
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProgramming in situ accelerated DNA walkers in diffusion-limited microenvironments†
Feng
Chen‡
a,
Jing
Xue‡
ab,
Min
Bai
a,
Jing
Qin
a and
Yongxi
Zhao
 *a
*a
aKey Laboratory of Biomedical Information Engineering of Ministry of Education, School of Life Science and Technology, Xi'an Jiaotong University, Xianning West Road, Xi'an, Shaanxi 710049, P. R. China. E-mail: yxzhao@mail.xjtu.edu.cn
bState Key Laboratory for Mechanical Behavior of Materials, School of Materials Science and Engineering, Xi'an Jiaotong University, Xianning West Road, Xi'an, Shaanxi 710049, P. R. China
First published on 22nd January 2019
Abstract
Macromolecule diffusion in cellular microenvironments dictates the kinetics of biochemical processes, yet inevitably limiting the assembly and operation of biomimetic motors. Herein we program in situ accelerated DNA walkers in diffusion-limited microenvironments such as molecularly crowded solutions and cytoplasm. All DNA components, including single-foot walkers, chemically damaged tracks and calibration elements, are anchored on individual gold nanoparticles. Two endogenous enzymes participating in base repair pathways are used to actuate on-particle walking via a base excision/hydrolyzation coupled reaction. The walkers are in situ driven without requiring external drivers and accelerated several times. They also avoid low-efficiency diffusion/assembly procedures and respond to heterogeneous cellular milieus with calibration function. We further regulated the walking kinetics via DNA densities and sets of enzymes, and demonstrated cytoplasmic behaviors of three kinds of walkers. They were utilized to profile DNA repair pathways and monitor enzyme catalysis in living cells.
Introduction
The diffusivity of nanosized objects in the cytoplasmic microenvironments of mammalian cells varies over orders of magnitude and dictates the kinetics of biochemical processes.1–4 However, it challenges the effective assembly and operation of nucleic acid modules as functional motors in living cells. Though building modules of sizes smaller than the cytoskeleton pore (50–75 nm) are not sterically hindered by excessive molecular crowding, their diffusive dynamics can still be decreased by non-specific biochemical (e.g., electrostatic) interactions with immobile or slowly moving intracellular components.1,2 To date, lots of DNA motors with life-like features (molecular robots, walkers, networks, etc.)5–19 have been designed, which behaved well in water solutions or other cell-free settings. As we know, DNA reactants freely diffuse in these environments owing to the low viscosity of water compared to the cytosolic fluid phase. Increasing the absolute concentration of reactants can accelerate reaction and improve yield, yet the concentration of nanosized objects in mammalian cells is limited by the internalization efficiency and biocompatibility.20–25 Therefore, these motors suffer from drawbacks such as slow multistep assembly and ineffective operation in cytoplasmic environments, hampering their translation into intracellular applications.To circumvent diffusion-limited assemblies and accelerate reaction dynamics, nature evolves supramolecular templates such as the cytoskeleton and cellular membranes for the recruitment of network components into higher-order complexes,26–28 thus increasing their effective concentrations instead of absolute ones. In addition, intracellular nucleic acid processes such as replication, transcription and damage repair recruit sets of associated enzymes to execute sequential manipulation of DNA or RNA. Inspired by these natural motors, we program in situ accelerated DNA motors that launch on-particle stochastic walking in diffusion-limited microenvironments such as the cytoplasm (Scheme 1). Two endogenous proteins, one DNA glycosylase and apurinic/apyrimidinic endonuclease 1 (APE1), involved in base-excision repair (BER) pathways are employed as actuator components. Their molecular weights are low, below 40 kDa, and they diffuse fast in the cytoplasm.1,2,29,30 All nucleic acid components (all-in-one design), including single-foot DNA walkers (DWs), dense DNA tracks (DTs) and calibration elements (CEs), are integrated on a single gold nanoparticle by Au–S chemistry pioneered by the Mirkin group.31–34 Compared to routine post-assembly, the proposed program avoids low-efficiency diffusion/assembly procedures and accelerates reaction dynamics. It can be in situ driven by endogenous enzymes without requiring exogenous drivers, and also integrates the calibration function for reliable response to heterogeneous environments of different cells.
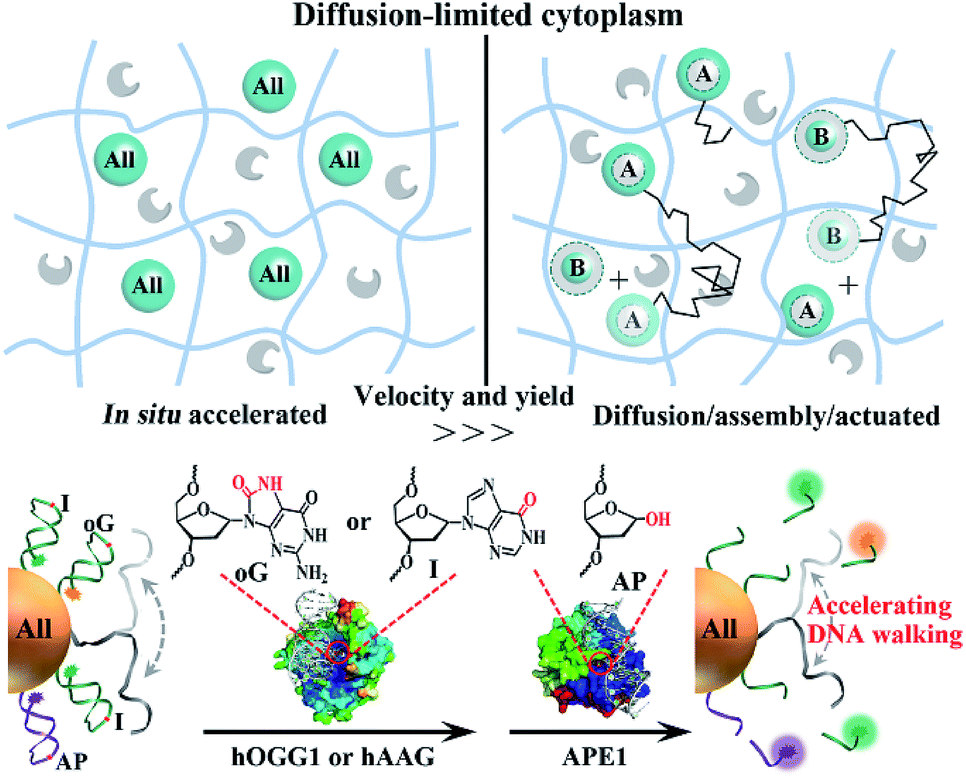 | ||
| Scheme 1 Schematic illustration of the comparison of in situ accelerated and routine post-assembly nanomotors in diffusion-limited microenvironments such as the cytoplasm. Nanosized modules (blue spheres) of size smaller than the cytoskeleton pore size (50–70 nm) may still diffuse (black polylines) slowly due to their non-specific interaction with immobile intracellular components. The endogenous proteins (gray shapes) are inert and can diffuse fast. ‘All’ highlighted spheres represent the integration of all modules into one system. In the post-assembly process, the motor can only be driven after the assembly of A and B modules. The proposed DNA walkers are powered by glycosylase and APE1. Crystal structures of chemically damaged (oG or AP site) DNA-bound human APE1 (PDB ID:1DEW) and hOGG1 (PDB ID:1EBM) were visualized and analyzed with PyMOL. The red circles indicate the locations of oG and AP sites in the crystal structures. The location of hAAG bound to I damage is not given. | ||
Results and discussion
In this work, human 8-oxoguanine glycosylase 1 (hOGG1) and alkyladenine glycosylase (hAAG) are used to initiate individual walkers. They can specifically bind to and excise damaged 8-oxoguanine (oG) or inosine (I) bases in dsDNA (Scheme 1, bottom). Two corresponding DTs, each containing oG or I, presents intramolecular stem–loop structures with terminal fluorescent labels. The DW is designed to form a damaged base-containing walker–track duplex with a DT. The CE is similar to the DT except for the different sequences and one AP site in the loop part. A few DWs and dozens of DTs together with CEs are anchored on one nanoparticle. Once the on-particle walker is internalized into the cells, endogenous hOGG1 or hAAG can straightly bind to walker–track duplexes and excise damaged bases to generate AP sites. Then, downstream APE1 in the BER pathway hydrolyzes the AP sites, inducing DT strand break and dissociation. The anchored DWs can form new walker–track duplexes with other DT molecules. Such a base excision/hydrolyzation reaction provides energy for anchored DWs to rapidly walk along on-particle DTs, and simultaneously unloads fluorophores to monitor DNA motion. The reference fluorophore in the CE can be released by APE1 due to its activity on single-stranded DNA, which can respond to the change of catalytic activity in different cells.We investigated the walking performance of proposed DNA walkers in different diffusion-limited microenvironments. Based on previous studies,35,36 a common macromolecular agent, Ficoll 400, was utilized to mimic the crowded conditions. One post-assembly nanosystem based on assembling two diffused modules (DT-anchored nanoparticles and free DWs) is designed as the negative control. First, DNA motion on immobilized nanoparticles was monitored by stimulated emission depletion (STED) microscopy (Fig. 1). The particles are adsorbed on chemically modified cover slides to limit their diffusion. DW walking-induced enzymatic digestion of DTs releases the fluorophores into solution, resulting in fluorescence loss of the focused single particles under STED. We initially observed fluorescent spots in the imaging areas of both all-in-one walkers and the post-assembly control (Fig. 1A and S1†). After adding two coupled enzymes, the bright spots in DNA motors showed dim fluorescence. Oppositely, most fluorescent spots of control experiments still maintain high brightness with the addition of enzymes and free DWs. A typical single-particle walking process was recorded in real time. We observed rapid fluorescence decrease in all-in-one DNA walkers compared to the post-assembly negative control. Then, we also demonstrated similar results in cytoplasm-like crowded and viscous solution environments. As shown in Fig. 1B and S2,† the initial walking velocities of both systems decrease with the increase of solution viscosity, revealing the restricted diffusion of nanosized objects in cytoplasmic environments. Even so, our DNA walker presents much higher values of velocity than those of the post-assembly control. All the above-mentioned results confirm that the proposed all-in-one design bypasses the diffusion-limited DNA assembly and increases local concentrations (Fig. 1C), and exhibits much faster reaction kinetics even in cytoplasm-like environments. In addition, we verified that the anchored DWs ramble along DTs on individual particles rather than preforming interparticle mobility (Fig. S3†). DWs with longer sequences may jump onto DTs of neighboring particles and even form unwanted particle aggregates.
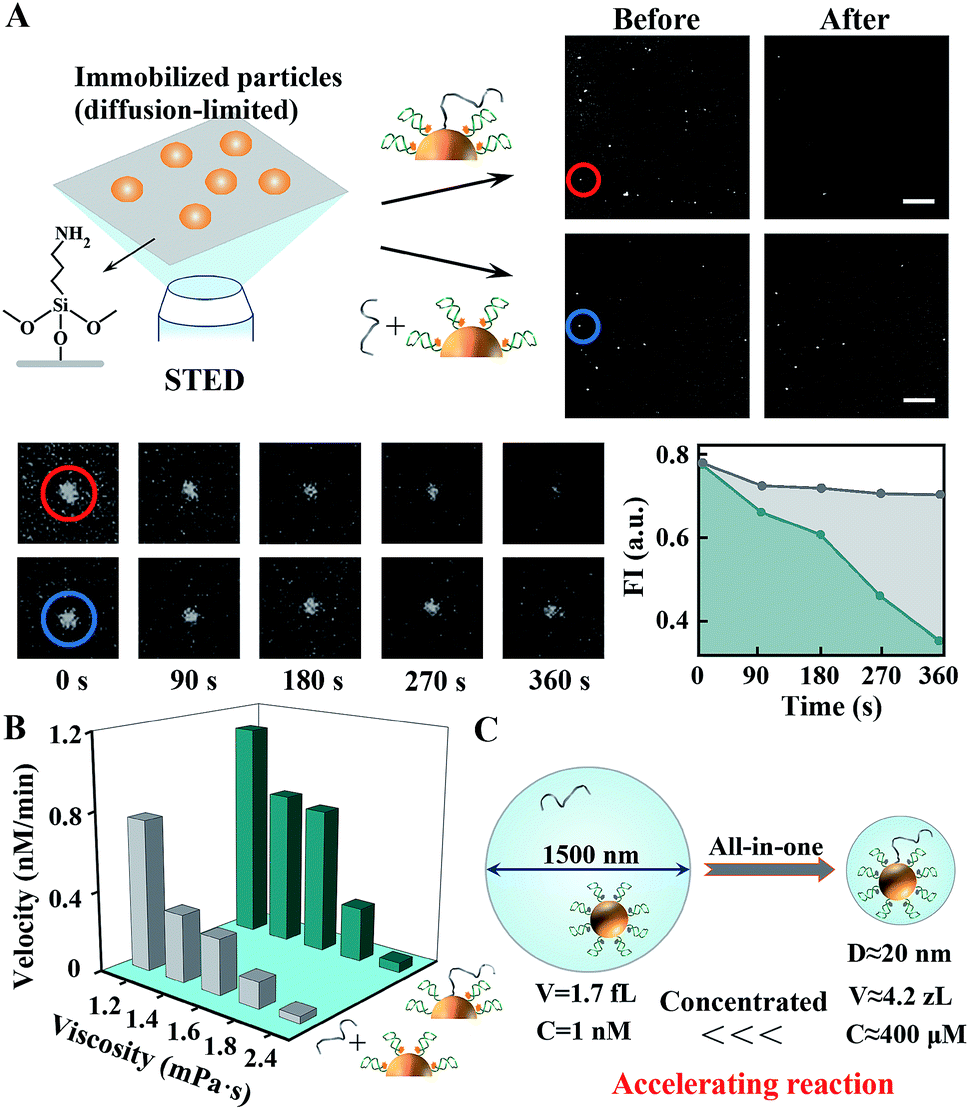 | ||
| Fig. 1 The operation of in situ accelerated DNA walkers in diffusion-limited microenvironments. (A) Single-particle fluorescence analysis using STED microscopy. Left top, schematics for STED imaging of DNA moving on particles fixed on cover slides; right top, STED images for DNA walkers and post-assembly negative control, scale bar = 5 μm. (B) Walking performance in cytoplasm-like viscous and crowded solutions. (C) Schematics for increasing local concentrations and reaction velocity using an all-in-one design. For 1 nM DT-anchored particles and free DWs, the volume of a sphere containing both objects is about 1.7 fL, and the corresponding diameter is 1500 nm. Anchoring DWs and DTs on the same nanoparticles brings them closer (<20 nm), which approximates 4.2 zL in volume and 400 μM in concentration. Yet the practical increase in concentration can be affected by many other conditions. | ||
The walking kinetics of different DNA walkers were then investigated. To simplify the reaction model, we only studied the effect of molecular density of on-particle DWs with fixed concentrations of other components. As shown in Fig. 2A and S4,† the walking velocity of single-track walkers controlled by hOGG1/APE1 gradually increases and slows down later with the increase of DW density. The inhibition induced by high DW density probably results from obstructive and electrostatic interference of each DW. Notably, systematic optimization of other components such as enzyme concentrations can further improve the walking performance. Moreover, a twin-track walker controlled by two pathways (hOGG1/APE1 and hAAG/APE1) is designed by anchoring both oG-DTs and I-DTs on the same particle. Its motion is similar to that of single-track ones, but exhibits more regulatory functions (Fig. 2B and S5†). Versatile walking performance can be achieved by modulating enzyme groups. We also investigate two BER pathway-powered mobility of free DWs between two kinds of particles only modified with oG-DTs or I-DTs. Free DWs as travellers are pre-hybridized with one of two particles. The fluorophores on the respective particles are randomly released (Fig. 2C), implying the interparticle travelling of free DNA. Such mobility takes longer time than anchored walking, again demonstrating the superiority of proposed walkers compared to post-assembly systems. Other repair proteins and nucleases fail to drive specialized DNA walkers (Fig. S6†), indicating their excellent resistance to non-specific enzymes. And CE-integrated DNA walkers can reflect the fluctuation of APE1 activity (Fig. S7†).
 | ||
| Fig. 2 The dynamic features of the proposed DNA walkers. (A) The effect of on-particle DW density on walking velocity of single-track walkers. The letter ‘d’ indicates distance. (B) Walking performance of the twin-track walkers controlled by two pathways (hOGG1/APE1 and hAAG/APE1). (C) Monitoring of free DWs travelling between two particles modified with oG-DTs or I-DTs. | ||
Finally, we studied the operation of the above-mentioned DNA walkers in the cytoplasmic milieu. Inconspicuous cytotoxicity of the DNA walkers on Hela cells was first confirmed (Fig. S8†). For single-track ones, two non-walking systems, one without DWs and the other with DWs of random sequences, are designed as negative controls. These on-particle integrated DNA systems are efficiently internalized into cells via receptor-mediated endocytosis pathways,25,37 eliminating the requirement of transfection reagents and asynchronous delivery of different modules. As shown in Fig. 3, neither of the two negative control systems induces obvious fluorescence signals in cells, whereas DNA walker-treated cells present high fluorescence responses (Fig. S9 and S10†). These results reveal that intracellular DNA walking is specifically powered by the corresponding BER pathways. Therefore, such DNA walkers can be used to profile DNA repair pathways, and even monitor the activity of relevant enzymes.
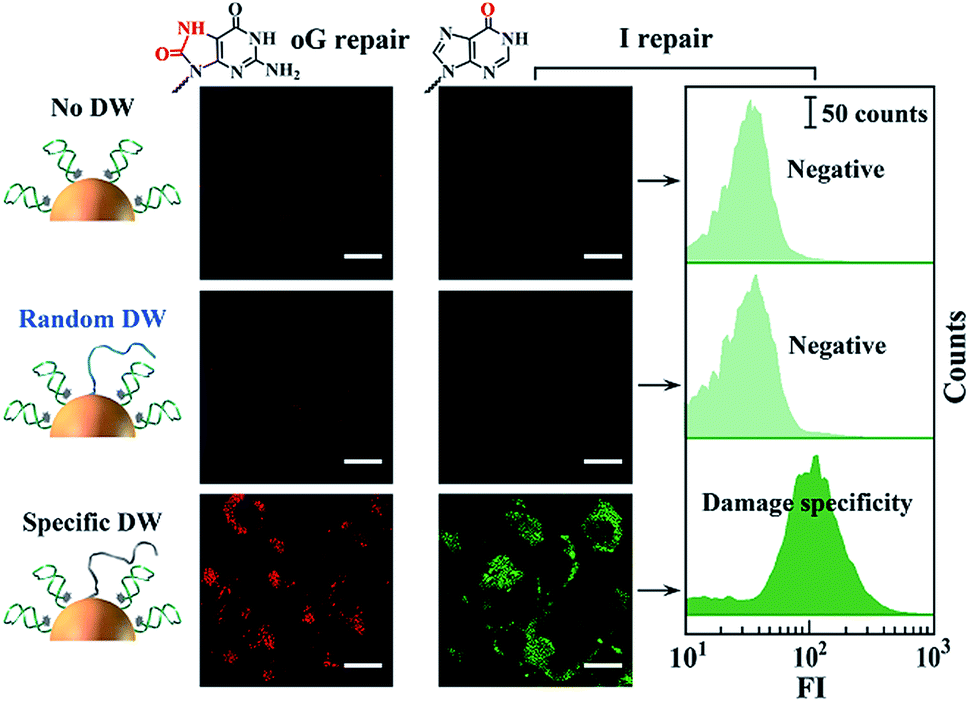 | ||
| Fig. 3 Monitoring of intracellular DNA damage repair pathways using corresponding DNA walkers. Left and right cell images each indicate the monitoring of guanine oxidation (G to oG) and adenosine deamination (A to I editing) damage repair. The scale bars for all cell images are 10 μm. Flow cytometry analysis (scale bar of 50 counts in all three histograms) is consistent with cell imaging. | ||
Specifically, hOGG1 activity was evaluated. Considering that the DNA walking is driven together by hOGG1 and APE1, we simplify such reaction as Pwalking = f(XhOGG1,XAPE1), where Pwalking represents two enzyme-controlled walking performance and XhOGG1 and XAPE1 indicate the activity of hOGG1 and APE1, respectively. Thus, we used CE-integrated DNA walkers to calibrate the APE1 change in different cells. The reference signal from the CE only depends on the activity of APE1, described as Rsignal = f(XAPE1). The potent and selective APE1 inhibitor CRT0044876 (7-nitroindole-2-carboxylic acid) is used to treat cells, obtaining cell samples with different APE1 activities. Both red and purple fluorescence signals, each representing Pwalking and Rsignal, are recorded (Fig. 4A and S11A†). We extracted the intensity information for each pixel of these cell images using MATLAB, and depicted the corresponding scatter plots that show the fluorescence distribution of the treated cells. It can be seen that Pwalking decreases with the decrease of Rsignal when treating the same cell line with increasing concentrations of the APE1 inhibitor. We reason that a positive correlation exists between Pwalking and APE1 or hOGG1 activity when hOGG1 or APE1 activity is maintained constant. Similar evaluation of intracellular hAAG activity is shown in Fig. S11B.† Moreover, we used CE-integrated twin-track walkers for simultaneous monitoring of activities of both hOGG1 and hAAG (Fig. 4B and S11C†). Reliable testing results are obtained with APE1-induced reference signals. These proposed DNA walkers can be applied to single cell analysis with microfluidic chips.
 | ||
| Fig. 4 Analysis of activities of intracellular glycosylases using in situ accelerated DNA walkers. (A) Monitoring hOGG1 in cell samples with different APE1 activities. Cell sample 2 and 3 are treated with 0.2 and 0.8 μM APE1 inhibitor, respectively. The intensity information (0–256) for each pixel in cell images (Fig. S11†) was extracted using MATLAB. Corresponding intensity distribution (vertical direction) plots are shown in the right panel. High scatter density and intensity values indicate a strong fluorescence response. The reference signals (purple) can indicate and adjust the change of APE1 activity in different cells. (B) Integrated monitoring of hOGG1 and hAAG in the same cell sample using twin-track walkers. | ||
To date, many DNA walkers have been well demonstrated with thermodynamic and even theoretical studies.10–13,38–40 Their operations in cytoplasmic environments have been rarely explored, probably due to complicated diffusion/assembly procedures and the lack of endogenous power modules. Recently, we reported intracellular assembly of active DNAzyme nanodevices for monitoring gene expression and regulation.41,42 In these studies, an exogenous fuel (divalent ion Mn2+ as a cofactor of DNAzyme) is generated by cytoplasmic reduction of MnO2 nanosheets, yet the diffusion/assembly efficiency is still limited in crowded and viscous environments. Notably, a few miRNA-initiated DNA walkers operating in living cells were rationally designed.43–45 They require the co-delivery of multiple DNA modules, complicated reaction procedures and/or exogenous power modules. Furthermore, the asynchronous cellular uptake of different building components can result in the loss of their distribution and stoichiometric information, and cause an unreliable response without a calibration component. Our DNA walkers avoid the low-efficiency diffusion/assembly procedures of multiple modules and are in situ driven by endogenous BER enzymes. Calibration components are also integrally designed for glycosylase monitoring via the response to heterogeneous APE1 expression in different cells, which is somewhat similar to the well-known function of housekeeping genes (e.g., GAPDH and β-actin) for gene expression analysis in PCR.
Conclusions
In summary, we have programmed in situ accelerated DNA walkers in living cells. All building components are decorated on a single nanoparticle. Two coupled enzymes participating in BER pathways actuate stochastic DNA walking via base excision/hydrolyzation reactions. They avoid low-efficiency diffusion/assembly procedures and accelerate reaction kinetics in a diffusion-limited milieu. Moreover, we regulated the walking dynamics using different sets of enzymes, and demonstrated intracellular behaviors of three kinds of DNA walkers. They were utilized to profile DNA damage repair and monitor enzyme catalysis with one integrated calibration component. Notably, other endogenous processes such as receptor-mediated signaling can also be utilized to design the above-mentioned DNA walkers with hormone response elements or aptamer probes. Overall, this work opens up new opportunities for programming next-generation DNA motors operating in the diffusion-limited and dynamically changing cytoplasm.Experimental section
Materials
Gold(III)chloride hydrate (HAuCl4·4H2O) 99% and trisodium citrate dehydrate (Na3C6H5O7·2H2O) 99% were purchased from Sigma-Aldrich (Oakville, ON, Canada). All the chemicals were of analytical grade and used as received without further purification. Milli-Q water (resistance > 18.2 MΩ) was used for solution preparation and reaction was RNase-free. Human 8-oxoguanine DNA glycosylase (hOGG1), human alkyladenine DNA glycosylase (hAAG), human apurinic/apyrimidinic endonuclease 1 (APE1), and CutSmart (50 mM KAc, 20 mM Tris-Ac, 10 mM Mg(Ac)2, 100 μg mL−1 BSA, pH 7.9) were purchased from New England Biolabs Inc. (Beverly, MA, U.S.A.). HPLC-purified oligonucleotides (Table S1†) were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China) and Takara Biotechnology Co. Ltd. (Dalian, China).Construction of in situ accelerated DNA walkers
Gold nanoparticles (AuNPs) were synthesized following a previously reported protocol. In brief, 1% (wt%) trisodium citrate solution was quickly added to a boiling, continuously stirred solution of HAuCl4. The solution was kept boiling and stirred for 30 min and slowly cooled to room temperature. The prepared AuNPs were stored at 4 °C for further use. Thiolated oligos DWs, DTs, and/or AP-R were activated using tris(2-carboxyethyl)phosphine (TCEP) prior to conjugation to the AuNPs. AuNPs, DWs and DTs or AP-R were mixed at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30:270. This solution was incubated overnight at room temperature. Aqueous NaCl (2.0 M, 10 mM PB) was then successively added to the solution to bring its total NaCl concentration to 0.3 M. After additional 12 h incubation, the resulting solution was centrifuged and the supernatant removed leaving a pellet of AuNPs at the bottom. The particles were then resuspended in 10 mM Tris–HCl buffer (pH 7.4). This washing process was repeated three times and the particles were stored at 4 °C until use.
:30:270. This solution was incubated overnight at room temperature. Aqueous NaCl (2.0 M, 10 mM PB) was then successively added to the solution to bring its total NaCl concentration to 0.3 M. After additional 12 h incubation, the resulting solution was centrifuged and the supernatant removed leaving a pellet of AuNPs at the bottom. The particles were then resuspended in 10 mM Tris–HCl buffer (pH 7.4). This washing process was repeated three times and the particles were stored at 4 °C until use.
Determination of the DNA density on the surface of AuNPs
The density of DNA strands on the surface of AuNPs was determined based on a published method.42 The conjugated DNA strands were first released by adding 2-mercaptoethanol. The solution was then centrifuged to precipitate the AuNPs and the supernatant containing the released substrate strands was analyzed using fluorescence measurements. Modified AuNPs mixed with 2-mercaptoethanol at a molar ratio of 1:1000 in 1× PBS solution were kept in the dark for 12 h. Then the solution was centrifuged at 12000g for 25 min to precipitate the AuNPs. 95% of the supernatant was removed and collected for fluorescence spectrum analysis. The concentrations of the track strands were obtained against a standard calibration curve of the fluorophore-labelled track strand. The average number of track strands per AuNP was then determined using the concentrations of AuNPs and track strands. The results showed that approximately 60 track DNA molecules were conjugated to each AuNP on average. On the basis of a molar ratio of 1:9 for the walker strand and the track strand during modification, about 7 walker sequences were conjugated to each AuNP. Therefore, the densities of DT and DW strands were about 1.0 × 10−1 and 1.0 × 10−2 nm−2 on AuNPs, respectively.
Fluorescence analysis of in situ accelerated DNA walkers
All the in vitro fluorescence measurements were performed using LightCycler 96 (Roche Applied Science, Mannheim, Germany) at 37 °C for 60 min. The concentrations of hOGG1, hAAG and APE1 were 4 U mL−1, 5000 U mL−1 and 2000 U mL−1, respectively. Cytoplasm-like crowded and viscous solutions were prepared by adding Ficoll 400 of different concentrations (1, 2.5, 5, and 6 wt%) to deionized water. The viscosity of the solutions was measured using a commercial viscometer (SNB-1, Fangrui, China).Cell culture and fluorescence imaging
HeLa cells were cultured in Dulbecco's modified Eagle medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% antibiotics penicillin/streptomycin (100 U mL−1) in a humidified incubator containing CO2 (5%) at 37 °C. HeLa cells were seeded in an 8-well chambered cover glass (Cellvis, USA) at 105 cells per well at 37 °C overnight and then incubated with 200 μL of serum-free culture media containing 20 μL DNA walking machine probe for 4 h. The cells were washed three times with 1× PBS before imaging. All fluorescence images were acquired using a Nikon A1 laser scanning confocal microscope. Three lasers (488, 561, and 633 nm) were used as the excitation sources.Flow cytometry assay
For the preparation of flow cytometry tests, HeLa cells were plated in a 12-well plate and grown for 12 h to reach 80–90% confluency. The cells in each well were incubated with 2 nM prepared DNA walking detector in 500 μL of fresh medium without FBS at 37 °C for 4 h and washed three times with 1× PBS. Then, the cells were detached from the 12-well plate using 0.25% trypsin for 3 min and suspended in PBS for flow cytometry. The samples were kept in the dark and on ice before tests. Flow cytometry assay was performed using a FACS Canto system (BD Biosciences, USA). In total 105 cells were counted for each sample.Single-particle fluorescence analysis using STED microscopy
The particle fluorescence imaging was carried out on the cover slides. The cover slides were first immersed in a mixture of 30% H2O2 and concentrated H2SO4 (1:3 v/v) for 30 min. After washing three times with deionized water and ethanol, respectively, the glass slides were immersed in a solution of APTES (10%, v/v) for 2 h for aminosilanization. The amino-coated glass slides were then sonicated in deionized water three times and dried using N2. The all-in-one designed particles and particles with DT only (for the routine post-assembly strategy) were deposited on the cover slides for 10 min, respectively, and washed with 1× Cutsmart buffer. Fluorescence images and videos of monitoring the two strategies were obtained using a commercial high resolution laser confocal microscope (TCS SP8 STED 3X, Leica). A 100× oil-immersion objective and 561 nm laser were used. hOGG1 and APE1 were added to the sample of all-in-one walkers as soon as the imaging started. For the routine post-assembly method, free DWs were added together with the above two enzymes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Science Foundation of China (No. 21475102, No. 31671013, No. 21705124 and No. 21874105), the China Postdoctoral Science Foundation (No. 2017M613102 and No. 2018T111032), the Fundamental Research Funds for the Central Universities and “Young Talent Support Plan” of Xi'an Jiaotong University.References
- F. Etoc, E. Balloul, C. Vicario, D. Normanno, D. Liße, A. Sittner, J. Piehler, M. Dahan and M. Coppey, Nat. Mater., 2018, 17, 740–746 CrossRef CAS PubMed.
- M. Delarue, G. Brittingham, S. Pfeffer, I. Surovtsev, S. Pinglay, K. Kennedy, M. Schaffer, J. Gutierrez, D. Sang and G. Poterewicz, Cell, 2018, 174, 338–349 CrossRef CAS PubMed.
- G. L. Lukacs, P. Haggie, O. Seksek, D. Lechardeur, N. Freedman and A. S. Verkman, J. Biol. Chem., 2000, 275, 1625–1629 CrossRef CAS PubMed.
- P. K. Verma, A. Kundu, J. H. Ha and M. Cho, J. Am. Chem. Soc., 2016, 138, 16081–16088 CrossRef CAS PubMed.
- K. Yehl, A. Mugler, S. Vivek, Y. Liu, Y. Zhang, M. Fan, E. R. Weeks and K. Salaita, Nat. Nanotechnol., 2016, 11, 184–190 CrossRef CAS PubMed.
- A. J. Thubagere, W. Li, R. F. Johnson, Z. Chen, S. Doroudi, Y. L. Lee, G. Izatt, S. Wittman, N. Srinivas and D. Woods, Science, 2017, 357, eaan6558 CrossRef PubMed.
- T. G. Cha, J. Pan, H. Chen, J. Salgado, X. Li, C. Mao and J. H. Choi, Nat. Nanotechnol., 2014, 9, 39–43 CrossRef CAS PubMed.
- T. Omabegho, P. S. Gurel, C. Y. Cheng, L. Y. Kim, P. V. Ruijgrok, R. Das, G. M. Alushin and Z. Bryant, Nat. Nanotechnol., 2018, 13, 34–40 CrossRef CAS PubMed.
- C. Jung, P. Allen and A. Ellington, Nat. Nanotechnol., 2016, 11, 157–163 CrossRef CAS PubMed.
- X. Yang, Y. Tang, S. D. Mason, J. Chen and F. Li, ACS Nano, 2016, 10, 2324–2330 CrossRef CAS PubMed.
- C. Jung, P. B. Allen and A. D. Ellington, ACS Nano, 2017, 11, 8047–8054 CrossRef CAS PubMed.
- D. Wang, C. Vietz, T. Schröder, G. P. Acuna, B. Lalkens and P. Tinnefeld, Nano Lett., 2017, 17, 5368–5374 CrossRef CAS PubMed.
- D. C. Khara, J. S. Schreck, T. E. Tomov, Y. Berger, T. E. Ouldridge, J. Doye and E. Nir, Nucleic Acids Res., 2017, 46, 1553–1561 CrossRef PubMed.
- J. Li, A. Johnson-Buck, Y. R. Yang, W. M. Shih, H. Yan and N. G. Walter, Nat. Nanotechnol., 2018, 13, 723–729 CrossRef CAS PubMed.
- N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2017, 3, 17068–17091 CrossRef.
- F. Hong, F. Zhang, Y. Liu and H. Yan, Chem. Rev., 2017, 117, 12584–12640 CrossRef CAS PubMed.
- X. He, R. Sha, R. Zhuo, Y. Mi, P. M. Chaikin and N. C. Seeman, Nat. Mater., 2017, 16, 993–997 CrossRef CAS PubMed.
- L. Hu, C.-H. Lu and I. Willner, Nano Lett., 2015, 15, 2099–2103 CrossRef CAS PubMed.
- S. H. Ko, Y. Chen, D. Shu, P. Guo and C. Mao, J. Am. Chem. Soc., 2008, 130, 17684–17687 CrossRef CAS PubMed.
- X. A. Wu, C. H. Choi, C. Zhang, L. Hao and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 7726–7733 CrossRef CAS PubMed.
- S. P. Narayan, C. H. J. Choi, L. Hao, C. M. Calabrese, E. Auyeung, C. Zhang, O. J. G. M. Goor and C. A. Mirkin, Small, 2015, 11, 4173–4182 CrossRef CAS PubMed.
- Z. Wu, H. Fan, N. S. R. Satyavolu, W. Wang, R. Lake, J. H. Jiang and Y. Lu, Angew. Chem., Int. Ed., 2017, 129, 8847–8851 CrossRef.
- L. Xu, Y. Gao, H. Kuang, L. M. Liz-Marzán and C. Xu, Angew. Chem., Int. Ed., 2018, 130, 10704–10708 CrossRef.
- M. Sun, L. Xu, J. H. Banhg, H. Kuang, S. Alben, N. A. Kotov and C. Xu, Nat. Commun., 2017, 8, 1847–1857 CrossRef PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- M. You, Y. Lyu, D. Han, L. Qiu, Q. Liu, T. Chen, W. C. Sam, L. Peng, L. Zhang and G. Bao, Nat. Nanotechnol., 2017, 12, 453–459 CrossRef CAS PubMed.
- M. C. Good, J. G. Zalatan and W. A. Lim, Science, 2011, 332, 680–686 CrossRef CAS PubMed.
- R. Peng, X. Zheng, Y. Lyu, L. Xu, X. Zhang, G. Ke, Q. Liu, C. You, S. Huan and W. Tan, J. Am. Chem. Soc., 2018, 140, 9793–9796 CrossRef CAS PubMed.
- C. Di Rienzo, V. Piazza, E. Gratton, F. Beltram and F. Cardarelli, Nat. Commun., 2014, 5, 5891–5899 CrossRef CAS PubMed.
- T. Kühn, T. O. Ihalainen, J. Hyväluoma, N. Dross, S. F. Willman, J. Langowski, M. Vihinen-Ranta and J. Timonen, PLoS One, 2011, 6, e22962 CrossRef PubMed.
- X. Zhang, M. R. Servos and J. Liu, J. Am. Chem. Soc., 2012, 134, 7266–7269 CrossRef CAS PubMed.
- B. Liu and J. Liu, J. Am. Chem. Soc., 2017, 139, 9471–9474 CrossRef CAS PubMed.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CrossRef CAS PubMed.
- R. Elghanian, J. J. Storhoff, R. C. Mucic, R. L. Letsinger and C. A. Mirkin, Science, 1997, 277, 1078–1081 CrossRef CAS PubMed.
- C. Alfano, D. Sanfelice, S. R. Martin, A. Pastore and P. A. Temussi, Nat. Commun., 2017, 8, 15428–15436 CrossRef CAS PubMed.
- M. M. K. Hansen, L. H. H. Meijer, E. Spruijt, R. J. M. Maas, M. V. Rosquelles, J. Groen, H. A. Heus and W. T. S. Huck, Nat. Nanotechnol., 2016, 11, 191–198 CrossRef CAS PubMed.
- D. A. Giljohann, D. S. Seferos, P. C. Patel, J. E. Millstone, N. L. Rosi and C. A. Mirkin, Nano Lett., 2007, 7, 3818–3821 CrossRef CAS PubMed.
- Y. Tian, Y. He, Y. Chen, P. Yin and C. Mao, Angew. Chem., Int. Ed., 2005, 117, 4429–4432 CrossRef.
- X. Qu, D. Zhu, G. Yao, S. Su, J. Chao, H. Liu, X. Zuo, L. Wang, J. Shi and L. Wang, Angew. Chem., Int. Ed., 2017, 56, 1855–1858 CrossRef CAS PubMed.
- H. Zhang, M. Lai, A. Zuehlke, H. Peng, X. F. Li and X. C. Le, Angew. Chem., Int. Ed., 2015, 54, 14326–14330 CrossRef CAS PubMed.
- F. Chen, M. Bai, K. Cao, Y. Zhao, J. Wei and Y. Zhao, Anal. Chem., 2018, 90, 2271–2276 CrossRef CAS PubMed.
- F. Chen, M. Bai, K. Cao, Y. Zhao, X. Cao, J. Wei, N. Wu, J. Li, L. Wang and C. Fan, ACS Nano, 2017, 11, 11908–11914 CrossRef CAS PubMed.
- H. Peng, X. F. Li, H. Zhang and X. C. Le, Nat. Commun., 2017, 8, 14378–14391 CrossRef PubMed.
- C. P. Liang, P. Q. Ma, H. Liu, X. G. Guo, B. C. Yin and B. C. Ye, Angew. Chem., Int. Ed., 2017, 129, 9205–9209 CrossRef.
- P. Q. Ma, C. P. Liang, H. H. Zhang, B. C. Yin and B. C. Ye, Chem. Sci., 2018, 9, 3299–3304 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05302b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |