
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand-induced reduction concerted with coating by atomic layer deposition on the example of TiO2-coated magnetite nanoparticles†
Sarai
García-García
 a,
Alberto
López-Ortega
*a,
Yongping
Zheng
b,
Yifan
Nie
b,
Kyeongjae
Cho
b,
Andrey
Chuvilin
ac and
Mato
Knez
*ac
a,
Alberto
López-Ortega
*a,
Yongping
Zheng
b,
Yifan
Nie
b,
Kyeongjae
Cho
b,
Andrey
Chuvilin
ac and
Mato
Knez
*ac
aCIC nanoGUNE, Tolosa Hiribidea 76, 20018 Donostia-San Sebastián, Spain. E-mail: lopezortega.alberto@gmail.com; m.knez@nanogune.eu
bDepartment of Materials Science and Engineering, University of Texas at Dallas, Richardson, Texas 75080, USA
cIKERBASQUE, Basque Foundation for Science, Maria Diaz de Haro 3, 48013 Bilbao, Spain
First published on 17th December 2018
Abstract
Atomic layer deposition is a chemical deposition technology that provides ultimate control over the conformality of films and their thickness, even down to Ångström-scale precision. Based on the marked superficial character and gas phase process of the technique, metal sources and their ligands shall ideally be highly volatile. However, in numerous cases those ligands corrode the substrate or compete for adsorption sites, well-known as side reactions of these processes. Therefore, the ability to control such side reactions might be of great interest, since it could achieve synchronous coating and alteration of a substrate in one process, saving time and energy otherwise needed for a post-treatment of the sample. Consequently, advances in this way must require understanding and control of the chemical processes that occur during the coating. In this work, we show how choosing an appropriate ligand of the metal source can unveil a novel approach to concertedly coat and reduce γ-Fe2O3 nanoparticles to form a final product composed of Fe3O4/TiO2 core/shell nanoparticles. To this aim, we envisage that appropriate design of precursors and selection of substrates will pave the way for numerous new compositions, while the ALD process itself allows for easy upscaling to large amounts of coated and reduced particles for industrial use.
Introduction
Atomic layer deposition (ALD)1–3 is a coating technology that in recent years became indispensable in microelectronics4,5 and it enjoys a rapidly growing interest for emerging applications in energy storage,6–9 medicine,10–12etc. The reason is that ALD provides ultimate control over the conformality of films and their thickness, even down to Ångström-scale precision.13 ALD relies on the temporal separation of two or more chemical precursors and their individual surface reactions on the substrate surface upon exposure. The reactions are self-saturating in each reaction stage, ensuring that one complete ALD cycle can only result in a maximum of one monolayer coating. Repeating an ALD cycle adds another layer and allows the coating thickness to be precisely controlled with the choice of the number of processing cycles.14 In contrast to CVD, which requires higher temperatures and simultaneous delivery of two or more precursors, ALD can be performed at comparatively lower temperatures and the deposition will be truly non-directional.15An important aspect of ALD is that it is a chemical deposition technology and as such requires understanding and control of the chemical processes that occur during the coating. Most considerations for the development of a process are directed towards the precursor chemistry, which is usually thoughtfully designed to enable a rapid process with ideally no impurities in the growing film and no corrosion or any other kind of harm to the coated substrate. Typically, metalorganic, organometallic or halide compounds are utilized as metal sources for coatings and the ligands of those compounds shall ideally be highly volatile.16–19 However, in numerous cases those ligands corrode the substrate or compete for adsorption sites,20,21 which are side reactions of such processes. Typically, such side reactions are unwanted, and precursors are chosen which will not release reactive ligands or intermediates. The ability to control such side reactions might, however, be of great interest, since one could achieve synchronous coating and alteration of a substrate in one process, thus saving time and energy otherwise needed for post-treatment of the sample. One such scenario refers to the oxidation state of a substrate material. Since most ALD processes operate with a counter precursor that contains oxygen, for example, water vapor, oxygen plasma or ozone, it becomes difficult to preserve or achieve a reduced state of a metal during the process, particularly in those cases where the metal is prone to easy oxidation. The choice of a precursor which contains ligands that upon release can reduce the substrate can be beneficial as it would avoid the need for post-processing, for example in the form of reductive annealing. In the case of powders, the applied ALD process itself allows for easy upscaling to large amounts of coated and reduced particles for industrial use. Nowadays, the most well-known reactors for this purpose are the so-called fluidized bed reactors and rotatory reactors. Such reactors provide conformal film deposition on large amounts of particles by ALD for emerging applications.22–27
The combination of TiO2/FeOx in the synthesis of core/shell nanoparticles and its induced chemical reduction reach a wide range of functionalities for various applied materials. Nanoscale titanium dioxide (TiO2) found applications as a UV light attenuator. The incorporation of this material in sunscreens, cosmetics, clothes and plastic containers has successfully been demonstrated.28 Besides, its photocatalytic activity is beneficial for the removal of persistent organic pollutants and thus for recycling waste water.29 The recollection of such nanoparticles from water, however, poses a problem. A way to address this is to combine TiO2 with magnetic materials, such as iron oxide, which allows a convenient recovery of the catalyst under an external magnetic field.30
The potential uses of such combined materials are not limited to this application. If one looks at the same materials from the perspective of magnetic properties, the great biocompatibility of titanium dioxide in combination with superparamagnetic particles opens the door to a wide range of applications such as, targeted drug delivery,31 magnetic resonance imaging,32 hyperthermia,33 magnetic separation of biomolecules,34 and sensing applications.35
Herein, we report the synthesis of Fe3O4/TiO2 core/shell nanoparticles by ALD at moderate temperatures. We apply an ALD process, which involves two independent and concerted chemical reactions. As a substrate we use commercial iron oxide nanoparticles and as precursors, Ti-based precursors and deionized water. We demonstrate how it is possible to coat and reduce, at the same time, iron oxide particles depending on the existing ligand in the selected metal-based precursor. In other words, while the ALD process to coat the particles with TiO2 takes place, the Fe3+ ions in the seed nanoparticles can be partially reduced to Fe2+, enforcing the transformation of γ-Fe2O3 to the well-known magnetite, Fe3O4, structure. The resulting core/shell particles have been systematically studied by X-ray diffraction, magnetometry, energy dispersive X-ray analysis and electron microscopy. The underlying mechanism of the reduction reaction was further modelled by density functional theory. Upon comparisons with various optional Ti-precursors, the study showed that the ligands of the titanium precursor tetrakis(dimethylamino)titanium(IV) (TDMATi) are responsible for the partial reduction of Fe3+ by forming tetramethyl hydrazine as a byproduct during the corresponding oxidation reaction. Both, simulation and experiment show that only this type of precursor enables the synchronous coating-reduction reaction, while the energy barriers for a reduction are too high in the case of alternative choices of precursors. They will only yield a coating of the particles. This finding opens the possibility to design novel ALD precursors for a combined one-step capping and reduction process for the synthesis of further material combinations.
Results and discussion
Commercial γ-Fe2O3 nanoparticles (sample FeOx) have been coated with TiO2 by ALD using, at a first stage, TDMATi and water as precursors (sample TDMATi–FeOx). Fig. 1(a) shows X-ray diffraction (XRD) patterns obtained from the samples before and after ALD coating. Both diffraction patterns are very similar showing characteristic peaks of the cubic spinel structure stemming from the iron oxide. It should be noted that no characteristic peaks from titanium oxide are seen, which is due to the amorphous character of the TiO2 deposited under the applied processing conditions.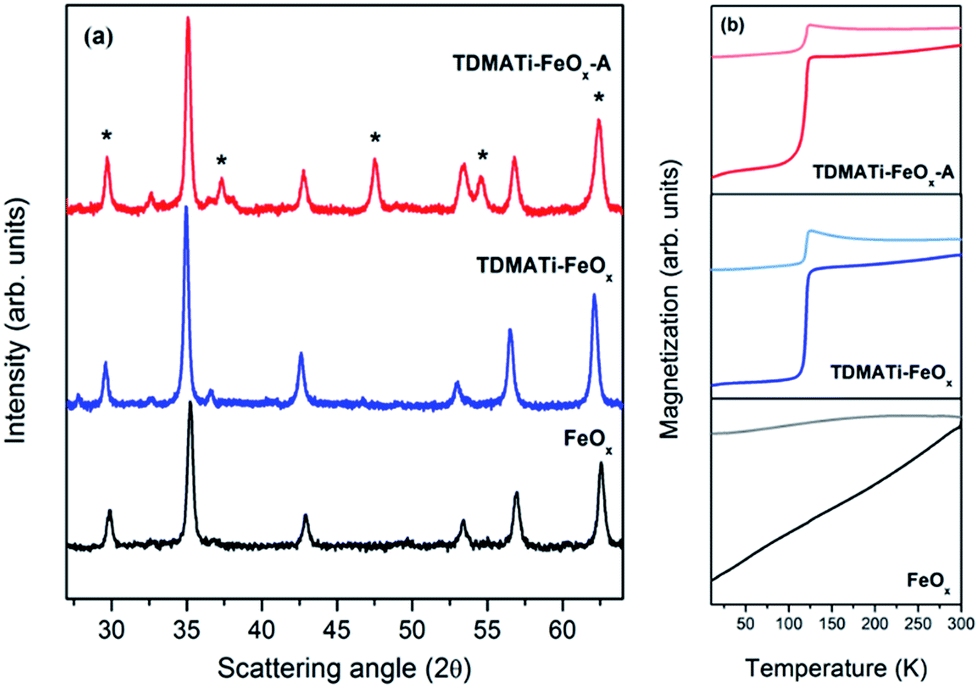 | ||
| Fig. 1 (a) XRD patterns of uncoated γ-Fe2O3 particles (FeOx) and TiO2-coated γ-Fe2O3 nanoparticles with TDMATi and H2O as precursors, before annealing (TDMATi–FeOx) and after annealing at 470 °C (TDMATi–FeOx-A). (b) Magnetization curves vs. temperature for the samples in (a). * denotes diffraction peaks originating from TiO2. | ||
However, the transmission electron micrographs show the formation of a core/shell structure. In Fig. 2 the core/shell construction of the nanoparticles is very obvious both from the contrast in scanning transmission electron microscopy (STEM) as well as the energy-dispersive X-ray spectroscopy (EDX) images, which show the involved elements in different colors. A more detailed analysis of the XRD patterns of samples FeOx and TDMATi–FeOx shows a clear alteration of the pristine iron oxide structure after the ALD process. While both patterns reveal the presence of a cubic spinel structure, the shift in their diffraction peaks and the concomitant variation of their cell parameter point towards the presence of two different iron oxide phases in the two samples.
 | ||
| Fig. 2 (a) STEM micrograph and (b) STEM-EDX map of Fe3O4 (red)/TiO2 (green) core/shell nanoparticles. | ||
The commercial particles have a cell parameter of 8.347(5) Å (see Table 1) which is characteristic of completely oxidized maghemite, γ-Fe2O3 (JCPDS card no. 39-1346), where Fe3+ cations occupy the octahedral and tetrahedral sites of the structure. In contrast, the sample TDMATi–FeOx shows an increase of the cell parameter to a value of 8.390(5) Å (see Table 1), which is characteristic of a partial Fe3+ → Fe2+ reduction of the octahedral cations to form the well-known magnetite, Fe3O4, structure (JCPDS card no. 19-629). Note that the crystal size of both samples is very similar with values of roughly 30 nm. The expected change of the volume of about 6% after the ALD process is not notable from Rietveld-based XRD measurements, since the resolution is not high enough for clearly confirming such small changes.
