
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect sequencing of 2′-deoxy-2′-fluoroarabinonucleic acid (FANA) using nanopore-induced phase-shift sequencing (NIPSS)†
Shuanghong
Yan‡
 ac,
Xintong
Li‡
*d,
Panke
Zhang
abc,
Yuqin
Wang
ac,
Hong-Yuan
Chen
abc,
Shuo
Huang
*abc and
Hanyang
Yu
*d
ac,
Xintong
Li‡
*d,
Panke
Zhang
abc,
Yuqin
Wang
ac,
Hong-Yuan
Chen
abc,
Shuo
Huang
*abc and
Hanyang
Yu
*d
aState Key Laboratory of Analytical Chemistry for Life Sciences, Nanjing University, 210023, Nanjing, China
bCollaborative Innovation Centre of Chemistry for Life Sciences, Nanjing University, 210023, Nanjing, China
cSchool of Chemistry and Chemical Engineering, Nanjing University, 210023, Nanjing, China. E-mail: shuo.huang@nju.edu.cn
dDepartment of Biomedical Engineering, College of Engineering and Applied Sciences, Nanjing University, 210023, Nanjing, China. E-mail: hanyangyu@nju.edu.cn
First published on 23rd January 2019
Abstract
2′-deoxy-2′-fluoroarabinonucleic acid (FANA), which is one type of xeno-nucleic acid (XNA), has been intensively studied in molecular medicine and synthetic biology because of its superior gene-silencing and catalytic activities. Although urgently required, FANA cannot be directly sequenced by any existing platform. Nanopore sequencing, which identifies a single molecule analyte directly from its physical and chemical properties, shows promise for direct XNA sequencing. As a proof of concept, different FANA homopolymers show well-distinguished pore blockage signals in a Mycobacterium smegmatis porin A (MspA) nanopore. By ligating FANA with a DNA drive-strand, direct FANA sequencing has been demonstrated using phi29 DNA polymerase by Nanopore-Induced Phase Shift Sequencing (NIPSS). When bound with an FANA template, the phi29 DNA polymerase shows unexpected reverse transcriptase activity when monitored in a single molecule assay. Following further investigations into the ensemble, phi29 DNA polymerase is shown to be a previously unknown reverse transcriptase for FANA that operates at room temperature, and is potentially ideal for nanopore sequencing. These results represent the first direct sequencing of a sugar-modified XNA and suggest that phi29 DNA polymerase could act as a promising enzyme for sustained sequencing of a wide variety of XNAs.
1. Introduction
Xeno-nucleic acids (XNAs) are a class of nucleic acid molecules with unnatural backbones or nucleobases.1,2 Due to their improved chemical diversity and biological stability, XNAs have demonstrated exciting applications in molecular medicine, synthetic biology and materials sciences.1–3 Similar to natural DNA and RNA, the functional properties of all XNA molecules such as aptamers and enzymes are dictated by their specific sequences. Although an XNA sequence could be deduced by copying XNA into DNA followed by conventional sequencing, this strategy inevitably overlooks the errors introduced during XNA reverse transcription.4,5 On the other hand, a direct sequence readout from any XNA polymer would provide invaluable information to the field of synthetic genetics,1,8 but this has never been reported due to the lack of appropriate polymerases compatible with current sequencing technologies.62′-deoxy-2′-fluoroarabinonucleic acid (FANA) is an RNA analogue in which the ribose ring has been replaced by a 2′-fluoroarabinose moiety (Fig. 1A).7 It has been demonstrated that FANA exhibits potent gene-silencing capability and a longer serum half-life.8,9 Recent advances in polymerase engineering have enabled in vitro evolution of functional FANA molecules with specific ligand-binding and catalytic activities.10,11 Although two engineered polymerases, such as D4K and RT521, could catalyze efficient information transfer between FANA and DNA,12 the fidelity of such an enzyme-mediated polymerization reaction has not been thoroughly examined due to the lack of direct FANA sequencing technologies. Though FANA may be sequenced by the approach of sequencing by hybridization using an Affymetrix chip,13–15 no related work has been reported to the best of our knowledge. Consequently, the development of a direct sequencing methodology for FANA, preferentially also applicable to other XNAs, is of great significance for the growth of the XNA field.
 | ||
| Fig. 1 Discrimination of FANA homopolymers via a static pore blockage. (A) The structure of 2′-deoxy-2′-fluoroarabinonucleic acid (FANA). (B) Schematic diagram of a static pore blockage event. A streptavidin molecule (purple sphere) is tethered to a piece of 3′-biotin (green triangle)-modified FANA. The FANA tethered in this way is electrophoretically driven into the MspA nanopore and held by streptavidin, generating a static, sequence dependent pore blockage current (pink region). (C) Representative pore blockage events from streptavidin-tethered FANA polyU (green), polyA (blue), and polyC (red) sequences. Io, open pore ionic current; Ib, blocked ionic current. (D) Event statistics of Ib/Io for different FANA homopolymers. Histogram peaks from different FANA homopolymers (polyU, green; polyA, blue; polyC, red) are Gaussian fitted. The experiments were carried out in the electrolyte buffered solution of 1 M KCl, 10 mM HEPES at pH 8.0. | ||
Nanopore sequencing, which recognizes DNA base identities16 and the corresponding epigenetic modifications17,18 from their unique physical or chemical properties,16,19,20 is an emerging single-molecule technology which is promising for direct XNA sequencing. In a typical nanopore measurement, the nanopore is the only access which permits the flow of the ionic current between two chambers containing electrolytic solutions. With an applied potential, charged analytes are electrically driven through the pore, and the analytical information could be readily recognized and distinguished from the current blockades. Nowadays, the MinION™ sequencer (Oxford Nanopore Technologies, UK), which offers advantages of speed, label-free and long read length, could sequence genomic DNA with an affordable cost in a palm sized device.21 However, though urgently needed, direct sequencing of xeno-nucleic acids has not been demonstrated to date.
For FANA to be directly sequenced by a nanopore, the pore blockage signals should show a detectable sequence dependence. As a proof of concept, a static pore blockage experiment assisted by a streptavidin stopper was performed with an engineered Mycobacterium smegmatis porin A (MspA) nanopore.22 Clearly resolvable signals are observed from homopolymer FANA (polyU, polyC and polyA, ESI Table S1†). As demonstrated with DNA sequencing using nanopores,19 the selection of a ratcheting enzyme (a helicase or a polymerase) which drives the strand to move along the pore restriction is critical for direct FANA sequencing. However, as a member of xeno-nucleic acids, FANA doesn′t possess a large archive of compatible enzymes that work at room temperature with a high processivity like phi29 DNA polymerase (DNAP).23,24 Here the processivity means an enzyme′s ability to catalyze “consecutive reactions without releasing its substrate”. As a compromise, the height of the octameric MspA nanopore25 could be utilized to perform “Nanopore Induced Phase-Shift Sequencing (NIPSS)”, which is the method first defined in this paper and is a universal nanopore sequencing method for a variety of biomacromolecules provided a DNA drive-strand could be chemically attached.
According to single molecule kinetics results observed from direct FANA sequencing using NIPSS and ensemble assays investigated by gel electrophoresis, the phi29 DNAP has been surprisingly shown to perform FANA templated DNA synthesis. Since FANA is a RNA analogue, the phi29 DNAP should thus be categorized as its reverse transcriptase. Although with a reduced efficiency against the opposing electric field during nanopore sequencing, phi29 DNAP, which is a highly processive enzyme that works at room temperature, is a promising candidate for protein engineering aiming for sustained nanopore sequencing of FANA. As reported, significant efforts have been made similarly to realize direct RNA sequencing using nanopores.26
To the best of our knowledge, this is the first report of single molecule FANA identification and direct sequencing using a nanopore sensor. Although preliminarily, the reported “NIPSS” method provides a universal means to sequence a variety of xeno-nucleic acids or even other biomacromolecules in a nanopore sequencing scheme, which could unambiguously discriminate between DNA, RNA or other XNA nucleotides even within a chimeric strand. The nanopore sequencing assay demonstrated in this paper could also be adapted to the screening of other XNA compatible motor proteins for sustained single molecule sequencing. The demonstrated results also add phi29 DNAP to the family of FANA reverse transcriptases as a promising model for more optimized and sustained FANA sequencing using nanopores.
2. Results and discussion
As an octameric ion channel protein, the Mycobacterium smegmatis porin A (MspA)22,25 nanopore, which has a short and narrow restriction, is ideal for nanopore sequencing with a high spatial resolution. As previously reported, an engineered MspA (D90N/D91N/D93N/D118R/E139K/D134R), which is designed to neutralize the original negative charges from its wildtype (PDB ID: 1uun),25 forms a stable octameric pore assembly and is free of spontaneous gating.22 This is the only nanopore used in this study, and this mutant is named MspA (Supporting Methods) throughout this paper for simplicity.2.1. Discrimination of FANA homopolymers via static pore blockages
To mimic a static stage during nanopore sequencing, pore blockage experiments were performed by first tethering a streptavidin molecule to a 3′-biotin modified nucleic acid strand. At +180 mV, the streptavidin tethered nucleic acid, which is electrophoretically driven into the MspA nanopore, generates a sequence specific blockage current (Ib). According to the crystal structure of the octameric MspA nanopore and previously published results,27 the pore restriction of the MspA simultaneously accommodates approximately 4 nucleotides.20 This leads to a blockage current dependent on the mean contribution from these 4 DNA bases (Fig. 1B). As demonstrated with ssDNA,20 this static pore blockage assay is an effective measure of the discriminatory capability of the pore for the nucleic acid of interest before nanopore sequencing is carried out.To evaluate potential FANA-pore restriction interactions28 and to probe whether FANA, which has an altered sugar backbone compared to DNA or RNA, could produce clear sequence specific blockage currents (Ib), three FANA homopolymers (FANA polyU, polyC and polyA) were designed (ESI Table S1,† Supporting Methods) and synthesized for use in the static pore blockage assay. FANA polyG was excluded to avoid complications from G-quadruplexes. Considering the high cost of the FANA monomer, three FANA homopolymers are designed in a chimeric DNA–FANA form (ESI Table S1†), where the nucleotides that are to be recognized by the pore restriction are still composed of FANA. When FANA, which is negatively charged, is electrophoretically driven into the pore at +180 mV, an instantaneous reduction in the ionic current (Ib) at the open pore level (Io) is observed. By evaluating different FANA homopolymers with this assay, which produces statistics for Ib from a programmed cycling voltage protocol, the value of Ib from different types of homopolymers follows the order Ib,polyU < Ib,polyA < Ib,polyC as demonstrated by their corresponding representative traces (Fig. 1C). By sequentially adding different types of homopolymer samples to the cis side during a continuous measurement with the same pore, the identity for each Ib value is judged by the order of appearance for the corresponding peak during statistics (ESI Fig. S1†). To minimize signal drifting from inevitable issues such as pore to pore variations or water evaporation during data acquisition, the value of Ib/Io is taken as a normalized percentage blockage amplitude. Histograms of the Ib/Io of FANA homopolymers and the corresponding Gaussian fittings are shown in Fig. 1D, which demonstrate the signal dispersion and peak separation. To form a statistical conclusion and to evaluate the reproducibility of the assay, three independent measurements were performed for each assay with different FANA homopolymers (ESI Table S2†).
As demonstrated, different FANA homopolymers can be readily distinguished by an MspA nanopore. According to previously published results, examination of DNA homopolymers shows a different order of Ib/Io (Ib,polyT < Ib,polyC < Ib,polyA) in contrast to the results from FANA (Ib,polyU < Ib,polyA < Ib,polyC), in which the order of Ib/Io from polyA and polyC is reversed. This order switch between polyA and polyC simply originates from the altered sugar backbone of FANA, which is a minor physical property variation sensed by the sharply restricting nanopore.
Since FANA polyU and polyA/C generate up to 6%/9% Ib/I0 values (ESI Table S2†), different sequence combinations in FANA are expected to result in detectable transitions in current values, presumably in a nanopore sequencing assay when a strand of FANA translocates through the restriction of MspA in a single nucleotide step.
2.2. Direct FANA sequencing via “Nanopore Induced Phase-Shift Sequencing (NIPSS)”
Similar to that of ssDNA29 or ssRNA,30 nanopore translocation of FANA results in resistive pulses too fast to resolve its sequence information (ESI Fig. S2†). Nanopore sequencing of DNA utilizes a ratcheting enzyme such as a phi29 DNA polymerase19 or an engineered helicase31 to reduce the DNA translocation speed down to a few tens of ms/base to fit the bandwidth (100 kHz) of a conventional patch clamp amplifier. The selection criteria of this ratcheting enzyme for nanopore sequencing is strictly limited by the requirement of high salt tolerance, low working temperature and high enzyme processivity.32 As a compromise between sequencing signal separation and enzyme efficiency, reported nanopore sequencing experiments were carried out in 0.3–0.5 M KCl buffer,33 which is still too high a salinity for most enzymes other than phi29 DNAP or Hel308 helicase.34 Besides, conventional nanopore recordings are performed at room temperature to avoid induced noise both from thermal vibrations and the Peltier heater itself. During nanopore sequencing, a ratcheting enzyme with a high processivity is critical for sequencing of a long read-length.Though limited, the selection of FANA compatible enzymes includes D4K, Deep Vent and RT521K polymerase,12,35 which may be adapted as motor enzymes in a nanopore sequencing assay. However, they are all designed to work at a higher temperature and they exhibit minimal FANA reverse transcriptase activity below 25 °C, at which routine nanopore sequencing is carried out. Although directed evolution might yield engineered FANA reverse transcriptases that are more temperature-tolerant, laborious efforts are required.
