
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron sulfur clusters in protein nanocages for photocatalytic hydrogen generation in acidic aqueous solutions†
Weijian
Chen
a,
Shuyi
Li
a,
Xiao
Li
a,
Chi
Zhang
b,
Xiantao
Hu
a,
Fan
Zhu
a,
Guosong
Shen
 a and
Fude
Feng
*a
a and
Fude
Feng
*a
aKey Laboratory of High Performance Polymer Material and Technology of Ministry of Education, Department of Polymer Science & Engineering, School of Chemistry & Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: fengfd@nju.edu.cn
bSchool of Chemistry & Chemical Engineering, Shangqiu Normal University, Shangqiu 476000, China
First published on 17th December 2018
Abstract
We took advantage of the iron binding affinity of apoferritin to immobilize iron–sulfur clusters into apoferritin up to 312 moieties per protein, with a loading rate as high as 25 wt%. The photocatalytic hydrogen generation activity in acidic aqueous solutions was achieved with TONs up to 31 (based on a single catalyst moiety) or 8.3 × 103 (based on a single protein) upon 3 h of visible light irradiation. The present study provides a versatile strategy to construct uniform protein/photocatalyst supramolecular systems with FeFe-H2ase activity.
Among some of the natural FeFe-hydrogenases (FeFe-H2ase), iron sulfur clusters containing Fe-bonding CO ligands play a key role in photocatalytic water splitting, with an activity to produce up to 6000–9000 molecules of H2 per second.1,2 Recently, increasing research studies have been focused on developing artificial hydrogenases, particularly based on Ni, Fe and Co metal complexes, for light driven water splitting purposes.3–5 Development of diiron dithiolate model complexes with water solubility becomes an important objective.
Self-assembly based biomimetic hydrogenase models are attractive due to their outstanding merits in uniformity, stability and efficacy.6,7 Generally, the active centers are located inside macromolecules or scaffolds. We briefly overviewed reported systems in Table S1† focused on the use of peptides/proteins,8 micelles,9 nanodots,10 metal–organic-frameworks (MOFs),11etc. Bioinspired hydrogenase mimics, especially protein-based systems immobilizing Fe–S clusters, provide unique microenvironments for synthetic catalysts.12 For example, Hayashi and coworkers reported a hydrogenase model system with an iron carbonyl cluster, Fe2(CO)9, coordinated to apocytochrome c for photochemical H2 evolution in aqueous solution.13 The single active center in this system was stabilized by covalently linking Fe–S clusters to cysteine residues at an accurate site, affording a turnover number (TON) up to 82. Alternatively, hierarchical protein scaffolds immobilizing multiple active sites have multiplied complexity in structure and mechanism, showing an advantage in photocatalytic H2 generation at a reduced cost of proteins while maintaining a high TON.14
Recent approaches have revealed that micelles can act as a microreactor and provide a high concentration of catalysts and photosensitizers to result in enhanced H2 production.15 It seems that crowded catalysts are able to maintain sufficient activity, but the mechanism behind this remains to be interpreted. Proteins can provide ideal scaffolds binding Fe–S clusters via specific interactions. Generally, natural Fe–S binding proteins such as cytochrome c and nitrobindin (β-barrel protein) bind Fe–S clusters via covalent interaction, however, lacking sufficient binding sites.12,13 We previously reported that horse spleen apoferritin (apo-HSF) has strong binding affinity toward iron–sulfur–nitrosyl cluster anions, with a high loading capacity up to 53 Roussin's black salt moieties per single protein.16 We speculate that the negative inner surface of apo-HSF plays a key role in iron binding due to the presence of abundant anionic amino acid residues in L-chains.17–19 In fact, diverse metal ions, metal complexes and organic compounds could be immobilized on the interior surface of apo-HSF.20,21 Turano et al. described a structural study on the Glu60, Glu61, and Glu64 residues from horse L-ferritins and found that these anionic residues could cooperatively bind iron in a trinuclear center which plays an important role in the biomineralization process.22 More recently, two identified Pt binding sites in the inner surface of apo-HSF were located in close proximity to His132 and His49 as revealed by an X-ray diffraction study, which might be the reason why a large amount of carboplatin could be accommodated inside the cage not just by coordination interaction.23 Similarly, Takafumi et al. reported that ferritin cages could serve as bioinorganic molecular templates to bind metal carbonyl compounds composed of Ru–CO or Mn–CO moieties for photoactivatable CO release, which was confirmed by the X-ray diffraction analysis of the crystal structures.24,25 Considering the abundant metal binding sites and hydrophobic microenvironment in the cavity,26 we expect that apo-HSF would be an ideal nano-platform for immobilizing [Fe2{(μ-SC2H4)(μ-SCH) (CH2)4COOH}(CO)6] (FeFe–COOH) in the cages (Fig. 1 and S1†).
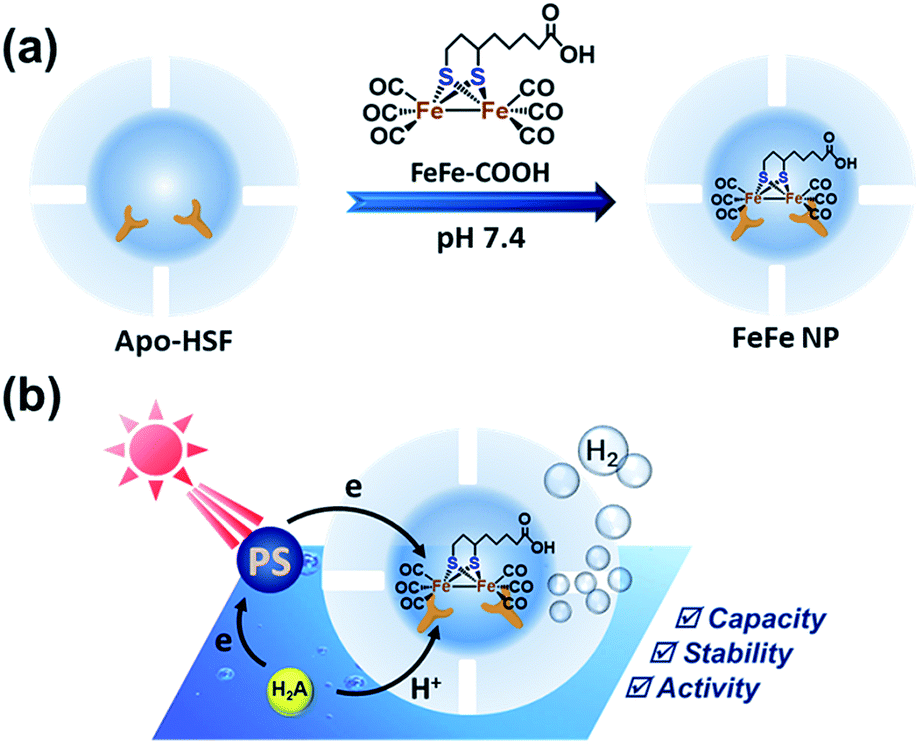 | ||
| Fig. 1 (a) Schematic illustration of artificial hydrogenase construction by encaging multiple Fe–S cluster-based catalysts into the apoferritin cavity. (b) Schematic representation of photocatalytic H2 evolution by FeFe nanoparticles (NPs) in aqueous media. PS and H2A denote the photosensitizer Ru(bpy)32+ and ascorbic acid, respectively. | ||
As a 24-mer natural protein, ferritin has a bundle of unique features, including self-assembly properties, precise cage alignment, a spacious cavity with metal binding sites, and a rigid and uniform nanostructure, which make ferritin cages widely used in biomedical applications such as drug delivery, optical imaging, cancer diagnosis and therapy, vaccine development, and artificial metalloenzymes.27–30 Due to the capability of accumulating metal complexes, ferritin cages are excellent candidates as catalyst vessels, without the need for additional organic ligands that require chemical modifications.31 Particularly, the strong diiron binding property of inner cage avoids scale-restricted biogenetic modification and minimizes the possibility of undesired catalyst leakage through the narrow pores of the cage shell in the course of the catalytic reaction. Compared to ferritin cages, other known protein cages or enzymes are dwarfed in iron accumulation. Moreover, the ferritin cages possess additional merits, such as robust structural stability, excellent water solubility and good durability. Hence, we chose ferritin cages as a carrier of diiron dithiolate-type photocatalysts and studied the hydrogenase activity of the assembled supramolecular structure in aqueous solutions.
The molecule FeFe–COOH (Fig. 1) was easily obtained by coupling the Fe-carbonyl compound and lipoic acid which is known as a cheap and natural anti-oxidant.32,33 FeFe–COOH was used as a catalyst and incorporated into apo-HSF at different feed ratios to form NPs. A pH alteration method that includes protein unfolding at pH 2 and refolding to the original structure at neutral or basic pH in the presence of a guest molecule for loading was widely applied.34 However, the loading of FeFe–COOH could be achieved at a constant pH of 7.4 without involving a protein disassembly process, similar to the previous example preparing Roussin's black salt@apo-HSF nanocomposites.16 The NPs were purified by successive dialysis against 10% acetonitrile–PBS (pH 7.4) and PBS, and finally on PD 10 desalting columns. According to the spectrophotometric analysis, the NPs contained 61 to 624 Fe atoms (equivalent to 30 to 312 FeFe–COOH moieties per single protein) with a loading efficiency (LE) up to 10% (Table 1), dependent on the feed ratios.
| Composite | Number of catalystsa | LEb, % | LCc, % |
|---|---|---|---|
| a The numbers represent the average amount of incorporated FeFe–COOH per apo-HSF. b The LEs of FeFe–COOH nanoparticles were calculated as the weight ratio of incorporated FeFe–COOH to fed FeFe–COOH. c The LCs of FeFe–COOH nanoparticles were calculated as the weight ratio of incorporated FeFe–COOH to FeFe NP. Apo-HSF has a molecular weight of approx. 450 kDa. | |||
| NP1 | 30 | 10.0 | 3.1 |
| NP2 | 59 | 9.8 | 6.0 |
| NP3 | 94 | 9.4 | 9.2 |
| NP4 | 169 | 8.4 | 15.4 |
| NP5 | 312 | 6.3 | 25.2 |
The resultant NPs, with NP4 prepared at a fixed molar feed ratio of 2000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as an example, exhibited two intense broad absorption bands in the UV-vis spectra at 300–400 nm and 450–600 nm originating from FeFe–COOH (Fig. S2†), and CO-stretching bands in the Fourier transform infrared (FT-IR) spectra that were identical to those of the unbound catalyst complexes in the 1950–2100 cm−1 range (Fig. 2a), thus confirming that the structure of FeFe–COOH was well preserved after encapsulation.12 Importantly, non-stained NPs were clearly visualized as uniform puncta under a high resolution transmission electron microscope (HR-TEM) (Fig. 2b), indicative of the iron-enriched core. The average diameter was estimated to be ∼8 nm, well matching the size of the inner cavity of apo-HSF, which suggests that the metal complexes were mainly located inside the protein cage. Despite the big number of entrapped metal complexes, the average hydrodynamic diameter (RH) was comparable to that of free apo-HSF (12.8 ± 2.1 nm versus 11.0 ± 0.8 nm) as determined by dynamic light scattering (DLS) (Fig. 2c and S3†). The secondary protein structures, in particular the α helices, were unaffected as evidenced by the constant peaks positively at 195 nm and negatively at 208/222 nm in the circular dichroism (CD) spectra which were measured before and after the encapsulation process (Fig. 2d).23,35 Meanwhile, the change of zeta-potential between apo-HSF and NP4 was negligible (−18.0 mV versus −17.5 mV). As compared to FeFe–COOH that seriously precipitated from aqueous solution at 51 μM due to the limited water solubility, NP4 was in a molecularly dispersed state in the aqueous solution as evidenced by the undetectable changes in UV-vis absorption, RH and CD spectra after one week of storage at 4 °C (Fig. S4 and S5†). These data were in good agreement with the notion that the NPs have an intact core–shell structure, with large numbers of FeFe–COOH encaged in the cavity. The loading capacity (LC) of NP4 was close to that of the protein/FeFe–COOH composite obtained by the common pH alteration method (337 versus 367 Fe atoms per single protein) at the same feed ratio (2000:1), which suggests that most FeFe–COOH moieties were accumulated in the inner surface.
:1 as an example, exhibited two intense broad absorption bands in the UV-vis spectra at 300–400 nm and 450–600 nm originating from FeFe–COOH (Fig. S2†), and CO-stretching bands in the Fourier transform infrared (FT-IR) spectra that were identical to those of the unbound catalyst complexes in the 1950–2100 cm−1 range (Fig. 2a), thus confirming that the structure of FeFe–COOH was well preserved after encapsulation.12 Importantly, non-stained NPs were clearly visualized as uniform puncta under a high resolution transmission electron microscope (HR-TEM) (Fig. 2b), indicative of the iron-enriched core. The average diameter was estimated to be ∼8 nm, well matching the size of the inner cavity of apo-HSF, which suggests that the metal complexes were mainly located inside the protein cage. Despite the big number of entrapped metal complexes, the average hydrodynamic diameter (RH) was comparable to that of free apo-HSF (12.8 ± 2.1 nm versus 11.0 ± 0.8 nm) as determined by dynamic light scattering (DLS) (Fig. 2c and S3†). The secondary protein structures, in particular the α helices, were unaffected as evidenced by the constant peaks positively at 195 nm and negatively at 208/222 nm in the circular dichroism (CD) spectra which were measured before and after the encapsulation process (Fig. 2d).23,35 Meanwhile, the change of zeta-potential between apo-HSF and NP4 was negligible (−18.0 mV versus −17.5 mV). As compared to FeFe–COOH that seriously precipitated from aqueous solution at 51 μM due to the limited water solubility, NP4 was in a molecularly dispersed state in the aqueous solution as evidenced by the undetectable changes in UV-vis absorption, RH and CD spectra after one week of storage at 4 °C (Fig. S4 and S5†). These data were in good agreement with the notion that the NPs have an intact core–shell structure, with large numbers of FeFe–COOH encaged in the cavity. The loading capacity (LC) of NP4 was close to that of the protein/FeFe–COOH composite obtained by the common pH alteration method (337 versus 367 Fe atoms per single protein) at the same feed ratio (2000:1), which suggests that most FeFe–COOH moieties were accumulated in the inner surface.
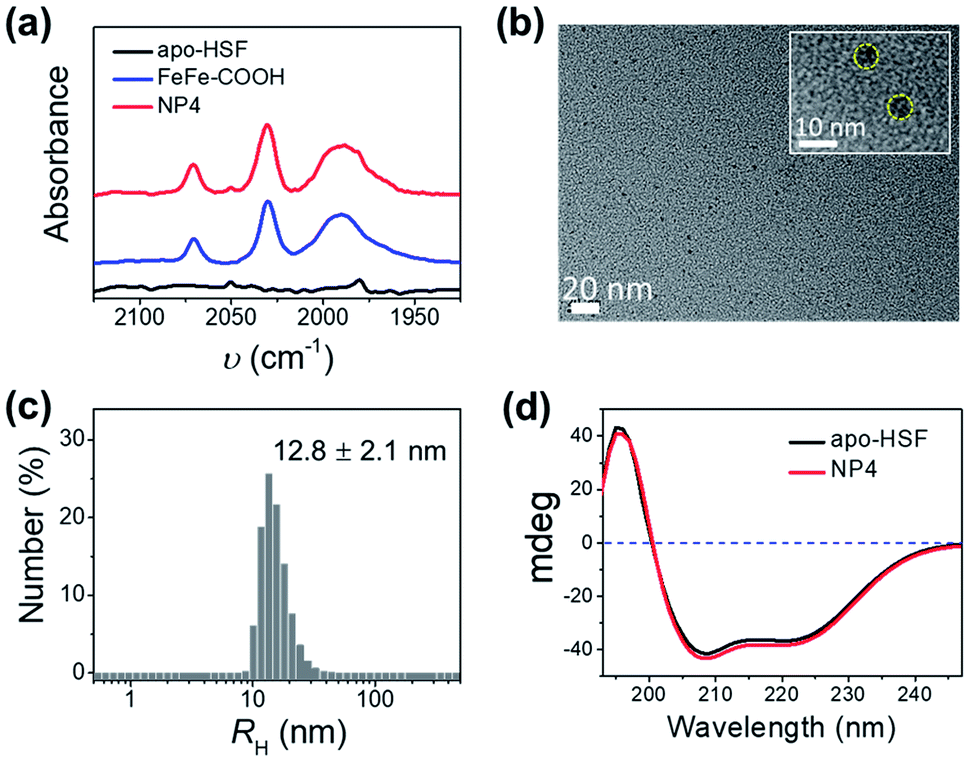 | ||
| Fig. 2 Characterization of NP4. (a) FT-IR spectra of apo-HSF, FeFe–COOH and NP4. (b) TEM image of non-stained NP4. Inset: HR-TEM of non-stained NP4. The yellow circle indicated the existence of a metal core. (c) DLS analysis of NP4. (d) CD spectra of apo-HSF and NP4 in PBS buffer. | ||
The occurrence of binding events associated with multiple coordination sites can be very complicated, as inferred from a quantity of previous reports on X-ray structural analysis of Fe ion-bound apoferritin.36–38 By raising the feed ratio to 5000:1 (NP5), we found that the loading capacity for FeFe–COOH was increased to 312 moieties (equivalent to 624 Fe atoms). This is an extremely large number for apo-HSF to accommodate an organometallic complex, with a loading rate as high as 25 wt% (Table 1). Assuming the protein cavity has a radius (R) of 4 nm and the crystal density (ρ) of FeFe–COOH is similar to [(μ-SCH2CH2CH2S)Fe2(CO)6] reported to be 1.887 g cm−3,39 the theoretical capacity (n) was roughly calculated according to eqn (1):
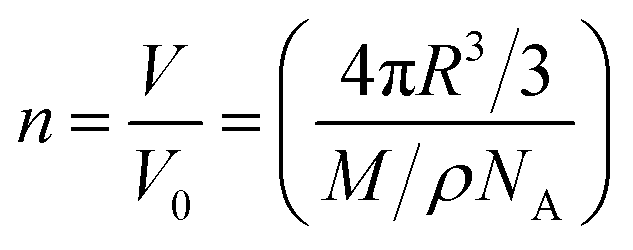 | (1) |
The mode of binding interactions between Fe2+ ions and apoferritin has been extensively studied, whereas the binding of metal complexes into apoferritin cages remains unclear. Obviously, the limited space of 3-fold pores and availability of cysteine residues potentially binding the diiron complex could not well interpret the large amount of complex incorporated. We hypothesized that the organic solvent treatment during preparation of NPs and the inner surface properties of apo-HSF that afford plenty of binding sites are crucial for transportation and accumulation of the diiron dithiolate complex.
We compared the loading number of FeFe–COOH at a fixed feed ratio of 2000:1 under different conditions: (1) NP4-1 was prepared in the absence of acetonitrile in both encapsulation and dialysis processes; (2) NP4 was prepared in PBS and dialysed in the presence of 10% acetonitrile–PBS, and (3) 10% acetonitrile was applied in both loading and dialysis processes for NP4-2 formulation. As a result (Table S2 and Fig. S6†), a maximum loading up to 203 catalyst moieties was achieved with NP4-2, in contrast to the notably reduced loading number (81 catalyst moieties) with NP4-1. The organic solvent treatment allows more catalyst molecules to transport into the inner cavity.
The inner wall of apoferritin is known to be highly negatively charged due to the abundant anionic amino acid residues such as glutamic acid.40,41 To mimic the anionic environment for assessing the binding affinity, we synthesized a water soluble peptide, poly(L-glutamic acid)42 (L-PGA) comprising 52 repeat units, by the ring-opening polymerization method (Fig. 3 and S7†), and found that L-PGA complexed with FeFe–COOH to form nanoparticles using the same encapsulation and dialysis protocols for NP4 preparation, as revealed by DLS and TEM analysis (Fig. S8†). The condensed nanostructure suggests the strong interaction between FeFe–COOH and L-PGA. As a result, the LE and LC of FeFe–COOH were estimated to be 19% and 29%, respectively, comparable to the binding of FeFe–COOH by apo-HSF. These data corroborate the assumption that the highly anionic environment is in favour of immobilizing FeFe–COOH, thanks to the coordination between the anionic peptide and diiron part.
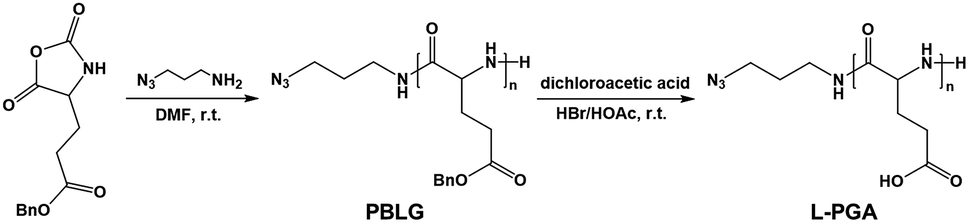 | ||
| Fig. 3 Synthetic route for L-PGA. | ||
To check the interaction between the incorporated FeFe–COOH and photosensitizer (PS) Ru(bpy)32+, we investigated the luminescence quenching of Ru(bpy)32+ at 629 nm by titrating NP4 into 5.0 μM Ru(bpy)32+ aqueous solution containing 0.1 M sodium ascorbate (NaHA) at pH 7.4, and achieved a Stern–Volmer quenching constant (KSV) of 5.2 × 103 M−1 according to the linear concentration-dependent quenching relationship (Fig. 4a), where I0 and Ip denote the Ru(bpy)32+ luminescence intensity in the absence and presence of incorporated FeFe–COOH at concentration Cincorporated FeFe–COOH, respectively. This KSV value was very close to that with the quenching of Ru(bpy)32+ luminescence by free FeFe–COOH (KSV 5.5 × 103 M−1) under the same conditions, which suggests that efficient electron transfer can take place between the incorporated FeFe–COOH and Ru(bpy)32+. According to a previous report on encapsulating ruthenium bipyridine complexes with apo-HSF, nonspecific embedding of Ru(bpy)32+ at the protein surface and penetration of Ru(bpy)32+ into the protein cavity were negligible,26 thus minimizing the possibility of PS contact with the caged FeFe–COOH. The approaching and accumulation of Ru(bpy)32+ to the protein surface is driven by the electrostatic attraction as apo-HSF has a very negative zeta potential,43 which is in favor of efficient electron transfer processes.44 This observation was similar to the long range electron transfer between Ru(bpy)32+ and the catalyst in amphiphilic polymeric micelles reported by Wu's group.9 In contrast, with a small KSV estimated to be 48 M−1, ascorbic acid (H2A) mediated quenching of Ru(bpy)32+ luminescence to a much lesser extent (Fig. S9†), ensuring that the excited state of PS was unaffected by H2A.
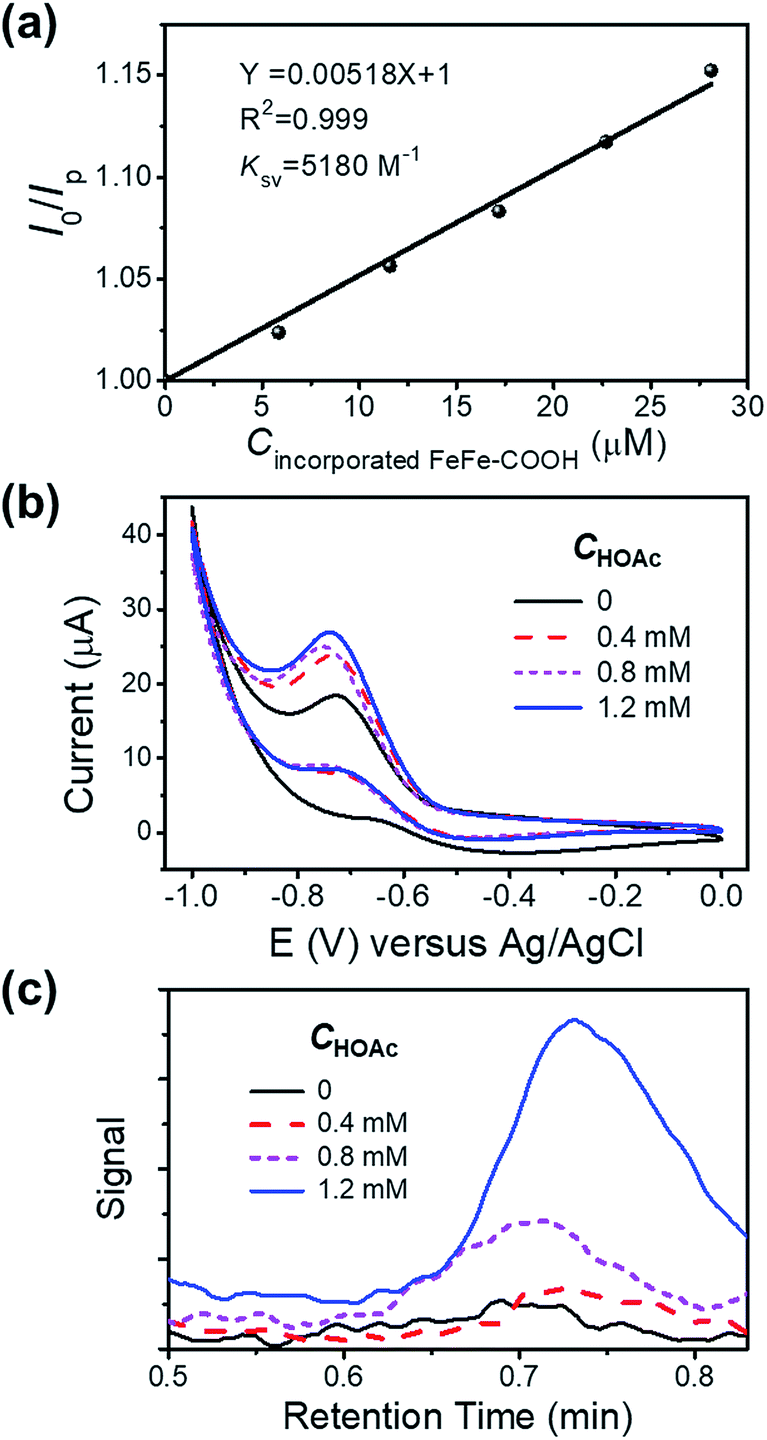 | ||
| Fig. 4 (a) The Stern–Volmer plot of I0/IP against Cincorporated FeFe–COOH by successive addition of NP4 to a solution of Ru(bpy)32+ (5 μM). Ru(bpy)32+ was excited at 453 nm and luminesced at 629 nm. (b) Cyclic voltammograms of NP4 at 51 μM incorporated FeFe–COOH in the presence of different concentrations of HOAc (0–1.2 mM) in 0.1 M Na2SO4 solution. (c) GC spectra of the gas phase collected during electrochemical reaction of NP4, with H2 detected at ∼0.7 min. | ||
To evaluate the proton reduction capability of FeFe–COOH and NPs in the presence of acetic acid (HOAc), we carried out cyclic voltammetry (CV) measurements in N2-saturated solution. The reduction peak was observed at −0.7 V versus Ag/AgCl to indicate the existence of FeIFe0 species after one electron transfer45 and intensified upon gradual addition of HOAc (Fig. S10a†), in accordance with an electrochemical catalytic proton reduction process.46,47 As compared to the free FeFe–COOH, the incorporated FeFe–COOH indicates a similar electrochemical response by transferring accepted electrons from the electrode to protons for H2 production (Fig. 4b). To verify the produced H2 in the electrochemical reaction, a fixed volume (1 mL) of gas above the reaction solution containing different concentrations of HOAc was extracted and subjected to gas chromatography (GC). As shown in Fig. 4c, in the GC spectra, H2 was detected at a retention time of ∼0.7 min with increasing signal as the HOAc concentration increased in the 0–1.2 mM range, which reveals that the incorporated FeFe–COOH perfectly retained its FeFe–H2ase activity in proteins similar to the free FeFe–COOH (Fig. S10b†).
To investigate the photocatalytic behavior of NPs in aqueous solution, we used Ru(bpy)32+ as PS and H2A as the proton source and the sacrificial electron donor. In the presence of Ru(bpy)32+ (1 mM) and ascorbic acid (50 mM) in aqueous solution (pH 5.3, 20 mL), NP4 (containing 51 μM incorporated FeFe–COOH) was irradiated by a 300 W xenon lamp (λ > 400 nm, 200 mW cm−2) under vacuum and the evolved H2 was detected by GC. A total amount of 695 μL (or 31 μmol) of H2 in 3 h was collected, resulting in a turnover number (TON) of 31 and 5.2 × 103 on the basis of FeFe–COOH moieties and single protein particles, respectively (Fig. 5a). In contrast, irradiation of free FeFe–COOH (51 μM) led to a low TON of 3.6 under the same conditions in the presence of Ru(bpy)32+ and ascorbic acid, which means protein encapsulation amplified H2 generation capability by a factor of 8.5. This amplification effect was comparable to that of most of the reported inorganic nano-platforms and assembled vesicles (Table S1†). The reduced H2 evolution for FeFe–COOH lacking protein encapsulation was partially attributed to the limited solubility in acidic solutions which induced formation of catalyst precipitates. We tested the photochemical H2 evolution performance of FeFe–COOH (51 μM) in acetonitrile/water (1:1, v/v) which well dissolved the catalyst, and found that the volume of evolved H2 only slightly increased (Fig. S11†). Although organic solvent prohibited the catalyst from precipitation, it was unfavorable in proton donating and did not substantially improve H2 production.9 No H2 evolution was detected without light or by light irradiation of FeFe–COOH, Ru(bpy)32+, and ascorbic acid separately (Fig. S12†), which are controls, confirming that each component is essential for photocatalytic H2 generation.
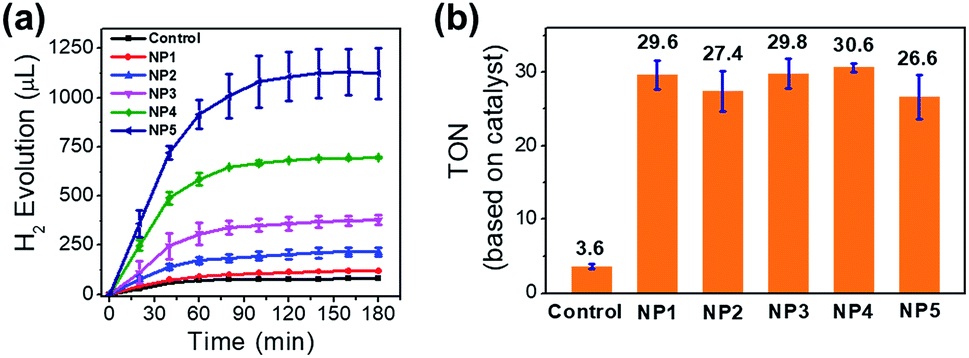 | ||
| Fig. 5 (a) Photocatalytic H2 evolution and (b) TON based on FeFe–COOH moieties for NP1–NP5, with free FeFe–COOH as a control. The concentration of incorporated FeFe–COOH was 9.0, 17.7, 28.2, 50.7 and 93.6 μM, respectively. The optical power was 200 mW cm−2. | ||
Factors, such as pH, PS concentration and catalyst feed ratio, have profound effects on the photocatalytic performance. With NP3 as an example, as the pH increased from 5.3 to 7.4, the evolved H2 volume in 3 h was decreased by 59%, owing to the lowered proton concentration that reduced the proton reduction rate (Fig. S13†). The pH-dependence also reflects that no barriers exist for proton transportation in the protein scaffold even though FeFe–COOH was caged inside. NP3 was stable at pH 5.3, as illustrated by the unchanged CD spectra in comparison to apo-HSF (Fig. S14†). In consideration of the isoelectric point of apoferritin, more acidic pH was not applied to avoid protein precipitation.48 As shown in Fig. S15,† lowering the Ru(bpy)32+ concentration from 1.0 mM to 0.2 mM gave rise to a decrease of the total amount of the evolved H2 in 3 h by 74%, due to the attenuated electron transfer from the excited state of Ru(bpy)32+ to the caged FeFe–COOH. Therefore, preferentially we performed photochemical reactions at pH 5.3 using 1 mM Ru(bpy)32+ for maximization of H2 productivity.
Increase of feed ratio in a range of 300:1 to 2000:1 (from NP1 to NP4) resulted in greater numbers (30 to 169) of incorporated catalysts per single protein (Table 1), while the LE decreased slightly from 10.0 to 8.4%. Interestingly, despite the higher feed ratio and more evolved H2 (Fig. 5a and b), the TON values (based on the FeFe–COOH moiety) upon 3 h of irradiation remained almost unchanged. This finding shows that the increase of local catalyst concentration had little impact on the activity of the incorporated catalyst. Interestingly, the catalyst incorporated in proteins showed enhanced activity, although it is separated from the PS situated at the outer protein surface by a 20–25 Å-thick protein shell. This means the electron transfer process was efficient over a long distance and did not need very close proximity between the catalyst and PS. Such a photoinduced long-distance electron transfer was not favored by a distance-limited superexchange mechanism, but was probably governed by a multistep electron tunneling pathway which is not rare in metalloproteins.49,50 For example, Knez et al. reported the bi-directional electron transport activity of L-chains across the shell of apo-HSF,51 supporting the possible mechanism of electron tunneling which was also suggested by Watt and coworkers in the study of long-distance electron transfer through the 20–25 Å shell of apo-HSF.49 This process was probably associated with redox active aromatic acids in the L-chains of apo-HSF which possesses 90–95% L-chains. Each L-chain contains 24 Phe, 6 Tyr and 1 Trp residues with redox potentials of ∼1 V versus NHE or more, and these relay amino acids were reported to act as stepping stones for the nearest neighbour hopping.52–54 In addition, α-helix is dominant in apo-HSF and the amino bonds along the α-helical backbones were electron hopping sites to allow for efficient long-distance electron transfer across the α-helix.55 The high-hierarchy and well-aligned structure of proteins and peptides enable broad electrical applications.56
Compared to the excited Ru(bpy)32+ with a higher redox potential (E2+*/3+ −0.83 versus SCE),57 the incorporated catalyst had a relatively low potential (Ered ∼ −0.7 V versus SCE), which is suitable for redox reactions. Besides, negatively charged apo-HSF facilitates accumulation of PS cations to its outer surface through electrostatic interaction and the enhanced local concentration of PS ensures sufficient availability of PS to interact with the incorporated catalyst. Hence, on the basis of the above information, we speculated that the core–shell structure of apo-HSF, not simply improving the solubility of the catalyst by a spacious cavity, should account for the enhancement of catalyst activity by facilitating long-distance electron hopping that traversed the protein shell.
After one week of storage of NP4 at 4 °C, in addition to the high stability of the protein structure (Fig. S5†), the activity of photocatalytic H2 evolution was perfectly preserved as compared to that of the freshly prepared sample (Fig. S16†). This observation suggests that the state of incorporated FeFe–COOH moieties was not changed as a result of stable immobilization.
The protein caging 312 FeFe–COOH moieties for NP5, although fewer than the theoretical 626, could be approx. solid. This huge number must exceed the quantity of iron anchoring sites. Under the same photocatalytic conditions, 3 h light illumination on 0.3 μM metalloprotein resulted in 1123 μL of evolved H2. The slightly decreased TON (26.6, based on the FeFe–COOH moiety) may reflect the influence on the catalytic activity from the changes of the microenvironment where the catalysts reside. Nevertheless, the TON based on a single protein reaches 8.3 × 103. In general, the single protein affords more H2 as more catalysts are caged.
It is noteworthy that the photochemical H2 evolution ceased after ∼100 min of light irradiation (Fig. 5a). According to the unaffected size and secondary structure as indicated by the DLS and CD spectra (Fig. S17†), the integrity of the protein shell was undisturbed before and after the photochemical reaction. To make sure whether the catalyst or PS led to reaction interruption, we added fresh NP4 or Ru(bpy)32+ to the reaction mixture after 3 h light irradiation and continued another run of photochemical reaction that was monitored by GC. As shown in Fig. 6a, H2 generation was undetectable with re-addition of Ru(bpy)32+, but resumed for another 100 min by re-addition of NP4. The experiment was repeated under the same conditions except for the replacement of NP4 with FeFe–COOH, and similar phenomena were observed (Fig. S18†). These results demonstrate that although the PS could last a long-term reaction, the catalyst was deactivated in a protein-independent manner as the reaction proceeded.
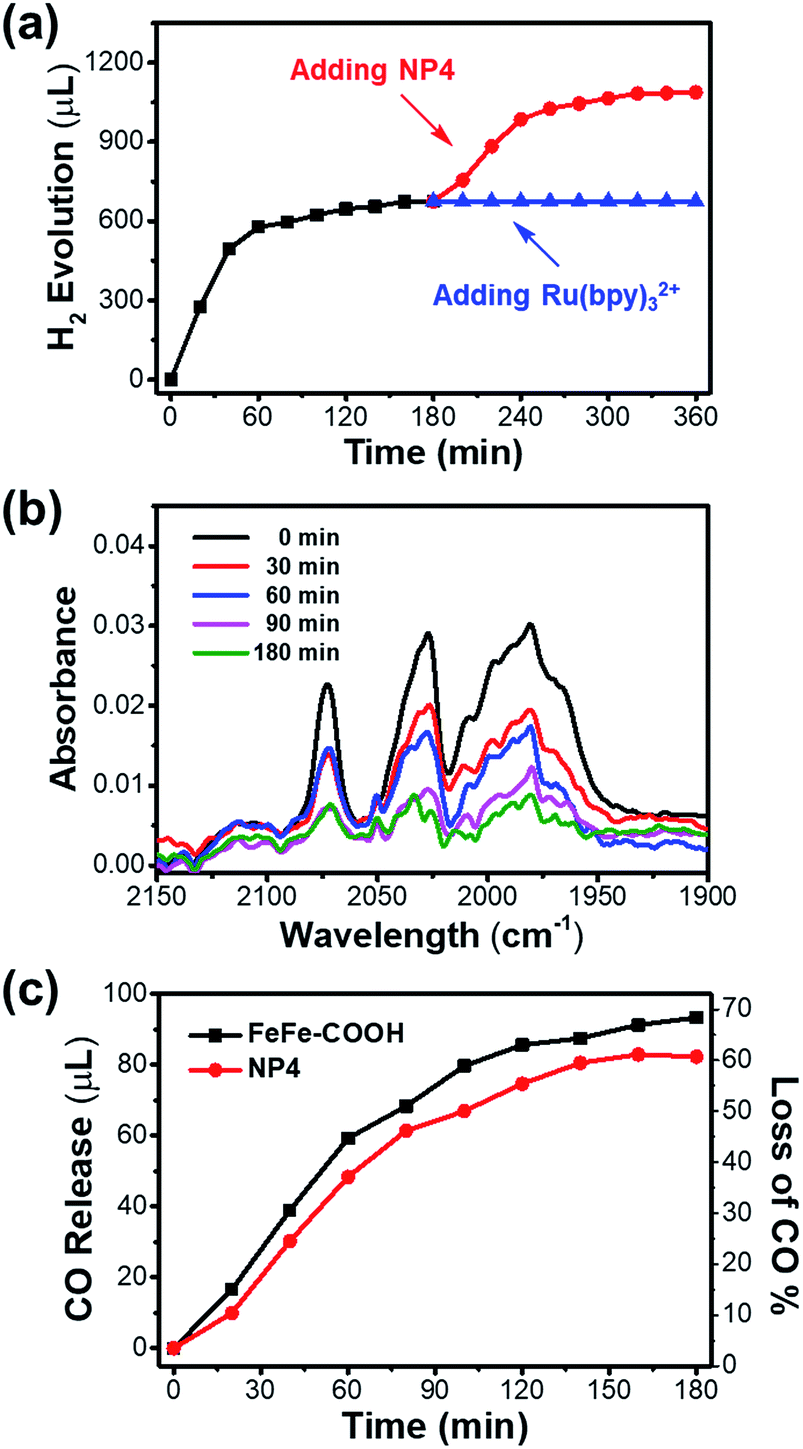 | ||
| Fig. 6 (a) Photocatalytic H2 evolution from solution (pH 5.3) containing Ru(bpy)32+ (1 mM), ascorbic acid (50 mM) and NP4 (51 μM incorporated FeFe–COOH). The optical power was 200 mW cm−2. Reaction was re-performed 3 h after adding another portion of Ru(bpy)32+ (1 mM) or NP4 (51 μM incorporated FeFe–COOH). (b) FT-IR spectroscopy of NP4 after different times of photochemical reaction under the same conditions described in (a). (c) The volumes of released CO and percentages of the loss of CO ligands after different times of photochemical reaction under the same conditions described in (a) and FeFe–COOH (51 μM) was used for comparison. CO was detected using an FID detector in GC analysis. | ||
We reasoned that decomposition of the catalyst may take place during the photochemical reaction as diiron dithiolate-type clusters were previously reported with the photostability issue.58,59 The FT-IR experiment results (Fig. 6b and S19†) showed that the absorption intensity of characteristic υCO bands for NP4 or FeFe–COOH samples at 1990, 2031, and 2071 cm−1 was gradually attenuated during 0–3 h of normal photochemical reaction. This evidence confirmed that the photoinduced dissociation of Fe–CO bonds occurred and thus affected the photocatalytic performance. The absence or presence of PS had little impact on the photostability of the catalyst (Fig. S20†).
To look close into the decomposition process of NP4 and FeFe–COOH, we employed a GC technique to quantitatively detect gaseous CO released from the reaction mixture. The release of CO in the presence of PS was relatively rapid in the first 100 min before a slowdown (Fig. 6c). Specifically, approx. 50% of the CO ligands of the incorporated catalyst in NP4 were dissociated after 100 min of light irradiation, at which point H2 evolution came to a complete cease. This result may suggest full deactivation of the catalyst after three Fe-bonding CO bonds were liberated. The loss of more CO ligands was rather slow, and reached only approx. 60% as the reaction extended to 3 h. In line with the FT-IR data, the PS and protein showed limited effect on the CO release rate (Fig. S21†), reflecting the irreversible photoinduced catalyst degradation, although the absorption of visible light by the catalyst was weak.
Conclusions
In summary, the incorporation of a synthetic iron–sulfur cluster, as a model photocatalyst, in apoferritin at neutral pH led to uniform nanoassemblies with a loading rate as high as 25 wt%. The intact protein nanocage structure with an outer diameter of ∼12 nm was well preserved. Under homogeneous acidic conditions, the photocatalysts in cages exhibited FeFe–hydrogenase activity to generate H2 with an enhancement of the TON up to 8.5-fold relative to the non-incorporated form upon visible light illumination. The enhanced activity of the incorporated catalyst was associated with the core–shell-shaped protein structure that improved the water solubility of the catalyst and facilitated long-distance electron transportation from photosensitizers to the catalyst across the protein shell. The strong iron binding affinity and tremendous iron loading capacity that have not been found in other protein cages, related to the anionic inner wall of the protein, make apoferritin an ideal scaffold for stable photocatalyst immobilization without compromising catalyst activity. The investigation on the photostability of the diiron dithiolate complex, arising from the metal–carbonyl ligand, will spur development of new catalysts with high activity for long-term photocatalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We're grateful to Prof. Wei Wang (Nanjing University) for help with DLS and zeta potential measurements, Prof. Dechen Jiang (Nanjing University) for help with CV measurements, and Prof. Weiyin Sun (Nanjing University) for help with GC measurements. We thank the National Basic Research Program of China (2015CB856300), the National Natural Science Foundation of China (21601119), the 1000 Young Talents Program, the Program for Changjiang Scholars and Innovative Research Team in University (IRT1252), Interdisciplinary Training for Graduate Students of Nanjing University (2016CL11), the Innovative Training Program for Undergraduate Students of Nanjing University (G201810284150), and the Fundamental Research Funds for the Central Universities (020514380140) for financial support.Notes and references
- M. Frey, ChemBioChem, 2002, 3, 153 CrossRef CAS.
- M. B. Wilker, K. E. Shinopoulos, K. A. Brown, D. W. Mulder, P. W. King and G. Dukovic, J. Am. Chem. Soc., 2014, 136, 4316 CrossRef CAS.
- L. M. Utschig, S. R. Soltau and D. M. Tiede, Curr. Opin. Chem. Biol., 2015, 25, 1 CrossRef CAS.
- Y. Shim, R. M. Young, A. P. Douvalis, S. M. Dyar, B. D. Yuhas, T. Bakas, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2014, 136, 13371 CrossRef CAS PubMed.
- T. Ueno, H. Tabe and Y. Tanaka, Chem.–Asian J., 2013, 8, 1646 CrossRef CAS PubMed.
- F. Wang, W. J. Liang, J. X. Jian, C. B. Li, B. Chen, C. H. Tung and L. Z. Wu, Angew. Chem., Int. Ed., 2013, 52, 8134 CrossRef CAS PubMed.
- T. Ueno, S. Abe, N. Yokoi and Y. Watanabe, Coord. Chem. Rev., 2007, 251, 2717 CrossRef CAS.
- Y. Sano, A. Onoda and T. Hayashi, J. Inorg. Biochem., 2012, 108, 159 CrossRef CAS.
- F. Wang, M. Wen, K. Feng, W. J. Liang, X. B. Li, B. Chen, C. H. Tung and L. Z. Wu, Chem. Commun., 2016, 52, 457 RSC.
- M. Wen, H. L. Wu, J. X. Jian, X. Z. Wang, X. B. Li, B. Chen, C. H. Tung and L. Z. Wu, ChemPhotoChem, 2017, 1, 260 CrossRef CAS.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997 CrossRef CAS PubMed.
- A. Onoda, Y. Kihara, K. Fukumoto, Y. Sano and T. Hayashi, ACS Catal., 2014, 4, 2645 CrossRef CAS.
- Y. Sano, A. Onoda and T. Hayashi, Chem. Commun., 2011, 47, 8229 RSC.
- P. C. Jordan, D. P. Patterson, K. N. Saboda, E. J. Edwards, H. M. Miettinen, G. Basu, M. C. Thielges and T. Douglas, Nat. Chem., 2016, 8, 179 CrossRef CAS PubMed.
- C. Orain, F. Quentel and F. Gloaguen, ChemSusChem, 2014, 7, 638 CrossRef CAS PubMed.
- X. Li, Y. Zhang, J. Sun, W. Chen, X. Wang, F. Shao, Y. Zhu, F. Feng and Y. Sun, ACS Appl. Mater. Interfaces, 2017, 9, 19519 CrossRef CAS.
- L. Banci, I. Bertini, K. L. Bren, M. A. Cremonini, H. B. Gray, C. Luchinat and P. Turano, J. Biol. Inorg Chem., 1996, 1, 117 CrossRef CAS.
- S. Zhang, J. Zang, H. Chen, M. Li, C. Xu and G. Zhao, Small, 2017, 13, 1701045 CrossRef.
- E. C. Theil, R. K. Behera and T. Tosha, Coord. Chem. Rev., 2013, 257, 579 CrossRef CAS.
- Z. Zhen, W. Tang, H. Chen, X. Lin, T. Todd, G. Wang, T. Cowger, X. Chen and J. Xie, ACS Nano, 2013, 7, 4830 CrossRef CAS PubMed.
- T. Ueno, M. Abe, K. Hirata, S. Abe, M. Suzuki, N. Shimizu, M. Yamamoto, M. Takata and Y. Watanabe, J. Am. Chem. Soc., 2009, 131, 5094 CrossRef CAS PubMed.
- C. Pozzi, S. Ciambellotti, C. Bernacchioni, F. Di Pisa, S. Mangani and P. Turano, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2580 CrossRef CAS PubMed.
- N. Pontillo, G. Ferraro, J. R. Helliwell, A. Amoresano and A. Merlino, ACS Med. Chem. Lett., 2017, 8, 433 CrossRef CAS PubMed.
- K. Fujita, Y. Tanaka, S. Abe and T. Ueno, Angew. Chem., Int. Ed., 2016, 55, 1056 CrossRef CAS.
- K. Fujita, Y. Tanaka, T. Sho, S. Ozeki, S. Abe, T. Hikage, T. Kuchimaru, S. Kizaka-Kondoh and T. Ueno, J. Am. Chem. Soc., 2014, 136, 16902 CrossRef CAS PubMed.
- X. Li, Y. Zhang, H. Chen, J. Sun and F. Feng, ACS Appl. Mater. Interfaces, 2016, 8, 22756 CrossRef CAS PubMed.
- G. Jutz, P. van Rijn, B. Santos Miranda and A. Böker, Chem. Rev., 2015, 115, 1653 CrossRef CAS PubMed.
- M. Truffi, L. Fiandra, L. Sorrentino, M. Monieri, F. Corsi and S. Mazzucchelli, Pharmacol. Res., 2016, 107, 57 CrossRef CAS.
- K. Fan, C. Cao, Y. Pan, D. Lu, D. Yang, J. Feng, L. Song, M. Liang and X. Yan, Nat. Nanotechnol., 2012, 7, 459 CrossRef CAS.
- Z. Wang, H. Gao, Y. Zhang, G. Liu, G. Niu and X. Chen, Front. Chem. Sci. Eng., 2017, 11, 633 CrossRef CAS.
- B. Maity, K. Fujita and T. Ueno, Curr. Opin. Chem. Biol., 2015, 25, 88 CrossRef CAS.
- X. Jiang, L. Long, H. Wang, L. Chen and X. Liu, Dalton Trans., 2014, 43, 9968 RSC.
- D. H. Kim, H. S. Hwang and K. Na, Biomacromolecules, 2018, 19, 3301 CrossRef PubMed.
- X. Wang and Z. Guo, Chem. Soc. Rev., 2013, 42, 202 RSC.
- A. K. Jones, B. R. Lichtenstein, A. Dutta, G. Gordon and P. L. Dutton, J. Am. Chem. Soc., 2007, 129, 14844 CrossRef CAS.
- L. Toussaint, L. Bertrand, L. Hue, R. R. Crichton and J. P. Declercq, J. Mol. Biol., 2007, 365, 440 CrossRef CAS.
- R. K. Behera and E. C. Theil, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7925 CrossRef CAS PubMed.
- G. R. Bakker and R. F. Boyer, J. Biol. Chem., 1986, 261, 13182 CAS.
- E. J. Lyon, I. P. Georgakaki, J. H. Reibenspies and M. Y. Darensbourg, Angew. Chem., Int. Ed., 1999, 38, 3178 CrossRef CAS.
- J. S. Jang, S. Yu, S. J. Choi, S. J. Kim, W. T. Koo and I. D. Kim, Small, 2016, 12, 5989 CrossRef CAS PubMed.
- T. Tosha, H. L. Ng, O. Bhattasali, T. Alber and E. C. Theil, J. Am. Chem. Soc., 2010, 132, 14562 CrossRef CAS.
- Z. Q. Wu, R. J. Ono, Z. Chen, Z. Li and C. W. Bielawski, Polym. Chem., 2011, 2, 300–302 RSC.
- R. Xing, X. Wang, C. Zhang, Y. Zhang, Q. Wang, Z. Yang and Z. Guo, J. Inorg. Biochem., 2009, 103, 1039 CrossRef CAS PubMed.
- K. M. Shin, J. W. Lee, G. G. Wallace and S. J. Kim, Sens. Actuators, B, 2008, 133, 393 CrossRef CAS.
- J. X. Jian, Q. Liu, Z. J. Li, F. Wang, X. B. Li, C. B. Li, B. Liu, Q. Y. Meng, B. Chen, K. Feng, C. H. Tung and L. Z. Wu, Nat. Commun., 2013, 4, 2695 CrossRef.
- J. F. Capon, F. Gloaguen, F. Y. Pétillon, P. Schollhammer and J. Talarmin, Coord. Chem. Rev., 2009, 253, 1476 CrossRef CAS.
- M. L. Singleton, D. J. Crouthers, R. P. Duttweiler III, J. H. Reibenspies and M. Y. Darensbourg, Inorg. Chem., 2011, 50, 5015 CrossRef CAS.
- L. Balejčíková, V. M. Garamus, M. V. Avdeev, V. I. Petrenko, L. Almásy and P. Kopčanský, Colloids Surf., B, 2017, 156, 375 CrossRef PubMed.
- G. D. Watt, D. Jacobs and R. B. Frankel, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 7457 CrossRef CAS.
- C. D. Bostick, S. Mukhopadhyay, I. Pecht, M. Sheves, D. Cahen and D. Lederman, Rep. Prog. Phys., 2018, 81, 026601 CrossRef PubMed.
- U. Carmona, L. Li, L. Zhang and M. Knez, Chem. Commun., 2014, 50, 15358 RSC.
- https://www.uniprot.org .
- J. G. Nathanael, L. F. Gamon, M. Cordes, P. R. Rable, T. Bally, K. M. Fromm, B. Giese and U. Wille, ChemBioChem, 2018, 19, 922 CrossRef CAS.
- N. L. Ing, M. Y. El-Naggar and A. I. Hochbaum, J. Phys. Chem. B, 2018, 122, 10403 CrossRef CAS.
- N. Amdursky, ChemPlusChem, 2015, 80, 1075 CrossRef CAS.
- S. S. Panda, H. E. Katz and J. D. Tovar, Chem. Soc. Rev., 2018, 47, 3640 RSC.
- Y. Na, P. Wei and L. Zhou, Chem.–Eur. J., 2016, 22, 10365 CrossRef CAS PubMed.
- A. I. Stewart, J. A. Wright, G. M. Greetham, S. Kaziannis, S. Santabarbara, M. Towrie, A. W. Parker, C. J. Pickett and N. T. Hunt, Inorg. Chem., 2010, 49, 9563 CrossRef CAS PubMed.
- I. Silaghi-Dumitrescu, T. E. Bitterwolf and R. B. King, J. Am. Chem. Soc., 2006, 128, 5342 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05293j |
| This journal is © The Royal Society of Chemistry 2019 |