
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen peroxide as a hydride donor and reductant under biologically relevant conditions†
Yamin
Htet
 ab,
Zhuomin
Lu
d,
Sunia A.
Trauger
c and
Andrew G.
Tennyson
*def
ab,
Zhuomin
Lu
d,
Sunia A.
Trauger
c and
Andrew G.
Tennyson
*def
aWyss Institute for Biologically Inspired Engineering, Harvard University, Cambridge, MA 02138, USA
bJohn A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, MA 02138, USA
cHarvard FAS Small Molecule Mass Spectrometry Facility, Harvard University, Cambridge, MA 02138, USA
dDepartment of Chemistry, Clemson University, Clemson, SC 29634, USA. E-mail: atennys@clemson.edu
eDepartment of Materials Science and Engineering, Clemson University, Clemson, SC 29634, USA
fCenter for Optical Materials Science and Engineering Technologies, Anderson, SC 29625, USA
First published on 14th December 2018
Abstract
Some ruthenium–hydride complexes react with O2 to yield H2O2, therefore the principle of microscopic reversibility dictates that the reverse reaction is also possible, that H2O2 could transfer an H− to a Ru complex. Mechanistic evidence is presented, using the Ru-catalyzed ABTS˙− reduction reaction as a probe, which suggests that a Ru–H intermediate is formed via deinsertion of O2 from H2O2 following coordination to Ru. This demonstration that H2O2 can function as an H− donor and reductant under biologically-relevant conditions provides the proof-of-concept that H2O2 may function as a reductant in living systems, ranging from metalloenzyme-catalyzed reactions to cellular redox homeostasis, and that H2O2 may be viable as an environmentally-friendly reductant and H− source in green catalysis.
Introduction
Hydrogen peroxide and its descendant reactive oxygen species (ROS) have historically been viewed in biological systems nearly exclusively as oxidants that damage essential biomolecules,1–3 but recent reports have shown that H2O2 can also perform essential signaling functions at low concentrations.4,5 Due to the damage caused by high ROS concentrations, significant efforts have been devoted to developing antioxidants that catalytically reduce ROS and other oxidizing radicals.6 Catalytic antioxidants require other species to serve as terminal reductants (ascorbate, glutathione, NADH, etc.) and, under certain conditions, depletion of endogenous reductants by a catalytic antioxidant can induce, rather than prevent, oxidative stress.7–9 A catalytic antioxidant that could harness H2O2 or other ROS as terminal reductants, akin to catalase and superoxide dismutase,10,11 would preclude adverse oxidative damage.Mechanistic studies on the Ru-catalyzed aerobic oxidation of alcohols have provided evidence that O2 can insert into a Ru–H bond and be subsequently released as H2O2 (Scheme 1A, red arrows).12,13 The principle of microscopic reversibility14 therefore dictates that it is mechanistically equivalent for H2O2 to react with a Ru complex and be subsequently released as O2 with concomitant formation of a Ru–H intermediate (Scheme 1A, blue arrows). In this reaction, H2O2 is oxidized to O2, and the 2e− liberated in this oxidation will be transferred to Ru in the form of a hydride (H−) ligand. The forward and reverse reactions, 1,2-insertion of O2 into Ru–H (red arrows) and β-hydride elimination (i.e., 1,2-deinsertion of O2)15 from Ru–OOH (blue arrows), proceed through a common transition state. The forward and reverse reactions in Ru–H + O2 ⇌ Ru–OOH could alternatively proceed via 1,1-insertion and 1,1-deinsertion of O2, respectively, but Ru–OOH would need to rearrange to a higher-energy species for this to be mechanistically feasible (vide infra).
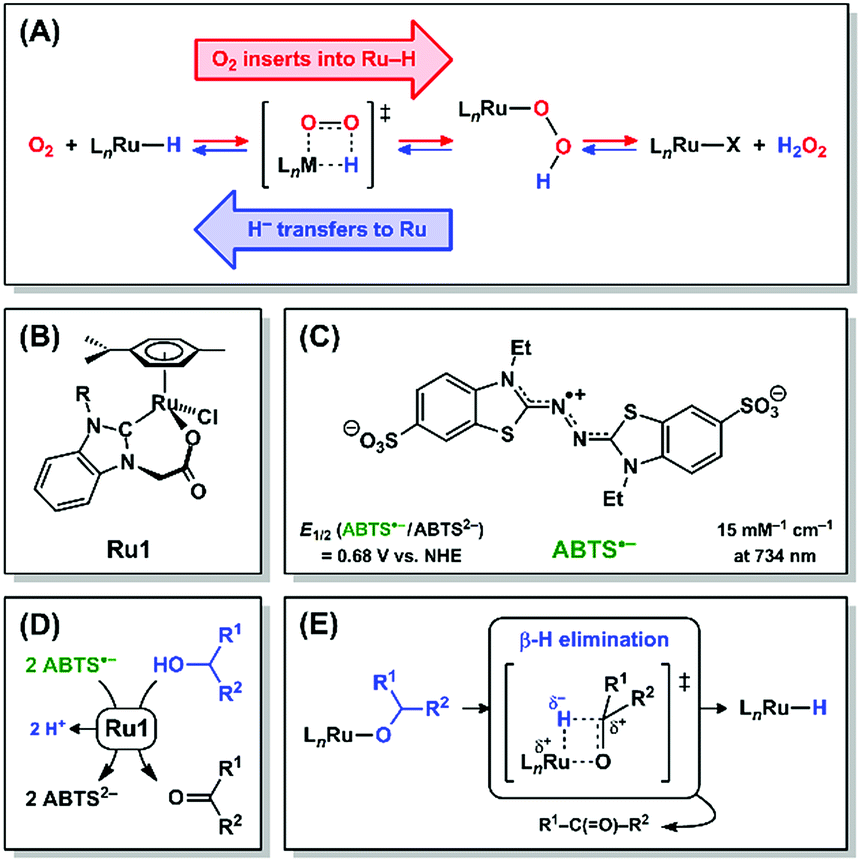 | ||
| Scheme 1 (A) 1,2-Insertion of O2 into Ru–H (red arrows), its microscopic reverse β-hydride elimination (i.e., 1,2-deinsertion of O2) from Ru–OOH (blue arrows), and their common transition state. Structures of (B) Ru1 and (C) ABTS˙−. Net reaction (D) and mechanism (E) for Ru1-catalyzed ABTS˙− reduction with an alcohol. | ||
Ruthenium complexes comprising H− and O2 ligands both bound to the same metal have been previously observed,16–20 which suggests that the formation of a Ru–H intermediate and O2 (Scheme 1A, blue arrows) can, under certain circumstances, be thermodynamically and/or kinetically more favorable than the insertion of O2 into the Ru–H bond (Scheme 1A, red arrows). Alternatively, if direct observation of a Ru–H intermediate produced by deinsertion of O2 from Ru–OOH is experimentally infeasible, due to existing at too low of a concentration or for too short of a lifetime, then the presence of Ru–H can be demonstrated inferentially via its chemical reactivity.
We recently reported a Ru complex (Ru1, Scheme 1B) that catalyzed the 1e− reduction of ABTS˙− (Scheme 1C) with biologically-relevant alcohols (ascorbate, glucose, NAD+, etc., Scheme 1D) as terminal reductants.21,22 Importantly, ABTS˙− undergoes 1e− reduction at a potential (E1/2 = +0.68 V vs. NHE) comparable to the ROS generated during oxidative stress,23–25 therefore the one-electron redox reactivity of ABTS˙− can thermodynamically approximate the corresponding reactivity of oxidizing species in living systems. Mechanistic studies by us26 provided evidence that ABTS˙− was reduced by a Ru–H intermediate formed via β-hydride elimination from a Ru–alkoxide (Scheme 1E). Because the individual steps leading up to Ru–H formation are well-understood and ABTS˙− concentration can be quantified at μM levels,27 we hypothesized that kinetic analysis of Ru1-catalyzed ABTS˙− reduction with H2O2 would reveal if any Ru–H intermediate had formed. Herein we demonstrate that H2O2 functions as a terminal reductant for Ru1-catalyzed ABTS˙− reduction in aerobic, aqueous solution. Moreover, we provide the first mechanistic evidence that H2O2 can function as an H− donor to generate the Ru–H intermediate that reduces ABTS˙−, in a manner consistent with β-hydride elimination (i.e., 1,2-deinsertion of O2) from Ru–OOH (Scheme 1A, blue arrows).
Results and discussion
Peroxide terminal reductant ability is unique to H2O2
By itself, Ru1 cannot reduce ABTS˙− to ABTS2− in phosphate buffered saline (PBS, pH 7.4),28 consistent with the fact that a catalyst cannot be consumed or produced by the net reaction (Fig. 1A(i)). Subsequent addition of H2O2 caused a decrease in radical absorbance at 734 nm (Fig. 1A(ii)) accompanied by an increase in absorbance at 340 nm, consistent with the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 conversion of ABTS˙− to ABTS2− (Fig. S1†). In the absence of Ru1, the addition of H2O2 afforded no change in ABTS˙− concentration, which demonstrated that the reactivity of H2O2 as a reductant was dependent on the catalyst being present. The oxidation of H2O2 to O2 (E°′ = −0.28 V at pH 7) by ABTS˙− (E1/2 = +0.68 V) is thermodynamically favorable,24,29 therefore the lack of reactivity between H2O2 and ABTS˙− in the absence of Ru1 demonstrated that the reduction of ABTS˙− with H2O2 is under kinetic control. When H2O2 was added to a PBS solution containing Ru1 and ABTS2−, no ABTS˙− formation was observed, which indicated that Ru1 does not exhibit peroxidase-like reactivity and does not convert H2O2 into other ROS capable of oxidizing ABTS2−.
:1 conversion of ABTS˙− to ABTS2− (Fig. S1†). In the absence of Ru1, the addition of H2O2 afforded no change in ABTS˙− concentration, which demonstrated that the reactivity of H2O2 as a reductant was dependent on the catalyst being present. The oxidation of H2O2 to O2 (E°′ = −0.28 V at pH 7) by ABTS˙− (E1/2 = +0.68 V) is thermodynamically favorable,24,29 therefore the lack of reactivity between H2O2 and ABTS˙− in the absence of Ru1 demonstrated that the reduction of ABTS˙− with H2O2 is under kinetic control. When H2O2 was added to a PBS solution containing Ru1 and ABTS2−, no ABTS˙− formation was observed, which indicated that Ru1 does not exhibit peroxidase-like reactivity and does not convert H2O2 into other ROS capable of oxidizing ABTS2−.
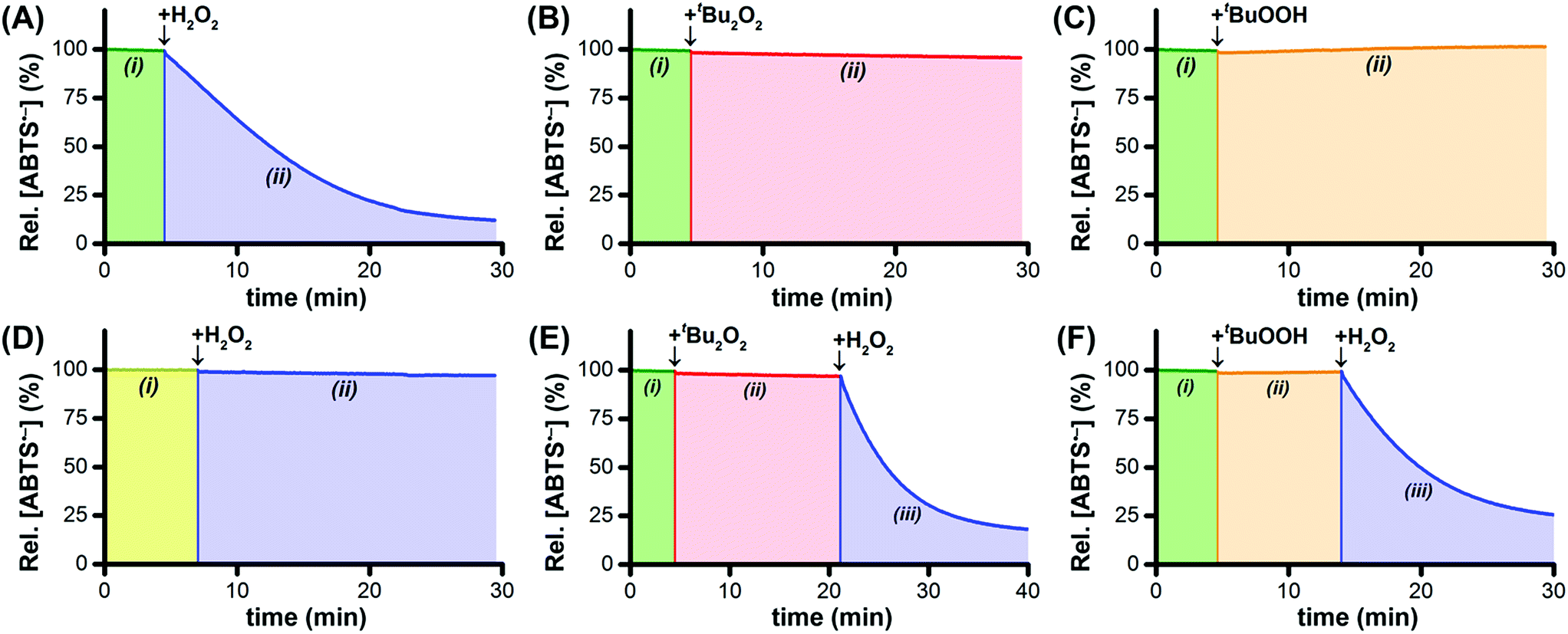 | ||
| Fig. 1 Plots of relative [ABTS˙−] vs. time which show that Ru1 by itself does not reduce ABTS˙− in aqueous solution (A–F, i). In PBS solutions containing Ru1 and ABTS˙−, the addition of H2O2 caused the radical absorbance to decrease (A, ii), but no ABTS˙− reduction occurred following the addition of t-Bu2O2 (B, ii) or t-BuOOH (C, ii). In contrast, the addition of H2O2 to a solution of Ru1 and ABTS˙− in pure water afforded no radical reduction (D, ii). Although neither t-Bu2O2 (E, ii) nor t-BuOOH (F, ii) enabled Ru1-catalyzed ABTS˙− reduction, the subsequent addition of H2O2 (E and F, iii) did produce decreases in radical absorbance. Conditions: [Ru1]0 = 5 μM, [ABTS˙−]0 = 50 μM, [ABTS2−]0 = 100 μM, [H2O2] or [t-Bu2O2] or [t-BuOOH] = 100 μM, PBS (pH 7.4), 25 °C. | ||
To test our hypothesis that H2O2 functioned as the terminal reductant for Ru1-catalyzed ABTS˙− reduction via β-hydride elimination from a Ru–OOH species (i.e., Scheme 1A, blue arrows), we explored the reactivity of other peroxides with this system. Di-tert-butyl peroxide (t-Bu2O2) cannot react with Ru1 to form a Ru–peroxo species and, if our hypothesis were correct, should therefore be incapable of serving as the terminal reductant. Gratifyingly, no ABTS˙− reduction occurred following the addition of t-Bu2O2 to Ru1 and ABTS˙− in PBS (Fig. 1B(ii)), which validated this expectation. Conversely, tert-butyl hydroperoxide (t-BuOOH) can form a Ru–peroxo species, i.e., Ru–OOR, but because the distal oxygen carries a tert-butyl group instead of a hydrogen atom, β-hydride elimination cannot occur, which would prevent oxidation of the peroxide and therefore preclude reduction of ABTS˙−. This expectation was validated with the observation (Fig. 1C(ii)) that radical absorbance did not decrease when a PBS solution containing Ru1 and ABTS˙− was treated with t-BuOOH, providing evidence that no Ru–H intermediate was generated. This result was also consistent with our previous findings that primary and secondary alcohols (e.g., EtOH, i-PrOH, etc.) could function as terminal reductants for Ru1-catalyzed ABTS˙− reduction, whereas a tertiary alcohol like t-BuOH could not,22,26 which reflected the ability (or inability) of the Ru–OR species to undergo β-hydride elimination (i.e., Scheme 1E).
Addition of H2O2 to Ru1 and ABTS˙− in pure H2O (instead of PBS) afforded no change in radical absorbance (Fig. 1D(ii)), which was consistent with our prior observations that a proton acceptor must be present for Ru1-catalyzed ABTS˙− reduction to occur and revealed that deprotonation of H2O2 was an essential step of the catalytic cycle leading up to the formation of the radical reducing species. Although neither t-Bu2O2 nor t-BuOOH afforded any decrease in radical absorbance (Fig. 1E(ii) and F(ii)), the subsequent addition of H2O2 to PBS solutions containing Ru1 and either t-Bu2O2 or t-BuOOH did result in ABTS˙− reduction (Fig. 1E(iii) and F(iii)). The absence of ABTS˙− reduction following the addition of t-Bu2O2 or t-BuOOH alone was therefore not due to catalyst deactivation, but was instead due to the fact that neither t-Bu2O2 nor t-BuOOH could function as terminal reductants. Collectively, these findings provided further evidence that, for ABTS˙− reduction to occur, a Ru–hydroperoxo species must first be formed, which in turn must be capable of undergoing β-hydride elimination to generate the Ru–H intermediate necessary for ABTS˙− reduction.
Reductant ability is not derived from H2O2 bond homolysis
Notably, the O–O bond dissociation energy (BDE) values for t-Bu2O2 (40.3 kcal mol−1) and t-BuOOH (46.1 kcal mol−1) are both significantly lower than the corresponding value of 49.5 kcal mol−1 for H2O2 (Fig. 2).30–32 Therefore, if the ability of H2O2 to reduce ABTS˙− derived from homolytic cleavage of its O–O bond at some point in the mechanism, then the addition of both t-Bu2O2 and t-BuOOH should have afforded a decrease in ABTS˙− concentration due to their lower activation barriers of O–O bond homolysis. Likewise, the O–H and O–C BDE values for t-BuOOH (84.2 and 73.6 kcal mol−1, respectively) were both lower than the value of 87.8 kcal mol−1 for the O–H BDE in H2O2.33–35 Consequently, if the mechanism of ABTS˙− reduction by H2O2 proceeded through an O–H bond homolysis step, then a decrease in radical absorbance should have occurred following the addition of t-BuOOH, given that homolytic cleavage of its O–H and O–C bonds would both have lower activation energies than O–H bond homolysis in H2O2.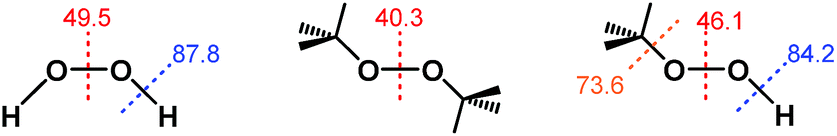 | ||
| Fig. 2 Bond-dissociation energies (in kcal mol−1) for H2O2, t-Bu2O2, and t-BuOOH. | ||
Furthermore, if homolysis of an O–O, O–H, or O–C bond in H2O2, t-Bu2O2, or t-BuOOH did occur, it would transiently generate one or more radical species of sufficiently strong oxidizing power (e.g., E°′ = +2.32 V for HO˙, +0.89 V for O2˙− derived from HO2˙ at pH 7, etc.) that oxidation of ABTS2− to ABTS˙− (E1/2 = +0.68 V) would occur.24,29 Similarly, although there are examples in the literature in which a Ru complex reacts with H2O2 or t-BuOOH to generate a Ru(IV)–, Ru(V)–, or Ru(VI)–oxo species,36–40 these high-valent Ru–oxo species undergo 1e− or 2e− reduction at potentials significantly higher than the ABTS˙−/ABTS2− redox couple.41–43 However, no ABTS˙− formation was observed when PBS solutions containing ABTS2− were treated with H2O2, t-Bu2O2, or t-BuOOH, either in the presence or absence of Ru1.
Collectively, the results from the experiments using H2O2, t-Bu2O2, and t-BuOOH provided strong evidence that the mechanism for the Ru1-catalyzed reduction of ABTS˙− with H2O2 does not involve any strongly oxidizing radicals or high-valent Ru–oxo species, but instead proceeds via heterolytic cleavage of the O–H bonds.
Reduction by H2O2 releases O2 gas
The volume of O2 gas evolved from these experiments was too small to measure directly because H2O2 was consumed from 3.00 mL reaction volumes. For example, reduction of 50 μM ABTS˙− to 50 μM ABTS2− would consume 25 μM H2O2 (each H− can reduce 2 ABTS˙−) and produce 75 nmol of O2 (25 μM × 3.00 mL), corresponding to 1.8 μL at 298 K and 1 atm.When the reaction volume was increased 1000-fold – when 100 μM H2O2 was added to a solution of 50 μM ABTS˙− and 5 μM Ru1 in 3.00 L of PBS – 1.8 ± 0.1 mL of O2 gas (72 ± 2 μmol) were collected, corresponding to a 96 ± 3% theoretical yield (Fig. S2†). UV/vis spectroscopic analysis of 3.0 mL aliquots taken from this reaction before and 30 min after the addition of H2O2 (Fig. S3†) revealed that [ABTS˙−] had decreased by 44 ± 1 μM (×3.00 L = 132 ± 3 μmol) and [ABTS2−] had increased by 42 ± 1 μM (×3.00 L = 126 ± 3 μmol).44 Thus, the consumption of 1.0 equiv. of ABTS˙− was accompanied by the formation of 0.95 ± 0.03 equiv. of ABTS2− and the generation of 0.54 ± 0.02 equiv. of O2 gas.
The evolution of O2 gas provided additional evidence that no O–O bond cleavage in H2O2 was occurring during the Ru1-catalyzed reduction of ABTS˙− with H2O2. Furthermore, the release of 1 equiv. of O2 for the reduction of every 2 equiv. of ABTS˙− demonstrated that each molecule of H2O2 functioned as a 2e− reductant, that both O-atoms from H2O2 ended up in O2, and that both H-atoms from H2O2 ended up (ultimately) as H+.
Ru1 and horseradish peroxidase compete for H2O2
Horseradish peroxidase (HRP) catalytically oxidizes ABTS2− to ABTS˙− using H2O2 as the terminal oxidant,45 which provided us with a convenient method to probe the concentrations of ABTS2− and H2O2 indirectly. To a solution of 5 μM Ru1 and 50 μM ABTS˙− in PBS (Fig. 3(i)) was added 20 μM H2O2, which produced a gradual decrease in [ABTS˙−] over the course of 45 min (Fig. 3(ii)). If our hypothesis were correct, that Ru1-catalyzes the reduction of ABTS˙− to ABTS2− using H2O2 as the terminal reductant, then this decrease in [ABTS˙−] over time should be accompanied by an increase in [ABTS2−] and a decrease in [H2O2]. The concentration of 20 μM H2O2 was therefore deliberately chosen to be insufficient to achieve quantitative ABTS˙− reduction, such that when the decrease in [ABTS˙−] had ceased, the solution would contain ABTS˙−, ABTS2−, and Ru1, but no H2O2. Addition of 10 nM HRP to this solution produced no increase in ABTS˙− absorbance (Fig. 3(iii)), which confirmed that all of the H2O2 had been consumed in the previous step. To determine if this lack of HRP-induced ABTS˙− formation was due to enzyme deactivation, a second 20 μM aliquot of H2O2 was then added (Fig. 3(iv)). The resulting gradual increase in [ABTS˙−] demonstrated that the lack of reactivity in the previous step was due to depletion of the terminal reductant and not enzyme deactivation. Furthermore, the formation of ABTS˙− in Fig. 3(iv) could only occur if there were ABTS2− present at the end of Fig. 3(iii). This, in turn, provided evidence that the decrease in ABTS˙− absorbance observed in Fig. 3(ii) was caused specifically by the one-electron reduction of ABTS˙− to ABTS2−. | ||
| Fig. 3 Plot of relative [ABTS˙−] vs. time following the sequential addition of 5 μM Ru1 (i), 20 μM H2O2 (ii), 10 nM HRP (iii), 20 μM H2O2 (iv), and then 50 mM EtOH (v). Conditions: [ABTS˙−]0 = 50 μM, PBS (pH 7.4), 25 °C. | ||
The concentration of H2O2 added in Fig. 3(iv) was equal to that added in Fig. 3(ii), therefore the HRP had access to a sufficient amount of terminal oxidant to oxidize all of the ABTS2− produced during Fig. 3(ii) and restore the concentration of ABTS˙− to the initial value in Fig. 3(i). However, the [ABTS˙−] in Fig. 3(iv) reached a plateau 12 min after the addition of H2O2 that was well below this initial value. The fact that the relative [ABTS˙−] did not increase back to 100% indicated that some other species was present that was competing with HRP for the H2O2. Likewise, the plateau in [ABTS˙−] indicated that ABTS˙− formation and reduction had both ceased, which was consistent with the H2O2 supply being depleted (i.e., no terminal oxidant available to HRP, no terminal reductant available to Ru1). Moreover, the rate of ABTS˙− formation with Ru1 present (i.e., Fig. 3(iv)) was significantly slower than when no Ru1 was present. Collectively, these results provided strong evidence that HRP-catalyzed ABTS2− oxidation, with H2O2 as the terminal oxidant, and Ru1-catalyzed ABTS˙− reduction, with H2O2 as the terminal reductant, were both occurring simultaneously and then both ceased when all the H2O2 had been consumed. After this plateau was reached, 50 mM EtOH was then added and produced an immediate decrease in radical absorbance that led to quantitative ABTS˙− reduction within 30 min (Fig. 3(v)), which demonstrated that Ru1 was still present and catalytically competent.
β-Hydride elimination from Ru–OOH is proposed mechanism
We propose the mechanism for Ru1-catalyzed ABTS˙− reduction with H2O2 is conserved with the previously reported mechanism in which EtOH is the terminal reductant.26 Addition of Ru1 to a solution of ABTS˙− and ABTS2− in PBS will result in rapid exchange46 of the Cl ligand with ABTS2−, ABTS˙−, and H2O (Scheme 2, orange arrows) to afford [LnRu-Ared]1−, [LnRu-Aox], and [LnRu-OH2]1+, respectively. Because ABTS2− inhibits Ru1-catalyzed ABTS˙− reduction by binding to Ru,26 kinetic experiments were performed with an excess of ABTS2− present to ensure reproducible data. Substitution of ABTS2− in [LnRu-Ared]1− (step 1) and ABTS˙− in [LnRu-Aox] (step 2) by H2O will also form [LnRu-OH2]1+. Exchange of H2O for H2O2 will afford [LnRu-(H2O2)]1+ (step 3), which will be converted into [LnRu-OOH] upon H+ dissociation to buffer (step 4). The OOH ligand will then undergo β-hydride elimination (viaTS5) to release O2 and generate [LnRu-H] (step 5). Although β-hydride elimination from a Ru–OOH species is unknown, the reverse reaction, insertion of O2 into a Ru–H bond, is known12,13 and proceeds via the same transition state structure (i.e., TS5). Alternatively, 1,1-deinsertion of O2 would also afford a Ru–H intermediate, but [LnRu-OOH] would first need to rearrange to a higher-energy species (vide infra). Computational studies of other [RuCl(L2)(η6-cymene)] complexes (L2 = a bidentate ligand) have shown that decreases in cymene hapticity to accommodate additional ligand binding have activation barriers below 19 kcal mol−1,47 which suggests that a similar hapticity decrease in TS5 would be thermally accessible. Once it has formed, [LnRu-H] will then be oxidized to [LnRu-H]1+ by ABTS˙−, affording ABTS2− (step 6). Dissociation of H+ from [LnRu-H]1+, 1e− oxidation of [LnRu] by ABTS˙−, and subsequent coordination of ABTS2− to [LnRu]1+ to restart the catalytic cycle (dashed arrow) will not influence the reaction rate or appear in the rate equation because they occur after the rate determining step. Although these transformations cannot be directly observed, literature precedents suggest that they are feasible under these reaction conditions.26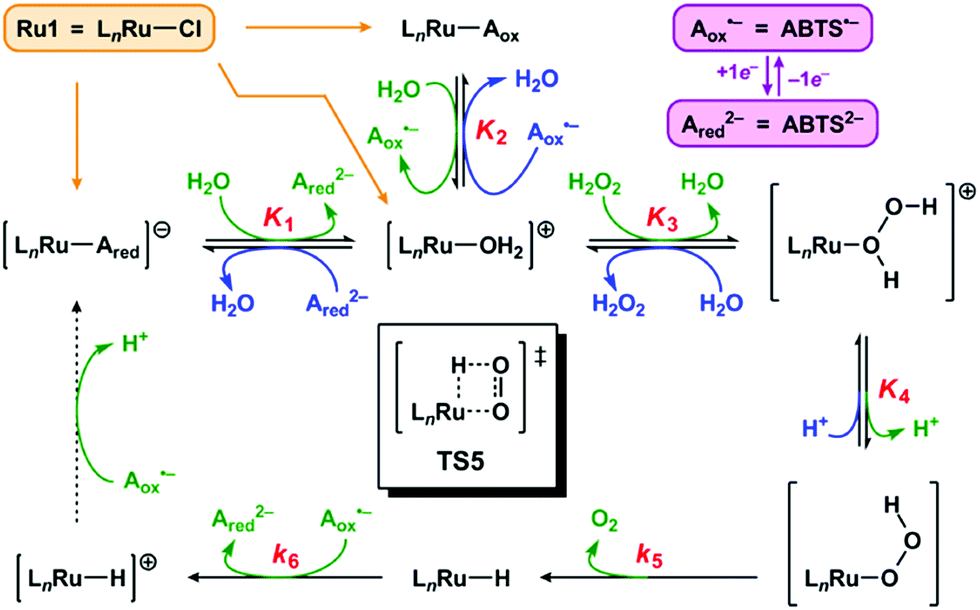 | ||
| Scheme 2 Proposed mechanism for Ru1-catalyzed ABTS˙− reduction with H2O2. Forward (clockwise) and reverse (counter-clockwise) reactions are colored in green and blue, respectively. Each Kn or kn corresponds to the equilibrium or rate constant, respectively, for the forward reaction in step “n” (i.e., step 1 has equilibrium constant K1, etc.). TS5 is the structure of the transition state for step 5. The dashed arrow includes multiple transformations that occur after the rate-determining steps (see ref. 26). All complexes shown above are in the Ru2+ oxidation state except for [LnRu-H]+, which is Ru3+. The spectator ligand set “Ln” comprises the η6-cymene and κ2-(C,O)-benzimidazolylidene–carboxylate ligands, but their hapticity/denticity may decrease to accommodate binding of additional ligands. | ||
If the proposed mechanism is valid, then ABTS˙− reduction can only occur if the Ru–H intermediate has formed, and this Ru–hydride intermediate, in turn, can only form if the OOH ligand on Ru has undergone β-hydride elimination. Within these constraints, an observation that [ABTS˙−] has decreased thus serves as an indirect indication that a Ru–OOH species has undergone β-hydride elimination and generated a Ru–H intermediate. Although β-hydride elimination from a Ru–OOH species (i.e., step 5) is unknown, the reverse reaction, insertion of O2 into a Ru–H bond, is known12,13 and proceeds via the same transition state (i.e., TS5). Furthermore, there are literature examples of Ru complexes that have both hydride and O2 ligands bound to the same metal center,16–20 which suggests that the formation of a Ru–H intermediate and O2 (i.e., step 5) can be more thermodynamically favorable and/or faster than the reverse reaction (insertion of O2 into the Ru–H bond to form a Ru–OOH species) under the appropriate experimental conditions.
Rate law evidence for proposed mechanism
To test the validity of our proposed mechanism for Ru1-catalyzed ABTS˙− reduction with H2O2, we derived the general rate law equation for the catalytic cycle presented in Scheme 2 as a function of the initial rate of ABTS˙− reduction (v0). If the proposed mechanism is valid, v0 should be equal to the product of the rate constant for step 6 (k6) times the concentrations of ABTS˙− and [LnRu-H] (eqn (1)). Utilizing the pre-equilibrium approximation allowed the initial rate of ABTS˙− reduction (v0) to be expressed as functions of [ABTS˙−]0, [ABTS2−]0, [H+]0, [H2O2]0, and [Ru1]0 (eqn (2)–(4); y = v0; x = concentration of independent variable; a, b, and c = constants; see eqn (S1)–(S8)† for full derivation). If the concentrations of all other species are held constant, the relationship between v0 and [ABTS˙−]0 will follow eqn (2), the relationship between v0 and [ABTS2−]0 as well as v0 and [H+]0 will follow eqn (3), and the relationship between v0vs. [H2O2]0 will follow eqn (4).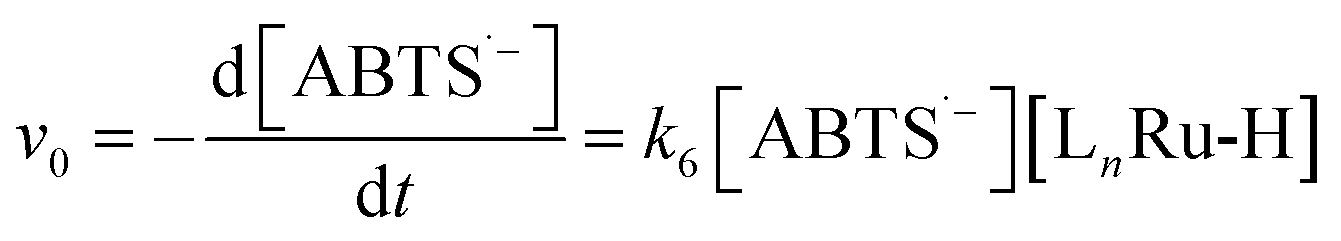 | (1) |
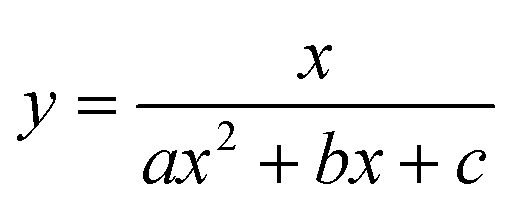 | (2) |
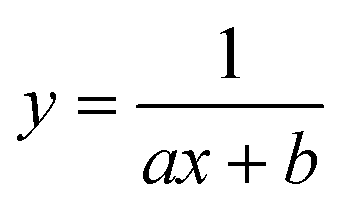 | (3) |
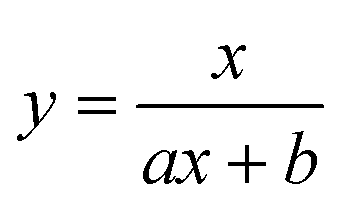 | (4) |
The v0 values for Ru1-catalyzed ABTS˙− reduction with H2O2 increased non-linearly as the initial ABTS˙− concentration increased, with v0 tapering off at higher values of [ABTS˙−]0 (Fig. 4A), and could be successfully fit using eqn (2). Although substrate binding saturation kinetics would produce a similar curve, this phenomenon could be ruled out because previous studies with EtOH as the terminal reductant instead revealed a linear relationship with [ABTS˙−]0.26 The non-linearity observed with H2O2 as the terminal reductant could be attributed to competitive binding between ABTS˙− and H2O2 (steps 2 and 3, Scheme 2), due to the significantly lower concentrations of terminal reductant employed in the current study (i.e., 100 μM for H2O2vs. 50 mM for EtOH) and its poorer Lewis basicity (pKa = 11.6 for H2O2vs. 15.7 for EtOH).48 With a sufficiently high terminal reductant concentration or metal binding ability, the contribution of step 2 to the mechanism becomes negligible and the overall rate equation simplifies to a linear form. Because the v0vs. [ABTS˙−] data could be fit using eqn (2), wherein the [ABTS˙−] term occurs in both the numerator and denominator, the mechanism of ABTS˙− reduction with H2O2 must involve (i) ABTS˙− dissociation from Ru before [LnRu-H] can form (consistent with step 2) and in a subsequent process (ii) bimolecular electron-transfer reaction to ABTS˙− from [LnRu-H] (consistent with step 6).
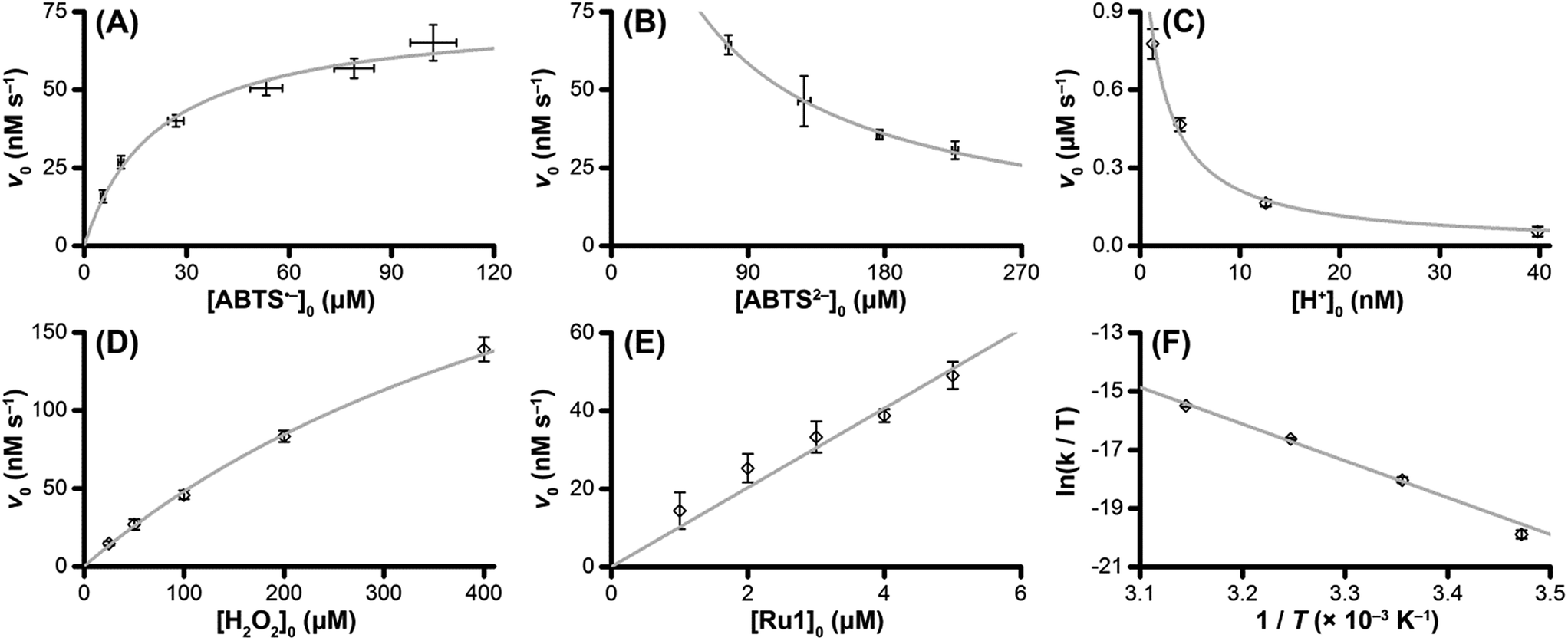 | ||
| Fig. 4 Dependence of the initial rate (v0) of Ru1-catalyzed ABTS˙− reduction with H2O2 on (A) [ABTS˙−]0 = 5, 10, 25, 50, 75, or 100 μM; (B) [ABTS2−]0 = 50, 100, 150, or 200 μM; (C) pH = 7.4, 7.9, 8.4 or 8.9; (D) [H2O2]0 = 25, 50, 100, 200 or 400 μM; (E) [Ru1]0 = 1, 2, 3, 4 or 5 μM; (F) T = 15, 25, 35 or 45 °C. The data points (◇) and error bars were determined from the average and standard deviation values obtained from 4 independent experiments performed on 4 different days, and the grey traces represent the model fits generated by eqn (2) (for ABTS˙−), eqn (3) (for ABTS2− and H+), and eqn (4) (for H2O2). Conditions: [Ru1]0 = 5 μM, [ABTS˙−]0 = 50 μM, [ABTS2−]0 = 100 μM, [H2O2]0 = 100 μM, PBS (pH 7.4), 25 °C. | ||
Plots of v0vs. [ABTS2−]0 and v0vs. [H+]0 (Fig. 4B and C) revealed that v0 decreased non-linearly with [ABTS2−]0 and [H+]0, respectively, and could each be fit by eqn (3). These results demonstrated that ABTS˙− reduction can only occur after ABTS2− dissociation from Ru (consistent with step 1) and H+ dissociation from H2O2 (consistent with step 4). The lack of ABTS˙− reduction in pure H2O (vide supra) provided additional support that H+ dissociation to solution is essential for reactivity. The plot of v0vs. [H2O2]0 (Fig. 4D) revealed a positive correlation and could be fit using eqn (4), whereby the deviation from linearity at higher concentrations indicated that H2O2 must bind to Ru at some point prior to ABTS˙− reduction (consistent with step 3). The linear relationship between v0 and [Ru1]0 (Fig. 4E) suggested that the observed reactivity was predominantly produced by a mononuclear species, consistent with our previous mechanistic studies.26
The Eyring–Polanyi plot (Fig. 4F) revealed a positive entropy of activation (ΔS‡ = 25.5 ± 1.9 cal mol−1 K−1), which indicated disorder was increasing during the rate determining step. This result was consistent with the proposed mechanism, in which a single intermediate, [LnRu-OOH], fragments into two separate molecules, [LnRu-H] and O2, via β-hydride elimination (step 5). Moreover, the ΔS‡ value observed for ABTS˙− reduction by Ru1 with H2O2 fell within the range of values observed for Ru1 with other terminal reductants (methanol, ethanol, isopropanol, and ethylene glycol, ΔS‡ = 11.4–32.8 cal mol−1 K−1) which have previously been shown to generate Ru–H intermediates via β-hydride elimination.26
Kinetic isotope effect evidence for proposed mechanism
Our prior mechanistic studies revealed a solvent kinetic isotope effect (KIE) of 1.74 for Ru1-catalyzed ABTS˙− reduction, reflecting the role of the solvent, H2O, as H+ acceptor (or D+ acceptor in the case of D2O) in step 4.26 When this solvent KIE was factored out, the ratio of observed rate constants (kobs) for Ru1-catalyzed ABTS˙− reduction with H2O2 in protio PBS vs. D2O2 in deutero PBS was determined to be 2.10 ± 0.24, which indicated significant O–H/O–D bond breakage was occurring during the rate determining step. Unfortunately, the individual contributions of step 4 (H+ dissociation) and step 5 (β-hydride elimination) could not be deconvoluted because D–O–O–H cannot be obtained in pure form and the pertinent Ru intermediates – LnRu–O(D)OH and LnRu–O(H)OD – will undergo H/D exchange in protio and deutero PBS on a faster timescale than Ru1-catalyzed ABTS˙− reduction. Therefore, the observed ratio of 2.10 ± 0.24 was treated as the product of the individual O–H/O–D KIE values for step 4 and step 5, and the square root of this ratio was calculated (1.45 ± 0.08) as an averaged approximation of how each step contributed to the overall mechanism (i.e., 1.45 for step 4 and 1.45 for step 5).Interestingly, this value was nearly identical to the O–H/D KIE of 1.45 measured with NAD+.21 Although H2O2 and NAD+ are structurally dissimilar, their O–H pKa values (11.6 for H2O2 and 11.8 for NAD+) are nearly identical,48,49 therefore their O–H bond polarizations will be highly conserved. As a result, H/D isotopic substitution in H2O2 should impact the transition state structure leading to O–H bond breakage to a similar extent as in NAD+. The fact that H2O2 and NAD+ afforded nearly identical O–H/O–D KIE values that aligned with their highly similar pKa values, despite their significant structural differences, provided further support for the proposed mechanism shown in Scheme 2.
Evidence for Ru–H intermediate
The range of suitable experimental conditions under which Ru1-catalyzed ABTS˙− reduction with H2O2 could still occur precluded direct detection of a Ru–H intermediate by 1H NMR or IR spectroscopy, or by ESI-MS. No radical reduction occurs in PBS solutions containing more than 20% CH3CN, and the maximum concentration attainable for stock solutions of Ru1 in CH3CN is 1 mM, therefore the highest concentration attainable for Ru1 in the ABTS˙− reduction experiments is 200 μM, which is well below the detection limit of IR spectroscopy.In addition, the proposed Ru–H intermediate is formed in the rate determining step, and there are 5 other possible Ru-containing species, and both of these factors would cause the concentration of any Ru–H intermediate to be significantly less than 200 μM. Furthermore, ABTS˙− is a paramagnetic species that can broaden or even suppress 1H NMR peaks via relaxation. It is therefore unsurprising that no Ru–H intermediate could be detected when Ru1-catalyzed ABTS˙− reduction with H2O2 was monitored by 1H NMR or IR spectroscopy.
Catalytic organic transformation reactions which proceed through Ru–H intermediates have been studied by mass spectrometry, however these reactions typically employ millimolar catalyst concentrations.50 Nonetheless, we sought to detect the Ru–H intermediate proposed for the Ru1-catalyzed ABTS˙− reduction with H2O2 using high-resolution Fourier transform mass spectrometry with electrospray ionization (ESI-MS). Because [LnRu-H] is neutral, it would need to acquire a charge to be detectable, such as by being converted to [{LnRu-H} + H]1+ (e.g., m/z = 503.1267) via protonation. Unfortunately, no peaks corresponding to this exact species were observed. One possibility is that the H− ligand of [LnRu-H] reacts with H+ to release H2 gas and thereby afford [LnRu]1+ (e.g., m/z = 501.1102), a species that was, in fact, observed in positive mode (Fig. S4†). However, this same species could also be produced by ligand dissociation from other intermediates, such as dissociation of ABTS2− from [LnRu-Ared]1−, ABTS˙− from [LnRu-Aox], H2O from [LnRu-OH2]1+, and H2O2 from [LnRu-(H2O2)]1+. Moreover, the ability of ESI-MS to detect the formation of [{LnRu-H} + H]1+ was severely hampered by the fact that the Ru1-catalyzed ABTS˙− reduction reaction solutions already contained both positively and negatively charged species that would not require ionization, and at much higher concentrations than any un-ionized [LnRu-H]. The presence of these charged species can cause severe ionization suppression in ESI-MS, significantly reducing the ability of this technique to ionize and detect low-concentration neutral molecules, such as the reaction intermediate [LnRu-H].
Although the 1H NMR, IR, and ESI-MS experiments did not lead to direct observation of [LnRu-H], the results did not exclude its formation and they were not inconsistent with the indirect evidence for the formation of [LnRu-H] provided by the UV/vis spectroscopic kinetic experiments. By itself, H2O2 was incapable of reducing ABTS˙− to ABTS2−, which could only occur if Ru1 was also present. In addition, the ABTS˙− reduction rate was linear with Ru1 concentration. Collectively, these results indicated that (1) a Ru-containing species functioned as the catalyst and (2) one or more mononuclear Ru-containing species were intermediates in the catalytic cycle.
No ABTS˙− reduction occurred with Ru1 by itself unless H2O2 was also present, which demonstrated that H2O2 was the terminal reductant. The inability of t-Bu2O2 and t-BuOOH to reduce ABTS˙− or to oxidize ABTS2− in the presence of Ru1 provided evidence that the terminal reductant ability of H2O2 did not involve O–O or O–H bond homolysis and that no oxidizing radicals or high-valent Ru–oxo or Ru–hydroxo species were generated during the catalytic cycle. The non-linear relationship between the ABTS˙− reduction rate and [H2O2] that gradually approached saturation at higher concentrations was consistent with H2O2 coordinating to Ru before the rate determining step. Likewise, the inverse relationship between the ABTS˙− reduction rate and [H+] indicated that one H-atom was lost from H2O2 as H+. Collectively, these results demonstrated that (3) a Ru(H2O2) species must be formed before the rate determining step, (4) H2O2 only underwent O–H bond breakage and only via heterolysis, and (5) that one O–H bond heterolyzed as O− and H+. The most likely product of this combination of processes would be a Ru–OOH species.
Others have demonstrated that H2O2 formation is responsible for the aerobic oxidation of alcohols12,13 by and chemotherapeutic activity51 of Ru-based catalysts, and H2O2 could only be produced by a Ru–H intermediate if it underwent insertion of O2 into the Ru–H bond, which would yield a Ru–OOH species. The microscopic reverse of this reaction is de-insertion of O2 from Ru–OOH, in which the H-atom is transferred to Ru as H− and the O–O single bond is converted to a double bond. The observation that 1 equiv. of O2 gas was released for every 2 equiv. of ABTS˙− reduced to 2 equiv. of ABTS2− provided evidence that deinsertion of O2 was indeed occurring. The large positive ΔS‡ value observed for Ru1-catalyzed ABTS˙− reduction with H2O2 demonstrated an increase in disorder during the rate determining step, which would be consistent with the fragmentation of one ligand into multiple ligands (regardless of whether these ligands remained bound to or subsequently dissociated from Ru).
It is important to note that de-insertion of O2 from Ru–OOH could proceed via 1,1-deinsertion of O2, rather than β-hydride elimination (i.e., 1,2-deinsertion of O2), and still yield a positive ΔS‡ value. For 1,1-deinsertion to occur, however, the atom connectivity in [LnRu-OOH] would need to rearrange to [LnRu-O(O)H]. The possible structures for [LnRu-O(O)H] (IVa and IVb) would be expected to be higher in energy than [LnRu-OOH]: both are formally charge-separated species, and IVb would contain a protonated O2 ligand (Fig. 5). As a result, any equilibrium between [LnRu-OOH] and IVa or IVb would be expected to favor [LnRu-OOH]. Similarly, dissociation of H+ from [LnRu(H2O2)] would be expected to occur preferentially at the OH group directly bound to Ru, which would yield [LnRu-OOH], rather than at the other OH group, which would yield IVa. Although 1,1-deinsertion and β-hydride elimination (i.e., 1,2-deinsertion) would both afford large, positive values for ΔS‡, we believe that β-hydride elimination is more probable than 1,1-deinsertion of O2 because it represents a lower-energy pathway.
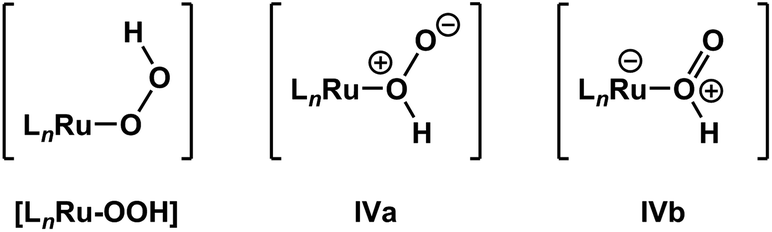 | ||
| Fig. 5 β-Hydride elimination (i.e., 1,2-deinsertion of O2) could proceed from [LnRu-OOH], whereas 1,1-deinsertion would first require rearrangement to a different form, such as IVa or IVb. | ||
The lack of reactivity with t-BuOOH provided evidence that the reduction of ABTS˙− with H2O2 did not involve either heterolytic or homolytic cleavage of the O–O bond, because the activation barriers for these transformations would be lower for t-BuOOH than for H2O2 and they would generate intermediates capable of oxidizing ABTS2−. Furthermore, the evolution of O2 gas provided additional evidence that the O–O bond in H2O2 is not broken heterolytically or homolytically. With these constraints, the only way an HOO− ligand on Ru could fragment into multiple species and be accompanied by the release of O2 gas would be via breakage of the O–H bond. Moreover, the only way this O–H bond could break that would be consistent with the fact that H2O2 must be oxidized to be able to serve as terminal reductant for Ru1-catalyzed ABTS˙− reduction would be if this O–H bond broke heterolytically to afford O2. Consequently, the fragmentation of an HOO− ligand to generate O2 could only occur if H− was also generated. Indeed, for every 2 equiv. of ABTS˙− reduced to 2 equiv. of ABTS2− by H2O2, 1 equiv. of O2 gas was released, indicating that H2O2 functions as a 2e− reductant in this reaction through its H-atoms. Because the two O-atoms are lost from H2O2 as O2 and one H-atom is lost as H+, the other H-atom must carry the 2e− for reducing 2 equiv. of ABTS˙−, which would most likely occur in the form of a hydride (H−).
Because Ru complexes comprising both H− and O2 ligands have been sufficiently stable to be characterizable by single-crystal X-ray diffraction,16–20 β-hydride elimination from an HOO− ligand can be sufficiently thermodynamically favorable to drive the conversion of Ru–OOH to Ru–H. Moreover, the ΔS‡ value observed with H2O2 fell within the range of values observed with non-tertiary alcohol-based terminal reductants that were also shown to proceed through β-hydride elimination transition states. Collectively, these findings provide evidence that (6) the oxidation of H2O2 to O2, which supplies the electrons necessary for ABTS˙− reduction, must occur via elimination of H− from an HOO− ligand. The most probable destination of any H− eliminated from an HOO− ligand bound to Ru would be that same metal center, the product of which would be a Ru–H intermediate.
Summary and conclusions
Our findings demonstrate that (i) a mononuclear Ru-containing species is the catalyst for ABTS˙− reduction, (ii) H2O2 is the terminal reductant, (iii) H2O2 is a 2e− reductant, (iv) H2O2 coordinates to Ru before the rate determining step, (v) the two O-atoms from H2O2 depart as O2 gas, and (vi) the two H-atoms from H2O2 depart as H+ and H−. Although the experimental constraints of the ABTS˙− reduction reaction were ultimately incompatible with direct observation of any Ru–H intermediate by 1H NMR, IR, or ESI-MS, the mechanism presented in Scheme 2 (with the caveat that the conversion of Ru–OOH to Ru–H could proceed via either β-hydride elimination or 1,1-deinsertion of O2) properly accounts for all of the aforementioned findings and provides a general rate law that accurately models all of the UV/vis spectroscopy kinetic data.This report constitutes, to the best of our knowledge, both (i) the first instance of H2O2 functioning as a terminal reductant under biologically-relevant conditions and (ii) the first instance of H2O2 functioning as a hydride donor. However, the ability of H2O2 to function as both an oxidant and reductant is not unprecedented and, in fact, serves as the basis for H2O2 fuel cells.52,53 Given the impressive advances using H2O2 as an oxidant in green catalysis,54–57 the newfound ability of H2O2 to function as an H− donor and reductant (the byproduct of which is O2) will lead to complementary advances using H2O2 as a green H− donor and reductant. Furthermore, establishing the proof-of-principle that H2O2 can act as an H− donor in a chemical reaction (i.e., reduction of ABTS˙− to ABTS2−) provides the foundation for future discoveries of biological reactions in which H2O2 acts as an H− donor in living systems. The ability of Ru1 to reduce oxidizing species using H2O2 as the terminal reductant under biologically-relevant conditions provides a strong impetus to investigate the therapeutic efficacy of Ru1 in maintaining cellular redox homeostasis or modulating essential cellular redox processes. The findings of our efforts in these areas will be detailed in a future report.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Science Foundation (DMR-1555224). We thank A. Mangalum for prior work with Ru1 and helpful discussions.Notes and references
- J. A. Imlay, Annu. Rev. Biochem., 2008, 77, 755–776 CrossRef CAS PubMed.
- J. A. Imlay, Annu. Rev. Microbiol., 2003, 57, 395–418 CrossRef CAS PubMed.
- R. Kohen and A. Nyska, Toxicol. Pathol., 2002, 30, 620–650 CrossRef CAS PubMed.
- H. J. Forman, M. Maiorino and F. Ursini, Biochemistry, 2010, 49, 835–842 CrossRef CAS PubMed.
- E. A. Veal, A. M. Day and B. A. Morgan, Mol. Cell, 2007, 26, 1–14 CrossRef CAS PubMed.
- S. Miriyala, I. Spasojevic, A. Tovmasyan, D. Salvemini, Z. Vujaskovic, D. St. Clair and I. Batinic-Haberle, Biochim. Biophys. Acta, 2012, 1822, 794–814 CrossRef CAS PubMed.
- A. Tovmasyan, R. S. Sampaio, M.-K. Boss, J. C. Bueno-Janice, B. H. Bader, M. Thomas, J. S. Reboucas, M. Orr, J. D. Chandler, Y.-M. Go, D. P. Jones, T. N. Venkatraman, S. Haberle, N. Kyui, C. D. Lascola, M. W. Dewhirst, I. Spasojevic, L. Benov and I. Batinic-Haberle, Free Radical Biol. Med., 2015, 89, 1231–1247 CrossRef CAS PubMed.
- M. K. Evans, A. Tovmasyan, I. Batinic-Haberle and G. R. Devi, Free Radical Biol. Med., 2014, 68, 302–314 CrossRef CAS PubMed.
- S. D. Amaral and B. P. Espósito, BioMetals, 2008, 21, 425–432 CrossRef PubMed.
- P. Chelikani, I. Fita and P. C. Loewen, Cell. Mol. Life Sci., 2004, 61, 192–208 CrossRef CAS PubMed.
- T. Fukai and M. Ushio-Fukai, Antioxid. Redox Signaling, 2011, 15, 1583–1606 CrossRef CAS PubMed.
- F. Nikaidou, H. Ushiyama, K. Yamaguchi, K. Yamashita and N. Mizuno, J. Phys. Chem. C, 2010, 114, 10873–10880 CrossRef CAS.
- K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2002, 41, 4538–4542 CrossRef CAS.
- D. G. Blackmond, Angew. Chem., Int. Ed., 2009, 48, 2648–2654 CrossRef CAS PubMed.
- “β-Hydride elimination from Ru–OOH” and “1,2-deinsertion of O2 from Ru–OOH” refer to the same process and can be used interchangeably in this context, but we primarily use the former in this manuscript to emphasize the reactivity of H2O2 as a hydride donor.
- A. E. W. Ledger, A. Moreno, C. E. Ellul, M. F. Mahon, P. S. Pregosin, M. K. Whittlesey and J. M. J. Williams, Inorg. Chem., 2010, 49, 7244–7256 CrossRef CAS PubMed.
- L. J. L. Häller, E. Mas-Marzá, A. Moreno, J. P. Lowe, S. A. Macgregor, M. F. Mahon, P. S. Pregosin and M. K. Whittlesey, J. Am. Chem. Soc., 2009, 131, 9618–9619 CrossRef.
- J. Matthes, S. Gründemann, A. Toner, Y. Guari, B. Donnadieu, J. Spandl, S. Sabo-Etienne, E. Clot, H.-H. Limbach and B. Chaudret, Organometallics, 2004, 23, 1424–1433 CrossRef CAS.
- M. Jiménez-Tenorio, M. C. Puerta and P. Valerga, Inorg. Chem., 1994, 33, 3515–3520 CrossRef.
- M. Jiménez-Tenorio, M. C. Puerta and P. Valerga, J. Am. Chem. Soc., 1993, 115, 9794–9795 CrossRef.
- Y. Htet and A. G. Tennyson, J. Am. Chem. Soc., 2016, 138, 15833–15836 CrossRef CAS PubMed.
- Y. Htet and A. G. Tennyson, Chem. Sci., 2016, 7, 4052–4058 RSC.
- Unless specified otherwise, all potentials are reported relative to NHE and have been adjusted to account for the effects of H+ concentration at pH 7.
- S. L. Scott, W.-J. Chen, A. Bakac and J. H. Espenson, J. Phys. Chem., 1993, 97, 6710–6714 CrossRef CAS.
- U. Jungwirth, C. R. Kowol, B. K. Keppler, C. G. Hartinger, W. Berger and P. Heffeter, Antioxid. Redox Signaling, 2011, 15, 1085–1127 CrossRef CAS PubMed.
- Y. Htet and A. G. Tennyson, Angew. Chem., Int. Ed., 2016, 55, 8556–8560 CrossRef CAS PubMed.
- R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, Free Radical Biol. Med., 1999, 26, 1231–1237 CrossRef CAS PubMed.
- All experiments were performed in PBS at pH 7.4 unless specified otherwise.
- P. M. Wood, Biochem. J., 1988, 253, 287–289 CrossRef CAS PubMed.
- F. Agapito, B. J. Costa Cabral and J. A. Martinho Simões, J. Mol. Struct.: THEOCHEM, 2005, 729, 223–227 CrossRef CAS.
- C. D. Wijaya, R. Sumathi and W. H. Green Jr, J. Phys. Chem. A, 2003, 107, 4908–4920 CrossRef CAS.
- R. D. Bach, J. Phys. Chem. A, 2016, 120, 840–850 CrossRef CAS PubMed.
- S. J. Blanksby, T. M. Ramond, G. E. Davico, M. R. Nimlos, S. Kato, V. M. Bierbaum, W. C. Lineberger, G. B. Ellison and M. Okumura, J. Am. Chem. Soc., 2001, 123, 9585–9596 CrossRef CAS PubMed.
- J. M. Simmie, G. Black, H. J. Curran and J. P. Hinde, J. Phys. Chem. A, 2008, 112, 5010–5016 CrossRef CAS PubMed.
- T. M. Ramond, S. J. Blanksby, S. Kato, V. M. Bierbaum, G. E. Davico, R. L. Schwartz, W. C. Lineberger and G. B. Ellison, J. Phys. Chem. A, 2002, 106, 9641–9647 CrossRef CAS.
- M.-Z. Wang, C.-Y. Zhou, M.-K. Wong and C.-M. Che, Chem.–Eur. J., 2010, 16, 5723–5735 CrossRef CAS PubMed.
- C. S. Yi, K.-H. Kwon and D. W. Lee, Org. Lett., 2009, 11, 1567–1569 CrossRef CAS PubMed.
- D. Chatterjee, A. Mitra and R. E. Shepherd, Inorg. Chim. Acta, 2004, 357, 980–990 CrossRef CAS.
- A. S. Goldstein, R. H. Beer and R. S. Drago, J. Am. Chem. Soc., 1994, 116, 2424–2429 CrossRef CAS.
- C.-M. Che, T.-F. Lai and K.-Y. Wong, Inorg. Chem., 1987, 26, 2289–2299 CrossRef CAS.
- A. Gerli, J. Reedijk, M. T. Lakin and A. L. Spek, Inorg. Chem., 1995, 34, 1836–1843 CrossRef CAS.
- C.-M. Che, V. W.-W. Yam and T. C. W. Mak, J. Am. Chem. Soc., 1990, 112, 2284–2291 CrossRef CAS.
- C.-M. Che, W.-T. Tang, W.-T. Wong and T.-F. Lai, J. Am. Chem. Soc., 1989, 111, 9048–9056 CrossRef CAS.
- ABTS˙− reduction was not quantitative after 30 min because the temperature of the 3.00 L solution was 19 °C, but the reduction was quantitative after 30 min at 25 °C (see Fig. S1†).
- L. Pitulice, I. Pastor, E. Vilaseca, S. Madurga, A. Isvoran, M. Cascante and F. Mas, Biocatal. Biotransform., 2013, 2, 1–5 Search PubMed.
- A. F. A. Peacock, M. Melchart, R. J. Deeth, A. Habtemariam, S. Parsons and P. J. Sadler, Chem.–Eur. J., 2007, 13, 2601–2613 CrossRef CAS PubMed.
- S. Dinda, K. L. Sebastian and A. G. Samuelson, Organometallics, 2010, 29, 6209–6218 CrossRef CAS.
- K. O. Christe, W. W. Wilson and E. C. Curtis, Inorg. Chem., 1979, 18, 2578–2586 CrossRef CAS.
- S. Sen, U. Pal and N. C. Maiti, J. Phys. Chem. B, 2014, 118, 909–914 CrossRef CAS PubMed.
- X. Wu, J. Liu, D. D. Tommaso, J. A. Iggo, C. R. A. Catlow, J. Bacsa and J. Xiao, Chem.–Eur. J., 2008, 14, 7699–7715 CrossRef CAS PubMed.
- Y. Fu, M. J. Romero, A. Habtemariam, M. E. Snowden, L. Song, G. J. Clarkson, B. Qamar, A. M. Pizarro, P. R. Unwin and P. J. Sadler, Chem. Sci., 2012, 3, 2485–2494 RSC.
- S. Fukuzumi, Joule, 2017, 1, 689–738 CrossRef CAS.
- S. Fukuzumi and Y. Yamada, ChemElectroChem, 2016, 3, 1978–1989 CrossRef CAS.
- M. S. Chen and M. C. White, Science, 2007, 318, 783–787 CrossRef CAS PubMed.
- S.-I. Murahashi, N. Komiya and H. Terai, Angew. Chem., Int. Ed., 2005, 44, 6931–6933 CrossRef CAS PubMed.
- R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977–1986 RSC.
- B. S. Lane and K. Burgess, Chem. Rev., 2003, 103, 2457–2474 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, rate law equation derivations, additional spectra, and digital images. See DOI: 10.1039/c8sc05418e |
| This journal is © The Royal Society of Chemistry 2019 |