As a compromise, direct FANA sequencing could be carried out by NIPSS as a proof of concept. Being a conically shaped nanopore with a finite height (∼10 nm), the restriction site of MspA, which reads the nucleic acid identity, is always at a fixed distance ahead of the reaction site of the motor protein. As has been reported,20 this fixed distance is equivalent in length to 14–15 nucleotides, which generates a fixed phase shift between electrochemical DNA reading and enzymatic DNA ratcheting. In principle, a limited length of any biomacromolecule could be sequenced by NIPSS as long as this chain-shaped polymer could be chemically tethered to a fragment of ssDNA, which is defined as the “drive-strand” during NIPSS.
Phi29 DNAP, which is widely used for isothermal gene amplification24 or nanopore sequencing,16 is a highly processive (>70 kilobase) enzyme with a strong strand displacement capacity that operates at room temperature. It is thus employed as the motor protein for phase-shift sequencing of FANA. To demonstrate its feasibility, a chimeric DNA–FANA template, which is composed of an FANA strand embedded within a DNA template, is custom synthesized and named FANAx (Fig. 2A, ESI Table S1, Fig. S3†). An abasic spacer, which is known to produce an abnormally large blockage current, is placed between the FANA and DNA as a signal marker to identify the beginning of sequencing signals from FANA. During nanopore sequencing, any plateau-shaped signal transition that appears after the signal from the abasic spacer represents successful direct FANA sequence reading.
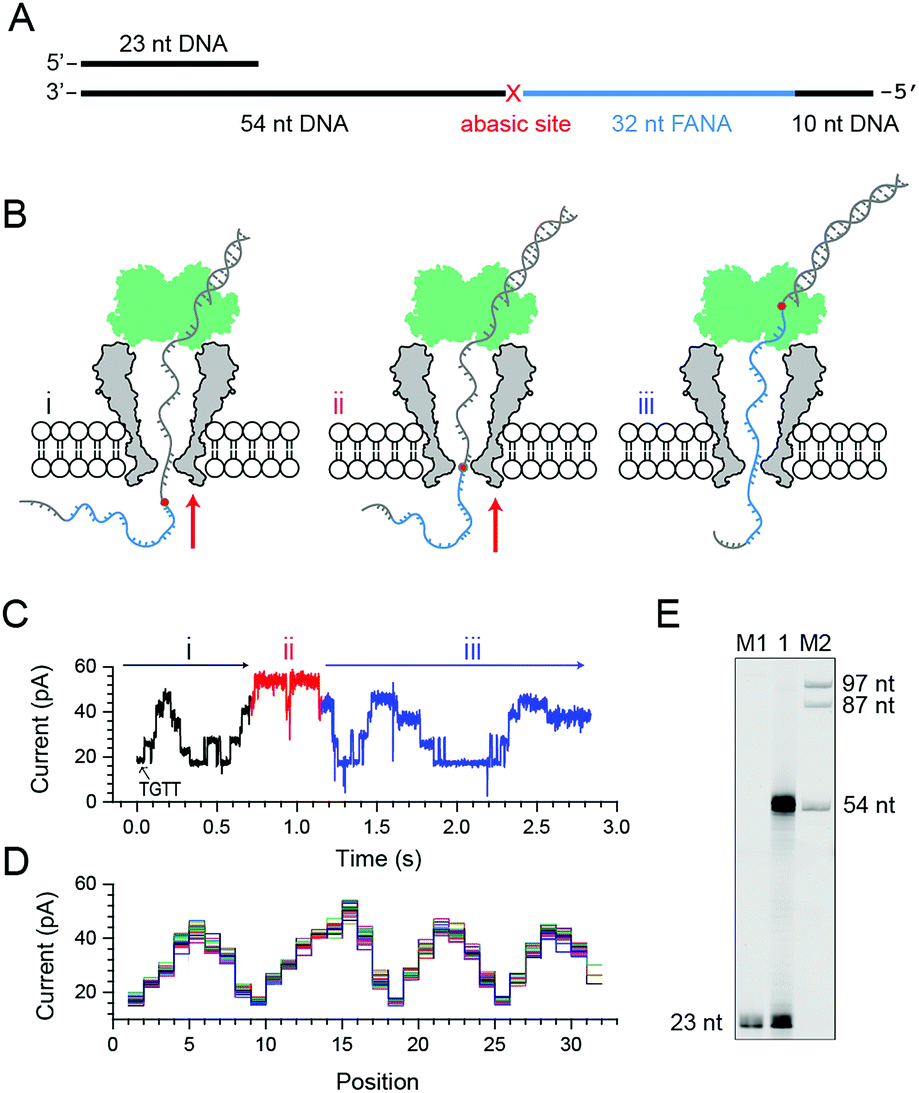 | ||
| Fig. 2 Nanopore sequencing of chimeric DNA–FANA with an abasic spacer (FANAx). (A) The FANAx template annealed with a primer (x: abasic site). The 54 nt DNA acts as the drive-strand for a primer extension. The abasic nucleotide (x) acts as a marker separating DNA and FANA. (B) Schematic diagram of nanopore sequencing of chimeric DNA (grey)-FANA (cyan) with an abasic spacer (red). During sequencing, the nucleic acid strand is directionally (red arrow) driven by a phi29 DNA polymerase (DNAP, green) via the primer extension. The recorded signal corresponds to the (i) DNA, (ii) abasic nucleotide and (iii) FANA sequence passing through the restriction site of MspA. Enzymatic ratcheting halts when the abasic site reaches the binding pocket of phi29 DNAP (ESI Fig. S4†). (C) A representative nanopore sequencing trace for FANAx. The sequencing trace corresponds to DNA (i, black), the abasic site (ii, red) and FANA (iii, blue) within the pore restriction. The step with an arrow is from the TGTT blockage. (D) Overlay of multiple time-normalized events using a level detection algorithm. (E) The phi29 DNAP-mediated FANAx reverse transcription assay analyzed by denaturing PAGE. The primer extension stops at the abasic site (54 nt). M1 and M2: DNA markers. | ||
Experimentally, direct FANA sequencing by NIPSS was carried out with an electrolyte buffer composed of 0.3 M KCl, 10 mM MgCl2, 10 mM (NH4)2SO4, 4 mM DTT and 10 mM HEPES at pH 7.5. After single pore insertion, the thermally annealed nanopore sequencing library (Supporting Methods) is added to the cis at a final concentration of 5 nM. The sequencing library (ESI Fig. S4†) is composed of three parts: the DNA–FANA chimera, primer and blocker (ESI Table S1†). The blocker protects the DNA–FANA chimera template from enzymatic extension in the solution. During continuous recordings at +180 mV, electrophoretic unzipping of the blocker strand triggers the initiation of the nanopore sequencing signal for FANAx when the phi29 DNAP based primer extension starts. During primer extension driven by the polymerase reaction from the phi29 DNAP, the nanopore reads DNA (Fig. 2B i), the abasic spacer (Fig. 2B ii) and FANA (Fig. 2B iii) sequentially.
From the published literature,16 the MspA reading of AGAA and TGTT (5′→3′ convention, if not otherwise stated) shows the highest and the lowest blockage level, respectively. As a model analyte with clearly distinguishable sequencing patterns, the FANAx is designed to possess a sequence repeat of AGAATGTT in the DNA part and AGAAUGUU in the FANA part (ESI Table S1†). The pore restriction simultaneously reads four nucleotides, and a sequential nanopore reading from TGTT to AGAA with a single nucleotide progression results in five plateaus linked by amplitude transitions between “TGTT, ATGT, AATG, GAAT and AGAA”, given that the nanopore sequencing reads following a 3′→5′ direction. The nanopore sequencing signal from the DNA part is thus expected to possess a triangular shape starting by reading TGTT. The DNA part of the nanopore sequencing signal is black colored in Fig. 2C, where 1.5 period of a triangular shape is recorded. Immediately after the DNA signal, nanopore reading of the abasic spacer results in an abnormally high blockage level higher than that from AGAA, which is colored red in the trace. Any step-shaped trace that appears after the blockage signal from the abasic spacer belongs to that from direct sequencing of FANA.
The FANA part of the signal, which is blue in the trace, shows a similar sequencing pattern in reference to its DNA counterpart. It can be seen that the blockage amplitude of AGAA and UGUU from FANA appears at about the same height as that of AGAA and TGTT from the DNA. However, only 4 plateau transitions are detected when reading UGUU to AGAA within the FANA part of the strand, which indicates that signal amplitude degeneracy exists among AUGU, AAUG and GAAU. To demonstrate the repeatability of the sequencing signal, 29 NIPSS events from FANAx were normalized and overlapped in Fig. 2D. DNA, abasic marker and FANA sequencing signals can be clearly recognized from their characteristic amplitudes (ESI Table S3†).
From single molecule NIPSS events, it can also be seen that the sequencing signal of FANAx always halts when the abasic site reaches the binding pocket of the phi29 DNAP as a result of the primer extension failure caused by the abasic site (ESI Fig. S5†). This phenomenon is also verified in the ensemble by a reverse transcription assay, which is reported by gel electrophoresis (Fig. 2E).
Although the read-length is currently limited to 14–15 bases, the first direct FANA sequencing using NIPSS has been successfully demonstrated with a piece of designed chimeric DNA–FANA strand using NIPSS. Similar to nanopore sequencing of DNA, nanopore sequencing of FANA showed clear sequence-specific pore blockage signals with distinguishable amplitude transitions assisted by the phi29 DNAP based primer extension. As demonstrated by FANAx, the maximum amplitude difference within the sequence currently being read could be more than 20 pA (ESI Table S3†).
2.3. Sequencing signal variation between DNA and FANA
As reported, DNA sequences could be deduced from raw nanopore sequencing data according to the previously published look-up table formed by 44 = 256 quadromeric combinations among 4 DNA bases as recognized by the restriction site of MspA.20 However, nanopore sequencing is extremely sensitive to minor variations in the chemical structures of the analyte as demonstrated from the NIPSS results of FANAx, where the DNA and FANA parts of the sequencing signal are only partially overlapped.To further investigate pore blockage amplitude variations between reading DNA and FANA during NIPSS, a chimeric DNA–FANA strand with a random FANA sequence was designed, synthesized and named FANA30 (Fig. 3A, ESI Table S1†). A DNA reference strand, which is composed of DNA nucleotides with an identical sequence to that of FANA30, was synthesized and named DNA30. Here, the uridine in FANA30 is replaced by thymidine in DNA30 (Fig. 3A, ESI Table S1†).
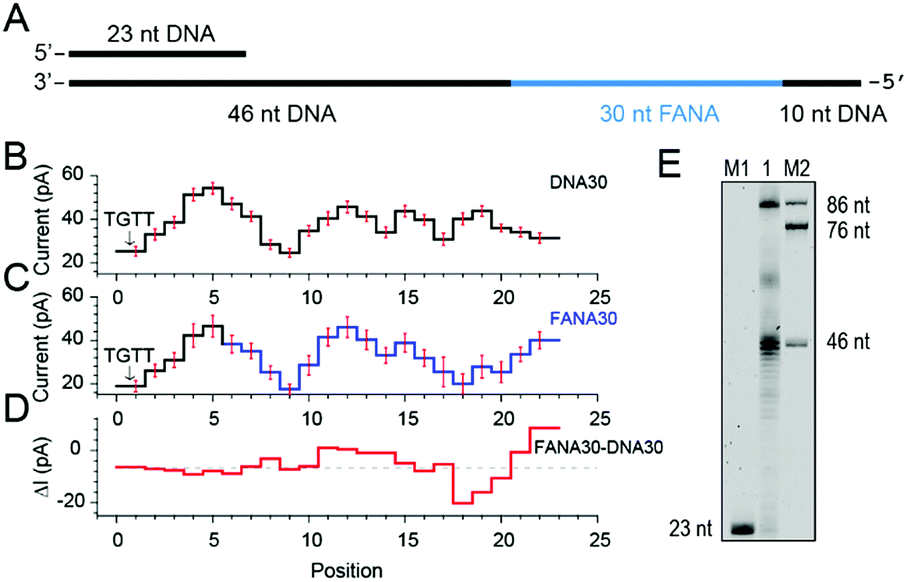 | ||
| Fig. 3 Discrimination between DNA and FANA via nanopore sequencing. (A) Diagram of the FANA30 template annealed with a primer. (B) Mean current levels of DNA30 (ESI Table S3†) extracted from multiple events (N = 22). Error bars (red) represent the corresponding standard deviations from different events. The step with an arrow is from the TGTT blockage. (C) Mean current levels of FANA30 extracted from multiple events (N = 22). Sequencing steps from DNA (black) and FANA (blue) are marked, respectively. Error bars (red) represent the corresponding standard deviations from different events. (ESI Table S3†) (D) Mean current level differences between FANA30 and DNA30. (E) The phi29 DNAP-mediated FANA30 reverse transcription assay analyzed by denaturing PAGE. The primer extension yields a detectable full-length product (86 nt) although a significant amount of the extension products stop at the DNA–FANA junction (46 nt). | ||
As extracted from nanopore sequencing signals from DNA30 and FANA30, sequence-specific mean current levels and the corresponding error bars from n = 22 NIPSS events are presented in Fig. 3B and C, respectively. Nanopore sequencing of FANA30 first generates five sequencing levels that originate from reading the DNA part of the strand. These five levels are marked in black in the event statistics of the signal and overlap almost completely with those from DNA30. The remaining signals, which are marked in blue, are from the phase-shift sequencing of the FANA (Fig. 3C). By subtracting the mean current signals of DNA30 from those of FANA30, the difference of signal amplitude between these two assays could be systematically evaluated (Fig. 3D). From these results, nanopore sequencing from the first five sequencing levels shows negligible differences between DNA30 and FANA30, considering that all these levels are from identical DNA nucleotides. The remaining signals show significant variations between DNA30 and FANA30 due to either the chemical structure variations from the 2′-fluoroarabinose and ribose sugars or from the difference in the base of uridine and thymidine. As shown in Fig. 3D, signal amplitude variations between DNA30 and FANA30 could reach up to 20 pA although chemical structure variations involve only a few atoms (ESI Table S3†).
Different from the NIPSS results of FANAx, where the abasic site prohibits further replication of FANA by the phi29 DNAP (ESI Fig. S5†), phi29 DNAP-based primer extension along the FANA30 in a NIPSS assay normally results in back and forth movement of the phi29 enzyme around the DNA–FANA interface (ESI Fig. S6†), where no abasic site remains. This single molecule phenomenon implies that the phi29 DNAP, which is a highly processive enzyme, attempts to replicate beyond the DNA–FANA interface as an FANA reverse transcriptase.
To further test this hypothesis, a phi29 DNAP-mediated reverse transcription assay for FANA30 was performed (ESI Fig. S7†) and analyzed by denaturing polyacrylamide gel electrophoresis (Fig. 3E). It was observed that a full-length reverse transcription product of 86 nucleotides formed, indicating that phi29 DNAP is capable of catalyzing FANA-templated DNA synthesis. Consistent with nanopore sequencing results, most primer extensions stop at the DNA–FANA junction, generating a 46 nt truncated product and suggesting that phi29 DNAP′s FANA reverse transcriptase activity is significantly reduced compared to its DNA polymerase activity.
Two chimeric DNA–FANA oligomers have been tested so far using NIPSS and confirm the new concept of direct FANA sequencing by the NIPSS strategy. As demonstrated with DNA, the restriction site of MspA simultaneously accommodates four DNA bases. This result indicates that by forming a look-up table composed of all 24 = 256 FANA sequence combinations, unknown FANA sequences could be deduced directly from the acquired nanopore sequencing data. To form an independent archive of FANA sequencing signals in the form of a look-up table, nanopore sequencing for FANA with a long read length is needed for efficient data acquisition. The unexpected reverse transcriptase activity from phi29 DNAP suggests that this DNA polymerase with a high processivity and low working temperature may also be compatible for sustained direct FANA sequencing beyond the DNA–FANA sequence interface.
2.4. Phi29 DNAP as an FANA reverse transcriptase
To quantitatively investigate the reverse transcriptase activity from the phi29 DNAP in single molecules, a third chimeric DNA–FANA oligomer containing a 42-nucleotide FANA sequence was synthesized and named FANA42 (Fig. 4A and ESI Table S1†). The FANA sequence within FANA42, which contains repeats of AGAAUGUU, is expected to show a nanopore sequencing pattern composed of a periodic triangular shape as demonstrated with FANAx (Fig. 2). This periodic pattern is designed to be asymmetric (Fig. 2), which helps to determine the relative position of the phi29 DNAP in reference to the chimeric DNA–FANA template during a NIPSS assay.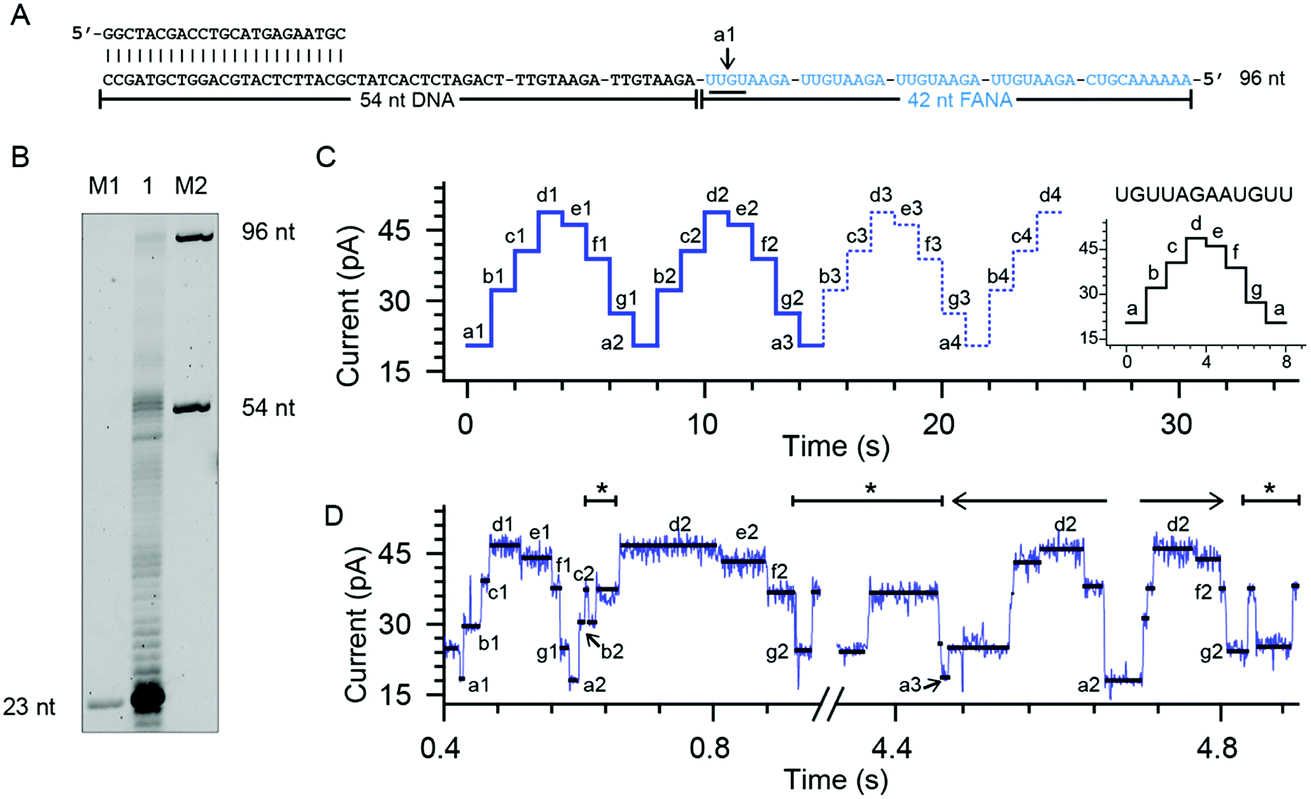 | ||
| Fig. 4 Phi29 DNAP as an FANA reverse transcriptase. (A) The FANA42 template annealed with a primer. (B) The phi29 DNAP-mediated FANA42 reverse transcription assay (lane 1) analyzed by denaturing PAGE. Extending of considerable amounts of primers stopped when encountering FANA residues in the template. A noticeable amount of primer was extended to the full-length product (96 nt). M1 and M2: DNA oligonucleotide markers with indicated lengths. (C) A predicted signal pattern from NIPSS recording of the FANA part of FANA42 based on its periodic signal pattern. The graph on the right represents one period of the NIPSS signal from UGUUAGAAUGUU. Blue dashed lines represent NIPSS signals that failed to appear due to reduced FANA reverse transcriptase activities against the opposing electric field. (D) A representative sequencing trace of FANA42. Characters from a to g represent different current levels obtained from (C); back and forth movement of the phi29 DNAP along the FANA template is marked with *. The 3′-5′ exonuclease activity, which is observed in single molecules during NIPSS reading of FANA42, is marked with a reverse arrow (←). The exonuclease activity-oriented reverse motion of the phi29 DNAP stopped at position a2, then followed by forward motion of the enzyme driven by DNAP reverse transcription, which is marked with a forward arrow (→). After that, back and forth movement between f2 and g2 starts again. | ||
In an ensemble reverse transcription assay investigated by denaturing polyacrylamide gel electrophoresis, primer extension for FANA42 mainly stops at the DNA–FANA junction although a noticeable amount of full length product (96 nucleotides) is detectable (Fig. 4B).
In the nanopore sequencing assay for FANA42, the DNA sequencing signal appears first and is followed by the NIPSS reading of the FANA (ESI Fig. S8†). Means and standard deviations of nanopore sequencing signals from FANA42 were extracted from N = 20 independent NIPSS events to generate statistics (Fig. 4C, ESI Fig. S9†). Within the statistics, a triangular shaped sequencing signal could be extracted and used as a reference with which the relative position of phi29 DNAP on the FANA42 can be detected (Fig. 4C). Within each cycle of FANA sequencing, a statistically normalized signal pattern with asymmetry could be extracted (image inset of Fig. 4C). Although back and forth movement of the enzyme is occasionally observed, the asymmetry of the sequencing signal serves to determine the position of the enzyme unambiguously. Within each cycle of FANA sequencing data, the pore blockage levels within each cycle are marked with characters from a to g for the sake of simplicity. Here, a1 means level “a” within the “1st” cycle.
The signal of interest starts from reading UGUU (a1), which is the 4 nucleotide combination that shows the lowest pore blockage level (a1, a2, a3, etc.) when read by the nanopore restriction (Fig. 2), and AGAA reports the highest pore blockage amplitude (d1, d3, d3, etc.) in contrast. This triangular shaped signal pattern from the nanopore sequencing assay can determine precisely the location of the enzyme in reference to the FANA template with an Å spatial resolution similar to that from nanopore tweezers, the SPRNT approach31 (ESI Table S3†).
According to the phase-shift sequencing data from FANAx, level f2 is achieved but the enzyme is unable to proceed further due to the abasic stopper within the binding pocket of the phi29 DNAP (ESI Fig. S5†). From nanopore sequencing of FANA42, it can be seen that the enzyme could proceed further to level a3, which is 2-nucleotides ahead of the DNA–FANA interface, and this is confirmed by measurements with FANAx (ESI Fig. S5†). Different from the results of FANAx, when the primer extension halts at level f2, nanopore sequencing of FANA42 shows frequent amplitude transitions within multiple levels (d2–a3). This single molecule phenomenon indicates that the phi29 DNAP attempts to proceed beyond the DNA–FANA interface but encounters difficulties (ESI Fig. S9†). Different from the spontaneous back and forth movement of the enzyme, which normally jumps between two sequencing levels, the observed amplitude transitions within multiple levels (Fig. 4D) are probably due to a dynamic balance between the reverse transcriptase and exonuclease activities which are both possessed by the phi29 DNAP. As shown in Fig. 4D, the phi29 DNAP moves backward from a3 to a2, which is a reverse movement of the enzyme with 7 nt steps, and this is immediately followed by a forward movement of the enzyme from a2 to g2. We can exclude the possibility that this backward movement of the enzyme is a result of electrophoretic unzipping, which is irreversible and would not be followed by any stepwise forward movement of the DNAP anymore. By acknowledging the pre-designed FANA42 sequence, which generates an asymmetric and periodic signal pattern during NIPSS, it is clear that this single molecule experiences enzymatic exonuclease/reverse transcriptase activity around the DNA–FANA interface.
Although the reverse transcriptase activity of phi29 DNAP is verified from ensemble assays (Fig. 3E and 4B), the DNA–FANA interface still appears to be a reverse transcription barrier for the phi29 DNAP according to the single molecule assay assisted by nanopore sequencing. It is suspected that the opposing electrophoretic force during nanopore measurements may be the origin of this reduced enzymatic efficiency, considering that both FANA30 and FANA42 show clear reverse transcription products in ensemble (Fig. 3E and 4B).
Still, in a practical nanopore sequencing assay of FANA by NIPSS as demonstrated in this paper, the FANA oligomer to be sequenced could be first enzymatically ligated with the DNA drive-strand on its 3′ end (ESI Fig. S3†). Although limited in read-length, phase shift sequencing of FANA is capable of decoding the sequence of the first 14–15 nucleotides on the 3′ end of the FANA analyte. Since short FANA polymers have been demonstrated to exhibit potent functional properties such as gene-silencing and catalytic activities,8,11 this read-length by NIPSS may be suitable for these FANA molecules, which have short lengths in nature. By introducing redundant materials above the pore, a NIPSS read-length of 30–40 bases should be technically feasible, which matches the read-length of early next generation sequencing platforms.36 In combination with a high-throughput nanopore array37 and bioinformatics tools for sequence decoding (ESI Fig. S10†), long FANA sequences may be re-assembled by fragmented sequence read-outs.
3. Conclusions
In summary, we have demonstrated a series of single molecule studies of FANA, a type of xeno-nucleic acid, using the MspA nanopore. According to the static blockage assay designed for FANA homopolymers, pore blockage amplitudes from different FANAs show distinguished sequence-specific signals with up to 30 pA signal separations (ESI Table S3†). We have further demonstrated the first direct FANA sequencing using “Nanopore Induced Phase-Shift Sequencing” (NIPSS). Though with a limited read-length for now, NIPSS is a universal single molecule sequencing technique for XNA or even other biomacromolecules as long as the analyte to be sequenced can be tethered to a DNA drive-strand. To further extend the read-length of NIPSS, pore engineering of MspA may also be performed by elevating the absolute height of the pore structure. This could be carried out by introducing redundant amino acids on the cis end of the pore vestibule. Alternatively, the discovery of other nanopore proteins with a more extended height than MspA, such as ClyA38 or FraC,39 could also be adapted for NIPSS. The phi29 DNAP, which shows reverse transcriptase activities for FANA templates at room temperature, is potentially suitable for nanopore sequencing or isothermal amplification of FANA. However, the phi29 DNAP doesn′t replicate FANA as efficiently as it replicates DNA due to its strong template discrimination against non-cognate substrate and its reduced efficiency against the opposing electrophoretic forces in a nanopore sequencing assay. Sustained nanopore sequencing of FANA may be achieved by protein evolution of the phi29 DNAP, which definitely leads to a promising future for a general sequencing method for the entire XNA family.Conflicts of interest
A patent related to nanopore based FANA sequencing has been filed.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 21327902, Grant No. 21675083, Grant No. 91753108, and Grant No. 21708018), Fundamental Research Funds for the Central Universities (Grant No. 020514380078, Grant No. 020514380120, and Grant No. 020514380142, 14380088 and 14380134), State Key Laboratory of Analytical Chemistry for Life Science (Grant No. 5431ZZXM1707 and Grant No. 5431ZZXM1804), 1000 Plan Youth Talent Program of China, and the Program for High-Level Entrepreneurial and Innovative Talents Introduction of Jiangsu Province. We gratefully acknowledge Prof. Philipp Holliger (MRC Laboratory of Molecular Biology) for the RT521K-expressing plasmid.References
- A. I. Taylor, S. Arangundy-Franklin and P. Holliger, Curr. Opin. Chem. Biol., 2014, 22, 79–84 CrossRef CAS PubMed.
- A. W. Feldmann and F. E. Romesberg, Acc. Chem. Res., 2018, 51, 394–403 CrossRef PubMed.
- V. B. Pinheiro and P. Holliger, Trends Biotechnol., 2014, 32, 321–328 CrossRef CAS PubMed.
- H. Y. Yu, S. Zhang, M. R. Dunn and J. C. Chaput, J. Am. Chem. Soc., 2013, 135, 3583–3591 CrossRef CAS PubMed.
- M. R. Dunn, C. Otto, K. E. Fenton and J. C. Chaput, ACS Chem. Biol., 2016, 11, 1210–1219 CrossRef CAS PubMed.
- S. Huang, Chin. Sci. Bull., 2014, 59, 4918–4928 CrossRef.
- J. K. Watts and M. J. Damha, Can. J. Chem., 2008, 86, 641–656 CrossRef.
- T. Dowler, D. Bergeron, A. L. Tedeschi, L. Paquet, N. Ferrari and M. J. Damha, Nucleic Acids Res., 2006, 34, 1669–1675 CrossRef CAS PubMed.
- J. K. Watts, A. Katolik, J. Viladoms and M. J. Damha, Org. Biomol. Chem., 2009, 7, 1904–1910 RSC.
- I. A. Ferreira-Bravo, C. Cozens, P. Holliger and J. J. DeStefano, Nucleic Acids Res., 2015, 43, 9587–9599 CAS.
- A. I. Taylor, V. B. Pinheiro, M. J. Smola, A. S. Morgunov, S. Peak-Chew, C. Cozens, K. M. Weeks, P. Herdewijn and P. Holliger, Nature, 2015, 518, 427–430 CrossRef CAS PubMed.
- V. B. Pinheiro, A. I. Taylor, C. Cozens, M. Abramov, M. Renders, S. Zhang, J. C. Chaput, J. Wengel, S. Y. Peak-Chew, S. H. McLaughlin, P. Herdewijn and P. Holliger, Science, 2012, 336, 341–344 CrossRef CAS PubMed.
- A. C. Pease, D. Solas, E. J. Sullivan, M. T. Cronin, C. P. Holmes and S. Fodor, Proc. Natl. Acad. Sci. U.S.A., 1994, 91, 5022–5026 CrossRef CAS.
- S. Drmanac, D. Kita, I. Labat, B. Hauser, C. Schmidt, J. D. Burczak and R. Drmanac, Nat. Biotechnol., 1998, 16, 54 CrossRef CAS PubMed.
- R. Drmanac, S. Drmanac, G. Chui, R. Diaz, A. Hou, H. Jin, P. Jin, S. Kwon, S. Lacy and B. Moeur, in Chip Technology, Springer, 2002, pp. 75–101 Search PubMed.
- A. H. Laszlo, I. M. Derrington, B. C. Ross, H. Brinkerhoff, A. Adey, I. C. Nova, J. M. Craig, K. W. Langford, J. M. Samson, R. Daza, K. Doering, J. Shendure and J. H. Gundlach, Nat. Biotechnol., 2014, 32, 829–833 CrossRef CAS PubMed.
- E. V. Wallace, D. Stoddart, A. J. Heron, E. Mikhailova, G. Maglia, T. J. Donohoe and H. Bayley, Chem. Commun., 2010, 46, 8195–8197 RSC.
- A. H. Laszlo, I. M. Derrington, H. Brinkerhoff, K. W. Langford, I. C. Nova, J. M. Samson, J. J. Bartlett, M. Pavlenok and J. H. Gundlach, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 18904 CrossRef CAS PubMed.
- G. M. Cherf, K. R. Lieberman, H. Rashid, C. E. Lam, K. Karplus and M. Akeson, Nat. Biotechnol., 2012, 30, 344–348 CrossRef CAS PubMed.
- E. A. Manrao, I. M. Derrington, A. H. Laszlo, K. W. Langford, M. K. Hopper, N. Gillgren, M. Pavlenok, M. Niederweis and J. H. Gundlach, Nat. Biotechnol., 2012, 30, 349–353 CrossRef CAS PubMed.
- J. Quick, N. J. Loman, S. Duraffour, J. T. Simpson, E. Severi, L. Cowley, J. A. Bore, R. Koundouno, G. Dudas, A. Mikhail, N. Ouédraogo, B. Afrough, A. Bah, J. H. J. Baum, B. Becker-Ziaja, J. P. Boettcher, M. Cabeza-Cabrerizo, Á. Camino-Sánchez, L. L. Carter, J. Doerrbecker, T. Enkirch, I. G. Dorival, N. Hetzelt, J. Hinzmann, T. Holm, L. E. Kafetzopoulou, M. Koropogui, A. Kosgey, E. Kuisma, C. H. Logue, A. Mazzarelli, S. Meisel, M. Mertens, J. Michel, D. Ngabo, K. Nitzsche, E. Pallasch, L. V. Patrono, J. Portmann, J. G. Repits, N. Y. Rickett, A. Sachse, K. Singethan, I. Vitoriano, R. L. Yemanaberhan, E. G. Zekeng, T. Racine, A. Bello, A. A. Sall, O. Faye, O. Faye, N. F. Magassouba, C. V. Williams, V. Amburgey, L. Winona, E. Davis, J. Gerlach, F. Washington, V. Monteil, M. Jourdain, M. Bererd, A. Camara, H. Somlare, A. Camara, M. Gerard, G. Bado, B. Baillet, D. Delaune, K. Y. Nebie, A. Diarra, Y. Savane, R. B. Pallawo, G. J. Gutierrez, N. Milhano, I. Roger, C. J. Williams, F. Yattara, K. Lewandowski, J. Taylor, P. Rachwal, D. J. Turner, G. Pollakis, J. A. Hiscox, D. A. Matthews, M. K. O. Shea, A. M. Johnston, D. Wilson, E. Hutley, E. Smit, A. Di Caro, R. Wölfel, K. Stoecker, E. Fleischmann, M. Gabriel, S. A. Weller, L. Koivogui, B. Diallo, S. Keïta, A. Rambaut, P. Formenty, S. Günther and M. W. Carroll, Nature, 2016, 530, 228–232 CrossRef CAS PubMed.
- T. Z. Butler, M. Pavlenok, I. M. Derrington, M. Niederweis and J. H. Gundlach, Proc. Natl. Acad. Sci. U.S.A., 2008, 105, 20647–20652 CrossRef CAS PubMed.
- L. Blanco and M. Salas, J. Biol. Chem., 1996, 271, 8509–8512 CrossRef CAS PubMed.
- F. B. Dean, J. R. Nelson, T. L. Giesler and R. S. Lasken, Genome Res., 2001, 11, 1095–1099 CrossRef CAS PubMed.
- M. Faller, M. Niederweis and G. E. Schulz, Science, 2004, 303, 1189–1192 CrossRef CAS PubMed.
- D. R. Garalde, E. A. Snell, D. Jachimowicz, B. Sipos, J. H. Lloyd, M. Bruce, N. Pantic, T. Admassu, P. James and A. Warland, Nat. Methods, 2018, 15, 201–206 CrossRef CAS PubMed.
- E. A. Manrao, I. M. Derrington, M. Pavlenok, M. Niederweis and J. H. Gundlach, PLoS One, 2011, 6, e25723 CrossRef CAS PubMed.
- M. Clamer, L. Höfler, E. Mikhailova, G. Viero and H. Bayley, ACS Nano, 2013, 8, 1364–1374 CrossRef PubMed.
- G. Maglia, M. R. Restrepo, E. Mikhailova and H. Bayley, Proc. Natl. Acad. Sci. U.S.A., 2008, 105, 19720–19725 CrossRef CAS PubMed.
- J. J. Kasianowicz, E. Brandin, D. Branton and D. W. Deamer, Proc. Natl. Acad. Sci. U.S.A., 1996, 93, 13770–13773 CrossRef CAS.
- I. M. Derrington, J. M. Craig, E. Stava, A. H. Laszlo, B. C. Ross, H. Brinkerhoff, I. C. Nova, K. Doering, B. I. Tickman, M. Ronaghi, J. G. Mandell, K. L. Gunderson and J. H. Gundlach, Nat. Biotechnol., 2015, 33, 1073–1075 CrossRef CAS PubMed.
- I. C. Nova, I. M. Derrington, J. M. Craig, M. T. Noakes, B. I. Tickman, K. Doering, H. Higinbotham, A. H. Laszlo and J. H. Gundlach, PLoS One, 2017, 12, e0181599 CrossRef PubMed.
- M. Jain, I. T. Fiddes, K. H. Miga, H. E. Olsen, B. Paten and M. Akeson, Nat. Methods, 2015, 12, 351–356 CrossRef CAS PubMed.
- H. Greenway and C. Osmond, Plant Physiol., 1972, 49, 256–259 CrossRef CAS.
- C. G. Peng and M. J. Damha, J. Am. Chem. Soc., 2007, 129, 5310–5311 CrossRef PubMed.
- J. C. Dohm, C. Lottaz, T. Borodina and H. Himmelbauer, Nucleic Acids Res., 2008, 36, e105 CrossRef PubMed.
- S. Huang, M. Romero-Ruiz, O. K. Castell, H. Bayley and M. I. Wallace, Nat. Nanotechnol., 2015, 10, 986–991 CrossRef CAS PubMed.
- M. Soskine, A. Biesemans, B. Moeyaert, S. Cheley, H. Bayley and G. Maglia, Nano Lett., 2012, 12, 4895–4900 CrossRef CAS PubMed.
- G. Huang, K. Willems, M. Soskine, C. Wloka and G. Maglia, Nat. Commun., 2017, 8, 935–946 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05228j |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |