analogues for comparison, supported by triamidoamines.
Here, we report the synthesis and characterisation of two triamidoamine uranium- and thorium-azides. Despite marginal differences in the covalent radii of these metals, the uranium complex is a rare example of an actinide molecular square whereas the thorium analogue is a molecular triangle. Reduction of the uranium-azide complex generates diuranium-nitrides, with two charge states of a UNU core being accessible, and interchangeable, with no evidence of C–H activation chemistry even under photolytic conditions. However, a ThNTh complex could not be isolated. Instead, the isolation of two ThN(H)Th complexes, which is an unprecedented linkage in thorium chemistry and rare in f-block chemistry generally,7c,d suggests that a dithorium-nitride complex is transiently formed but activates any available C–H bonds, be they part of arene solvent or in the absence of arenes the triamidoamine ligand. The ThNTh unit therefore appears to be highly reactive in a triamidoamine ligand environment, and its instability contrasts to ThPTh and ThAsTh congeners and UNU analogues and also suggests a systematic pattern of metal oxidation state-dependent C–H activation reactivity for actinide-nitrides. Surprisingly, computational studies reveal a σ > π energy ordering for the bridging nitride linkages in this study, a phenomenon so far only found in terminal uranium-nitrides and uranyl complexes with very short U–N/–O distances.
Results and discussion
Preparation of uranium- and thorium-azide complexes
In order to prepare MNM linkages we pursued a M–N3 reduction approach using the TrenDMBS {N(CH2CH2NSiMe2But)3}3− ligand as this was anticipated to be sterically open enough to allow any nitrides to bridge, whereas the bulkier TrenTIPS {N(CH2CH2NSiPri3)3}3− variant stabilises ThPTh and ThAsTh linkages but terminal UN for uranium. Accordingly, treatment of [M(TrenDMBS)(I)] (M = U, 1; Th, 2)17 with excess NaN3 or KN3 affords [{M(TrenDMBS)(μ-N3)}n] (M = U, n = 4, 3; Th, n = 3, 4) as green-yellow and colourless crystalline solids after work-up in isolated yields of 35 and 86%, respectively, Schemes 1 and 2.18 The combined characterisation data support the formulations of 3 and 4, in particular the ATR-IR spectra of 3 and 4 both exhibit strong absorptions at 2131 cm−1, which is characteristic of actinide-bound bridging-azide ligands.10b,c,19 The magnetic moment of 3, Fig. 1,18 in the solid-state at 298 K is 5.52 μB per molecule decreasing smoothly to 0.78 μB at 2 K (3.29 and 0.41 μB per uranium ion in 3, respectively) and tending to zero as expected for a tetrametallic UIV complex, since UIV usually has a magnetic singlet ground state at low temperature.2,20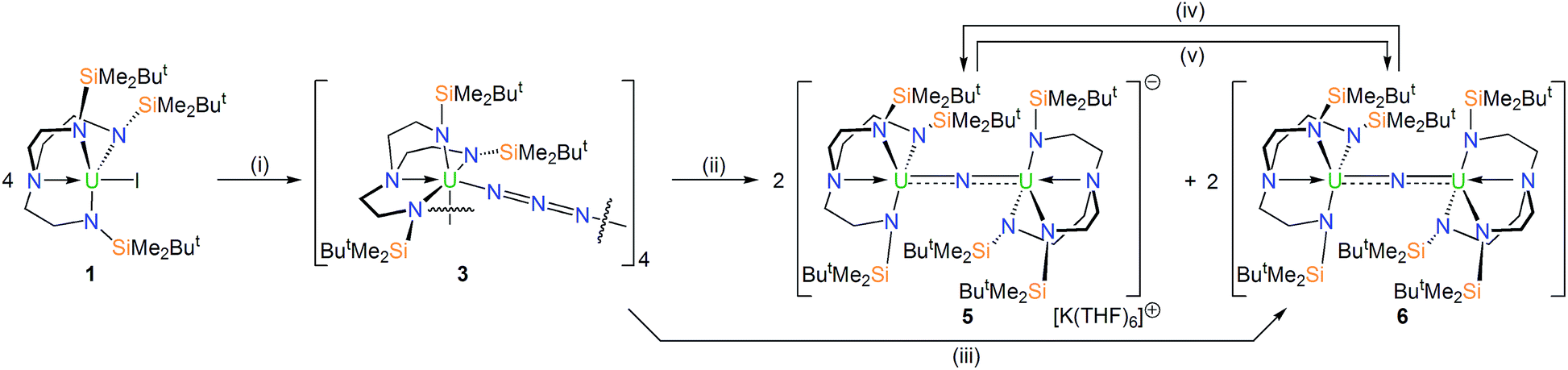 | ||
