
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and folding behaviour of poly(p-phenylene vinylene)-based β-sheet polychromophores†
Elizabeth
Elacqua
 ab,
Geoffrey T.
Geberth
c,
David A.
Vanden Bout
*c and
Marcus
Weck
*a
ab,
Geoffrey T.
Geberth
c,
David A.
Vanden Bout
*c and
Marcus
Weck
*a
aMolecular Design Institute, Department of Chemistry, New York University, New York, NY 10003, USA. E-mail: marcus.weck@nyu.edu
bDepartment of Chemistry, The Pennsylvania State University, University Park, PA 16802, USA
cDepartment of Chemistry, University of Texas at Austin, Austin, TX 78712, USA. E-mail: davandenbout@mail.utexas.edu
First published on 20th December 2018
Abstract
This contribution describes the design and synthesis of β-sheet-mimicking synthetic polymers comprising distinct poly(p-phenylene vinylene) (PPV) and poly(norbornene) (PNB) backbones with multiple turns. The rod–coil–coil–rod tetrablock copolymers, synthesized using ring-opening metathesis polymerization (ROMP) and featuring orthogonal face-to-face π–π stacking and phenyl/perfluorophenyl interactions, show persistent folding both in bulk and at the single molecule level, irrespective of the number of β-turns. Single molecule polarization studies reveal that the copolymers are more anisotropic than the corresponding homopolymers. Examination of the spectral signatures of the single molecules shows a dominant emissive chromophore in the linked materials compared to the homopolymer. The lack of significant spectral changes of the folded materials along with the existence of a dominant emission spectrum supports the proposed structure of well-aligned, minimally-interacting chromophores. Utilization of this reliably folding, phenyl/perfluorophenyl functionality could provide an extremely useful tool in future functional materials design.
Introduction
The structure of biopolymers is governed by a vast array of individually simplistic, yet collaboratively complex orthogonal noncovalent interactions that give rise to a 3D folded architecture. The design of such ‘foldamers’ from a synthetic perspective is largely accomplished by inclusion of peptidic and/or proteinic residues1,2 that drive self-assembly using a variety of complementary hydrogen bonding elements in combination with hydrophobic interactions and charges3–5 to dictate both shape and folding sequence and ultimately function.6 Despite the wealth of synthetic polymer backbones and expansive knowledge of directional orthogonal self-assembly,7 the engineering of exclusively synthetic folding architectures reminiscent of proteins remains largely terra incognita. In the synthetic realm, helical polymers represent a well-established field that most closely realizes biological motifs (i.e., α-helices);3,8–12 sheet-like materials based upon synthetic polymers still remain underdeveloped.13,14Considerable strides have been made to engineer synthetic polymers with a high degree of control over secondary structures. Selective-point folding strategies15,16 featuring the pre-positioning of noncovalent motifs along a polymer backbone, or intra-chain covalent chemistry, have been reported, wherein reversible folding was achieved using directional interactions, such as hydrogen bonding,17–20 metal coordination,21 and host-guest complexation with cyclodextrins.22 Orthogonal single-chain folding comprising two hydrogen bonding motifs23–26 or combined hydrogen bonding and metal coordination or host-guest chemistry have been used to fabricate more complex patterns. Covalent crosslinking strategies,27–29 such as intra-chain anthracene photodimerization,30 radical polymerization,31,32 and post-polymerization Diels–Alder chemistry,33 have also been reported. Huc and coworkers reported β-sheet foldamers using pre-organized aromatic oligoamides.14,34 While the oligoamides represent the most structurally complex synthetic system to date, possessing up to four β-turns, they are oligomers realized through iterative syntheses. We have described the synthesis of conjugated polymer-based β-sheet-like mimics sustained through orthogonal face-to-face π–π stacking and phenyl/perfluorophenyl interactions.13 Our scaffold is derived from ring-opening metathesis polymerization (ROMP), wherein a pair of poly(p-phenylenevinylene) (PPV) strands were co-polymerized with a flexible poly(norbornene) (PNB) coil between them. While the PPV–PNB–PPV block copolymer was sequenced with helical and coil-like building blocks to engineer complex supramolecular multiblock copolymers,13 the engineering of synthetic polymers featuring multiple β-turns remains elusive. Key to our design is the use of a Hoveyda–Grubbs II initiator in a single iterative living ROMP to install up to 16 consecutive polymer blocks. The activity of the carbene is preserved throughout the duration of the polymerization, as evidenced by 1H NMR spectroscopy. The resultant polymers are characterized in solution through 1H NMR and fluorescence spectroscopies, as well as in the solid state as films through wide-angle X-ray scattering (WAXS). To elucidate individual polymer chain folding modes, we investigated a series of these polymers, containing up to five discrete turns, using single-molecule polarization studies, wherein each PPV segment acts as an individual chromophore and can be used to probe the folding behaviour.
Folded polymeric materials have been studied using single molecule excitation polarization spectroscopy in an effort to measure the extent to which specific conformational and photophysical behaviours could be designed at the single molecule level.35 Here, we take a similar approach by comparing a PPV homopolymer with five sequentially larger folded materials. The conformations of these polymers are studied at the single molecule level using polarization spectroscopy to examine the alignment of the chromophore units within the polymer. The emission spectra demonstrate that the polymer structures maintain a large separation between chromophore units, preventing electronic interactions and chromophore aggregation effects. This combination of anisotropy measurements and fluorescence spectra strongly suggests that the phenyl/perfluorophenyl functionality reliably and robustly facilitates β-sheet-type structures even at the single molecule level, thus opening a new set of tools in the realm of conjugated polymer design.
Results and discussion
Synthetic β-sheet design
A retrosynthetic approach to attain a synthetic β-sheet would denote the major disconnection point between the ‘strands’ and the ‘hairpin turn’. For the synthetic polymer chemist, this either necessitates clever post-polymerization or iterative strategies to link the two ‘synthons’ together, or a design amenable to introducing rigid turns within a continuous pattern that can have molecular recognition elements installed within. Our retrosynthetic analysis identified PPVs as model strands within the β-sheet framework, interwoven with a flexible polymer that would be predisposed to intrachain collapse (Fig. 1).36 Intrachain collapse has been commonly studied in the context of single-chain polymer nanoparticles, often being orchestrated by both covalent capture and dynamic noncovalent chemistry, wherein solvent is used, in part, to help drive the self-assembly process.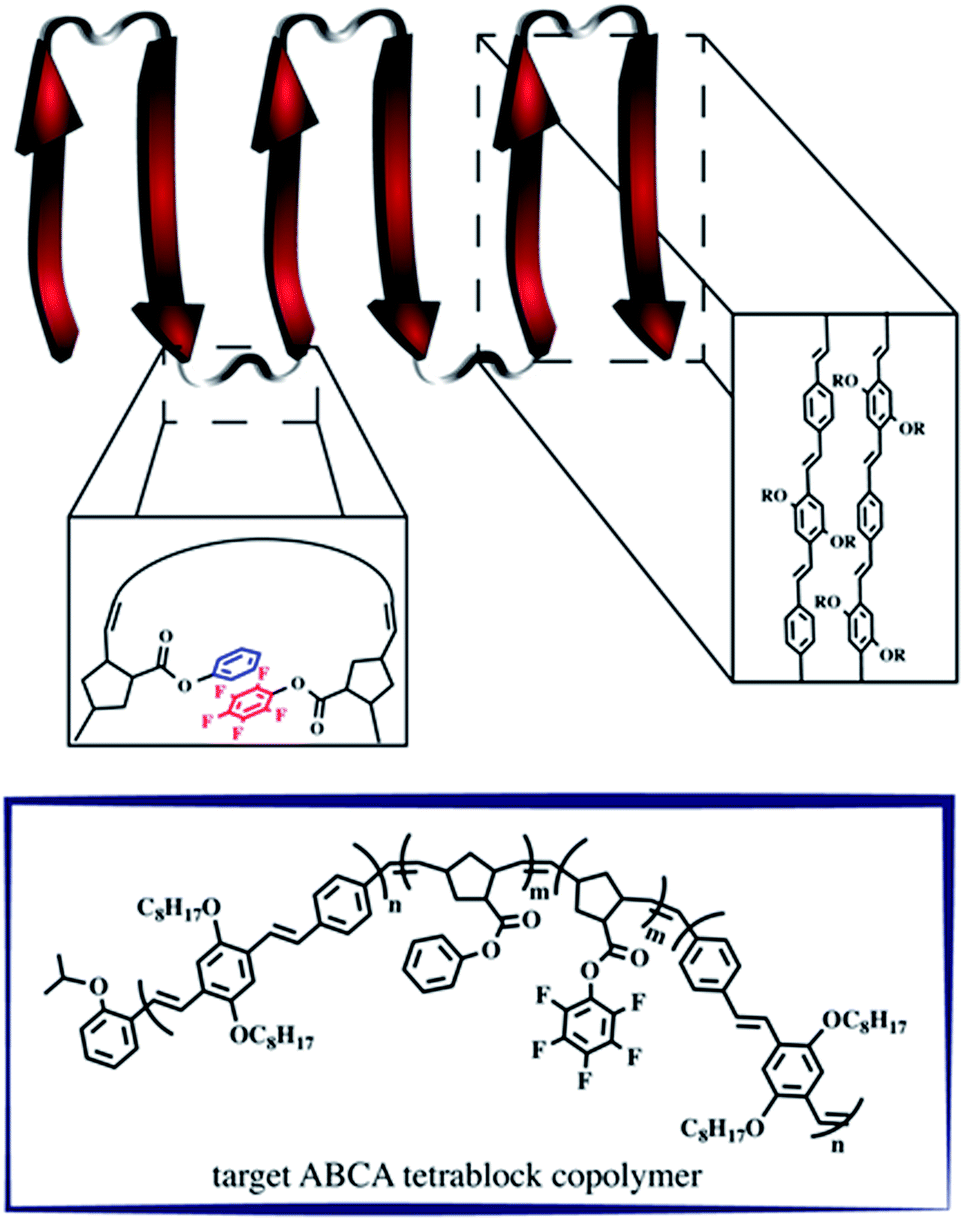 | ||
| Fig. 1 Schematic depicting the supramolecular design strategy to engineer synthetic β-sheet polymers using iterative ROMP and targeted model tetra block copolymers. | ||
Since PPVs aggregate in solvents such as THF and toluene, the design required the use of an interaction known to afford chloroform-stable stacks that would allow for the PPV chains to remain stable and present fewer aggregative tendencies, while remaining synthetically accessible. Quadrupolar interactions based upon phenyl/perfluorophenyl π-stacking do not hinder the ROMP and have been used to organize solvent-specific β-hairpin turns in polymers through stacking of pendant chains.36 Additional use of phenyl/perfluorophenyl interactions is observed in materials relying on preorganization of molecular components such as organic co-crystals and liquid crystals.37–40
PPVs are well-known to engage in face-to-face π–π stacking,41 and are accessible through ROMP.42–45 To connect the PPV strands, we polymerize norbornenes containing phenyl and perfluorophenyl46 pendant groups that participate in quadrupole interactions. The resultant tetrablock copolymer framework is soluble in common organics (e.g., CHCl3, THF, toluene) and can trigger organized folding in chloroform, such that the PPV stacks resemble strands within an antiparallel β-sheet framework.
Single-turn β-sheet synthesis and characterization
The tetrablock copolymers are synthesized using three distinct strained cyclic monomers in the presence of Hoveyda–Grubbs second generation initiator (H–GII), with the monomer consumption being monitored using 1H NMR spectroscopy. First, dioctyloxy[2.2]paracyclophane-1,9-diene (pCpd) is polymerized to completion, followed by exo-phenyl norbornene-5-carboxylate (NB-Ph). Upon completion of the NB-Ph polymerization, exo-pentafluorophenyl norbornene-5-carboxylate (NB-PFP) is added and polymerized to completion. The target tetrablock containing one designed folding segment is completed with a final addition and polymerization of pCpd (Scheme 1).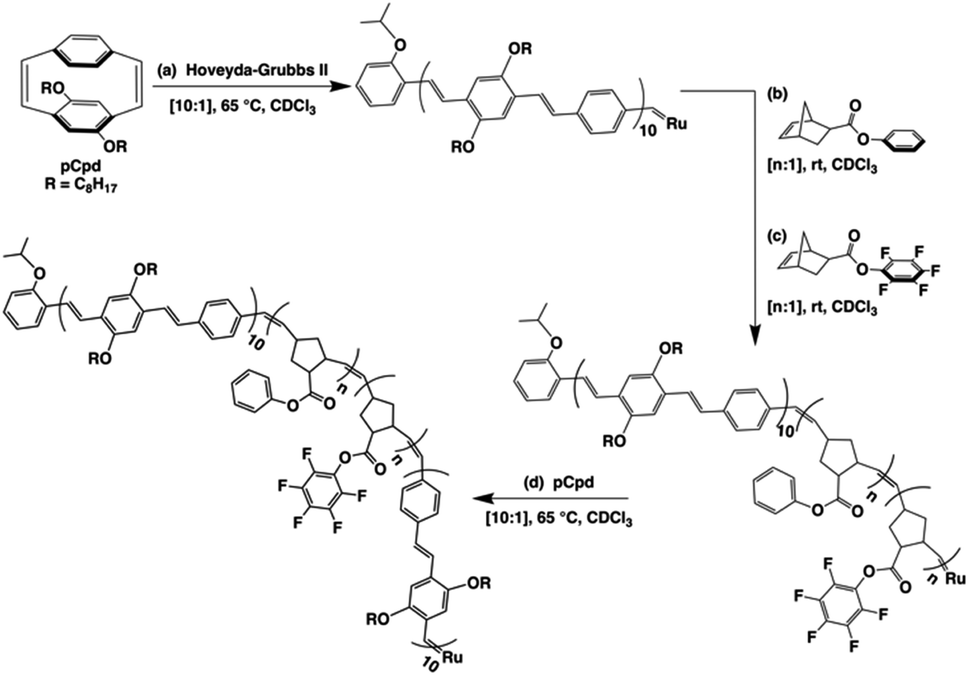 | ||
| Scheme 1 Iterative ROMP strategy to achieve folded PPV–(PNB–PNB–PPV)n architectures. The final active carbene is either quenched to afford the 1T sample, or subsequent iterations of steps (b)–(d) are performed to achieve higher folded structures. All PPV block lengths are approximately ten repeat units, while PNB block lengths of 20–25 units were targeted. | ||
Throughout the polymerization, we monitored monomer consumption as well as block lengths via1H NMR spectroscopy (Fig. 2). Signals related to both initiating and propagating end groups are not only prominent, but also located in unobstructed areas. Specifically, resonances related to the initiating isopropoxy C–H proton and the growing carbene are observed at 4.6 and 16.2 ppm, respectively (Fig. 2a and b). The relative integration ratio of the two remains 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 throughout the duration of the experiment as expected for highly controlled polymerizations, and thus, can be used to provide accurate block lengths. Upon complete polymerization of block 1 (PPV), we calculate a Dp of 10 based on the integration of the end groups relative to the –OCH2 signals of the polymer side-chains centered at 3.6 and 4.0 ppm, which corresponds to the feed ratio (Fig. 2a, bottom).
:1 throughout the duration of the experiment as expected for highly controlled polymerizations, and thus, can be used to provide accurate block lengths. Upon complete polymerization of block 1 (PPV), we calculate a Dp of 10 based on the integration of the end groups relative to the –OCH2 signals of the polymer side-chains centered at 3.6 and 4.0 ppm, which corresponds to the feed ratio (Fig. 2a, bottom).
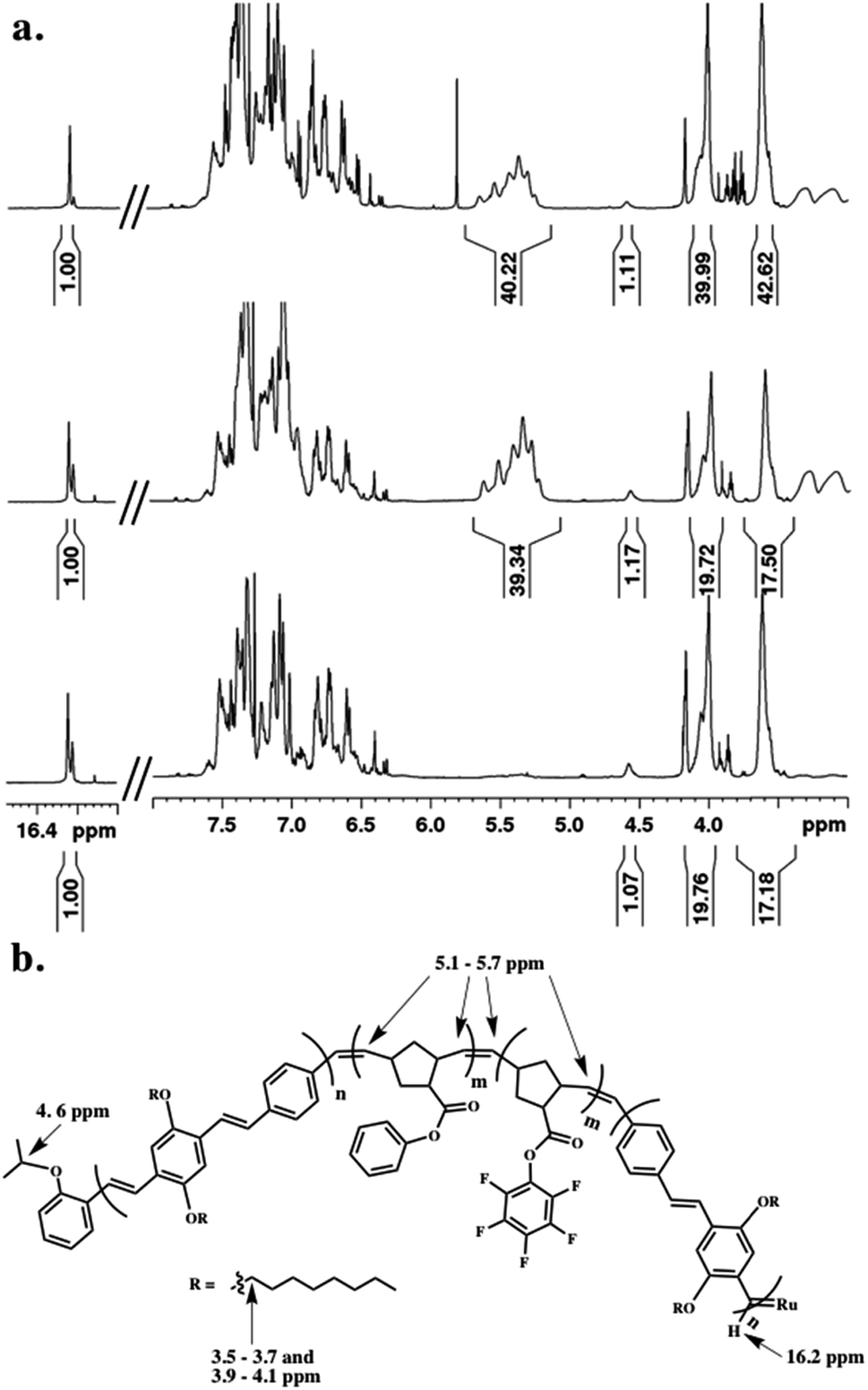 | ||
| Fig. 2 (a) 1H NMR spectral overlay during the polymerization of (bottom) first PPV block, (middle) combined PNB blocks, and (top) final PPV block incorporated prior to quenching, and (b) key protons related to block length analysis highlighted on final polymer structure prior to quenching. | ||
We then added NB-Ph to the reaction vessel and polymerized at room temperature for 20 minutes, followed by NB–PFP, which was similarly polymerized for 20 minutes. The unsaturated PNB backbone signals converge in an area that is devoid of PPV related signals. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C multiplet appears between 5.1–5.7 ppm (Fig. 2a, middle). The block length can be confirmed by referencing to the aforementioned end-group and PPV signals. While we do not monitor the norbornene monomer consumption between the two block additions, we can determine the individual block lengths through the difference in HAr protons of the PPV backbone and PNB-Ph side-chains. A last equivalent of pCpd was added and polymerized until completion (or until relative target block length is achieved) (Fig. 2a, top). The dispersity (Đ) and molecular weight (Mn) of the final PPV–PNB–PNB–PPV sample was 1.22 and 1.96 × 104 g mol−1, respectively. As PPV molecular weights are overestimated by GPC analysis, block lengths were analyzed throughout the polymerization, and confirmed through 1H NMR spectroscopic analyses both during polymerization and after purification.
C multiplet appears between 5.1–5.7 ppm (Fig. 2a, middle). The block length can be confirmed by referencing to the aforementioned end-group and PPV signals. While we do not monitor the norbornene monomer consumption between the two block additions, we can determine the individual block lengths through the difference in HAr protons of the PPV backbone and PNB-Ph side-chains. A last equivalent of pCpd was added and polymerized until completion (or until relative target block length is achieved) (Fig. 2a, top). The dispersity (Đ) and molecular weight (Mn) of the final PPV–PNB–PNB–PPV sample was 1.22 and 1.96 × 104 g mol−1, respectively. As PPV molecular weights are overestimated by GPC analysis, block lengths were analyzed throughout the polymerization, and confirmed through 1H NMR spectroscopic analyses both during polymerization and after purification.
Multiple-turn β-sheet synthesis and characterization
Intrigued by the prominence of the propagating carbene signal throughout the duration of the polymerization,47 we synthesized and examined the properties stemming from a series of PPV–(PNB–PNB–PPV)n tetrablock copolymers wherein n is equal to the number of designed turn segments. We targeted a maximum of n = 5 with the caveat that this would require the propagating ROMP carbene to maintain stability for approximately one week. We utilized an iterative ROMP strategy to prepare the desired β-sheet-like frameworks. To ensure consistency within the series of polymers, we used one pot, wherein upon completion of each turn, a predetermined amount of polymerized material was drawn up and separately quenched, while the remaining polymerization mixture was sequentially fed more of the appropriate norbornene monomers, followed by pCpd (see ESI† for details).48 Evidenced by in situ1H NMR spectral analysis, the iterative polymerization proceeds with full fidelity and control over both end groups (Fig. 3).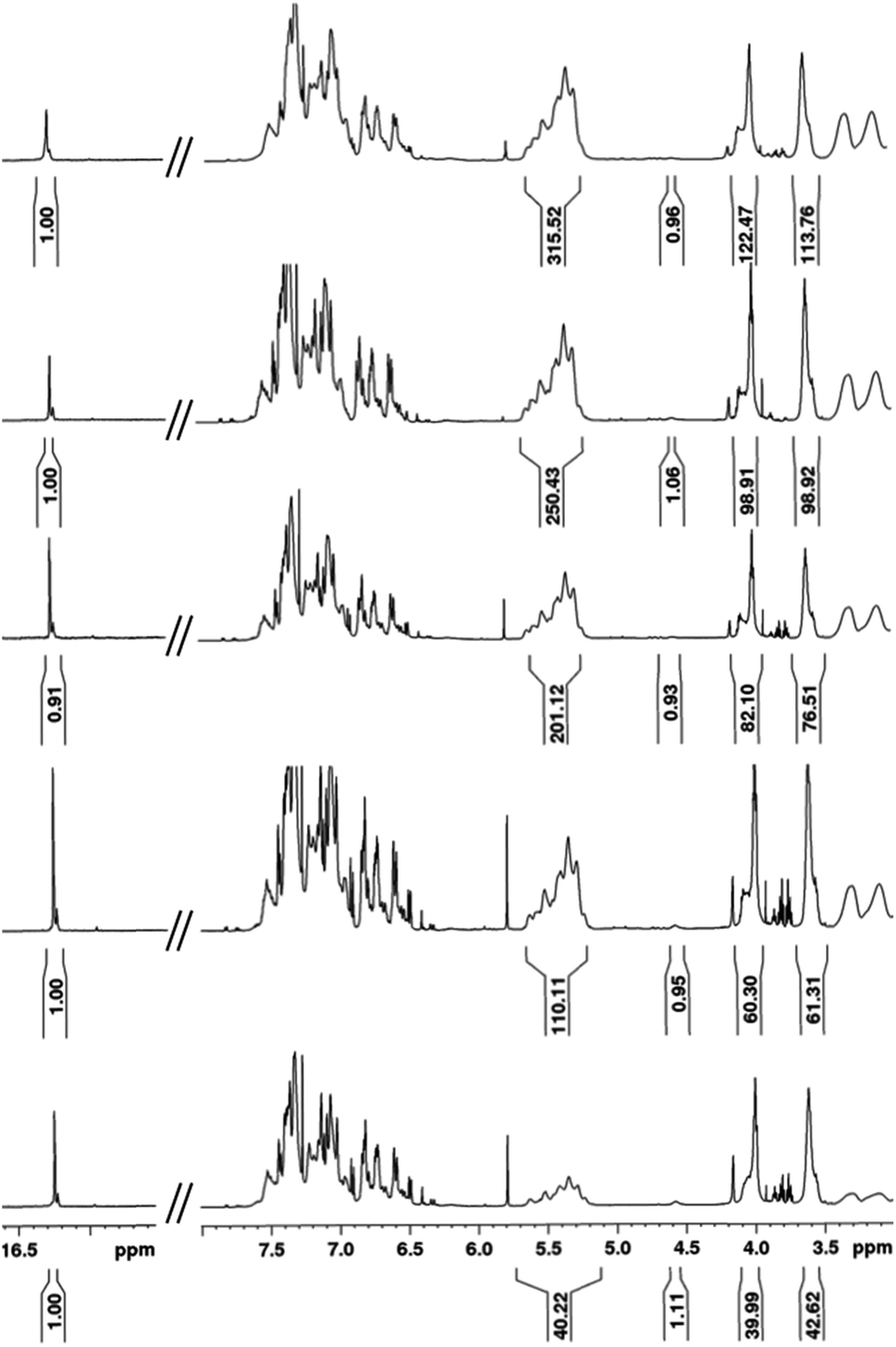 | ||
| Fig. 3 1H NMR spectral overlay depicting different points during one-pot iterative polymerization of (bottom to top): 1T, 2T, 3T, 4T, and 5T polymers. | ||
All polymers were quenched and then purified through sequential reprecipitation into methanol and THF. After purification, the resultant polymers (PPV–(PNB–PNB–PPV)n wherein n = 1–5, denoted 1T through 5T) were dissolved in chloroform and subjected to UV-irradiation (λ = 360 nm) to promote isomerization of the cis-, trans-microstructure. 1H NMR spectroscopy was used to characterize all polymers and to provide accurate insights into molecular weight through end-group analysis (Table 1).
| Polymer | M n | M w | Đ | M n |
|---|---|---|---|---|
| a Determined by SEC (CHCl3) using poly(styrene) standards. b Determined using 1H NMR spectroscopic end-group analysis. | ||||
| PPV | 7600 | 11000 |
1.41 | 4800 |
| PPV–(PNB–PNB)–PPV | 20900 |
24600 |
1.17 | 15000 |
| PPV–(PNB–PNB–PPV)2 | 48700 |
56600 |
1.16 | 28000 |
| PPV–(PNB–PNB–PPV)3 | 56000 |
80300 |
1.43 | 44000 |
| PPV–(PNB–PNB–PPV)4 | 65200 |
90500 |
1.39 | 56000 |
| PPV–(PNB–PNB–PPV)5 | 75600 |
96000 |
1.27 | 70000 |
We turned to WAXS to gain more insight into the folded structure, and the PPV strand conformation within it. PPV blocks are crystalline and well-defined, exhibiting signature peaks corresponding to inter-backbone alkyloxy chain separation (on the order of 18 Å) and face-to-face π–π stacking (d = 4.2–4.6 Å), as well as intra-backbone monomer repeat units (d = 6.2 Å).49 PNBs are generally amorphous;49 yet, a block copolymer comprised of solely the interior PNB blocks exhibits an intense Bragg reflection at d = 4.8 Å, owing to the pendant phenyl–perfluorophenyl interactions. The diffractogram of the tetrablock copolymer (Fig. 4, red) displayed broad features at d = 21.1 Å, 12.9 Å, and 4.7 Å (with a shoulder at 3.95 Å), suggesting the signature face-to-face π–π stacking and alkyl–alkyl interactions are maintained in the sheet–coil–sheet framework. Within the ABCA framework, some side-chains exhibit an extended conformation (d = 21.1 Å), while others suggest the presence of more twisting and interdigitation (d = 13.0 Å).
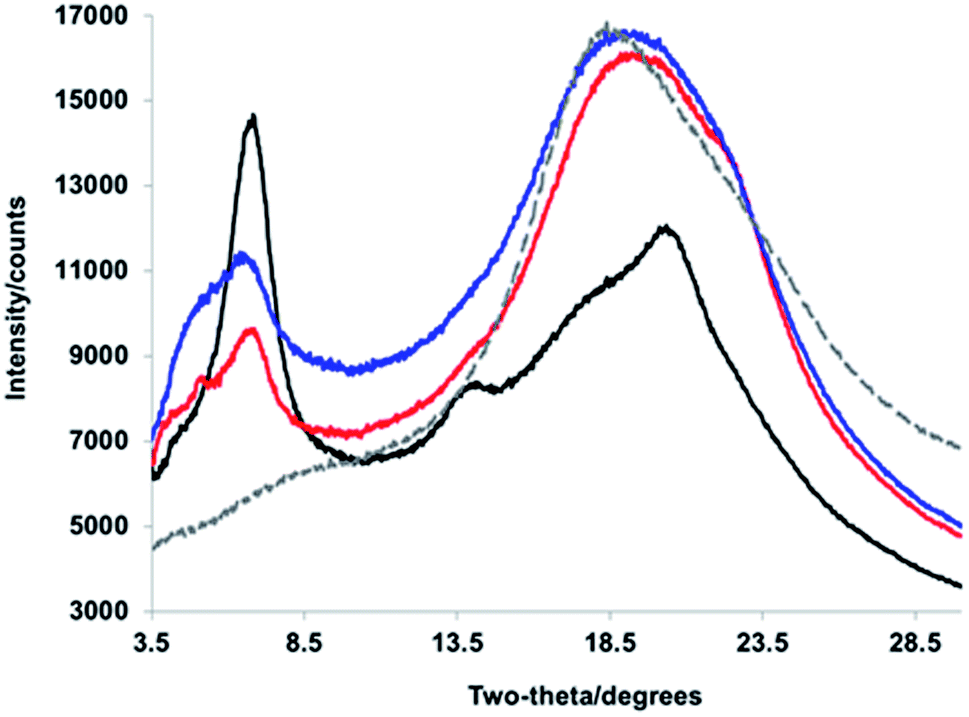 | ||
| Fig. 4 WAXS diffractograms corresponding to (black) PPV–(PNB–PPV)5, (blue) PPV–(PNB–PPV)3, (red) PPV–PNB–PPV, and (gray, dashed) PNB interior blocks. | ||
As the block copolymer lengthens (e.g., from PPV–PNB–PPV to PPV–(PNB–PPV)3 and PPV–(PNB–PPV)5), one significant change lies in the relative intensities of signals related to the alkyl–alkyl conformations and π–π stacking. Specifically, as more designed β-sheet-like turns were installed, the intensity of the inter-backbone alkyl–alkyl signal increased relative to that of the π–π stacking (Fig. 4), likely owing to the side-chains demonstrating a prominent role in self-assembly of the block copolymer. In addition, the broadened nature of the alkyl–alkyl side-chain-related signals appeared as a more prominent Bragg reflection indicative of a heightened degree of side-chain interdigitation within the final folded structures. This suggests that the folding is less reliant on π–π stacking of the chromophores, consistent with the formation of either side-on folded or face-to-face sheets with a larger distance between the PPV chromophores.
Effect of self-assembled β-turns and chromophore count on single-chain folding order
To investigate the role of the β-turn in these polymers, the electronic spectra of the PPV homopolymer (HP) and the series of PPV–(PNB–PNB–PPV)n (1T–5T) were measured in bulk solutions. The absorption of the PPV homopolymer exhibited little structure and peaked at a wavelength of 465 nm (Fig. 5). The folded PPV–(PNB–PNB–PPV)n polymers demonstrated similar behaviour. A slight shift to lower energies was evidenced by a shoulder at 532 nm in the three highest molecular weight samples (3T–5T), likely resulting from some aggregation. In contrast to the absorption, the emission spectra were virtually identical, exhibiting an emission wavelength of 524 nm. They also demonstrated a clear vibronic progression with spacing of ∼280000 cm−1. The ratio of the first two peaks (I0–0/I0–1) produced a value of 1.6. The aggregation observed in the folded 3T–5T polymers did not play a significant role in the emission given that all of the spectra exhibited identical λmax and vibronic progressions. The parent PPV exhibited a slightly broader emission spectrum compared to the folded materials and shows a slightly increased emission from the first vibronic peak. These changes are small and likely the result of the homopolymer having longer stretches of PPV repeat units (more than 25) compared to the heteropolymers (10–15). Based on aggregation models, it would be expected that electronic interactions between the chromophores would lead to both spectral shifts and changes in the ratio of the 0–1 peak to the 0–0 peak.50 Since neither of these effects were observed, we concluded that the active chromophores are maintained despite the presence of the flexible non-conjugated PNB blocks and that there is limited, if any, electronic coupling between the chromophore strands.
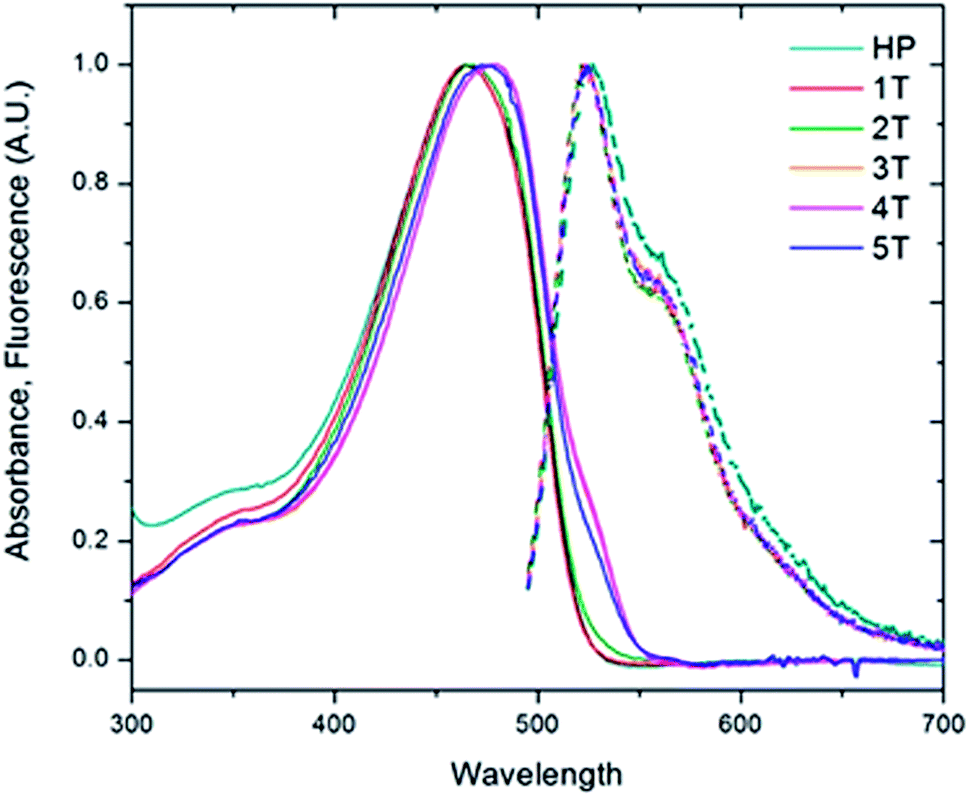 | ||
| Fig. 5 Solution absorption and fluorescence emission spectra of the homopolymer (HP, teal) and the folded PPV–(PNB–PNB–PPV)n polymer samples (1T–5T) in chloroform. | ||
To examine the folded functionality on the structure of the polymers, we performed single molecule excitation polarization spectroscopy.51 Ultra-dilute solutions of the polymer samples were embedded in a non-emissive poly(methyl methacrylate) matrix and excited by a rotating linearly polarized laser source. The emission intensity, I, of each individual molecule was then fitted as a function of the angle of rotation, θ,
| I(θ) ∝ 1 + Mcos2(θ − ϕ) |
Histograms were constructed using the M values of approximately 1000 molecules for each of the different polymer samples (Fig. 6). The modulation depths were binned by rounding to the nearest 0.1 for the M value. This histogram served as a visualization to ascertain both the average folding order and approximate how many different conformations a material adopted, and how prevalent the conformations were based on the broadness or narrowness of the histograms, or if any skews were present in the typically Gaussian curves. The PPV homopolymer (HP) displayed an average M value of 0.67, which is comparable to other conjugated polymer systems, such as high molecular weight MEH-PPV51,52 or regioregular poly(3-hexylthiophene).53 Even with the relatively high average in the modulation depth value, the distribution for the homopolymer was relatively broad. Moreover, the peak in the distribution was actually at the lower value of 0.5–0.6. The homopolymer histogram appeared somewhat broader than the folded polymer histograms and is shifted slightly to lower M values, indicating a wider variety of possible conformations, particularly less ordered ones. The introduction of the interior PNB block led to a slight increase in the modulation depth distribution. All of the folded polymers exhibited slightly higher mean M values of ∼0.7, with the exception of the largest sample, 5T, which exhibited a mean M value of 0.68. As the number of β-turns increased in the 1T–4T materials, the distribution started to narrow and shift slightly to higher M values. While there is a small shift in the mean, it is more relevant to look at the shape of the overall distribution. As the number of β-turns increases up to four, the distribution skews to higher M values and narrows. In the polymer sample comprising the most folds, 5T, however, the distribution appeared to broaden again and resemble the homopolymer distribution. This resulted in a shift in the mean M value down to 0.68. The slight lowering observed in the 5T polymer suggested that the heterogeneities inherent in both the chromophores and the internal PNB functionalities started to disrupt the clean folding of the polymer at higher molecular weights. For the 1T–4T polymers, the tighter M values skewed to the more ordered end of the distribution, implying that the polymers displayed a less heterogeneous distribution of conformations that is more anisotropic in nature. The distributions of the modulation depths were reproducible, indicating that the folding is extremely robust.
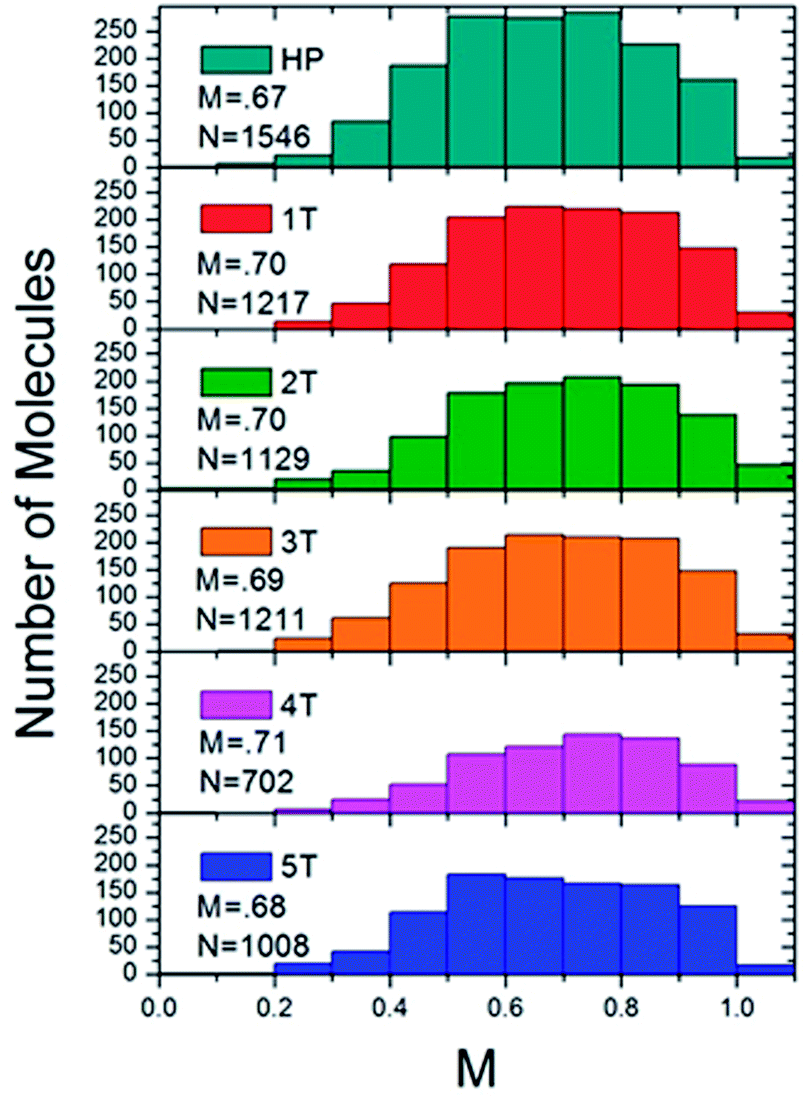 | ||
| Fig. 6 Histograms of the ensemble of fluorescence intensity modulation depth, M, of the homopolymer (top) and the folded PPV–(PNB–PNB–PPV)n polymer samples. | ||
Effect of folding on electronic properties
To determine the effect of the folded phenyl/perfluorophenyl poly(norbornene) (PNB) functionality on the polymer photophysical behaviour, we examined the spectra of single polymer molecules. These spectra were then compared both to each other, and the bulk solution spectra. In solution, when the chains are assumed to adopt unfolded, non-interacting conformations, the fluorescence spectra of all of the polymers followed the same vibronic progression with a 0–0 transition maximum at 524 nm (Fig. 5). The bulk spectra from solution can be compared to ensemble spectra of the single molecules taken in the solid state (Fig. 7). Each of the folded materials demonstrated a 0–0 transition energy fairly close to the bulk solution maximum with a nearly identical lineshape. The 4T polymer displayed a slight deviation from this pattern, having shifted slightly to the red compared to both the solution and the other materials. The largest variation is observed for the homopolymer, which exhibited an ensemble spectrum very different from both the other materials and the bulk solution. The homopolymer single molecule ensemble spectrum is red shifted by approximately 20 nm. In addition, it has a broader fluorescence envelope with less distinct vibronic features. This suggests that the homopolymer single molecules have a broader distribution of contributing chromophores compared to the folded PPV–(PNB–PNB–PPV)n materials.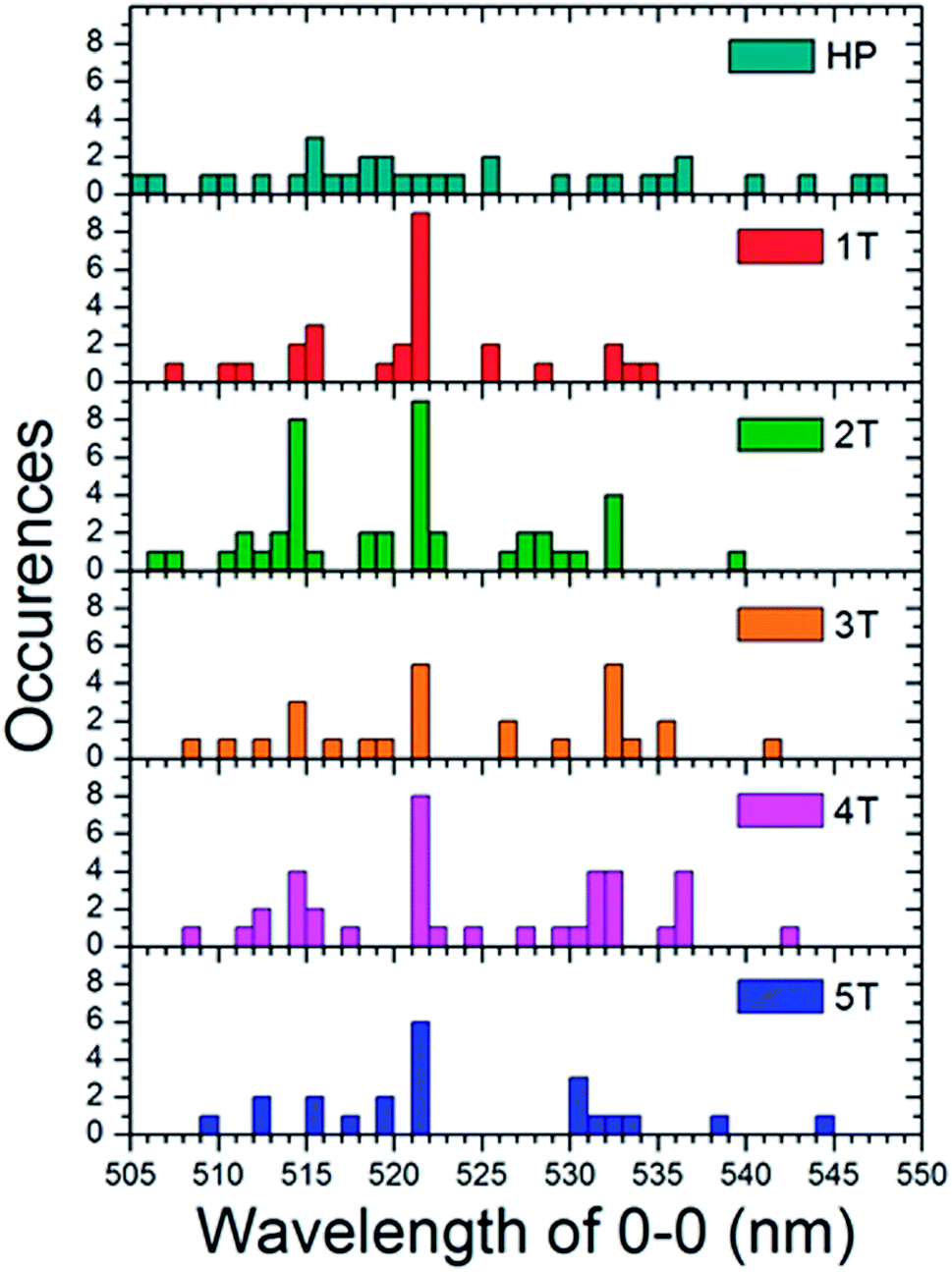 | ||
| Fig. 7 Histograms of the 0–0 vibronic transition in the single molecule spectra for both the homopolymer (HP) and each of the folded PPV–(PNB–PNB–PPV)n polymer samples. | ||
To comprehend the underlying distribution of spectra, we engineered histograms (Fig. 7) of the peak 0–0 emission energies for each individual single molecule spectrum. These were taken at the center wavelength of the most prominent higher energy transition, which was presumed to be the 0–0 peak for each sample. This data indicates that each folded material is dominated by single molecules with a peak at 521 nm which is similar to the bulk maximum at 524 nm. The folded samples also show sub-ensembles of molecules that emit ∼514 nm and ∼532 nm. This abundance of dominant 521 nm emitting molecules suggests that in the majority of the polymers there is little to no electronic interactions and that the chromophores behave much as they do in the solution state. The slight difference in the wavelength could be due to the different instruments used for the two experiments or subtle changes in environment in going from chloroform to a PMMA matrix.
Given that the interior PNB functionality should hold the chromophores in a folded ordered conformation sustained by phenyl/perfluorophenyl interactions, the lack of strong changes in the emission spectra suggests that they position the PPV chains at distances far enough away from one another that they are not electronically coupled. In contrast, the homopolymer is distinctly different. The individual molecules display a wide variety of fluorescence maxima ranging from 505 nm to nearly 550 nm with no emerging dominant species. This is representative of a wide array of conformations and conjugation lengths of the chromophores. This distribution of spectral positions is consistent with the broadening and shoulders exhibited in the generated ensemble spectrum of the homopolymer as well as the solution spectrum (Fig. 8).
 | ||
| Fig. 8 Ensemble spectra generated by summing all of the individual single molecule spectra acquired for the homopolymer (HP) and the folded PPV–(PNB–PNB–PPV)n polymer samples. | ||
To further examine the predominate form of the polymer that emits at 521 nm, we created sub-ensemble spectra by combining all the spectra that exhibited a 521 nm peak emission (Fig. 9). Between the five folded samples, all sub-ensemble spectra are effectively identical to one another, demonstrating identical breadth and vibronic progressions. Additionally, the vibronic progression is the same as that observed in the bulk solution, where the chains are assumed to be unfolded, yielding the same I0–0/I0–1 ratio of ∼1.6 for all copolymer samples. Given that even weak electronic coupling should result in changes in the vibronic progressions upon aggregation,50 it appears that the polymer folds into structures in which the chromophores are sufficiently far apart to avoid electronic interactions. This is true even as the number of chromophores increases up to 6.
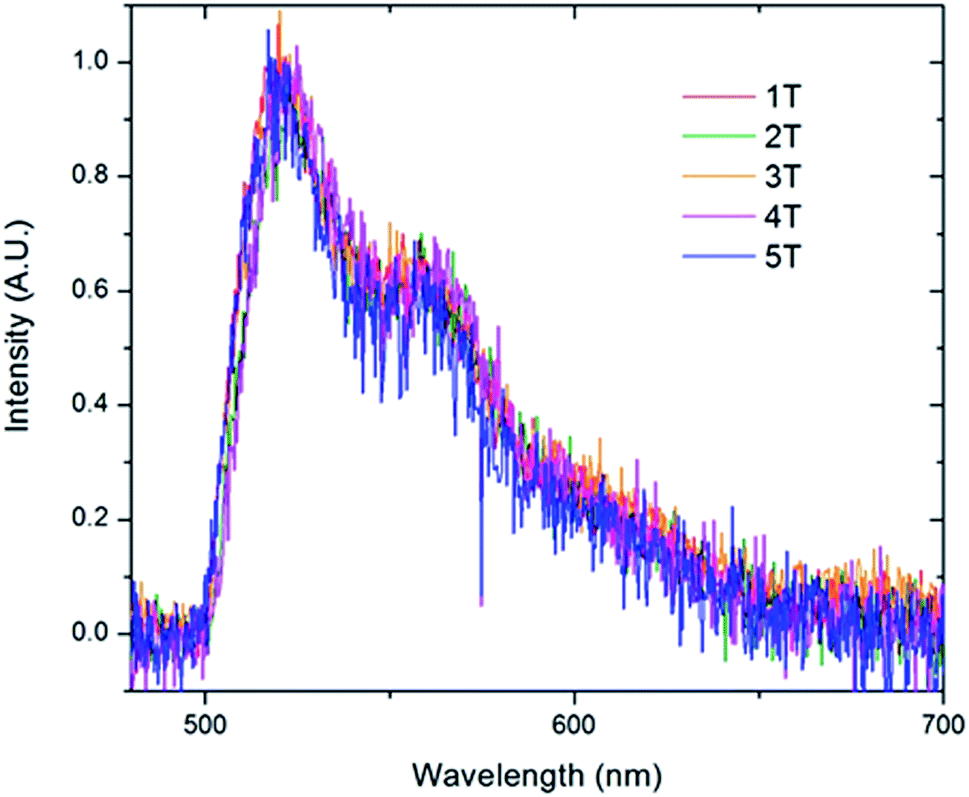 | ||
| Fig. 9 Sub-ensemble spectra generated by summing all of the individual single molecule spectra that exhibited the 0–0 transition at 521 nm acquired for the folded PPV–(PNB–PNB–PPV)n polymer samples. | ||
Conclusions
We have synthesized a series of polychromophoric β-sheet foldamers using an iterative ROMP strategy. Directional phenyl/perfluorophenyl interactions were used to engineer synthetic β-sheet folding of PPVs comprising up to five engineered β-hairpin turns through chain collapse in the presence of a good solvent. We investigated the folding behaviour of the polymers in both solution and in the solid state. In solution, the folded polymer samples behave similarly to the homopolymer; the absorption and emission maxima were around 465 nm and 524 nm, respectively, signifying that the folded chromophores do not participate in any added electronic communication aided by the folded flexible PNB segments. In the solid-state, the polymer series (1T–5T) engages in more side-chain interdigitation as the number of folds increases, suggesting the polymer folding motif is less dependent on the π–π stacking of the chromophores, consistent with the formation of either side-on folded or face-to-face sheets with a larger distance between the β-strands.We probed the folded polymer series at the single chain level, wherein the PPV–(PNB–PNB–PPV)n series is observed to behave more anisotropically than the PPV homopolymer, and demonstrates nearly identical spectra in solution and at the single molecule level. The polymers were found to fold consistently in the same manner, regardless of the molecular weight or the variable PNB lengths within the β-turns, demonstrating the robust nature of the phenyl/perfluorophenyl interaction to achieve synthetic β-sheets.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors acknowledge financial support from the National Science Foundation (MW, NSF) under award number CHE-1506890. D. A. V. B. acknowledges support from the National Science Foundation (CHE-1310222). We also acknowledge the National Institutes of Health (NIH) for the purchase of the Advance III-600 CPTCI-cryoprobe head (S10 grant, OD016343).Notes and references
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219–3232 CrossRef CAS.
- D. Haldar, H. Jiang, J. M. Leger and I. Huc, Angew. Chem., Int. Ed., 2006, 45, 5483–5486 CrossRef CAS.
- L. Guo, Y. Chi, A. M. Almeida, I. A. Guzei, B. K. Parker and S. H. Gellman, J. Am. Chem. Soc., 2009, 131, 16018–16020 CrossRef CAS.
- S. De, D. Koley and S. Ramakrishnan, Macromolecules, 2010, 43, 3183–3192 CrossRef CAS.
- K. Goto and J. S. Moore, Org. Lett., 2005, 7, 1683–1686 CrossRef CAS PubMed.
- E. Elacqua, D. S. Lye and M. Weck, Acc. Chem. Res., 2014, 47, 2405–2416 CrossRef CAS PubMed.
- J. F. Reuther, D. A. Siriwardane, O. V. Kulikov, B. L. Batchelor, R. Campos and B. M. Novak, Macromolecules, 2015, 48, 3207–3216 CrossRef CAS.
- Y.-X. Lu, Z.-M. Shi, Z.-T. Li and Z. Guan, Chem. Commun., 2010, 46, 9019–9021 RSC.
- E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102–6211 CrossRef CAS PubMed.
- E. Yashima, K. Maeda and Y. Furusho, Acc. Chem. Res., 2008, 41, 1166–1180 CrossRef CAS PubMed.
- E. R. Gillies, F. Deiss, C. Staedel, J.-M. Schmitter and I. Huc, Angew. Chem., Int. Ed., 2007, 46, 4081–4084 CrossRef CAS.
- E. Elacqua, K. B. Manning, D. S. Lye, S. K. Pomarico, F. Morgia and M. Weck, J. Am. Chem. Soc., 2017, 139, 12240–12250 CrossRef CAS PubMed.
- L. Sebaoun, V. Maurizot, T. Granier, B. Kauffmann and I. Huc, J. Am. Chem. Soc., 2014, 136, 2168–2174 CrossRef CAS PubMed.
- A. M. Hanlon, C. K. Lyon and E. B. Berda, Macromolecules, 2016, 49, 2–14 CrossRef CAS.
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2016, 37, 29–46 CrossRef CAS.
- E. B. Berda, E. J. Foster and E. W. Meijer, Macromolecules, 2010, 43, 1430–1437 CrossRef CAS.
- T. Mes, R. van der Weegen, A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2011, 50, 5085–5089 CrossRef CAS PubMed.
- P. J. M. Stals, M. A. J. Gillissen, T. F. E. Paffen, T. F. A. Greef, P. Lindner, E. W. Meijer, A. R. A. Palmans and I. K. Voets, Macromolecules, 2014, 47, 2947–2954 CrossRef CAS.
- T. Terashima, T. Mes, T. F. A. De Greef, M. A. J. Gillissen, P. Besenius, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 4742–4745 CrossRef CAS PubMed.
- J. Willenbacher, O. Altintas, P. W. Roesky and C. Barner-Kowollik, Macromol. Rapid Commun., 2014, 35, 45–51 CrossRef CAS PubMed.
- J. Willenbacher, B. V. Schmidt, D. Schulze-Suenninghausen, O. Altintas, B. Luy, G. Delaittre and C. Barner-Kowollik, Chem. Commun., 2014, 50, 7056–7059 RSC.
- O. Altintas, P. Krolla-Sidenstein, H. Gliemann and C. Barner-Kowollik, Macromolecules, 2014, 47, 5877–5888 CrossRef CAS.
- O. Altintas, E. Lejeune, P. Gerstel and C. Barner-Kowollik, Polym. Chem., 2012, 3, 640–651 RSC.
- N. Hosono, M. A. J. Gillissen, Y. C. Li, S. S. Sheiko, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2013, 135, 501–510 CrossRef CAS PubMed.
- N. Hosono, P. J. M. Stals, A. R. A. Palmans and E. W. Meijer, Chem.–Asian J., 2014, 9, 1099–1107 CrossRef CAS PubMed.
- A. Prasher, C. M. Loynd, B. T. Tuten, P. G. Frank, D. M. Chao and E. B. Berda, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 209–217 CrossRef CAS.
- D. M. Chao, X. T. Jia, B. Tuten, C. Wang and E. B. Berda, Chem. Commun., 2013, 49, 4178–4180 RSC.
- B. T. Tuten, D. M. Chao, C. K. Lyon and E. B. Berda, Polym. Chem., 2012, 3, 3068–3071 RSC.
- P. G. Frank, B. T. Tuten, A. Prasher, D. Chao and E. B. Berda, Macromol. Rapid Commun., 2014, 35, 249–253 CrossRef CAS.
- C. K. Lyon, E. O. Hill and E. B. Berda, Macromol. Chem. Phys., 2016, 213, 501–508 CrossRef.
- J. P. Cole, J. J. Lessard, C. K. Lyon, B. T. Tuten and E. B. Berda, Polym. Chem., 2015, 6, 5555–5559 RSC.
- O. Altintas, J. Willenbacher, K. N. R. Wuest, K. K. Oehlenschlaeger, P. Krolla-Sidenstein, H. Gliemann and C. Barner-Kowollik, Macromolecules, 2013, 46, 8092–8101 CrossRef CAS.
- L. Sebaoun, B. Kauffmann, T. Delclos, V. Maurizot and I. Huc, Org. Lett., 2014, 16, 2326–2329 CrossRef CAS PubMed.
- B. Shao, X. Zhu, K. N. Plunkett and D. A. Vanden Bout, Polym. Chem., 2017, 8, 1188–1195 RSC.
- J. Lu, N. ten Brummelhuis and M. Weck, Chem. Commun., 2014, 50, 6225–6227 RSC.
- R. Xu, W. B. Schweizer and H. Fraunenrath, J. Am. Chem. Soc., 2008, 130, 11437–11445 CrossRef CAS PubMed.
- M. Weck, A. R. Dunn, K. Matsumoto, G. W. Coates, E. B. Lobkovsky and R. H. Grubbs, Angew. Chem., Int. Ed., 1999, 38, 2741–2745 CrossRef CAS PubMed.
- G. W. Coates, A. R. Dunn, L. M. Henling, J. W. Ziller, E. B. Lobkovsky and R. H. Grubbs, J. Am. Chem. Soc., 1998, 120, 3641–3649 CrossRef CAS.
- G. W. Coates, A. R. Dunn, L. M. Henling, D. A. Dougherty and R. H. Grubbs, Angew. Chem., Int. Ed., 1997, 37, 248–251 CrossRef.
- S. H. Chen, A. C. Su, S. R. Han, S. A. Chen and Y. Z. Lee, Macromolecules, 2004, 37, 181–186 CrossRef CAS.
- F. Menk, M. Mondeshki, D. Dudenko, S. Y. Shin, D. Schollmeyer, O. Ceyhun, T. L. Choi and R. Zentel, Macromolecules, 2015, 48, 7435–7445 CrossRef CAS.
- E. Elacqua and M. Weck, Chem.–Eur. J., 2015, 21, 7151–7158 CrossRef CAS PubMed.
- D. Maker and U. H. Bunz, Macromol. Rapid Commun., 2014, 35, 2096–2100 CrossRef PubMed.
- C. Y. Yu, M. Helliwell, J. Raftery and M. L. Turner, Chem.–Eur. J., 2011, 17, 6991–6997 CrossRef CAS PubMed.
- F. Menk, S. Shin, K. O. Kim, M. Scherer, D. Gehrig, F. Laquai, T. L. Choi and R. Zentel, Macromolecules, 2016, 49, 2085–2095 CrossRef CAS.
- The total polymerization time of blocks A, B, and C (combined with NMR spectroscopic monitoring) is on the order of 22–24 hours.
- While all PPV block lenghts were approximately 10 units (i.e., 20 phenylenevinylene units) and PNB block lenghts were targeted to be between 20–25 units, the exact PNB lengths did vary as the polymerization proceeded. We attribute this to the accuracy in the amount of polymer drawn out of the pot and quenched upon each subsequent ‘turn’.
- E. Elacqua, A. Croom, K. B. Manning, D. Lye, S. K. Pomarico, L. Young and M. Weck, Angew. Chem., Int. Ed., 2016, 55, 15873–15878 CrossRef CAS PubMed.
- F. C. Spano and S. Silva, Annu. Rev. Phys. Chem., 2014, 65, 447–500 CrossRef PubMed.
- T. Adachi, J. Brazard, P. Chokshi, J. C. Bolinger, V. Ganesan and P. F. Barbara, J. Phys. Chem. C, 2010, 114, 20896–20902 CrossRef CAS.
- J. C. Bolinger, M. C. Traub, J. Brazard, P. F. Barbara and D. A. Vanden Bout, Acc. Chem. Res., 2012, 45, 1992–2001 CrossRef CAS PubMed.
- T. Adachi, J. Brazard, R. J. Ono, B. Hanson, M. C. Traub, Z.-Q. Wu, Z. B. Li, J. C. Bolinger, V. Ganesan, C. W. Bielawski, D. A. Vanden Bout and P. F. Barbara, J. Phys. Chem. Lett., 2011, 2, 1400–1404 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05111a |
| This journal is © The Royal Society of Chemistry 2019 |