| Sample | Composition | ALD process | Phase | Lattice parameter (Å) | Crystal size (nm) | Verwey transition |
|---|---|---|---|---|---|---|
| FeOx | Commercial FeOx | — | γ-Fe2O3 | 8.347(5) | 32.5(5) | No |
| TDMATi–FeOx | TiO2/FeOx | TDMATi + H2O (150 °C) | Fe3O4 | 8.390(5) | 31(5) | Yes |
| TDMATi–FeOx-A | TiO2/FeOx, annealed (470 °C) | TDMATi + H2O (150 °C) | Fe3O4 | 8.388(5) | 32(5) | Yes |
| TiO2 | 3.782(5)/9.483(5) | 23(5) | ||||
| FeOx-A | Commercial FeOx, annealed (470 °C) | — | α-Fe2O3 | 5.031(5)/13.738(5) | 70(5) | No |
| TiCl4–FeOx | TiO2/FeOx | TiCl4 + H2O (150 °C) | γ-Fe2O3 | 8.339(5) | 28(5) | No |
| TDMASn–FeOx | SnO2/FeOx | TDMASn + H2O (150 °C) | γ-Fe2O3 | 8.351(5) | 23(5) | No |
| TDMASn–FeOx-250 | SnO2/FeOx | TDMASn + H2O (250 °C) | Fe3O4 | 8.380(5) | 30(5) | Yes |
| TDMAHf–FeOx | HfO2/FeOx | TDMAHf + H2O (150 °C) | Fe3O4 | 8.382(5) | 44(5) | Yes |
In order to gain more insight into the phase transformation observed after the ALD process we compared the magnetic properties of both samples by measuring their magnetization temperature curves under both field cooling (FC) and zero field cooling (ZFC) conditions (see Fig. 1(b)). Both samples depict the expected behavior of nanostructured magnetic nanoparticles where the FC and ZFC curves are distinct at low temperatures and tend to converge at the so-called blocking temperature (TB).36 Indeed, for such large iron oxide particles (i.e., 30 nm) this transition was observed at temperatures above RT.37 The sample TDMATi–FeOx shows a similar trend to the untreated sample, but with an extrapolated TB at higher temperatures. More interesting is, however, another magnetic transition that can be clearly observed at 120 K and can be ascribed to the Verwey temperature (TV) of Fe3O4. This transition is characteristic of Fe3O4 which undergoes a transformation from cubic (T > TV) to monoclinic (T < TV) symmetry.38 Such transformation is related to modifications in the crystal symmetry and the cation ordering and is accompanied by dramatic changes in electrical conductivity and heat capacity. Given the results of the magnetic characterization, it can be stated that the iron in the commercial particles (FeOx) suffered a transformation from a completely oxidized state, typical of maghemite (γ-Fe2O3), to a partially reduced state as it is typical of magnetite during the ALD process.
For crystallographic confirmation of the deposition of titanium oxide over the iron oxide, the particles have been annealed at 470 °C (sample TDMATi–FeOx-A). Fig. 1(a) shows the evolution of anatase-type TiO2 (JCPDS card no. 21-1272) after annealing, which resulted from the crystallization of the deposited film. The peaks of the cubic spinel structure of Fe3O4 are not affected by the heat treatment, preserving the structure obtained after the ALD process. The magnetization measurement of the particles after annealing further confirms the presence of magnetite showing its characteristic Verwey transition (see Fig. 1(b)). It is interesting to note that no traces of any further iron oxide crystal structure can be found, and the crystal size of the phase is very similar to that of the untreated sample (i.e., 30 nm). Both effects demonstrate that the TiO2 coating avoids intimate contact between the particles and thus their agglomeration and phase transformation to more stable iron oxide phases (i.e., α-Fe2O3), even if the temperature is increased to 470 °C.39
A further experiment was performed to compare the thermal stability of the material. Uncoated commercial γ-Fe2O3 nanoparticles were annealed using the same conditions as those of the sample TDMATi–FeOx-A (470 °C – 3 h). The results showed that the particles without TiO2 coating became α-Fe2O3 and their particle size increased from 30 nm to 70 nm due to the partial sintering of the nanoparticles after the thermal treatment as shown in Table 1. Besides, no reduction of particles was observed in the latter case. Similar structural transformation is expected for the Fe3O4 structure as it has already been demonstrated for thin films and nanoparticles.40,41
The TiO2 coating of iron oxide particles by ALD using TDMATi as the precursor not only protects the nanostructures from oxidation and agglomeration, but also induces a partial reduction of pristine γ-Fe2O3 and a concomitant phase transformation. Such a transformation may be triggered by either the metal or the ligand present in the ALD precursor.
Therefore, a series of experiments have been performed in order to identify the source. Firstly, the growth of TiO2 has been carried out using an alternative titanium precursor (titanium chloride, TiCl4) in order to identify a potential effect of the titanium ion. In the second stage, the effect of the (CH3)2N− ligand has been evaluated by performing ALD coating with another TDMA-based metal precursor, tetrakis(dimethylamino)tin(IV) (TDMASn).
The sample TiCl4–FeOx has been obtained after an ALD process of the same commercial iron oxide particles using TiCl4 and demineralized water as precursors at 150 °C. From the Rietveld analysis of the X-ray diffraction data in Fig. 3, it can be observed that after the TiO2 deposition the cell parameter of the iron oxide core remains similar to that of the pristine sample (γ-Fe2O3) (see Table 1). The magnetometric curves show no signs of Verwey transition. Therefore, the titanium cation appears not to be responsible for the partial reduction of the γ-Fe2O3 particles, which is reasonable from the chemical perspective as it is already present in the oxidation state +4 and further oxidation would not be possible under the given conditions.
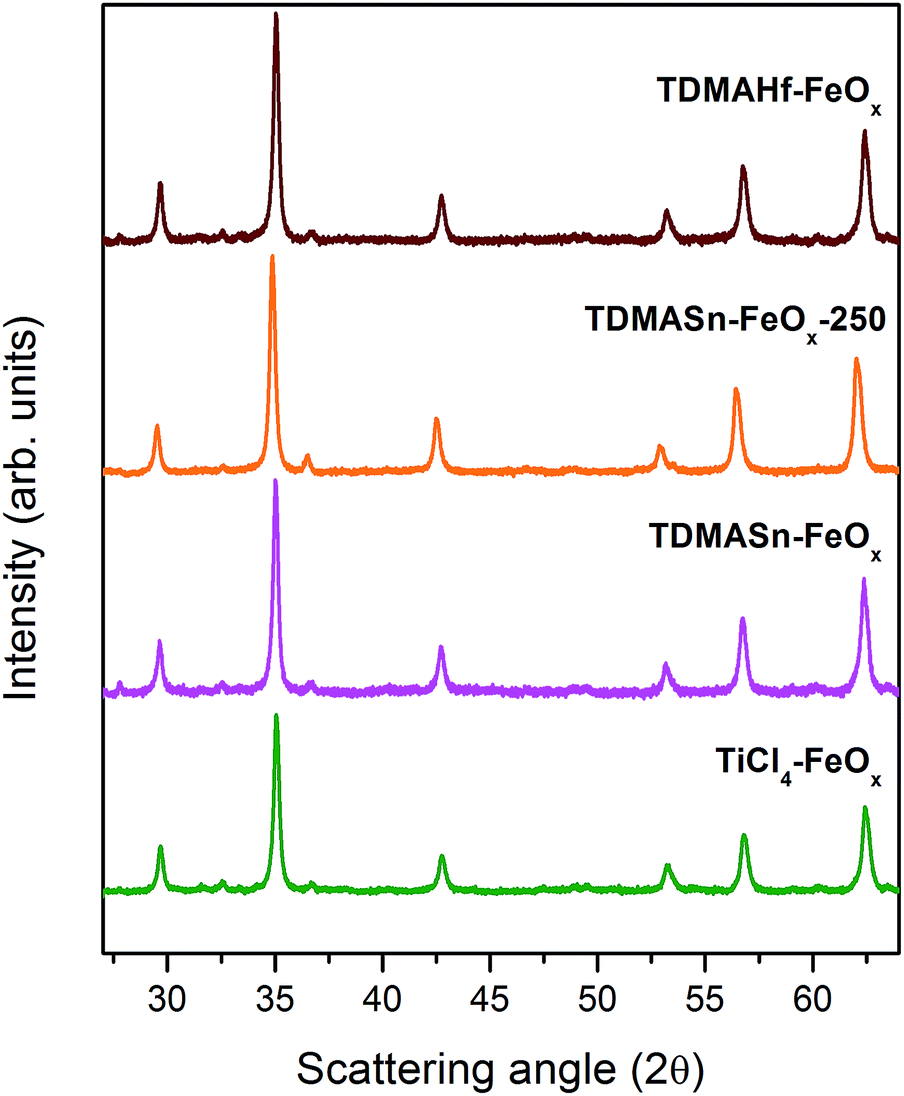 | ||
| Fig. 3 XRD patterns of TiO2-coated γ-Fe2O3 nanoparticles with TiCl4 and H2O ALD precursors (TiCl4–FeOx), SnO2-coated γ-Fe2O3 nanoparticles with TDMASn and H2O ALD precursors, at processing temperatures of 150 °C (TDMASn–FeOx) and 250 °C (TDMASn–FeOx-250) and HfO2-coated γ-Fe2O3 nanoparticles with TDMAHf and H2O ALD precursors (TDMAHf–FeOx). | ||
For investigating the ligand ((CH3)2N−) as a potential reducing agent, TDMASn and demineralized water were used as precursors in an ALD process at 150 °C (sample TDMASn–FeOx). Similar to the TiCl4 ALD process, the crystal structure of the iron oxide particles after using TDMASn seems not to be affected. The particles largely maintain their cell parameters (Table 1) and no presence of Verwey transition is observed.
However, considering the different Pauling electronegativities of Ti and Sn (i.e., 1.54 and 1.96, respectively), the reactivity of the ligand may be strongly affected by the metal cation present in the precursor. Assuming that the energy required for the ligand dissociation from the coordination sphere of the metal in TDMASn is higher than in the case of TDMATi, the ALD process with the tin precursor has been repeated at higher temperatures, namely, at 250 °C (sample TDMASn–FeOx-250). As a result, the iron oxide nanostructure is affected in a similar manner to that with TDMATi. The cell parameter is increased (Table 1) and the Verwey transition appears in the magnetization curves confirming the Fe3+ → Fe2+ partial reduction and, thus, the Fe3O4 formation.
Finally, in order to further confirm that the TDMA ligand is responsible for the reduction, a further ALD process with tetrakis(dimethylamino)hafnium(IV) (TDMAHf), sample TDMAHf–FeOx, as a precursor has been performed. Demineralized water was used as a counter precursor. Hf has an electronegativity similar to that of Ti (i.e., 1.3), which implies that upon correct assumption of the mechanism, similar results to those with TDMATi can be obtained. A partial reduction of γ-Fe2O3 may take place already at lower temperatures. Indeed, the Rietveld analysis of the samples TDMAHf–FeOx shows that the cell parameter is increased after the TDMAHf process to permit the formation of the Fe3O4 structure. In the magnetization curves the Verwey transition is clearly observed. Another possible precursor combination involves tetrakis(dimethylamino)zirconium(IV) (TDMAZr) and demineralized water. Zr has an electronegativity of 1.33, a value between the Ti and Hf electronegativities (1.54 and 1.3, respectively). Dennis M. Hausmann et al. successfully performed ALD depositions of ZrO2 with those precursors at 150 °C, under the same process conditions as those used here for TDMA-based precursors. Supposing that the ligand separation and reduction is a function of the Pauling electronegativity and, in consequence, the process temperature, both ZrO2 deposition and reduction of iron oxide particles are expected from this precursor combination as well.42,43
The results show that the anion of the precursor, ((CH3)2N−), is responsible for the partial reduction of iron oxide and the concomitant structural modification of the initial seed nanoparticles. In the first step of the ALD process, the TDMATi precursor loses the amino groups while anchoring to the surface of the nanoparticles. Those ligands can lose one electron each and recombine to form gas phase (CH3)2NN(CH3)2 (tetramethylhydrazine), which is the oxidation product as the γ-Fe2O3 substrate is being reduced.
We performed a DFT modelling of the reaction energies for such a case and compared it to the model of reactions with different precursors with varying metals and ligands (Fig. 4). Indeed, the calculations show that TDMAHf and TDMAZr have negative reaction energies (−0.37 eV and −0.25 eV, respectively), indicating an excellent capability of reducing γ-Fe2O3 to Fe3O4. TDMATi also energetically favours the reduction with an estimated activation barrier of 0.85 eV. However, with increasing the Pauling electronegativity of the central metal atom, as in the case of TDMASn and TDMAIr, the reduction reaction becomes energetically less favourable with larger activation barriers of 1.12 eV and 2.16 eV, respectively, indicating that higher reaction temperatures are needed to induce the reduction. On the other hand, the DFT-calculated reaction models of titanium precursors with different ligands such as titanium isopropoxide Ti(OiPr)4 and TiCl4 show much higher reaction barriers of 4.08 eV and 4.45 eV, respectively, meaning that no reduction is expected with those precursor families.
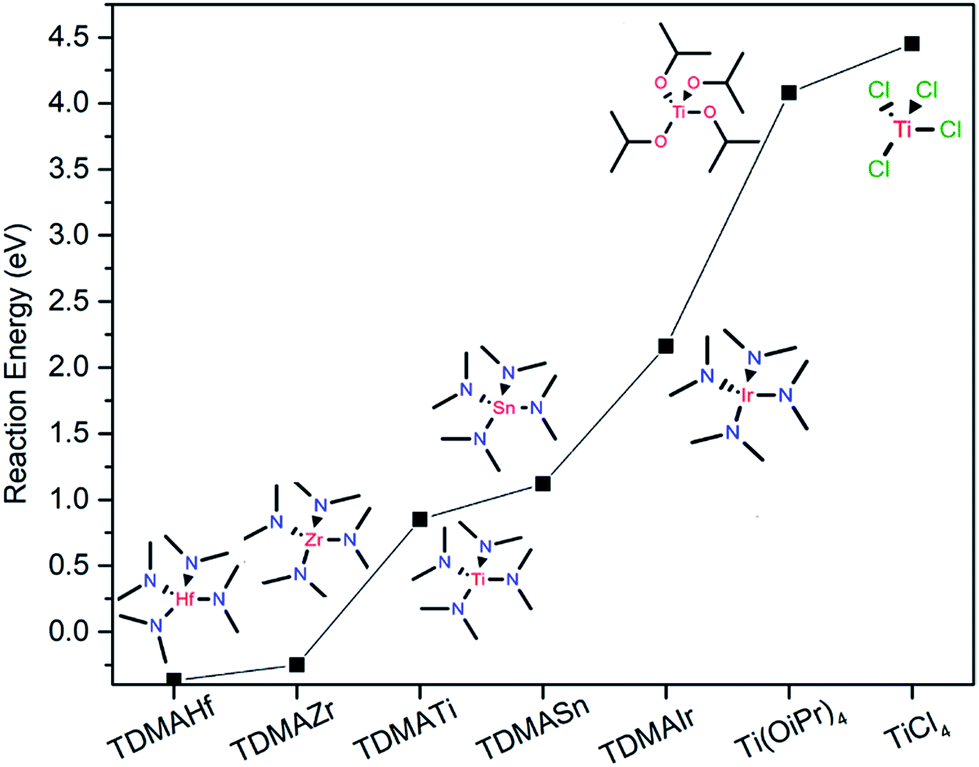 | ||
| Fig. 4 Comparison of the DFT-calculated reaction energies of reducing Fe2O3 to Fe3O4 by using different ALD precursors. | ||
These theoretical results well explain the phenomena observed experimentally and support the importance of precursor ligands for such concerted reduction-coating processes. When an ALD process is carried out, the electrons originating from the amino ligands of TDMATi create a chemical potential gradient at the nanoparticle surface triggering the reduction process along the whole iron oxide structure. This hypothesis is supported by the magnetometric curves measured after different ALD cycle numbers and the visual color change of the particles after the ALD process (Fig. 5). The characteristic TV of the Fe3O4 structure appears already after the first ALD cycle and becomes more pronounced as the number of cycles increases, which is very likely related to a mediated diffusion and growth mechanism.44 Therefore, even though the experiments were carried out with nanoparticles of approximately 50 nm in size, it can be assumed that the reduction process, which affects only the surface in contact with the precursors, will be to a large extent independent of the particle size. Therefore, the concerted process presented in this work can be envisaged for larger particle sizes and thin films as well. The porosity of the substrate plays a role in the process as it supports the diffusion of the precursors and the reduction of the substrate. Besides, it is possible to appreciate visually the color change from orange, in commercial γ-Fe2O3, to black, after the reduction to Fe3O4 (see Fig. 5).
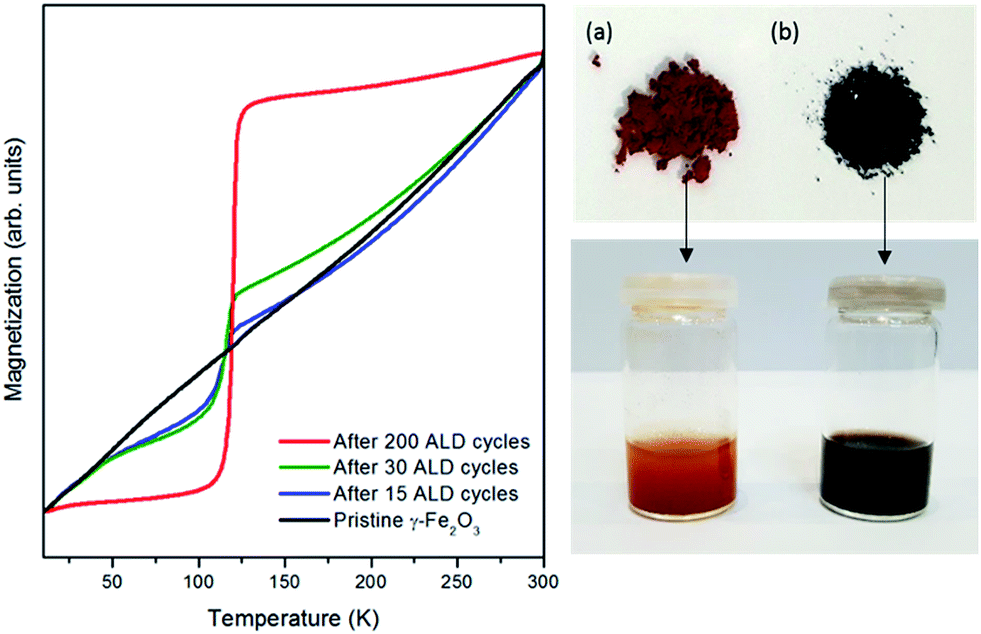 | ||
| Fig. 5 Left panel: magnetization vs. temperature curves of Fe3O4/TiO2 core/shell nanoparticles after various numbers of ALD coating cycles and a concerted reduction of the γ-Fe2O3 nanoparticles to Fe3O4. Right panel: visual color change of the particles in powder form (top) and dispersed in water (bottom); (a) untreated commercial γ-Fe2O3 nanoparticles, and (b) the same particles after applying various ALD cycles, forming Fe3O4/TiO2 core/shell nanoparticles. The color change from orange to black is indicative of a transformation from γ-Fe2O3 to magnetite. | ||
Experimental
Sample preparation
All samples were prepared following the same procedure. Commercial maghemite (γ-Fe2O3) nanoparticles with a particle size less than 50 nm, (CAS: 1309-37-1) from Sigma Aldrich, were dispersed in ethanol and sonicated for 15 minutes for better dispersion. Thereafter, the nanoparticles were spread over fused quartz substrates and dried for 24 hours. Prior to loading, the fused quartz substrates were cleaned in an ultrasonicator using acetone and then ethanol and blown dry using nitrogen gas.45ALD process
The coating of the nanoparticles was performed in a commercial ALD reactor (Savannah S100, Cambridge NanoTech Inc). The processes were carried out under a constant nitrogen gas flow rate of 20 standard cubic centimeters per minute (sccm).Various metal-containing ALD precursors have been used for the coating of the nanoparticles, including tetrakis(dimethylamino)titanium(IV) (TDMATi, [(CH3)2N]4Ti), titanium chloride (TiCl4), tetrakis(dimethylamino)tin(IV) (TDMASn, [(CH3)2N]4Sn) and tetrakis(dimethylamino)hafnium(IV) (TDMAHf, [(CH3)2N]4Hf), all of which were obtained from Sigma Aldrich. Demineralized water was used as the oxygen source. The coated nanoparticles were prepared following a typical ALD timing sequence expressed as t1–t2–t3–t4, where t1 is the exposure time to the first precursor, t2 is the purge time following the first exposure, t3 is the exposure time to the second precursor, and t4 is the purge time following the exposure to the second precursor. The times corresponding to t1–t2–t3–t4 were 20–25–20–25, all given in seconds. The pulsing times were a function of the vapor pressure of the precursor. For TDMA-based precursors (titanium, tin and hafnium) 0.5 s at 150 °C process temperature was used and 0.2 s was the pulsing time only for the comparative case, at 250 °C process temperature. For the chloride precursor 0.05 s was used, and for demineralized water in all the cases 0.05 s was the pulsing time.45 The process temperature in all cases was 150 °C. One further process was carried out at 250 °C with TDMASn in order to evaluate the thermal impact of the ALD process on the substrate. For all coatings, a thickness of approximately 30 nm was targeted. In order to obtain this thickness, 440 ALD cycles were performed for a TiO2 deposition with TDMATi, calculated from the obtained growth rate of 0.73 Å per cycle. For the TiCl4 process, 500 ALD cycles were applied, resulting in a growth rate of 0.66 Å per cycle. For the SnO2 process, 380 ALD cycles (growth rate: 0.79 Å per cycle) at 150 °C and 200 cycles (growth rate: 1.50 Å per cycle) at 250 °C were applied. For the HfO2 process, 400 ALD cycles were performed (growth rate: 0.82 Å per cycle).
After the ALD coating process (TiO2 generation with TDMATi and demineralized water), the coated, TDMATi–FeOx sample, and uncoated γ-Fe2O3 nanoparticles were subjected to ex situ annealing in order to crystalize the deposited film and make comparisons with the untreated particles. Both thermal treatments were carried out in air at atmospheric pressure and at 470 °C for 3 hours.46
Structural characterization
Scanning transmission electron microscopy (STEM) images were acquired with a FEI Titan operating at 300 keV in scanning mode with a coupled energy-dispersive X-ray spectrometer (STEM-EDX). The samples were dissolved in ethanol and drop cast onto carbon coated copper grids. Powder X-ray diffraction (XRD) patterns were recorded with a PANalytical XPert Pro X-ray diffractometer using Cu Kα radiation (λ = 1.5405980 Å) operating at 45 kV/40 mA in the 25–65° 2θ range with a scan speed of 0.00144°·s−1. Quantitative analysis of the XRD data was performed with a full pattern fitting procedure based on the fundamental parameter approach (Rietveld method) using MAUD software (http://maud.radiographema.eu/).The magnetic properties of the nanoparticles were measured from tightly packed powder samples using a vibrating sample mode magnetometer (Quantum Design SQUID-VSM overcool) with a 70 kOe maximum field. Magnetization versus temperature measurements were performed under zero-field cooled (ZFC) and field cooled (FC) conditions with 50 Oe probe fields.
Computational methods
All the calculations in this work were performed using the density functional theory (DFT) method implemented in the Vienna ab initio simulation package (VASP)47,48 with the projector-augmented wave (PAW) method.49 The generalized gradient approximation (GGA) with the semilocal Perdew–Burke–Ernzerhof (PBE)50 function was adopted to describe the exchange correlation interactions. The cutoff energy was set to be 450 eV. The structures were optimized with criteria of convergence for energy and force as 10−4 eV and 10−2 eV Å−1, respectively. On-site Coulomb repulsion of Fe 3d electrons was treated by Hubbard U correction51 with an effective U value Ueff = 4 eV according to previous theoretical reports.52,53The reaction tendency of reducing Fe2O3 to Fe3O4 with different ALD precursors was determined by the reaction energies of the following reaction:
| 6Fe2O3 + M–L4 → 4Fe3O4 + MO2 + 2L2 |
The molecular precursors and oxidation products are modelled in a 20 Å × 20 Å × 20 Å supercell using the standard lattice parameters of all oxides.
Conclusions
In this work, we demonstrate that the ALD process not only coats nanoparticles in a controlled way, but also depending on the ligand of the metallic precursor, the substrate material is modified with respect to its final chemical and/or structural properties. γ-Fe2O3 nanoparticles have been coated with TiO2 and concertedly reduced to magnetite. As a result, Fe3O4/TiO2 core/shell nanoparticles have been generated by atomic layer deposition at moderate temperatures. The deposited coatings prevent agglomeration of the nanoparticles and re-oxidation to γ-Fe2O3 even at high temperatures, allowing for the use of post-process annealing and thus crystallization of the amorphous TiO2 to anatase, a beneficial characteristic which has an important role in many industrial and bio-related applications.The reduction of γ-Fe2O3 to Fe3O4 is a function of the applied precursor and the processing temperature. The ligand of the precursor (CH3)2N− can become oxidized and recombine to form tetramethylhydrazine, while acting as a reducing agent for Fe3+. The cation of the precursor also plays an important role. The more electronegative the cation is, the more energy is necessary to release the ligands, which is important for the recombination. Therefore, the temperature required for ALD processes to reduce the sample is higher for TDMASn than for TDMATi and TDMAHf.
The use of an ALD process to concertedly coat and reduce nanomaterials simplifies numerous attempts to generate multifunctional materials for emerging applications, such as energy storage or medicine. The appropriate design of precursors and selection of substrates will pave the way for numerous new compositions, while the ALD process itself allows for easy upscaling to large amounts of coated and reduced particles for industrial use.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work has been performed with financial support of the Spanish Ministry of Economy and Competitiveness (MINECO) within grant agreement no. MAT2016-77393-R, including FEDER funds, and the Maria de Maeztu Units of Excellence Programme – MDM-2016-0618. The authors acknowledge networking support within the COST action MP1402. A. L.-O. acknowledges the Juan de la Cierva Program (MINECO IJCI-2014-21530). Y. Z. acknowledges International Energy Joint R & D Program (No. 20168510011350) of the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Ministry of Knowledge Economy.Notes and references
- G. N. Parsons, S. M. George and M. Knez, MRS Bull., 2011, 36, 865–871 CrossRef CAS.
- S. M. George, Chem. Rev., 2009, 110, 111–131 CrossRef PubMed.
- M. Knez, Mater. Matters, 2011, 3, 2–7 Search PubMed.
- R. W. Johnson, A. Hultqvist and S. F. Bent, Mater. Today, 2014, 17, 236–246 CrossRef CAS.
- F. Lee, S. Marcus, E. Shero, G. Wilk, J. Swerts, J. W. Maes, T. Blomberg, A. Delabie, M. Gros-Jean and E. Deloffre, ASMC Advanced Semicond. Manuf. Conf. Proc., 2007, pp. 359–365 Search PubMed.
- H. C. M. Knoops, M. E. Donders, M. C. M. van de Sanden, P. H. L. Notten and W. M. M. Kessels, J. Vac. Sci. Technol., A, 2012, 30, 010801 CrossRef.
- Y. S. Jung, A. S. Cavanagh, L. A. Riley, S. H. Kang, A. C. Dillon, M. D. Groner, S. M. George and S. H. Lee, Adv. Mater., 2010, 22, 2172–2176 CrossRef CAS.
- B. Yan, X. Li, Z. Bai, X. Song, D. Xiong, M. Zhao, D. Li and S. Lu, J. Power Sources, 2017, 338, 34–48 CrossRef CAS.
- W. Niu, X. Li, S. K. Karuturi, D. W. Fam, H. Fan, S. Shrestha, L. H. Wong and A. I. Y. Tok, Nanotechnology, 2015, 26, 064001 CrossRef CAS PubMed.
- S. A. Skoog, J. W. Elam and R. J. Narayan, Int. Mater. Rev., 2013, 58, 113–129 CrossRef CAS.
- R. J. Narayan, S. P. Adiga, M. J. Pellin, L. A. Curtiss, S. Stafslien, B. Chisholm, N. A. Monteiro-Riviere, R. L. Brigmon and J. W. Elam, Mater. Today, 2010, 13, 60–64 CrossRef CAS.
- M. Knez, A. Kadri, C. Wege, U. Gösele, H. Jeske and K. Nielsch, Nano Lett., 2006, 6, 1172–1177 CrossRef CAS PubMed.
- W.-S. Choi, Transactions on Electrical and Electronic Materials, 2009, 10, 200–202 CrossRef.
- R. P. Chaukulkar and S. Agarwal, J. Vac. Sci. Technol., A, 2013, 31, 031509 CrossRef.
- J. Leem, I. Park, Y. Li, W. Zhou, Z. Jin, S. Shin and Y. Min, Bull. Korean Chem. Soc., 2014, 35, 1195–1201 CrossRef CAS.
- R. L. Puurunen, J. Appl. Phys., 2005, 97, 121301 CrossRef.
- J. H. Yum, T. Akyol, D. A. Ferrer, J. C. Lee, S. K. Banerjee, M. Lei, M. Downer, T. W. Hudnall, C. W. Bielawski and G. Bersuker, J. Vac. Sci. Technol., A, 2011, 29, 061501 CrossRef.
- C. H. Hou, M. C. Chen, C. H. Chang, T. B. Wu, C. D. Chiang and J. J. Luo, J. Electrochem. Soc., 2008, 155, G180–G183 CrossRef CAS.
- M. Tallarida, C. Adelmann, A. Delabie, S. Van Elshocht, M. Caymax and D. Schmeisser, Appl. Phys. Lett., 2011, 99, 10–13 CrossRef.
- C. K. Ande, H. C. M. Knoops, K. de Peuter, M. van Drunen, S. D. Elliott and W. M. M. Kessels, J. Phys. Chem. Lett., 2015, 6, 3610–3614 CrossRef CAS PubMed.
- B. J. O'Neill, D. H. K. Jackson, J. Lee, C. Canlas, P. C. Stair, C. L. Marshall, J. W. Elam, T. F. Kuech, J. A. Dumesic and G. W. Huber, ACS Catal., 2015, 5, 1804–1825 CrossRef.
- A. P. Didden, J. Middelkoop, W. F. A. Besling, D. E. Nanu and R. Van De Krol, Rev. Sci. Instrum., 2014, 85, 013905 CrossRef PubMed.
- J. A. McCormick, B. L. Cloutier, A. W. Weimer and S. M. George, J. Vac. Sci. Technol., A, 2007, 25, 67–74 CrossRef CAS.
- W. J. Kim, E. Jang and T. J. Park, Appl. Surf. Sci., 2017, 419, 159–164 CrossRef CAS.
- J. D. Ferguson, K. J. Buechler, A. W. Weimer and S. M. George, Powder Technol., 2005, 156, 154–163 CrossRef CAS.
- E. Jang, K. Sridharan, Y. M. Park and T. J. Park, Chem.–Eur. J., 2016, 22, 12022–12026 CrossRef CAS PubMed.
- K. Sridharan, E. Jang, Y. M. Park and T. J. Park, Chem.–Eur. J., 2015, 21, 19136–19141 CrossRef CAS PubMed.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS.
- M. E. Borges, M. Sierra, J. Méndez-Ramos, P. Acosta-Mora, J. C. Ruiz-Morales and P. Esparza, Sol. Energy Mater. Sol. Cells, 2016, 155, 194–201 CrossRef CAS.
- W. Wu, X. Xiao, S. Zhang, F. Ren and C. Jiang, Nanoscale Res. Lett., 2011, 6, 1–15 Search PubMed.
- N. H. Cho, T. C. Cheong, J. H. Min, J. H. Wu, S. J. Lee, D. Kim, J. S. Yang, S. Kim, Y. K. Kim and S. Y. Seong, Nat. Nanotechnol., 2011, 6, 675–682 CrossRef CAS PubMed.
- G. P. Yan, L. Robinson and P. Hogg, Radiography, 2007, 13, e5–e19 CrossRef.
- M. Ohtake, M. Umemura, I. Sato, T. Akimoto, K. Oda, A. Nagasako, J. H. Kim, T. Fujita, U. Yokoyama, T. Nakayama, Y. Hoshino, M. Ishiba, S. Tokura, M. Hara, T. Muramoto, S. Yamada, T. Masuda, I. Aoki, Y. Takemura, H. Murata, H. Eguchi, N. Kawahara and Y. Ishikawa, Sci. Rep., 2017, 7, 1–12 CrossRef.
- J. Nam, H. Huang, H. Lim, C. Lim and S. Shin, Anal. Chem., 2013, 85, 7316–7323 CrossRef CAS.
- M. Pumera, Mater. Today, 2011, 14, 308–315 CrossRef CAS.
- C. P. Bean and J. D. Livingston, J. Appl. Phys., 1959, 30, S120–S129 CrossRef.
- D. Caruntu, G. Caruntu and C. J. O'Connor, J. Phys. D: Appl. Phys., 2007, 40, 5801–5809 CrossRef CAS.
- F. Walz, J. Phys.: Condens. Matter, 2002, 14, R285 CrossRef CAS.
- W. Promnopas, S. Promnopas, T. Phonkhokkong, T. Thongtem, D. Boonyawan, L. Yu, O. Wiranwetchayan, A. Phuruangrat and S. Thongtem, Surf. Coat. Technol., 2016, 306, 69–74 CrossRef CAS.
- A. Jafari, S. Farjami Shayesteh, M. Salouti and K. Boustani, J. Magn. Magn. Mater., 2015, 379, 305–312 CrossRef CAS.
- F. Genuzio, A. Sala, T. Schmidt, D. Menzel and H.-J. Freud, J. Phys. Chem. C, 2014, 118, 29068–29076 CrossRef CAS.
- D. M. Hausmann, E. Kim, J. Becker and R. G. Gordon, Chem. Mater., 2002, 14, 4350–4358 CrossRef CAS.
- J. Provine, P. Schindler, J. Torgersen, H. J. Kim, H.-P. Karnthaler and F. B. Prinz, J. Vac. Sci. Technol., A, 2016, 34, 01A138 CrossRef.
- A. López-ortega, E. Lottini, G. Bertoni, C. D. J. Fernández and C. Sangregorio, Chem. Mater., 2017, 29, 1279–1289 CrossRef.
- A. B. F. Martinson, M. J. DeVries, J. A. Libera, S. T. Christensen, J. T. Hupp, M. J. Pellin and J. W. Elam, J. Phys. Chem. C, 2011, 115, 4333–4339 CrossRef CAS.
- C.-P. Lin, H. Chen, A. Nakaruk, P. Koshy and C. C. Sorrell, Energy Procedia, 2013, 34, 627–636 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Furthmiiller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 3865–3868 CrossRef CAS PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- F. Kong, R. C. Longo, D. Yeon, J. Yoon, J. Park, C. Liang, S. Kc, Y. Zheng, S. Doo and K. Cho, J. Phys. Chem. C, 2015, 119, 21904–21912 CrossRef CAS.
- F. Zhou, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 235121–235151 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Magnetization vs. temperature graphics of comparison samples. List of possible reaction schemes and DFT calculated reaction energies for reducing Fe2O3 to Fe3O4 by using different ALD precursors. XRD patterns of TiO2-coated (TDMATi–FeOx) and uncoated FeOx nanoparticles before and after the annealing process. See DOI: 10.1039/c8sc04474k |
| This journal is © The Royal Society of Chemistry 2019 |