| Scheme 1 Synthesis of 3, 5 and 6 from 1. Reagents and conditions: (i) 4NaN3, THF, –4NaI; (ii) 4KC8, THF, –4C8, –2KN3, –2N2; (iii) hν, 125 W Hg-lamp, 7 h, toluene, –5N2; (iv) KC8, THF, –C8; (v) AgBPh4, toluene, –KBPh4, –Ag0, –6THF. | ||
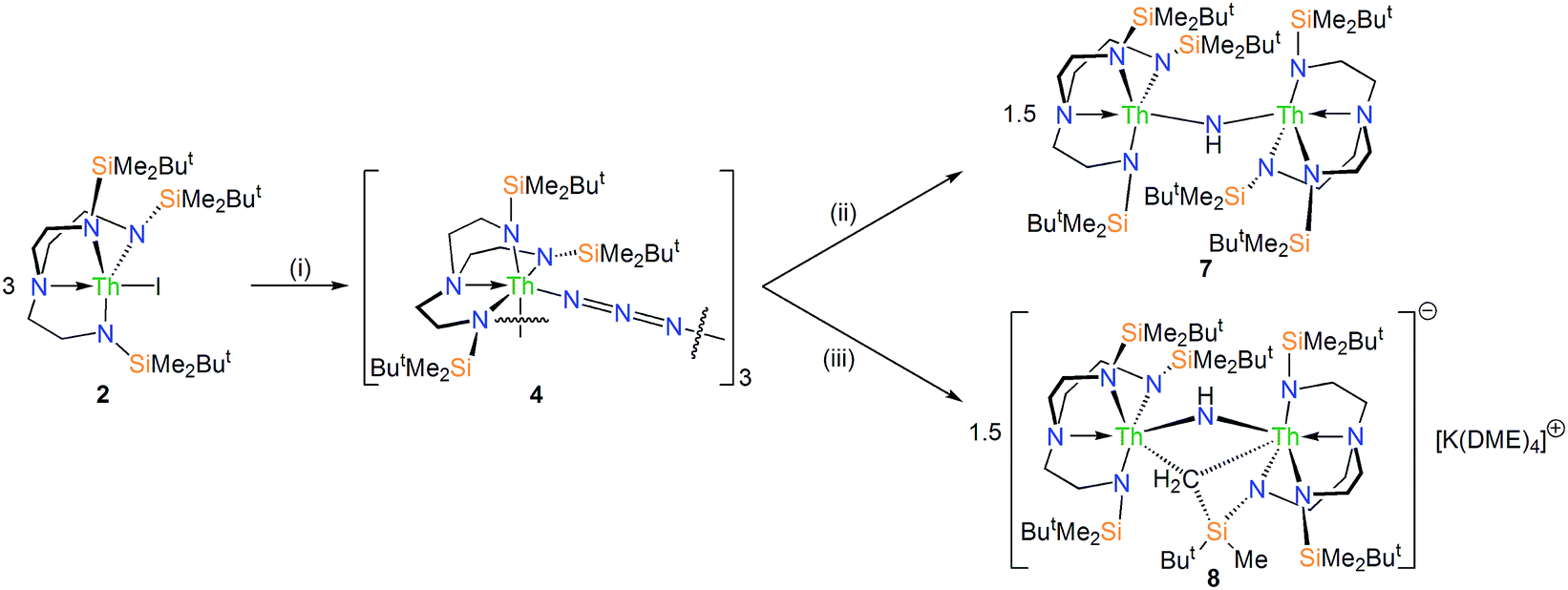 | ||
| Scheme 2 Synthesis of 4, 7 and 8 from 2. Reagents and conditions: (i) 3KN3, THF, –3KI; (ii) 3KC8, benzene or toluene, –3C8, –1.5 KN3, –1.5N2, –KCH2Ph or –KC6H5; (iii) 3KC8, THF or DME, –1.5KN3, –1.5N2. | ||
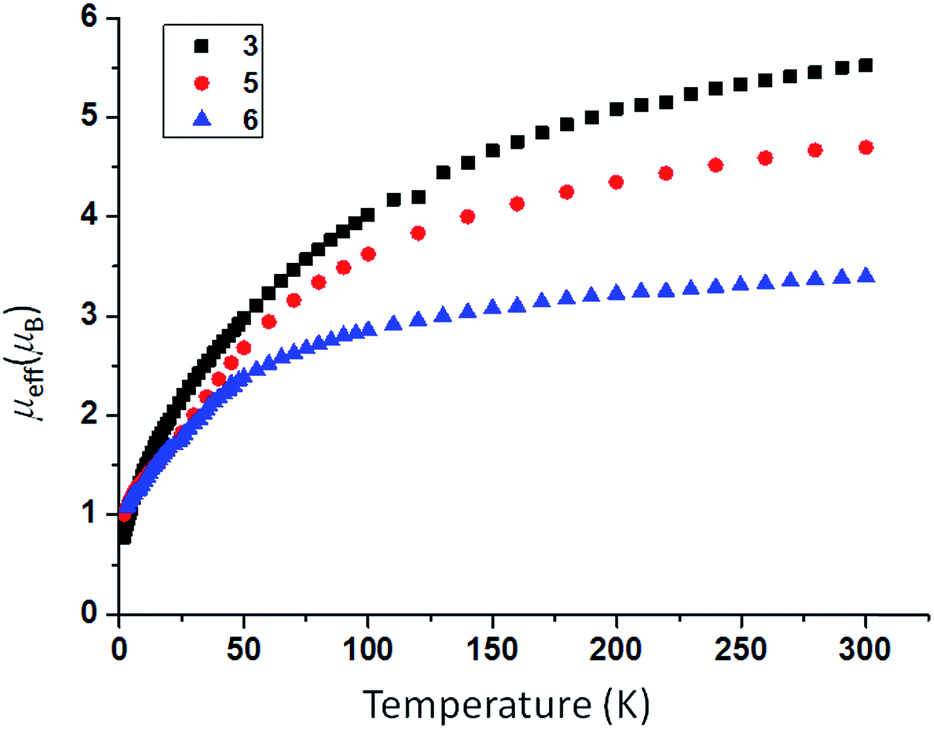 | ||
| Fig. 1 Variable temperature SQUID magnetic moment data for 3 (black squares), 5 (red circles), and 6 (blue triangles) over the range 2–300 K. | ||
In order to confirm the formulations of 3 and 4 we determined their solid-state structures, Fig. 2a and 3a. Surprisingly, although 1 and 2, and chloride analogues, are monomers, 3 and 4 are tetrameric and trimeric in the solid-state. Such molecular squares and triangles21 are relatively rare motifs in actinide chemistry,10b,c,22 since polymeric formulations tend to dominate19f as there is not usually a strong orbitally-driven geometric preference for M–L angles that generate squares or triangles. However, it would appear that when the C3v symmetry of TrenDMBS is lowered to Cs the cleft that opens up allows two azides to enter the coordination sphere of uranium in 3 at an approximate right angle (∼85°) whereas for the larger thorium in 4 the azides approach at a slightly more acute N–Th–N angle (∼79°), which seems to be enough to switch from tetramer to trimer. It would seem that the N–Th–N angle can close at the larger metal without as much inter-azide clashing due to longer Th–N bonds placing the azides further apart from one another, which accounts for the aggregation states of 3 and 4. The U– and Th–Nazide distances in 3 and 4 are longer than in terminal azide complexes,19 and we note that they are longer when trans to a TrenDMBS amide centre (3, 2.540(5); 4, 2.609(8) Å av.) than amine centre (3, 2.425(5); 4, 2.478(7) Å av.), possibly implying a trans-influence. All other bond lengths are within normal ranges and do not suggest any strong activation of the azides.
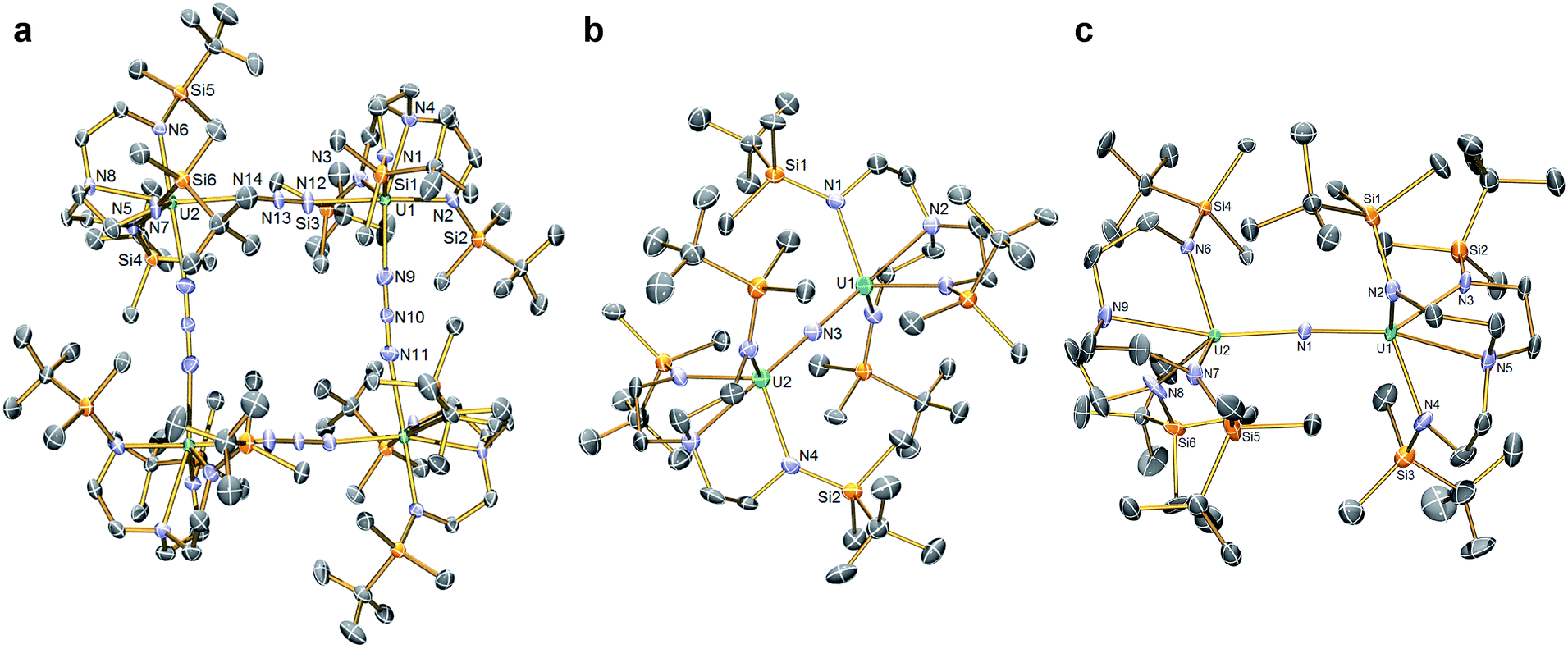 | ||
| Fig. 2 Molecular structures of (a) 3, (b) the anion component of 5, (c) 6. Structures are depicted with selective symmetry-unique labels, 40% probability displacement ellipsoids, and hydrogen atoms, minor disorder components, and lattice solvent molecules are omitted for clarity. | ||
 | ||
| Fig. 3 Molecular structures of (a) 4, (b) 7, (c) the anion component of 8. Structures are depicted with selective labels, 40% probability displacement ellipsoids and hydrogen atoms (except N–H and H2CSi), minor disorder components, and lattice solvent molecules are omitted for clarity. | ||
Uranium-azide reductions and characterisation of nitride products
With 3 and 4 in-hand we investigated their reduction chemistries. The reaction of 3 with KC8 always produces the diuranium(IV/IV)-nitride [{U(TrenDMBS)}2(μ-N)][K(THF)6] (5) and the mixed-valence diuranium(IV/V)-nitride [{U(TrenDMBS)}2(μ-N)] (6) in ∼42% overall yield (total yield by uranium content, 28% 5 and 14% 6 estimated from integration of 1H NMR data), with concomitant elimination of N2 and KN3.18 The ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 varies from 77:23 to 50:50 as the KC8 ratio is varied from 5 to 3 equivalents but is independent of the solvent used (benzene, toluene, THF, DME). Other KC8 ratios gave intractable product mixtures. Complexes 5 and 6 can be separated by fractional crystallisation, however we find that 6 can be cleanly prepared in 45% isolated crystalline yield by photolysis of 3 with a 125 W Hg-lamp for 7 hours. Gratifyingly, 6, or a known mixture of 5 and 6, can be reduced with KC8 to give solely 5, and 5 or a known mixture of 5 and 6 can be oxidised with AgBPh4 to give exclusively 6, Scheme 1. Interestingly, 5 and 6 are always formed in the respective reduction and oxidation reactions, irrespective of the amounts of KC8 (2–8 equiv.) and AgBPh4 (2–3 equiv.) used, and we found no evidence for further reductions or oxidations, respectively.
:6 varies from 77:23 to 50:50 as the KC8 ratio is varied from 5 to 3 equivalents but is independent of the solvent used (benzene, toluene, THF, DME). Other KC8 ratios gave intractable product mixtures. Complexes 5 and 6 can be separated by fractional crystallisation, however we find that 6 can be cleanly prepared in 45% isolated crystalline yield by photolysis of 3 with a 125 W Hg-lamp for 7 hours. Gratifyingly, 6, or a known mixture of 5 and 6, can be reduced with KC8 to give solely 5, and 5 or a known mixture of 5 and 6 can be oxidised with AgBPh4 to give exclusively 6, Scheme 1. Interestingly, 5 and 6 are always formed in the respective reduction and oxidation reactions, irrespective of the amounts of KC8 (2–8 equiv.) and AgBPh4 (2–3 equiv.) used, and we found no evidence for further reductions or oxidations, respectively.
As expected, the characterisation data for 5 and 6 are distinct, reflecting their UIV/UIV and UIV/UV formulations, respectively. The 1H NMR spectra of 5 and 6 span the ranges +96 to −34 and +25 to −13 ppm, respectively, reflecting their 5f2/2vs. 5f2/1 natures. The solution Evans method (298 K) gives magnetic moments of 4.0 and 3.5 μB per molecule of 5 and 6, whereas the solid-state magnetic moments, Fig. 1,18 are 4.70 and 3.39 μB, respectively. These values decrease to 1.00 and 1.07 μB at 2 K, respectively. For 5 the respective values per uranium ion are 3.39 (298 k) and 0.74 μB (2 K), which per ion are slightly higher than the corresponding values for 3 reflecting their nitride and azide formulations.16b,23 The data for 6 are consistent with its UIV/UV combination,2,20 where the UV ion has a magnetic doublet ground state at all temperatures, and anti-ferromagnetic U–U coupling is suggested by a maximum at ∼60 K in the χ vs. T plot of 5.2,24,25 The presence of UV in 6 is unequivocally confirmed by EPR spectroscopy (S- and X-bands) at 5 K, Fig. 4,18 which reveals two similar sets of rhombic g-values with geff = 3.13, 0.95, 0.50, and 2.70, 0.74, 0.43; these data reflect the presence of two conformational isomers in the solid-state structure of 6 due to positional disorder of three of the six SiMe2But groups, and we note that the effective g-values of spin–orbit doubles are extremely sensitive to small changes in structure.26 An electrochemical irreversible one-electron process at E1/2 −1.4 V (vs. [Fc(Cp)2]0/+1) for the [UIV/UV]/[UIV/UIV]− redox couple is found, Fig. 5, which contrasts to 3 and 4 which do not exhibit any electrochemical events in the solvent-accessible window of 2.5 to −3.0 V. The chemical inter-conversion of 5 and 6 suggests the presence of robust UNU cores, as was found for [{U(NBut[3,5-Me2C6H3])3}2(μ-N)]n (n = +1, 0, −1) which can exist in three charge states,10f but the irreversible electrochemical behaviour may reflect structural changes in the UNU angles of 5 compared to 6 (see below).
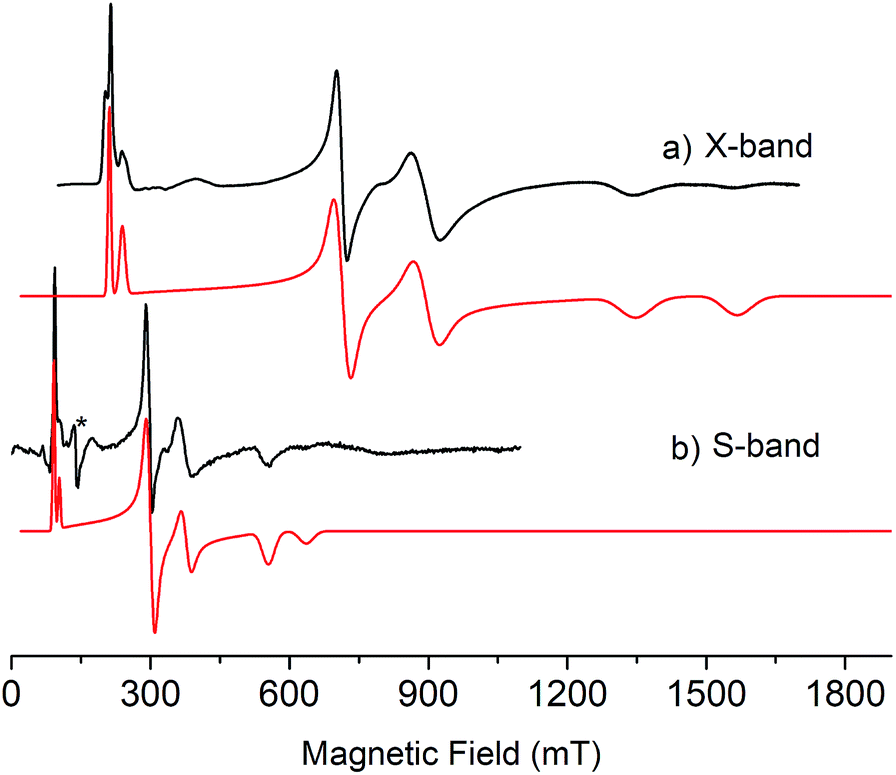 | ||
| Fig. 4 (a) X-band (9.39 GHz) and (b) S-band (3.87 GHz) EPR spectra of polycrystalline samples of 6 at 5 K. Black lines are experimental data and red lines are simulations. * = cavity signal. | ||
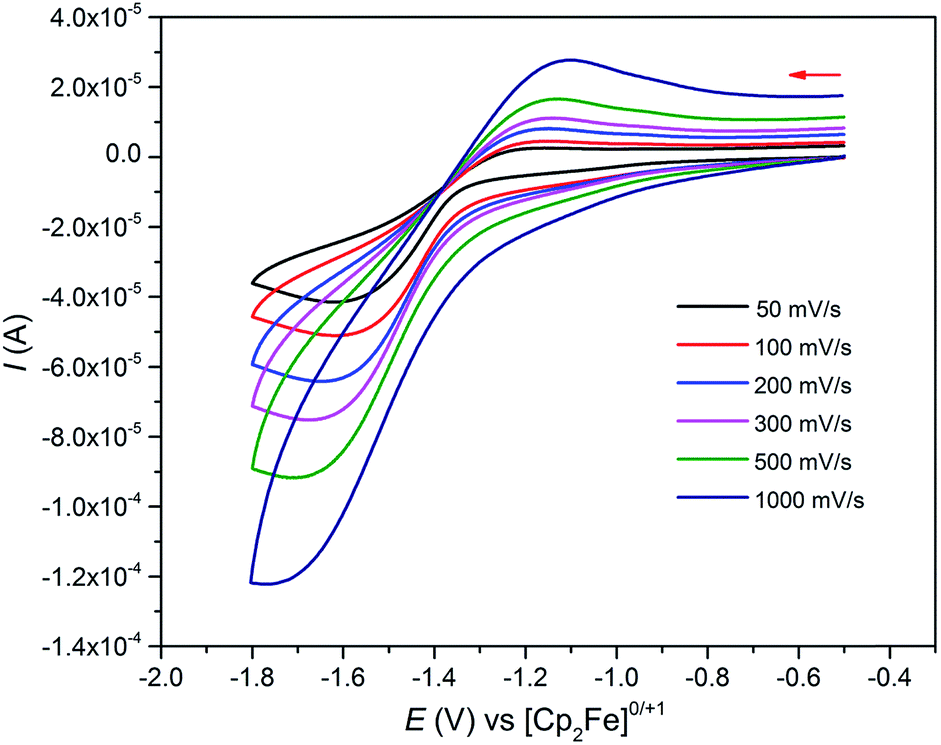 | ||
| Fig. 5 Cyclic voltammogram of 0.1 M 6 in THF at selected sweep rates (0.5 M [N(nBu)4][BArF4] supporting electrolyte) vs. [Fe(Cp)2]+/0 showing a single irreversible redox process. Test solutions of 5 or 6 in THF with [N(nBu)4][BArF4] under identical conditions show no evidence of stability issues. | ||
The solid-state structures of 5 and 6 were determined, Fig. 2b and c, revealing structural differences reflecting their different oxidation state formulations. The anion component of 5 resides on a crystallographic 3-fold rotation axis and therefore the U–N–U and Nnitride–U–Namine angles are rigorously 180°, however in 6 the U–N–U angle is bent at 161.2(2)°. In 5 the U1/2–N nitride, amide, and amine distances are 2.0648(2), 2.343(3), and 2.733(5) Å, respectively; these distances reflect the bridging nature of the nitride, that is consistent with other UNU distances,10 the anionic, charge rich nature of this component, since the amide distances are longer than usual for Tren-UIV complexes, and possibly a strong trans-influence from the nitride since the amine distances are quite long like in related ThPTh and ThAsTh complexes,16c,d,e but in contrast to the situation found in a related terminal Tren-uranium(VI)-nitride.11b In 6 the U–Nnitride distances are now inequivalent at 2.081(5) and 2.136(5) Å, and the U–Namide and U–Namine distances (av. 2.287(5) and 2.649(5) Å) are now shorter than in 5, presumably reflecting the neutral formulation of 6 and a reduced nitride trans-influence since the Nnitride–U–Namine angles are now ∼159°.
Thorium-azide reductions and characterisation of imido products
The reduction of 4, in contrast to 3, gives two distinct products, in addition to N2 and KN3, that are exclusive to the solvent media, Scheme 2.18 When aromatic solvents (benzene, toluene) are used instead of securing [{Th(TrenDMBS)}2(μ-N)][K] the parent imido [{Th(TrenDMBS)}2(μ-NH)] (7) is isolated in 52% crystalline yield. When ethereal solvents (THF, DME) are used the cyclometallated tuck-in-tuck-over,27 parent imido [{Th(TrenDMBS)}{Th(N[CH2CH2NSiMe2But]2CH2CH2NSi[μ-CH2]MeBut)}(μ-NH)][K(DME)4] (8) is isolated in 46% crystalline yield. The 1H NMR spectrum of 7 reveals a resonance at 5.55 ppm that corresponds to one N–H proton; this resonance disappears when the reaction is conducted in D8-toluene, suggesting the source of H is aromatic solvent with K–C6H5/–CH2Ph as by-products. In-line with this, 7 does not react with benzyl potassium. The presence of the N–H group is confirmed by a broad absorption at 3390 cm−1 in the ATR-IR spectrum of 7. The 1H NMR spectrum of 8 is now more complicated due to the desymmetrisation of one of the TrenDMBS ligands, but the N–H proton resonance can be observed at 5.39 ppm.The molecular structures of 7 and 8 were determined, Fig. 3b and c. In 7 the Th–Nimido–Th angle is 145.96(19)° and the imido adopts a trigonal planar geometry in contrast to ThP(H)Th and ThAs(H)Th linkages16c,d that are T-shaped reflecting a sp2-NH dianion but p-orbital-dominated bonding of PH and AsH dianions. The Th–Nimido distances of 2.331(4) and 2.312(4) Å are similar to the Th–Namide distances (∼2.330 Å) and ∼0.3 Å longer than ThNR terminal imido bonds.13 In 8 the Th–Nimido–Th angle is 120.9(7)°, reflecting the presence of the tuck-in-tuck-over cyclometallate enforcing a constrained C–Th–N–Th four-membered ring. Despite this, the Th–Nimido distances of 2.309(15) and 2.264(15) Å are essentially the same as those in 7. The Th–C distances of 2.88(2) and 2.78(2) Å are long, as observed in other Th–TrenDMBS cyclometallates.28
Discussion of the contrasting nature of uranium- and thorium-nitride reactivities
The formation and isolation of 5 and 6, especially the latter under photolytic conditions, is significant because photolysis of terminal uranium(VI)-nitrides10i,11b and a uranium(IV)-nitride generated transiently by reduction6b have all resulted in C–H activation of ancillary ligands to produce alkyl-amides or a parent imido-alkyl, respectively. However, 5 and 6 contain quite robust, redox inter-convertible UNU cores, and when N2 is eliminated from 3 the nitride secures stabilisation by two uranium Lewis acid centres rather than instigating C–H activation reactions.Although reduction of 4 does not lead to the isolation of dithorium-nitrides, the isolation of 7 and 8 is instructive. Like 5 and 6, reduction of an azide precursor, 4, results in the formation of a ThNTh unit, except in both cases this is protonated. For 7, the potassium from reduction has been exchanged for a proton suggesting that a transient nitride [{Th(TrenDMBS)}2(μ-N)]− (9−) has C–H activated arene solvents, and we note the yield of 7 does not vary when changing the solvent from benzene to toluene. Likewise, in the formation of 8 a μ-NH unit forms again, but this time in the absence of deprotonatable arene solvent, and with the potassium cation sequestered by ethereal solvent, the transient nitride has C–H activated the TrenDMBS to form a tuck-in-tuck-over cyclometallate27,28 liberating the requisite proton to form the imido group. We deduce that the transient ThNTh unit is highly reactive, certainly as reactive as Th–CR3 species that also cyclometallate Tren-ligands due to proximity and entropy effects,28 and more reactive than the stable UNU units in 5 and 6. Indeed, DFT calculations18 on [{U(TrenDMBS)}2(μ-N)]− (5−) and 9− suggest that the latter contains more polar and ionic metal-nitride linkages, but importantly the frontier molecular orbitals that principally comprise the ThNTh bonding interactions in 9− are destabilised by ∼1.1 (σ) and ∼0.4 (π) eV compared to the corresponding UNU orbitals of 5−. This results in a more basic, effectively superbasic, nucleophilic nitride in 9−, as experimentally inferred by the isolation of 5versus7 and 8, and shown computationally where the anion of 8 is found to be 19.8 kcal mol−1 more stable than 9− and formed via a transition state with an experimentally accessible barrier of 16.4 kcal mol−1.18
The formation of 8 parallels the reactivity of a transiently formed uranium(IV)-nitride that undergoes ligand C–H activation to give a cyclometallated (alkyl) ligand and a uranium(IV) parent imido functionality by 1,2-addition,6b since the alternative of 1,1-insertion to give an alkyl-amide as observed for uranium(VI)-nitrides10i,11b would, formally, implicate the likely very unfavourable formation of sub-valent thorium ions. This suggests that there may be a systematic pattern of C–H activation reactions for MVIvs. MIV ions in this context generally, Fig. 6.
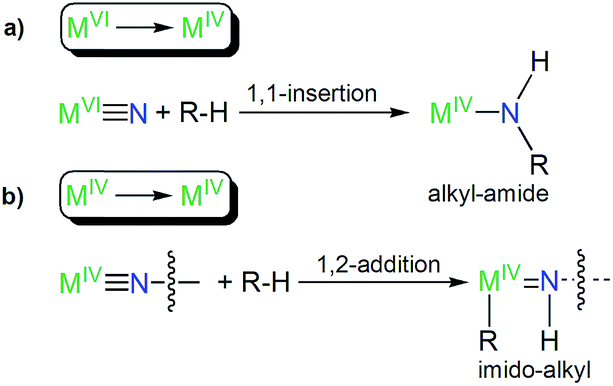 | ||
| Fig. 6 Systematic types of emerging reactivity patterns of actinide-nitrides suggested by this work and ref. 6b, 10i and 11b. (a) C–H activation of a R–H bond by a metal(VI)-nitride produces a metal(IV)-alkyl-amide by 1,1-insertion. (b) C–H activation of a R–H bond by a terminal or bridging metal(IV)-nitride produces a bridging or terminal metal(IV)-imido-alkyl by 1,2-addition; the R group can be coordinated to the metal(IV) ion or be eliminated with a group 1 metal. | ||
Lastly, we note that the σ-bonding component of the UIVNUIV and ThIVNThIV (9− is depicted in Fig. 7) bonds reported here are higher in energy than the two π-contributions.18 This scenario is usually only observed in terminal uranium nitrides and uranyl with short UN and UO bonds.9,11 Why this is the case here, and whether this is related to the formation of 7 and 8, is currently unclear and work is on-going to rationalise this observation.
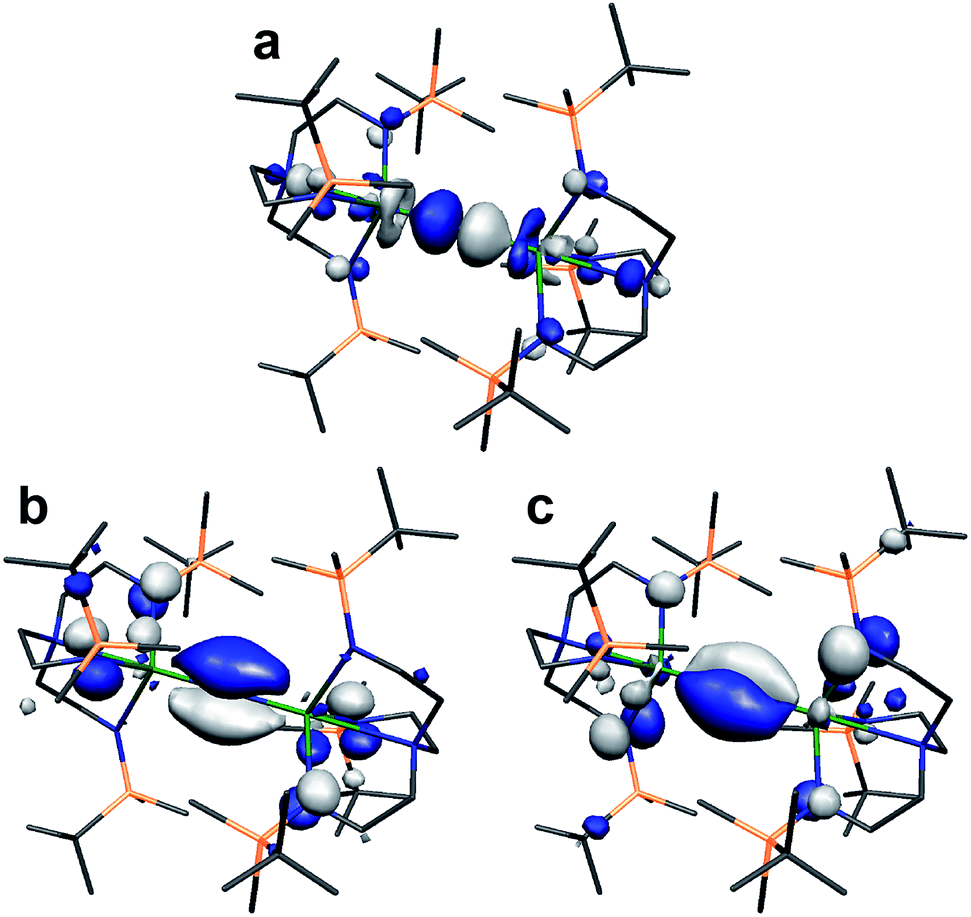 | ||
| Fig. 7 Kohn Sham molecular orbital representations of the principal frontier orbitals of 9− with hydrogen atoms omitted for clarity. (a) HOMO (365a, −1.884 eV); (b) HOMO−7 (358a, −2.598 eV); (c) HOMO−8 (357a, −2.599 eV). The computed thorium-nitride distances are 2.1373 and 2.1373 Å and the computed Th–Nnitride–Th angle is 180°. | ||
Conclusions
In conclusion, the synthesis of two uranium- and thorium-azide complexes has provided rare examples of actinide molecular square and triangle complexes. We have prepared two diuranium-nitride complexes in different charge states; these UNU complexes are quite robust, and do not engage in C–H activation chemistry, even under photolytic conditions, unlike terminal uranium(IV/VI)-nitrides. Attempts to prepare a dithorium-nitride complex resulted in the isolation of two parent imido complexes, in-line with the paucity of isolable molecular thorium-nitrides to date. However, the two dithorium-imido products suggest for the first time that reduction of thorium-azides can generate nitrides, and provides evidence that a transient and highly reactive dithorium-nitride is formed, but that this linkage is highly basic and nucleophilic so is capable of activation C–H bonds of arene solvent or the supporting TrenDMBS ligand. The contrasting stabilities of UNU and putative ThNTh units reported here may be related to the general tendency of uranium to engage in more covalent bonding than thorium, on a like-for-like basis. The results here suggest a general pattern of actinide-nitride reactivity where metal(IV)-nitrides, bridging or terminal, activate C–H bonds to produce imido-alkyl combinations, whereas metal(VI)-nitrides produce alkyl-amide linkages, which can be related to the range of accessible oxidation states of these ions during reactions. Lastly, computational studies surprisingly reveal a σ > π energy ordering for the bridging nitride linkages in this study, a phenomenon so far only found in terminal uranium-nitrides and uranyl complexes with very short U–N/–O distances.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge funding and support from the UK Engineering and Physical Sciences Research Council (grants EP/K024000/1, EP/M027015/1, and EP/P001386/1), European Research Council (grant CoG612724), Royal Society (grant UF110005), the National EPSRC UK EPR Facility, The Universities of Manchester and Nottingham, and the UK National Nuclear Laboratory. The X-ray crystallographic coordinates for structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 1868610–1868615.† All other data are available from the corresponding authors upon request.Notes and references
- (a) T. W. Hayton, Dalton Trans., 2010, 39, 1145 RSC; (b) T. W. Hayton, Chem. Commun., 2013, 49, 2956 RSC; (c) M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137 CrossRef CAS PubMed; (d) H. S. La Pierre and K. Meyer, Prog. Inorg. Chem., 2014, 58, 303 CAS.
- S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 8604 CrossRef CAS PubMed.
- (a) T. W. Hayton, J. M. Boncella, B. L. Scott, P. D. Palmer, E. R. Batista and P. J. Hay, Science, 2005, 310, 1941 CrossRef CAS PubMed; (b) T. W. Hayton, J. M. Boncella, B. L. Scott, E. R. Batista and P. J. Hay, J. Am. Chem. Soc., 2006, 128, 10549 CrossRef CAS PubMed; (c) T. W. Hayton, J. M. Boncella, B. L. Scott and E. R. Batista, J. Am. Chem. Soc., 2006, 128, 12622 CrossRef CAS PubMed; (d) L. P. Spencer, P. Yang, B. L. Scott, E. R. Batista and J. M. Boncella, J. Am. Chem. Soc., 2008, 130, 2930 CrossRef CAS PubMed.
- (a) N. H. Anderson, S. O. Odoh, Y. Yao, U. J. Williams, B. A. Schaefer, J. J. Kiernick, A. J. Lewis, M. D. Goshert, P. E. Fanwick, E. J. Schelter, J. R. Walensky, L. Gagliardi and S. C. Bart, Nat. Chem., 2014, 6, 919 CrossRef CAS PubMed; (b) N. H. Anderson, H. Yin, J. J. Kiernicki, P. E. Fanwick, E. J. Schelter and S. C. Bart, Angew. Chem., Int. Ed., 2015, 54, 9386 CrossRef CAS PubMed.
- N. H. Anderson, J. Xie, D. Ray, M. Zeller, L. Gagliardi and S. C. Bart, Nat. Chem., 2017, 9, 850 CrossRef CAS PubMed.
- (a) D. M. King, J. McMaster, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake and S. T. Liddle, J. Am. Chem. Soc., 2014, 136, 5619 CrossRef CAS PubMed; (b) K. C. Mullane, H. Ryu, T. Cheisson, L. N. Grant, J. Y. Park, B. C. Manor, P. J. Carroll, M.-H. Baik, D. J. Mindiola and E. J. Schelter, J. Am. Chem. Soc., 2018, 140, 11335 CrossRef CAS PubMed.
- (a) M. Falcone, L. Chatelain and M. Mazzanti, Angew. Chem., Int. Ed., 2016, 55, 4074 CrossRef CAS PubMed; (b) M. Falcone, C. E. Kefalidis, R. Scopelliti, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed., 2016, 55, 12290 CrossRef CAS PubMed; (c) M. Falcone, L. Chatelain, R. Scopelliti, I. Živković and M. Mazzanti, Nature, 2017, 547, 332 CrossRef CAS PubMed; (d) M. Falcone, L. N. Poon, F. Fadaei Tirani and M. Mazzanti, Angew. Chem., Int. Ed., 2018, 57, 3697 CrossRef CAS PubMed.
- (a) W. J. Evans, S. A. Kozimor and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 14264 CrossRef CAS PubMed; (b) N. A. Siladke, K. R. Meihaus, J. W. Ziller, M. Fang, F. Furche, J. R. Long and W. J. Evans, J. Am. Chem. Soc., 2012, 134, 1243 CrossRef CAS PubMed.
- D. M. King and S. T. Liddle, Coord. Chem. Rev., 2014, 266–267, 2 CrossRef CAS.
- (a) I. Korobkov, S. Gambarotta and G. P. A. Yap, Angew. Chem., Int. Ed., 2002, 41, 3433 CrossRef CAS; (b) W. J. Evans, S. A. Kozimor and J. W. Ziller, Science, 2005, 309, 1835 CrossRef CAS PubMed; (c) W. J. Evans, K. A. Miller, J. W. Ziller and J. Greaves, Inorg. Chem., 2007, 46, 8008 CrossRef CAS PubMed; (d) G. Nocton, J. Pécaut and M. Mazzanti, Angew. Chem., Int. Ed., 2008, 47, 3040 CrossRef CAS PubMed; (e) A. R. Fox and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 5716 CrossRef CAS PubMed; (f) A. R. Fox, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 3250 CrossRef CAS PubMed; (g) S. Fortier, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 6888 CrossRef CAS PubMed; (h) T. K. Todorova, L. Gagliardi, J. R. Walensky, K. A. Miller and W. J. Evans, J. Am. Chem. Soc., 2010, 132, 12397 CrossRef CAS PubMed; (i) R. K. Thomson, T. Cantat, B. L. Scott, D. E. Morris, E. R. Batista and J. L. Kiplinger, Nat. Chem., 2010, 2, 723 CrossRef CAS PubMed; (j) C. Camp, J. Pécaut and M. Mazzanti, J. Am. Chem. Soc., 2013, 135, 12101 CrossRef CAS PubMed; (k) N. Tsoureas, A. F. R. Kilpatrick, C. J. Inman and F. G. N. Cloke, Chem. Sci., 2016, 7, 4624 RSC.
- (a) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717 CrossRef CAS PubMed; (b) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2013, 5, 482 CrossRef CAS PubMed; (c) P. A. Cleaves, D. M. King, C. E. Kefalidis, L. Maron, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 10412 CrossRef CAS PubMed; (d) D. M. King, P. A. Cleaves, A. J. Wooles, B. M. Gardner, N. F. Chilton, F. Tuna, W. Lewis, E. J. L. McInnes and S. T. Liddle, Nat. Commun., 2016, 7, 13773 CrossRef CAS; (e) P. A. Cleaves, C. E. Kefalidis, B. M. Gardner, F. Tuna, E. J. L. McInnes, W. Lewis, L. Maron and S. T. Liddle, Chem.–Eur. J., 2017, 23, 2950 CrossRef CAS PubMed.
- (a) N. Kaltsoyannis, Inorg. Chem., 2013, 52, 3407 CrossRef CAS PubMed; (b) M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2014, 257, 394 CrossRef.
- (a) A. Haskel, T. Straub and M. S. Eisen, Organometallics, 1996, 15, 3773 CrossRef CAS; (b) W. Ren, G. Zi, D.-C. Fang and M. D. Walter, J. Am. Chem. Soc., 2011, 133, 13183 CrossRef CAS; (c) W. Ren, G. Zi and M. D. Walter, Organometallics, 2012, 31, 672 CrossRef CAS; (d) N. L. Bell, L. Maron and P. L. Arnold, J. Am. Chem. Soc., 2015, 137, 10492 CrossRef CAS PubMed; (e) M. E. Garner, S. Hohloch, L. Maron and J. Arnold, Organometallics, 2016, 35, 2915 CrossRef CAS.
- (a) G. P. Kushto, P. F. Souter and L. Andrews, J. Chem. Phys., 1998, 108, 7121 CrossRef CAS; (b) M. Zhou and L. Andrews, J. Chem. Phys., 1999, 111, 11044 CrossRef CAS; (c) X. Wang and L. Andrews, Dalton Trans., 2009, 9260 RSC.
- I. Korobkov, S. Gambarotta and G. P. A. Yap, Angew. Chem., Int. Ed., 2003, 42, 4958 CrossRef CAS PubMed.
- (a) B. M. Gardner, G. Balázs, M. Scheer, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 4484 CrossRef CAS PubMed; (b) B. M. Gardner, G. Balázs, M. Scheer, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2015, 7, 582 CrossRef CAS PubMed; (c) E. P. Wildman, G. Balázs, A. J. Wooles, M. Scheer and S. T. Liddle, Nat. Commun., 2016, 7, 12884 CrossRef CAS PubMed; (d) E. P. Wildman, G. Balázs, A. J. Wooles, M. Scheer and S. T. Liddle, Nat. Commun., 2017, 8, 14769 CrossRef CAS PubMed; (e) T. M. Rookes, B. M. Gardner, G. Balázs, M. Gregson, F. Tuna, A. J. Wooles, M. Scheer and S. T. Liddle, Angew. Chem., Int. Ed., 2017, 56, 10495 CrossRef CAS PubMed.
- P. Roussel, N. W. Alcock, R. Boaretto, A. J. Kingsley, I. J. Munslow, C. J. Sanders and P. Scott, Inorg. Chem., 1999, 38, 3651 CrossRef CAS.
- See the ESI† for further details.
- (a) N. A. Budantseva, G. B. Andreev, A. M. Fedoseev and M. Y. Antipin, Russ. J. Coord. Chem., 2003, 29, 265 CrossRef CAS; (b) I. Castro-Rodriguez and K. Meyer, J. Am. Chem. Soc., 2005, 127, 11242 CrossRef CAS PubMed; (c) S. Fortier, G. Wu and T. W. Hayton, Dalton Trans., 2010, 39, 352 RSC; (d) O. Bénaud, J.-C. Berthet, P. Thuéry and M. Ephritikhine, Inorg. Chem., 2011, 50, 12204 CrossRef PubMed; (e) W. Ren, E. Zhou, B. Fang, G. Hou, G. Zi, D.-C. Fang and M. D. Walter, Angew. Chem., Int. Ed., 2014, 53, 11310 CrossRef CAS; (f) M. J. Monreal, L. A. Seaman, G. S. Goff, R. Michalczyk, D. E. Morris, B. L. Scott and J. L. Kiplinger, Angew. Chem., Int. Ed., 2016, 55, 3631 CrossRef CAS PubMed.
- (a) I. Castro-Rodríguez and K. Meyer, Chem. Commun., 2006, 1353 RSC; (b) D. R. Kindra and W. J. Evans, Chem. Rev., 2014, 114, 8865 CrossRef CAS PubMed.
- P. H. Dinolfo, S.-S. Sun and J. T. Hupp, Encyclopedia of Supramolecular Chemistry, ed. J. L. Atwood and J. W. Steed, CRC Press, 2004, p. 395 Search PubMed.
- (a) J. M. Manriquez, P. J. Fagan, T. J. Marks, S. H. Vollmer, C. S. Day and V. W. Day, J. Am. Chem. Soc., 1979, 101, 5075 CrossRef CAS; (b) P. C. Leverd, T. Arliguie, M. Ephritikhine, M. Nierlich, M. Lance and J. Vigner, New J. Chem., 1993, 17, 769 CAS; (c) D. L. Clark, J. C. Gordon, J. C. Huffman, J. G. Watkin and B. D. Zwick, New J. Chem., 1995, 19, 495 CAS; (d) J. L. Kiplinger, J. A. Pool, E. J. Schelter, J. D. Thompson, B. L. Scott and D. E. Morris, Angew. Chem., Int. Ed., 2006, 45, 2036 CrossRef CAS PubMed; (e) C. P. Larch, F. G. N. Cloke and P. B. Hitchcock, Chem. Commun., 2008, 82 RSC; (f) M. J. Monreal, S. I. Khan, J. L. Kiplinger and P. L. Diaconescu, Chem. Commun., 2011, 47, 9119 RSC.
- D. P. Halter, H. S. La Pierre, F. W. Heinemann and K. Meyer, Inorg. Chem., 2014, 53, 8418 CrossRef CAS PubMed.
- UIV–UIV magnetic exchange coupling is rare: (a) J. D. Rinehart, T. D. Harris, S. A. Kozimor, B. M. Bartlett and J. R. Long, Inorg. Chem., 2009, 48, 3382 CrossRef CAS PubMed; (b) B. S. Newell, A. K. Rappé and M. P. Shores, Inorg. Chem., 2010, 49, 1595 CrossRef CAS PubMed; (c) O. P. Lam, F. W. Heinemann and K. Meyer, Chem. Sci., 2011, 2, 1538 RSC; (d) B. M. Gardner, J. C. Stewart, A. L. Davis, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9265 CrossRef CAS PubMed; (e) B. M. Gardner, D. M. King, F. Tuna, A. J. Wooles, N. F. Chilton and S. T. Liddle, Chem. Sci., 2017, 8, 6207 RSC.
- R. K. Rosen, R. A. Andersen and N. M. Edelstein, J. Am. Chem. Soc., 1990, 112, 4588 CrossRef CAS.
- A. Formanuik, A.-M. Ariciu, F. Ortu, R. Beekmeyer, A. Kerridge, F. Tuna, E. J. L. McInnes and D. P. Mills, Nat. Chem., 2017, 9, 578 CrossRef CAS PubMed.
- B. M. Gardner, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, J. Am. Chem. Soc., 2009, 131, 10388 CrossRef CAS PubMed.
- B. M. Gardner, W. Lewis, A. J. Blake and S. T. Liddle, Organometallics, 2015, 34, 2386 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental and computational details. CCDC 1868610–1868615. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05473h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |