
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorogenic hydrogen sulfide (H2S) donors based on sulfenyl thiocarbonates enable H2S tracking and quantification†
Yu
Zhao
 ,
Matthew M.
Cerda
and
Michael D.
Pluth
*
,
Matthew M.
Cerda
and
Michael D.
Pluth
*
Department of Chemistry and Biochemistry, Institute of Molecular Biology, Materials Science Institute, University of Oregon, Eugene, OR 97403, USA. E-mail: pluth@uoregon.edu
First published on 10th December 2018
Abstract
Hydrogen sulfide (H2S) is an important cellular signaling molecule that exhibits promising protective effects. Although a number of triggerable H2S donors have been developed, spatiotemporal feedback from H2S release in biological systems remains a key challenge in H2S donor development. Herein we report the synthesis, evaluation, and application of caged sulfenyl thiocarbonates as new fluorescent H2S donors. These molecules rely on thiol cleavage of sulfenyl thiocarbonates to release carbonyl sulfide (COS), which is quickly converted to H2S by carbonic anhydrase (CA). This approach is a new strategy in H2S release and does not release electrophilic byproducts common from COS-based H2S releasing motifs. Importantly, the release of COS/H2S is accompanied by the release of a fluorescent reporter, which enables the real-time tracking of H2S by fluorescence spectroscopy or microscopy. Dependent on the choice of fluorophore, either one or two equivalents of H2S can be released, thus allowing for the dynamic range of the fluorescent donors to be tuned. We demonstrate that the fluorescence response correlates directly with quantified H2S release and also demonstrate the live-cell compatibility of these donors. Furthermore, these fluorescent donors exhibit anti-inflammatory effects in RAW 264.7 cells, indicating their potential application as new H2S-releasing therapeutics. Taken together, sulfenyl thiocarbonates provide a new platform for H2S donation and readily enable fluorescent tracking of H2S delivery in complex environments.
Introduction
Hydrogen sulfide (H2S) is an important gaseous molecule that plays critical roles in living systems.1–3 Endogenous H2S is primarily produced from cysteine (Cys) and homocysteine (Hcy) by four main enzymes, including cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), 3-mercaptopyruvate sulfur transferase (3-MST), and cysteine aminotransferase (CAT).4–8 Due to its bioregulatory and protective roles, H2S is considered as an important cellular signaling molecule, much like nitric oxide (NO) and carbon monoxide (CO).8–11Although many H2S-related biological functions have been discovered in the past two decades, many investigations have been limited due to the lack of controllable and refined H2S delivery systems (H2S donors). Inorganic sulfide salts, such as sodium sulfide (Na2S) and sodium hydrosulfide (NaHS), are widely used in H2S investigations, but they release H2S quickly in aqueous media, thus failing to mimic the slow and well-regulated in vivo H2S generation.12 By contrast, the H2S release from GYY4137, a phosphorodithioate-based H2S donor, is slow in aqueous buffer, and the low H2S releasing efficiency remains as a major concern when applying it to the living systems.13 Building from these donor scaffolds, a series of synthetic H2S donors have been developed in the last decade.14–19 These donors are activated by different triggers, such as enzymes,20,21 cellular thiols,22–27 pH modulation,28,29 and photo activation,30–33 and the released H2S exhibits promising activities in different physiological and pathological processes.17,18
To expand the library of H2S donors, we recently developed the carbonyl sulfide (COS)-based platform for H2S donation. In this approach, caged-thiocarbamates are activated and the resultant intermediates undergo a cascade reaction to release COS, which is quickly converted to H2S by the ubiquitous enzyme carbonic anhydrase (CA) (Fig. 1a).34 Expanding on this initial report, we, as well as others, have applied the similar caged-COS systems to include a series of triggerable COS-based H2S donors which can be activated by different mechanisms, such as reactive oxygen species (ROS),35–37 esterase,38,39 cellular nucleophiles,40 click chemistry,41 light,42–44 and Cys,45 to release H2S under various conditions.45,46
 | ||
| Fig. 1 (a) General design of previous self-immolative COS/H2S donors. (b) COS/H2S release from fluorogenic caged-sulfenyl thiocarbonate donor motifs. | ||
Although H2S release from these donors has been measured using different methods, the real-time tracking of donor activation and H2S delivery in living systems remains a key challenge due to the inherent limitations of current H2S detection methods. For example, the colorimetric methylene blue (MB) assay has been widely used to measure H2S levels, but requires strongly acidic conditions, which may trigger H2S release from acid labile sulfide pools. Additionally, MB is an end-point assay which destroys experimental samples, thus making it not feasible for in vivo H2S detection.47 Similarly, H2S-selective electrodes are most often used in bulk measurements rather than non-homogenized biological samples.47 By contrast, H2S fluorescent probes have attracted much attentions due to their high sensitivities and have been used to sense and visualize H2S in biological samples.48 Albeit promising, H2S fluorescent probes are prone to react with reactive cellular species, such as Cys or glutathione (GSH), which results in either probe consumption or false positive signals. In addition, all of the above methods consume the H2S being measured. Recently, we reported a series of γ-ketothiocarbamates (γ-KetoTCM) that release p-nitroaniline as a colorimetric indicator; however, although the colorimetric response enables monitoring of COS/H2S release in cuvettes, the direct tracking in live-cell environments remains an un-met need.49 Taken together, a key advancement compatible with live-cell and tissue experiments would be the development of H2S donors that deliver H2S with a concomitant fluorescence response to enable tracking of H2S delivery by common microscopy techniques.
Aligned with these needs, we report here the development of a new H2S-releasing strategy based on caged sulfenyl thiocarbonates and apply this donor platform to access fluorescent turn-on H2S donors. In this approach, cellular thiols (i.e. Cys and GSH) activate the sulfenyl thiocarbonates through thiol-mediated disulfide reduction to release COS, which is quickly converted to H2S by CA (Fig. 1b). To the best of our knowledge, the sulfenyl thiocarbonate reduction strategy provides a new activation pathway that has not been used to trigger COS/H2S release from donor platforms. Unlike currently-available donors that function through initial COS release, the sulfenyl thiocarbonate system does not generate reactive electrophile byproducts upon activation, which provides a significant advance in the field. In addition, we leverage this new donor strategy to access systems in which a concomitant fluorescence turn-on occurs upon donor activation, thus allowing for real-time monitoring and quantification of H2S release using fluorescence spectroscopy.
Results and discussion
To test our hypothesis that caged-sulfenyl thiocarbonates could serve as thiol-triggered fluorescent COS/H2S donors, four donors (FLD-1–4) were prepared by reacting fluorescein or 3-O-methylfluorescein with ((benzyl)dithio)carbonyl chloride 1 or ((phenol)dithio)carbonyl chloride 2 in the presence of DIPEA (Scheme 1a). A caged thiocarbonate (TCN-1) was synthesized as a triggerless control compound, which is expected to be stable toward thiol activation due to the lack of a disulfide functional group (Scheme 1b).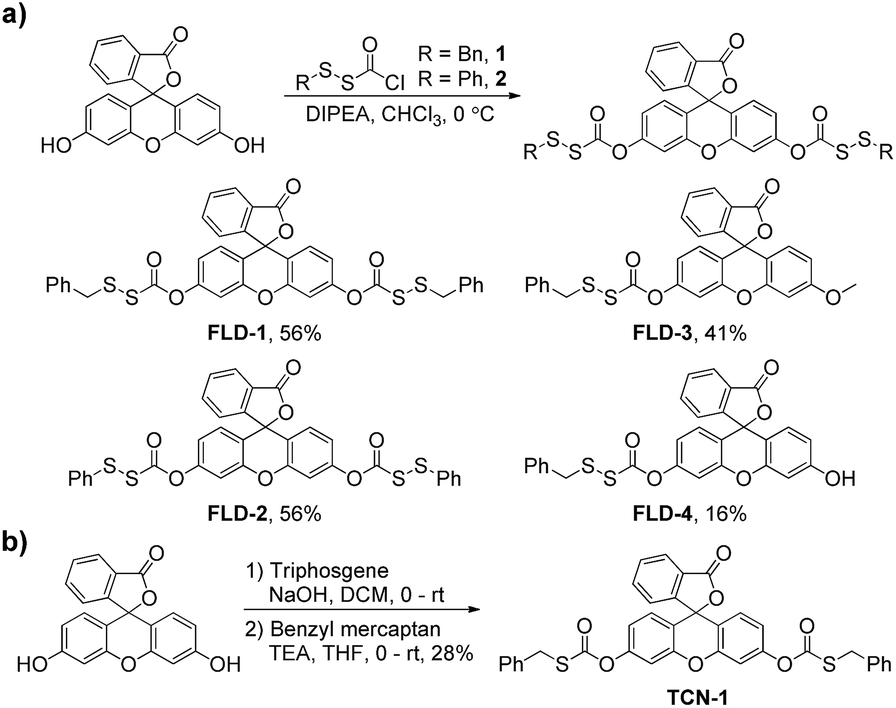 | ||
| Scheme 1 Synthesis of fluorescent COS/H2S donors (a) and thiocarbonate control compound (b). | ||
With these compounds in hand, we first evaluated their spectroscopic properties in PBS buffer (pH 7.4, 10 mM). As expected, FLD-1–3 and TCN-1 are not absorptive in the visible region and are all nonfluorescent because the fluorescein unit is locked in the closed lactone form. By contrast, FLD-4 shows a prominent absorbance band in the visible region (λmax = 449 nm, ε = 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 ± 2500 M−1 cm−1) with measurable fluorescence (λem = 514 nm, Φ = 0.11 ± 0.01) due to the free hydroxyl group (Table S1†). Based on the promising spectroscopic properties, large dynamic range, and efficient release of two equivalents of COS/H2S, we chose to use FLD-1 as the model donor for initial reactivity and selectivity evaluations.
300 ± 2500 M−1 cm−1) with measurable fluorescence (λem = 514 nm, Φ = 0.11 ± 0.01) due to the free hydroxyl group (Table S1†). Based on the promising spectroscopic properties, large dynamic range, and efficient release of two equivalents of COS/H2S, we chose to use FLD-1 as the model donor for initial reactivity and selectivity evaluations.
To test the reactivity of FLD-1 towards Cys-induced activation, FLD-1 (10 μM) was incubated with Cys (100 μM) in PBS buffer (pH 7.4, 10 mM) containing physiologically-relevant concentrations of CA (25 μg mL−1), and the fluorescence intensity was measured using a fluorescence spectrometer. As expected, Cys successfully activated FLD-1 and resulted in a 500-fold fluorescence turn on over 2 h, demonstrating the release of the fluorescein upon FLD-1 activation (Fig. 2a). Fluorescein formation was also confirmed by UV-vis spectroscopy under the identical conditions (Fig. S1†). A Cys-dependent fluorescence enhancement was observed when treating FLD-1 (10 μM) with increasing concentrations of Cys (1–20 equiv.), indicating a high sensitivity of FLD-1 towards Cys. No fluorescent signal was observed in the absence of Cys, suggesting that FLD-1 is stable in aqueous buffer, and that it is not hydrolyzed to provide false signals (Fig. 2b). In addition, to confirm the H2S delivery from FLD-1, we treated FLD-1 (10 μM) with Cys (100 μM) in PBS containing CA (25 μg mL−1) and quantified H2S release using the MB assay. In this experiment, 15 μM of H2S was detected (75% releasing efficiency), which is consistent with our hypothesis that 2 equivalents of COS/H2S would be released upon FLD-1 activation (Fig. S2†). Taken together, these experiments demonstrate that FLD-1 is highly responsive to Cys activation.
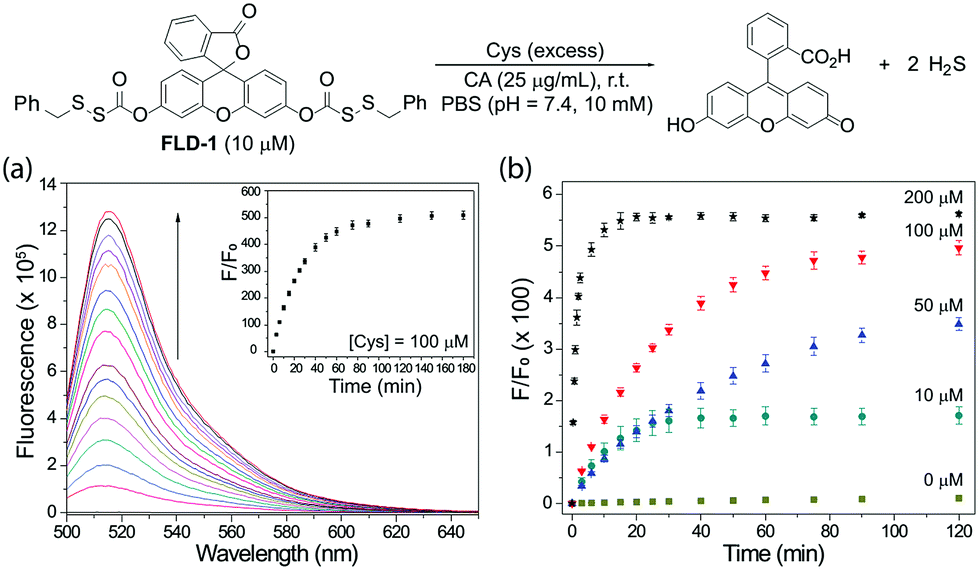 | ||
| Fig. 2 (a) Time-dependent fluorescence spectra of FLD-1 (10 μM) in PBS (pH 7.4, 10 mM) containing Cys (100 μM) and CA (25 μg mL−1). (b) Cys-dependent (0–200 μM) fluorescence turn on of FLD-1 (10 μM) in PBS. λex = 490 nm, λem = 500–650 nm, and slit width = 0.3 mm. The experiments were performed in triplicate and results are expressed as mean ± SD (n = 3). | ||
To determine whether H2S release correlated directly with the observed fluorescence response, we measured the fluorescent response from FLD-3 in the presence of Cys and CA and quantified H2S release using the MB assay. We chose to use FLD-3 as the model compound for these investigations because it only contains one sulfenyl thiocarbonate moiety, and therefore should simplify the reaction kinetics. In comparison, FLD-1 contains two sulfenyl thiocarbonate groups, and the cleavage of one sulfenyl thiocarbamate would generate FLD-4 as a reaction intermediate, which exhibits moderate fluorescence (see Fig. 4). Incubation of FLD-3 (10 μM) with Cys (100 μM) resulted in a rapid fluorescence response with 96% of the H2S release measured by MB. At extended time points, we observed a slight decrease in measured H2S, possibly due to volatilization of H2S in the headspace of the closed system or adventitious oxidation of released H2S. Negligible H2S was detected in the absence of CA, indicating that Cys-triggered H2S delivery from FLD-3 proceeds through intermediate COS formation (Fig. 3a). Importantly, the strong linear correlation between the measured fluorescence and H2S measured from the MB method detection (first 25 min, R2 = 0.988) demonstrates that fluorescent readouts can serve as reliable optical tools to track COS/H2S release from FLD donors with temporal resolution (Fig. 3b). Moreover, this linear correlation suggests that choice of other fluorophores with different brightnesses and photophysical properties could be used to access different dynamic ranges of H2S release, thus enabling this approach to be translated to different types of experimental designs.
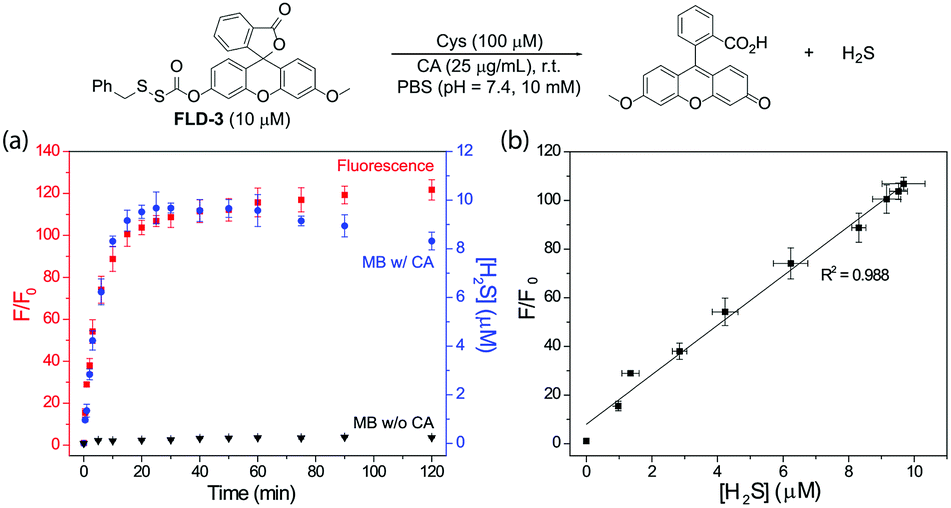 | ||
| Fig. 3 (a) Time-dependent fluorescence turn on (red) and H2S release (blue) upon FLD-3 (10 μM) activation in PBS (pH 7.4, 10 mM) containing Cys (100 μM) and CA (25 μg mL−1). No H2S was detected in the absence of CA (black). λex = 454 nm, λem = 500–650 nm, and slit width = 0.3 mm. (b) Correlation between fluorescence measurement and MB detection. The experiments were performed in triplicate. The results are expressed as mean ± SD (n = 3). | ||
Having confirmed the efficiency of Cys-triggered fluorescence turn on and COS/H2S release, we next evaluated the reactivity of other FLD donors towards Cys activation. Treating FLD-1–3 (10 μM) with Cys (100 μM) in PBS buffer (pH 7.4, 10 mM) resulted in a 120–500-fold fluorescence turn on over 2 h. We attribute the faster response of FLD-2 to the more electrophilic phenyl sulfenyl thiocarbonate in comparison to the less electrophilic benzyl sulfenyl thiocarbonate in FLD-1. FLD-4, however, provided minimal fluorescence enhancement due to its strong background fluorescence. No fluorescence response from TCN-1 (10 μM) was observed under the identical conditions, which demonstrates the stability of the thiocarbonate group in the presence of Cys (Fig. 4).
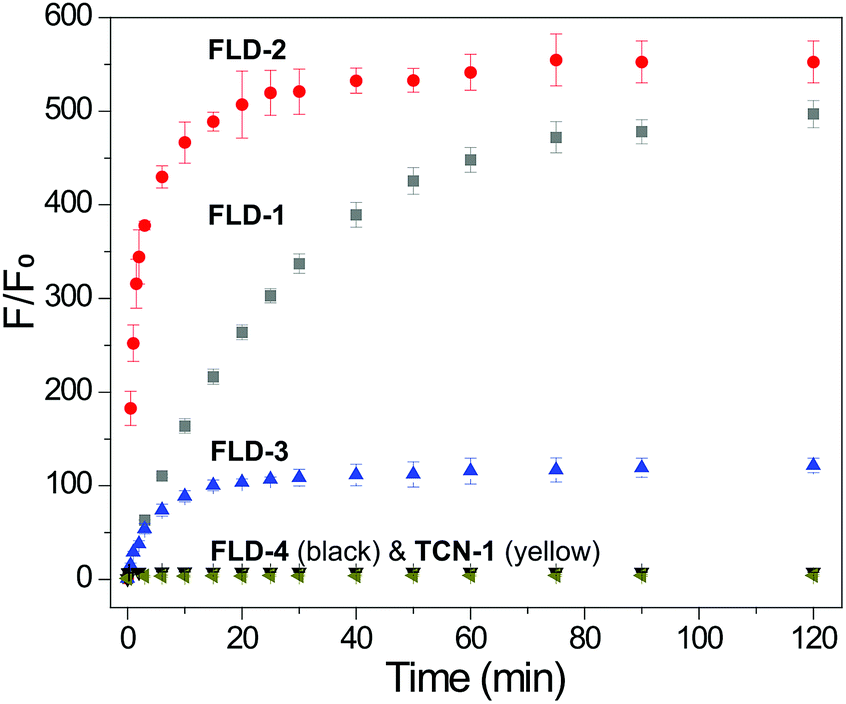 | ||
| Fig. 4 Fluorescence turn on of FLD-1 (gray), FLD-2 (red), FLD-3 (blue), FLD-4 (black), and TCN-1 (dark yellow) (10 μM) in PBS (pH 7.4, 10 mM) containing Cys (100 μM). λex = 490 nm for FLD-1, 2, 4, and TCN-1, λex = 454 nm for FLD-3, λem = 500–650 nm, and slit width = 0.3 mm. The experiments were performed in triplicate. The results are expressed as mean ± SD (n = 3). | ||
To further support our proposed activation mechanism in Fig. 1b, as well as the activation of the sulfenyl thiocarbonate group by other thiols, we incubated FLD-1 (10 μM) in PBS (pH 7.4, 10 mM) with 10 equivalents of benzyl mercaptan (100 μM) for 1 h and analyzed the reaction products by HPLC (Fig. S3†). Consistent with our proposed activation mechanism, we observed FLD-1 consumption and the formation of both benzyl disulfide and fluorescein, which supports the proposed mechanism.
In addition to Cys and BnSH, other cellular thiols and biological nucleophiles were tested to determine whether they resulted in donor activation. In these experiments, FLD-1 (10 μM) was incubated with GSH, homocysteine (Hcy), N-acetyl cysteine (NAC), penicillamine (PEN), or bovine serum albumin (BSA) (100 μM), in PBS buffer (pH 7.4, 10 mM) containing CA (25 μg mL−1) and the fluorescent intensity was measured after 2 h. As expected, FLD-1 is stable in PBS at physiological pH in the absence of thiols (Fig. 5 bar 1). Incubation of FLD-1 with Cys, NAC, GSH, and Hcy, however, led to a significant fluorescence enhancement, indicating successful donor activation and COS/H2S release (Fig. 5 bars 2–5 and S4†). In addition, these results demonstrate that the sulfenyl thiocarbonate group is responsive to different types of thiols. In comparison, PEN resulted in only minimal fluorescence response and BSA failed to activate the donor presumably due to the bulkiness of these two thiol species, which hindered their reactions with FLD-1 (Fig. 5 bars 6 and 7). Additionally, N-ethylmaleimide (NEM) pretreatment of Cys samples significantly reduced the fluorescence enhancement from FLD-1, confirming the necessity of the thiol-induced reduction for the donor activation (Fig. 5 bar 8).
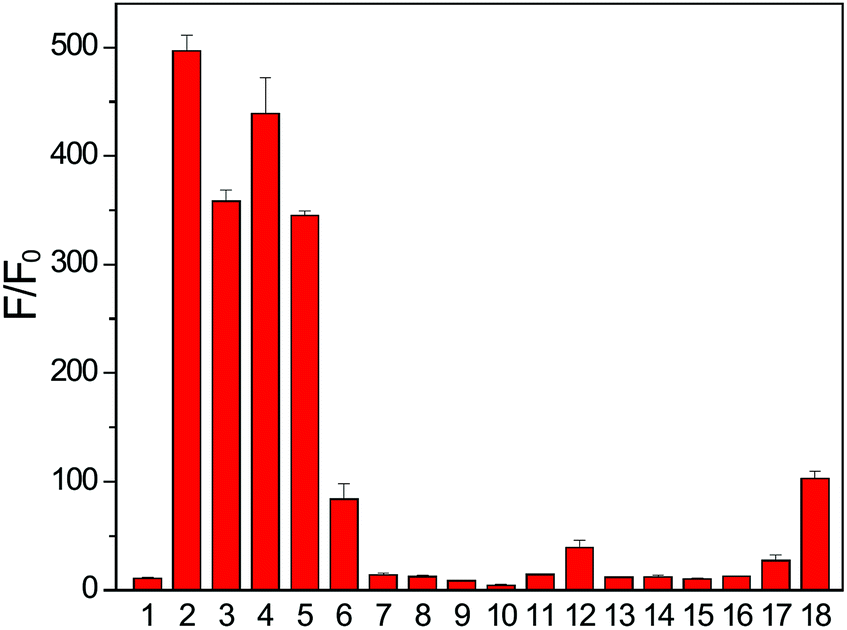 | ||
| Fig. 5 Fluorescence turn on of FLD-1 (10 μM) in the presence of cellular RSONs (100 μM): (1) FLD-1 only, (2) Cys, (3) NAC, (4) GSH, (5) Hcy, (6) PEN, (7) BSA, (8) Cys + NEM, (9) H2O2, (10) ClO−, (11) O2−, (12) TBHP, (13) Ser, (14) Lys, (15) Gly, (16) GSSG, (17) GSNO, and (18) PLE (1 U mL−1). Fluorescence intensity was measured after 2 h incubation. The experiments were performed in triplicate. The results were expressed as mean ± SD (n = 3). | ||
We also tested the response of the FLD donors in the presence of other cellular reactive sulfur, oxygen, and nitrogen species (RSONs). FLD-1 (10 μM) was incubated with RSONs (100 μM), such as hydrogen peroxide (H2O2), hyperchlorite (ClO−), superoxide (O2−), tert-butyl hydroperoxide (TBHP), serine (Ser), lysine (Lys), glycine (Gly), oxidized glutathione (GSSG), and S-nitrosoglutathione (GSNO), and COS/H2S release was monitored by tracking the fluorescein formation. As expected, minimal fluorescence was observed in these experiments, confirming the stability of FLD-1 to common RSONs (Fig. 5 bars 9–17). Because the carbonate functional group may be sensitive to esterase-catalyzed hydrolysis, we also tested the esterase stability of the sulfenyl thiocarbonate group by incubating FLD-1 (10 μM) with porcine liver esterase (PLE, 1 U mL−1). Although a slight fluorescence turn on was observed after a 2 h incubation, the observed response was much lower than that from thiol activation (Fig. 5 bar 18). Taken together, these studies demonstrate that FLD-1 is highly responsive and selective to thiol activation and common cellular RSONs do not trigger FLD-1 to release COS/H2S.
After confirming the thiol-triggered COS/H2S release from FLD-1 in aqueous buffer, we next investigated the H2S delivering capacity of FLD-1 in cellular environment. In these experiments, HeLa cells were treated with FLD-1 (50 μM) or TCN-1 (50 μM) and H2S release was monitored using C7-Az, a H2S-responsive fluorescent probe.50,51 Incubation of HeLa cells with C7-Az (50 μM) alone resulted in a negligible fluorescence response, indicating minimal endogenous H2S (Fig. 6 top row). Treatment with TCN-1 also failed to provide a fluorescence signal, suggesting that the thiocarbonate group was stable and did not decompose to generate false signals in cellular environments (Fig. 6 middle row). By contrast, a strong C7-Az fluorescent signal was observed when incubating HeLa cells with FLD-1, suggesting that H2S release was successfully triggered by endogenous thiols. In addition, a strong fluorescence signal was also observed from activated FLD-1 in the fluorescein channel, confirming the fluorescence response upon donor activation (Fig. 6 bottom row). Taken together, these results demonstrate that the FLD donors not only function as efficacious H2S donors in live cells, but also provide a fluorescence signal that enables observing H2S release.
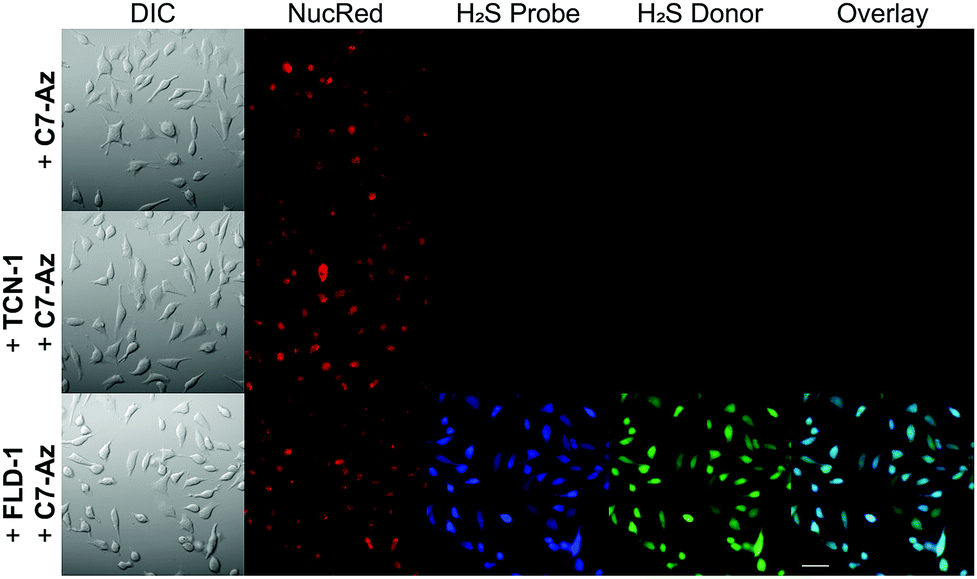 | ||
| Fig. 6 H2S delivery from FLD-1 in HeLa cells. HeLa cells were treated with NucRed nuclear dye and C7-Az (50 μM) in DMEM only (top row) or DMEM containing TCN-1 (50 μM) (middle row) or FLD-1 (50 μM) (bottom row) for 30 min. Cells were then washed with PBS and cell images were taken in PBS using a fluorescent microscope. Bar scale: 50 μm. | ||
To further demonstrate the H2S-releasing fidelity of the FLD donors we also investigated the anti-inflammatory activities of FLD-1. We pretreated macrophage RAW 264.7 cells with FLD-1 (0–25 μM) for 2 h, followed by a 24 h incubation with lipopolysaccharide (LPS, 0.5 μg mL−1) to trigger the inflammatory response. The inflammation event usually results in the NO generation, which can be monitored by measuring nitrite (NO2−) accumulation. We chose to use concentrations of FLD-1 up to 25 μM because these concentrations did not induce cytotoxicity (Fig. S6†).
As expected, the pretreatment of RAW 264.7 cells with FLD-1 showed a dose-dependent inhibition of NO2− accumulation, indicating anti-inflammatory activity from FLD-1. Although GYY4137 has shown anti-inflammatory effects at higher concentration and longer incubation time (i.e. 100–1000 μM and 24 h incubation),52 such cytoprotection was not observed at the 25 μM concentration used for comparison, highlighting the efficacious H2S release from FLD-1 in the cellular environment (Fig. 7). To further confirm that the observed effects were due to H2S rather than other components of donor activation, we treated cells with 25 μM of TCN-1, fluorescein or benzyl mercaptan and measured NO2− production. None of these compounds attenuated NO2− generation, confirming that the anti-inflammatory activities of FLD-1 is due to H2S release (Fig. S7†). Overall, these studies demonstrate that FLD-1 releases COS/H2S in complex cellular environment and exhibits promising anti-inflammatory protections, indicating potential applications of FLD-1 as H2S-releasing therapeutics.
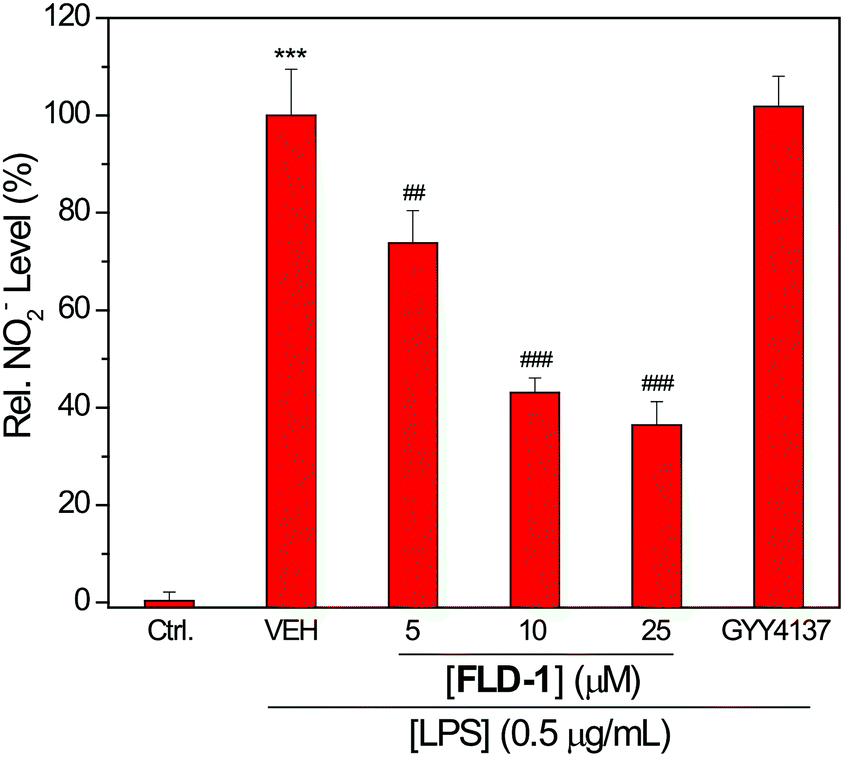 | ||
| Fig. 7 Effects of FLD-1 on LPS-induced NO2− accumulation. RAW 264.7 cells were pretreated with FLD-1 (5–25 μM) or GYY4137 (25 μM) for 2 h, followed by a 24 h treatment of LPS (0.5 μg mL−1). Results are expressed as mean ± SD (n = 4). ***P < 0.001 vs. the control group; ##P < 0.01 vs. vehicle-treated group; ###P < 0.001 vs. vehicle-treated group. | ||
Conclusions
In conclusion, we prepared and evaluated a class of caged sulfenyl thiocarbonates as new controllable and fluorescent COS/H2S donors. Thiol-triggered COS/H2S release from these molecules has been detected and visualized in both aqueous buffer and in live cells. Importantly, the concomitant release of a fluorescein reporter after H2S release enables the real-time monitoring of H2S release dynamics. In addition, we demonstrate that FLD-1 exhibits a dose-dependent inhibition of the LPS-induced NO formation, which is consistent with anti-inflammatory activities of H2S. Taken together, caged-sulfenyl thiocarbonates are promising new class of COS/H2S donors with high potential for accessing H2S-related protective activities, and the developed fluorescent donors provide new methods for monitoring H2S release in real-time in complex environments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research reported in this publication was supported by the NIH (R01GM113030) and Dreyfus Foundation. NMR, fluorescence microscopy, and MS instrumentation in the UO CAMCOR facility is supported by the NSF (CHE-1427987, CHE-1531189, and CHE-1625529).Notes and references
- C. Szabo, Nat. Rev. Drug Discovery, 2007, 6, 917–935 CrossRef PubMed.
- R. Wang, Physiol. Rev., 2012, 92, 791–896 CrossRef PubMed.
- M. R. Filipovic, J. Zivanovic, B. Alvarez and R. Banerjee, Chem. Rev., 2018, 118, 1253–1337 CrossRef PubMed.
- O. Kabil and R. Banerjee, Antioxid. Redox Signaling, 2014, 20, 770–782 CrossRef PubMed.
- S. Bruce King, Free Radical Biol. Med., 2013, 55, 1–7 CrossRef PubMed.
- L. F. Hu, M. Lu, P. T. Hon Wong and J. S. Bian, Antioxid. Redox Signaling, 2011, 15, 405–419 CrossRef PubMed.
- B. D. Paul and S. H. Snyder, Nat. Rev. Mol. Cell Biol., 2012, 13, 499–507 CrossRef PubMed.
- J. M. Fukuto, S. J. Carrington, D. J. Tantillo, J. G. Harrison, L. J. Ignarro, B. A. Freeman, A. Chen and D. A. Wink, Chem. Res. Toxicol., 2012, 25, 769–793 Search PubMed.
- K. R. Olson, Antioxid. Redox Signaling, 2012, 17, 32–44 Search PubMed.
- C. Szabo, Antioxid. Redox Signaling, 2012, 17, 68–80 Search PubMed.
- L. Li, P. Rose and P. K. Moore, Annu. Rev. Pharmacol. Toxicol., 2011, 51, 169–187 CrossRef PubMed.
- E. R. DeLeon, G. F. Stoy and K. R. Olson, Anal. Biochem., 2012, 421, 203–207 CrossRef PubMed.
- L. Li, M. Whiteman, Y. Y. Guan, K. L. Neo, Y. Cheng, S. W. Lee, Y. Zhao, R. Baskar, C. H. Tan and P. K. Moore, Circulation, 2008, 117, 2351–2360 CrossRef PubMed.
- M. D. Pluth, T. S. Bailey, M. D. Hammers, M. D. Hartle, H. A. Henthorn and A. K. Steiger, Synlett, 2015, 26, 2633–2643 CrossRef.
- C. Szabo and A. Papapetropoulos, Pharmacol. Rev., 2017, 69, 497–564 CrossRef PubMed.
- M. D. Hartle and M. D. Pluth, Chem. Soc. Rev., 2016, 45, 6108–6117 RSC.
- Y. Zhao, T. D. Biggs and M. Xian, Chem. Commun., 2014, 50, 11788–11805 RSC.
- Y. Zhao, A. Pacheco and M. Xian, Handb. Exp. Pharmacol., 2015, 230, 365–388 Search PubMed.
- C. R. Powell, K. M. Dillon and J. B. Matson, Biochem. Pharmacol., 2018, 149, 110–123 CrossRef.
- P. Shukla, V. S. Khodade, M. SharathChandra, P. Chauhan, S. Mishra, S. Siddaramappa, B. E. Pradeep, A. Singh and H. Chakrapani, Chem. Sci., 2017, 8, 4967–4972 RSC.
- Y. Zheng, B. Yu, K. Ji, Z. Pan, V. Chittavong and B. Wang, Angew. Chem., Int. Ed., 2016, 55, 4514–4518 CrossRef.
- J. C. Foster, C. R. Powell, S. C. Radzinski and J. B. Matson, Org. Lett., 2014, 16, 1558–1561 CrossRef.
- Y. Zhao, S. Bhushan, C. Yang, H. Otsuka, J. D. Stein, A. Pacheco, B. Peng, N. O. Devarie-Baez, H. C. Aguilar, D. J. Lefer and M. Xian, ACS Chem. Biol., 2013, 8, 1283–1290 CrossRef.
- Y. Zhao, H. Wang and M. Xian, J. Am. Chem. Soc., 2011, 133, 15–17 CrossRef.
- Y. Zhao, C. Yang, C. Organ, Z. Li, S. Bhushan, H. Otsuka, A. Pacheco, J. Kang, H. C. Aguilar, D. J. Lefer and M. Xian, J. Med. Chem., 2015, 58, 7501–7511 CrossRef.
- M. M. Cerda, M. D. Hammers, M. S. Earp, L. N. Zakharov and M. D. Pluth, Org. Lett., 2017, 19, 2314–2317 CrossRef.
- Y. Zhao, J. Kang, C. M. Park, P. E. Bagdon, B. Peng and M. Xian, Org. Lett., 2014, 16, 4536–4539 CrossRef.
- J. Kang, Z. Li, C. L. Organ, C. M. Park, C. T. Yang, A. Pacheco, D. Wang, D. J. Lefer and M. Xian, J. Am. Chem. Soc., 2016, 138, 6336–6339 CrossRef.
- J. Wu, Y. Li, C. He, J. Kang, J. Ye, Z. Xiao, J. Zhu, A. Chen, S. Feng, X. Li, J. Xiao, M. Xian and Q. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 27474–27481 CrossRef.
- N. O. Devarie-Baez, P. E. Bagdon, B. Peng, Y. Zhao, C. M. Park and M. Xian, Org. Lett., 2013, 15, 2786–2789 CrossRef CAS.
- N. Fukushima, N. Ieda, K. Sasakura, T. Nagano, K. Hanaoka, T. Suzuki, N. Miyata and H. Nakagawa, Chem. Commun., 2014, 50, 587–589 RSC.
- Y. Venkatesh, J. Das, A. Chaudhuri, A. Karmakar, T. K. Maiti and N. D. P. Singh, Chem. Commun., 2018, 54, 3106–3109 RSC.
- Z. Y. Xiao, T. Bonnard, A. Shakouri-Motlagh, R. A. L. Wylie, J. Collins, J. White, D. E. Heath, C. E. Hagemeyer and L. A. Connal, Chem.–Eur. J., 2017, 23, 11294–11300 CrossRef CAS.
- A. K. Steiger, S. Pardue, C. G. Kevil and M. D. Pluth, J. Am. Chem. Soc., 2016, 138, 7256–7259 CrossRef CAS.
- Y. Zhao and M. D. Pluth, Angew. Chem., Int. Ed., 2016, 55, 14638–14642 CrossRef CAS.
- Y. Zhao, H. A. Henthorn and M. D. Pluth, J. Am. Chem. Soc., 2017, 139, 16365–16376 CrossRef CAS.
- P. Chauhan, S. Jos and H. Chakrapani, Org. Lett., 2018, 20, 3766–3770 CrossRef CAS PubMed.
- P. Chauhan, P. Bora, G. Ravikumar, S. Jos and H. Chakrapani, Org. Lett., 2017, 19, 62–65 CrossRef CAS.
- A. K. Steiger, M. Marcatti, C. Szabo, B. Szczesny and M. D. Pluth, ACS Chem. Biol., 2017, 12, 2117–2123 CrossRef CAS.
- C. R. Powell, J. C. Foster, B. Okyere, M. H. Theus and J. B. Matson, J. Am. Chem. Soc., 2016, 138, 13477–13480 CrossRef CAS.
- A. K. Steiger, Y. Yang, M. Royzen and M. D. Pluth, Chem. Commun., 2017, 53, 1378–1380 RSC.
- Y. Zhao, S. G. Bolton and M. D. Pluth, Org. Lett., 2017, 19, 2278–2281 CrossRef CAS.
- A. K. Sharma, M. Nair, P. Chauhan, K. Gupta, D. K. Saini and H. Chakrapani, Org. Lett., 2017, 19, 4822–4825 CrossRef CAS.
- P. Štacko, L. Muchová, L. Vítek and P. Klán, Org. Lett., 2018, 20, 4907–4911 CrossRef.
- Y. Zhao, A. K. Steiger and M. D. Pluth, Chem. Commun., 2018, 54, 4951–4954 RSC.
- P. Bora, P. Chauhan, K. A. Pardeshi and H. Chakrapani, RSC Adv., 2018, 8, 27359–27374 RSC.
- B. Peng and M. Xian, Asian J. Org. Chem., 2014, 3, 914–924 CrossRef CAS.
- V. S. Lin, W. Chen, M. Xian and C. J. Chang, Chem. Soc. Rev., 2015, 44, 4596–4618 RSC.
- Y. Zhao, A. K. Steiger and M. D. Pluth, Angew. Chem., Int. Ed., 2018, 57, 13101–13105 CrossRef CAS.
- B. Chen, W. Li, C. Lv, M. Zhao, H. Jin, J. Du, L. Zhang and X. Tang, Analyst, 2013, 138, 946–951 RSC.
- M. K. Thorson, T. Majtan, J. P. Kraus and A. M. Barrios, Angew. Chem., Int. Ed., 2013, 52, 4641–4644 CrossRef CAS.
- M. Whiteman, L. Li, P. Rose, C. H. Tan, D. B. Parkinson and P. K. Moore, Antioxid. Redox Signaling, 2010, 12, 1147–1154 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: H2S release curves, cytotoxicity data, NMR spectra. See DOI: 10.1039/c8sc05200j |
| This journal is © The Royal Society of Chemistry 2019 |