
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A retro Baeyer–Villiger reaction: electrochemical reduction of [60]fullerene-fused lactones to [60]fullerene-fused ketones†
Chuang
Niu
 a,
Dian-Bing
Zhou
a,
Yong
Yang
a,
Zheng-Chun
Yin
a and
Guan-Wu
Wang
*ab
a,
Dian-Bing
Zhou
a,
Yong
Yang
a,
Zheng-Chun
Yin
a and
Guan-Wu
Wang
*ab
aHefei National Laboratory for Physical Sciences at Microscale, CAS Key Laboratory of Soft Matter Chemistry, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), Center for Excellence in Molecular Synthesis of CAS, Department of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: gwang@ustc.edu.cn
bState Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou, Gansu 730000, P. R. China
First published on 16th January 2019
Abstract
A highly efficient electrochemical reduction of [60]fullerene-fused lactones to [60]fullerene-fused ketones, a formal process of retro Baeyer–Villiger reaction, has been achieved for the first time. The electrochemically generated dianionic [60]fullerene-fused lactones can be transformed into [60]fullerene-fused ketones in the presence of acetic acid in 85–91% yields. Control experiments have been performed to elucidate the reaction mechanism. The products have been characterized with spectroscopic data and single-crystal X-ray analysis. Moreover, the electrochemical properties have also been investigated.
Introduction
The Baeyer–Villiger oxidation is one of the most important transformations in organic synthesis, because valuable esters and lactones can be obtained directly from the corresponding ketones (Scheme 1a).1 However, to the best of our knowledge, the retro Baeyer–Villiger reaction, that is, the direct reduction of esters/lactones to ketones accompanied by the elimination of only one oxygen atom via either a deoxygenative (–O) or dehydrative (–H2O) pathway, has never been reported and remains a challenging task.2,3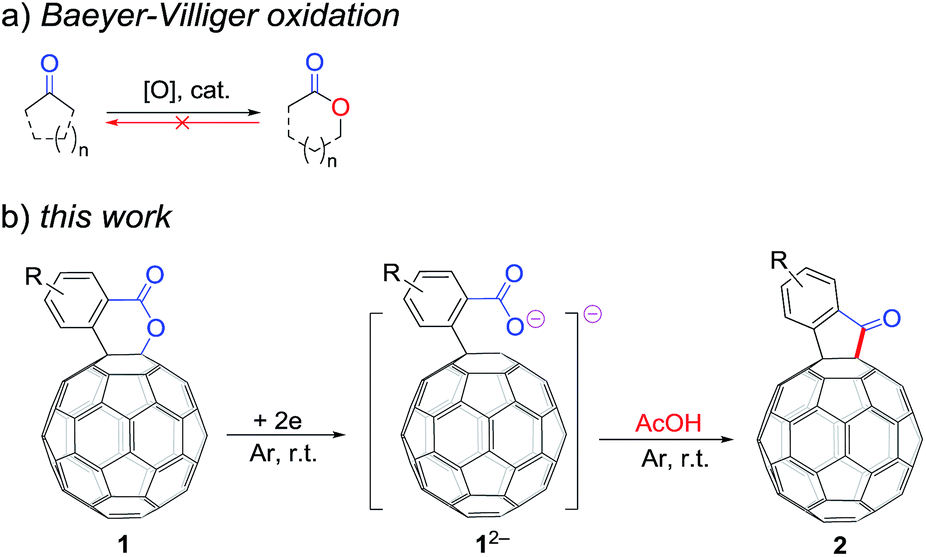 | ||
| Scheme 1 (a) Baeyer–Villiger oxidation. (b) Retro Baeyer–Villiger reaction of C60-fused lactones. | ||
Over the past few decades, fullerene derivatives have attracted much attention due to their potential for application in the fields of biomedical and materials science.4 Therefore, a great diversity of synthetic protocols for functionalizing fullerenes have been developed by chemists.5,6 Among the numerous methods, electrosynthesis has been demonstrated as a novel and efficient strategy due to its mild reaction conditions, good regioselectivity, and relatively high yields.6
It has been shown that electrochemically generated fullerene anions, especially singly bonded fullerene dianions, can be readily prepared and used as building blocks in the regioselective synthesis of fullerene derivatives with novel addition patterns.6 In an attempt to protonate dianionic [60]fullerene (C60)-fused lactones with acetic acid (AcOH), C60-fused ketones 2,7 rather than the expected tetrahydrofullerenes,6i can be surprisingly obtained in high yields (Scheme 1b). This is the first time the direct reduction of lactones to ketones, which is a formal retro reaction of Baeyer–Villiger oxidation, has been realized. Herein, we report this unprecedented retro Baeyer–Villiger reaction of C60-fused lactones by the electrochemical approach.
Results and discussion
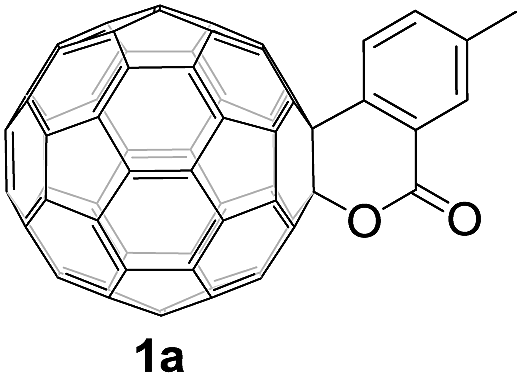
The employed C60-fused lactone 1a was synthesized according to our previous procedure.8 Cyclic voltammetry (CV) of 1a in o-dichlorobenzene (ODCB) containing 0.1 M tetra-n-butylammonium perchlorate (TBAP) showed that the first redox was an irreversible one-electron transfer process with an Epc of −0.60 V (A) versus a saturated calomel electrode (SCE), and the second redox was chemically quasi-reversible on the CV timescale with Epc at −1.14 V (B) (Fig. 1a), indicating that the compound underwent a chemical reaction process after receiving one electron. The heterolytic cleavage of the C60–O bond occurred to provide the ring-opened radical anion 1a˙−, in which the negative charge and unpaired electron were distributed on the fullerene skeleton and/or the carbonyl group, respectively (vide infra), once 1a acquired one electron. Upon acceptance of the second electron, a singly bonded dianionic species 1a2−, in which one negative charge was located at the carboxylate group and another one was distributed on the fullerene cage, was formed.9 These ring-opened structures were further confirmed by the visible/near-infrared (Vis/NIR) study of 1a˙− and 1a2−, which were obtained by controlled potential electrolysis (CPE) at −0.90 V and −1.34 V, respectively. The Vis/NIR spectra of 1a˙− and 1a2− (Fig. 1b) showed strong absorption bands at λ = 986 and 652 nm, which were in excellent agreement with those of the singly bonded anions of a C60-fused oxazoline (λ = 963, and 645 nm),9c a C60-fused sultone (λ = 983 and 648 nm),9d and a C60-fused indoline (λ = 966 and 648 nm).9e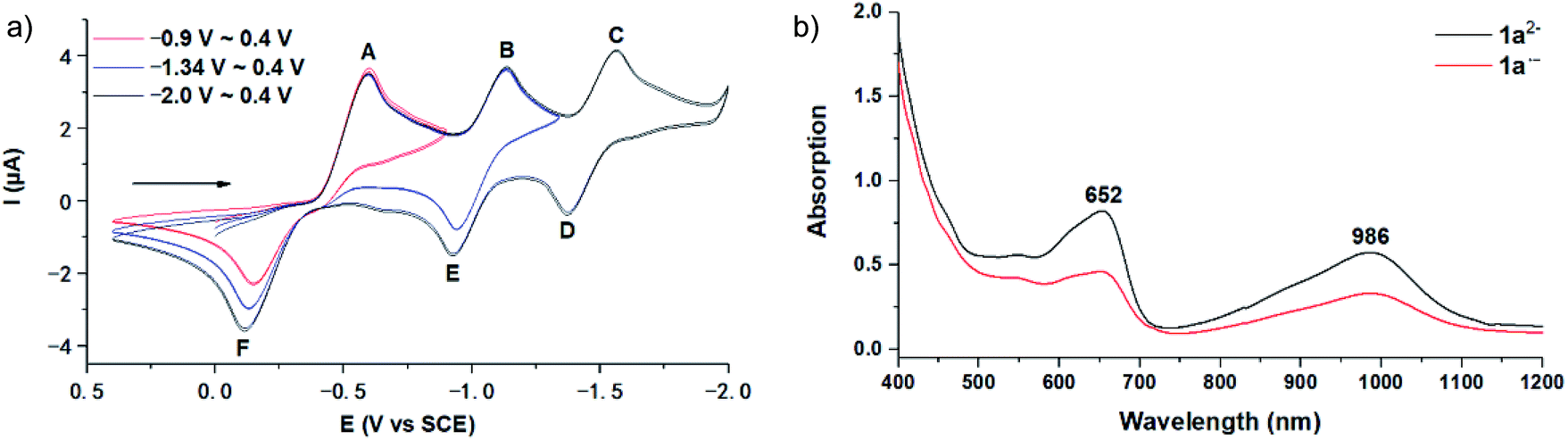 | ||
| Fig. 1 (a) Cyclic voltammograms of compound 1a (1.0 mM) shown within different potential windows. The CVs recorded in ODCB containing 0.1 M TBAP starting from 0.0 V toward the negative potential with a scan rate of 50 mV s−1. The arrows indicate the scan direction for the cyclic voltammetric measurements. (b) Vis/NIR spectra of 1a˙− (red) and 1a2− (black) in ODCB (0.25 mM). | ||
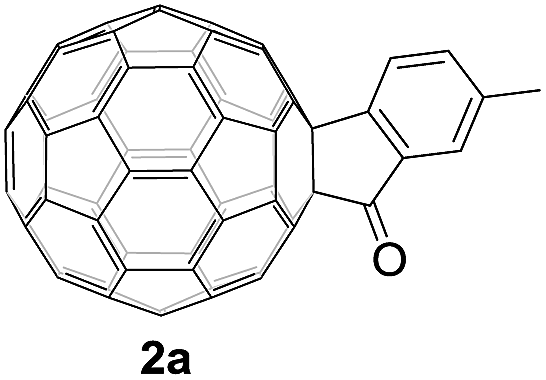
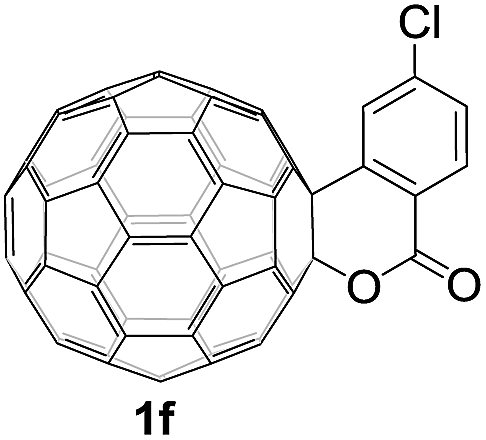
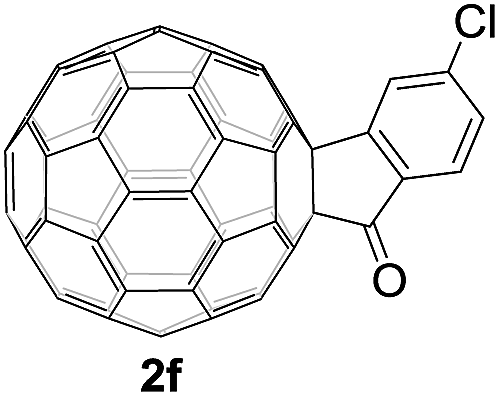
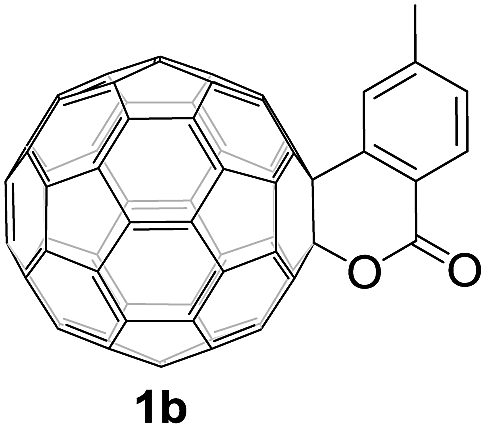
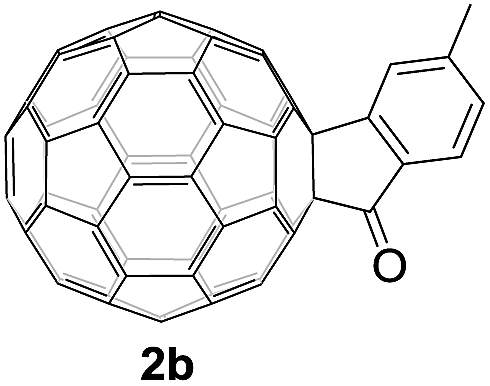
Controlled potential electrolysis of 1a (0.015 mmol) in 15.0 mL of anhydrous ODCB solution containing 0.1 M TBAP was carried out at −1.34 V to obtain 1a2− under an argon atmosphere at ambient temperature (∼25 °C). With an aim to protonate 1a2−, AcOH (10 equiv.) was added, and the reaction mixture was stirred at room temperature for 30 min. To our surprise, an intriguing product, C60-fused ketone 2a, was obtained in 91% yield. Importantly, this unexpected dehydrative retro Baeyer–Villiger reaction could be extended to other C60-fused lactones, and the results are summarized in Table 1. C60-fused lactones with electron-donating groups including the methyl and methoxy groups as well as electron-withdrawing groups such as the chloro and carbonyl groups at different positions of the aromatic ring afforded 2a–g in excellent yields of 86–91%. Detailed comparisons of these results showed that the electronic properties (entries 1–4 vs. entries 5–7) and locations (entry 1 vs. entry 2, entry 3 vs. entry 4, and entry 5 vs. entry 6) of the substituents on the phenyl ring had little effect on the product yields, indicating that the ring-closure of 1a–g2− to afford 2a–g was a highly efficient process. In addition, when the di-substituted substrate with two methoxy groups was employed, the corresponding product 2h could also be obtained smoothly in 85% yield. Finally, C60-fused lactone 1i with no substituent on the phenyl ring gave the simplest C60-fused ketone 2i in 90% yield.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | C60-fused lactone 1 | Potential (V) | Product 2 | Yieldb (%) | Entry | C60-fused lactone 1 | Potential (V) | Product 2 | Yieldb (%) |
| a All the reactions were performed with 0.015 mmol of 1a–i2− and 0.150 mmol of acetic acid at room temperature (∼25 °C) for 30 min under an argon atmosphere. b Isolated yield. | |||||||||
| 1 |
|
−1.34 |
|
91 | 6 |
|
−1.31 |
|
90 |
| 2 |
|
−1.38 |
|
90 | 7 |
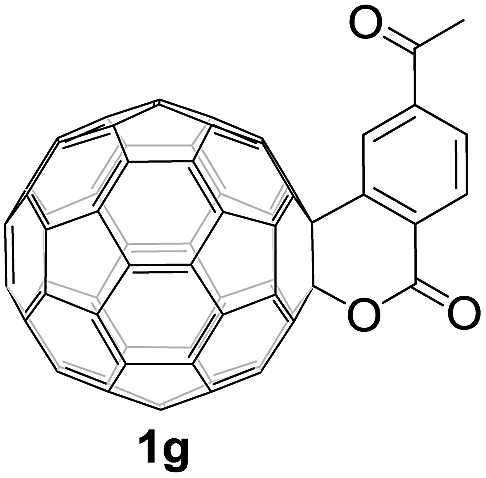
|
−1.38 |
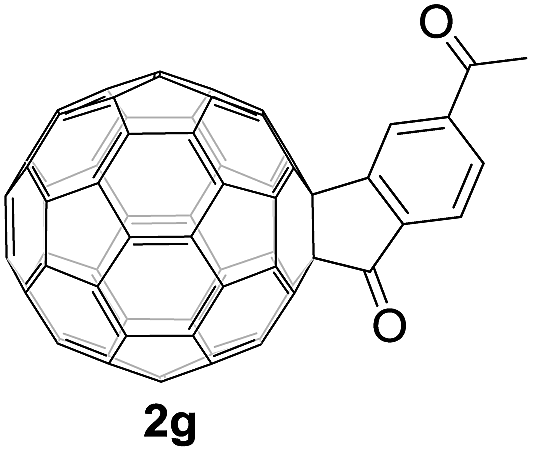
|
88 |
| 3 |
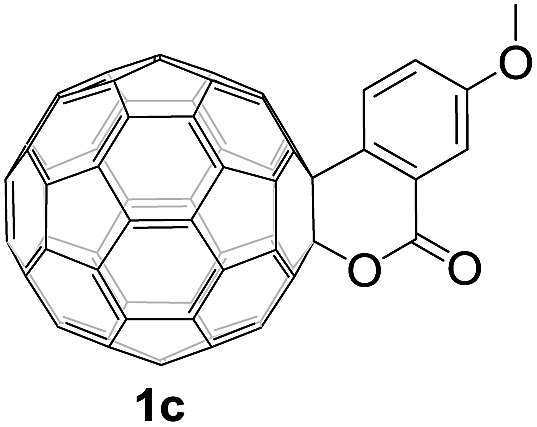
|
−1.38 |
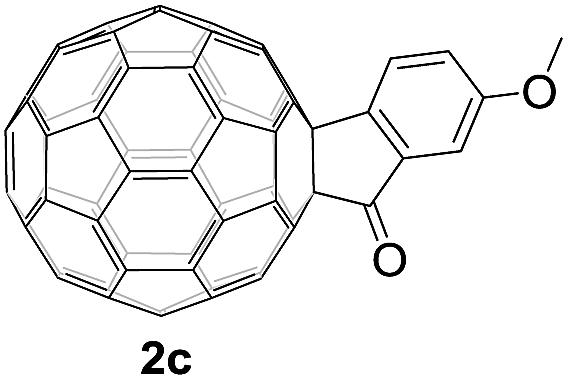
|
89 | 8 |
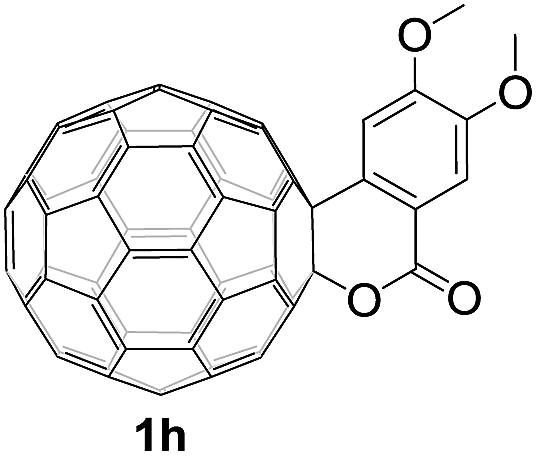
|
−1.36 |
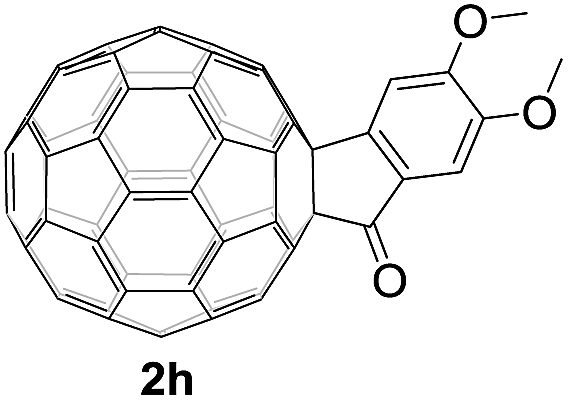
|
85 |
| 4 |
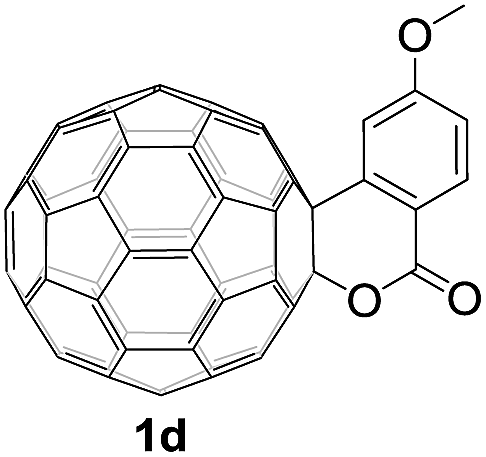
|
−1.38 |
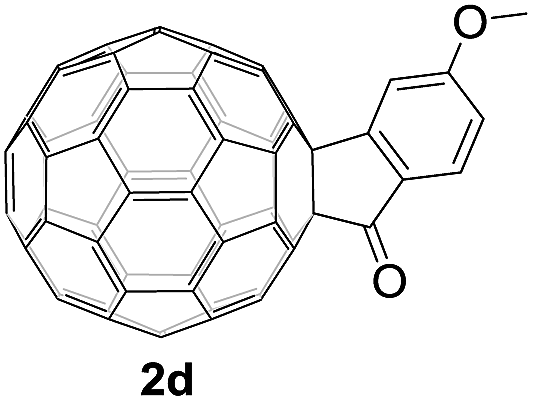
|
91 | 9 |
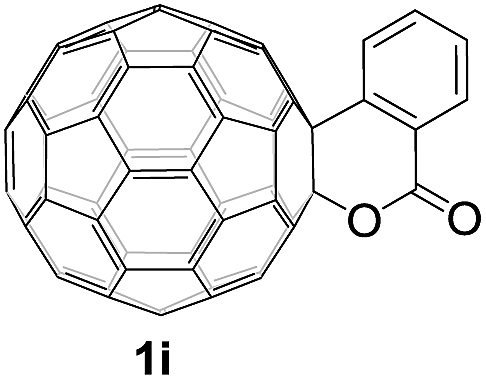
|
−1.36 |
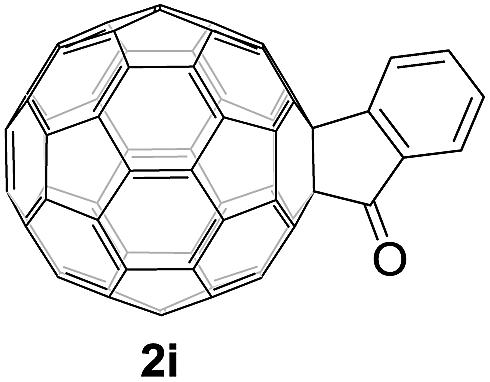
|
90 |
| 5 |
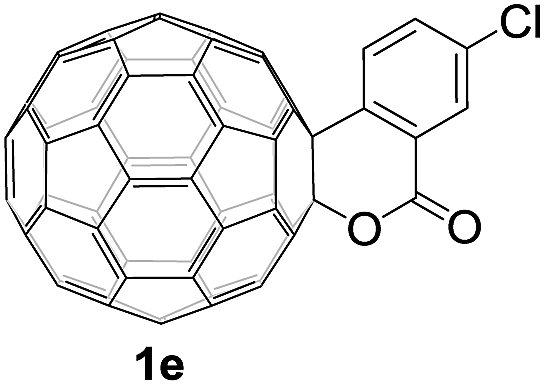
|
−1.38 |
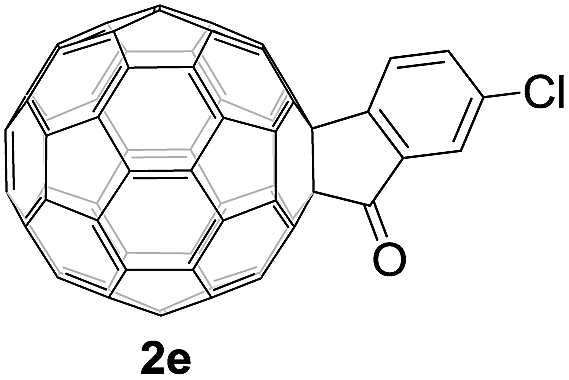
|
86 | |||||
The structures of products 2a–i were unambiguously characterized by MALDI-TOF MS, 1H NMR, 13C NMR, FT-IR, and UV-vis spectrometry. All mass spectra of these products exhibited the correct [M]+ peaks. Their 1H NMR spectra displayed the expected chemical shifts as well as the splitting patterns for all protons. The 13C NMR spectra of 2a–i exhibited no more than 30 peaks in the range of 135–159 ppm for the 58 sp2-carbons of the fullerene cage and two peaks at 70–80 ppm for the two sp3-carbons of the fullerene skeleton, consistent with the Cs symmetry of their molecular structures. Their UV-vis spectra exhibited a peak at 430–432 nm, which corresponds to the diagnostic absorption of 1,2-adducts of C60 at the [6,6]-junction. The structures of products were unambiguously confirmed by the single-crystal X-ray diffraction analysis of 2f as an example (Fig. 2).10
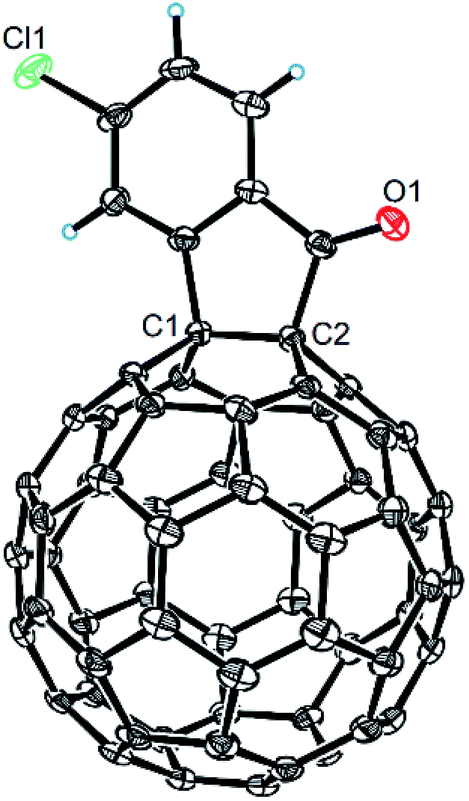 | ||
| Fig. 2 ORTEP diagram for one enantiomer of 2f with thermal ellipsoids shown at 50% probability. The toluene molecule is omitted for clarity. | ||
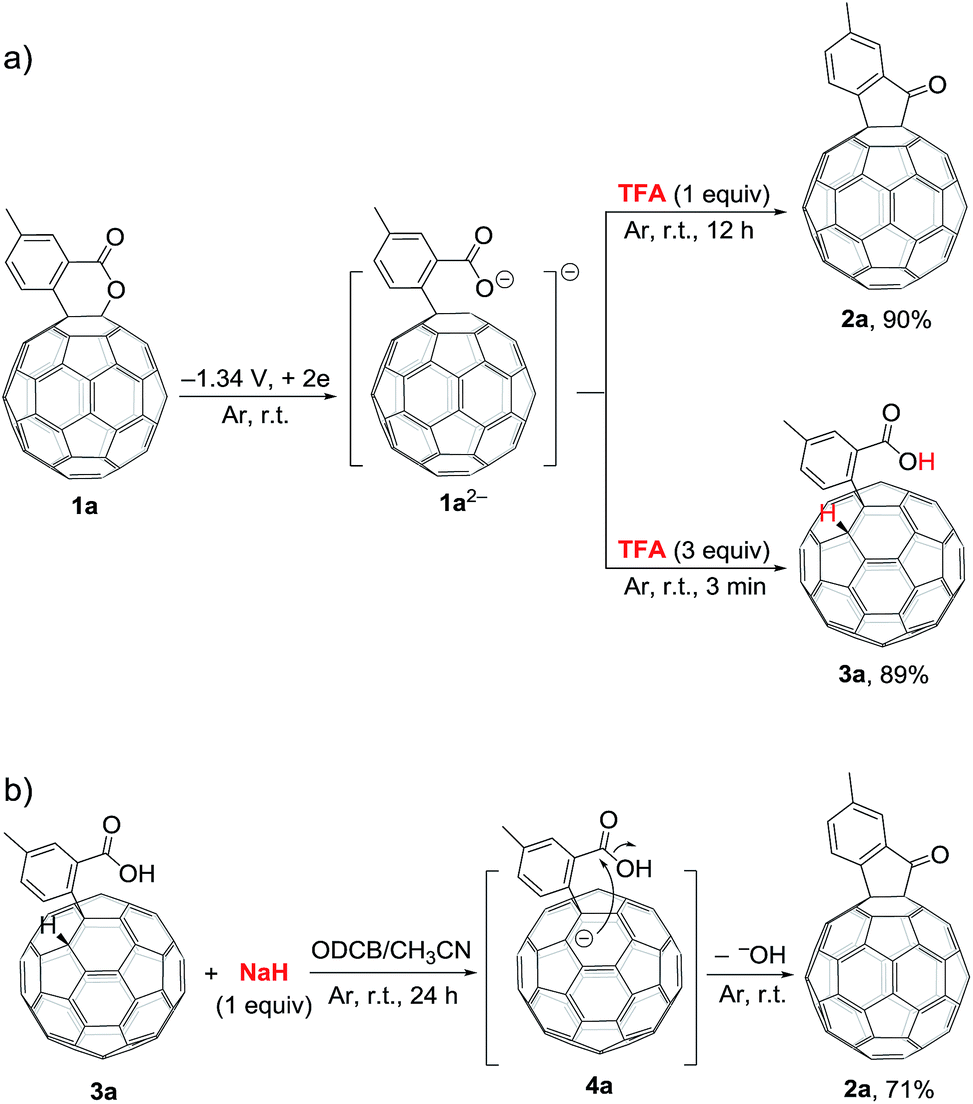
During the screening of the added acids, it was intriguingly found that different amounts of trifluoroacetic acid (TFA) afforded different products. When 1a2− was treated with 1 equiv. of TFA, 2a could also be obtained in 90% yield, but required a long reaction time of 12 h. However, when 1a2− was reacted with 3 equiv. of TFA for only 3 min, hydrofullerene 3a was obtained in 89% yield (Scheme 2a). The structure of 3a was established by its spectral data, particularly the singlet at δH = 6.89 ppm for the diagnostic fullerenyl proton in its 1H NMR spectrum.6f,i,j,9a,e,11 Additional control experiments showed that treatment of 3a with 1 equiv. of sodium hydride (NaH) in a mixture of ODCB and CH3CN (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at room temperature under an argon atmosphere provided 2a in 71% yield (Scheme 2b). The reported pKa values of TFA, t-BuC60H, PhCO2H, and AcOH in DMSO were 3.45, 5.7, 11.1, and 12.3, respectively.12 Although their corresponding pKa values in ODCB or a mixture of ODCB and CH3CN are unavailable, it is reasonable to assume that the relative pKa values of the same order are retained in these solvent systems. Therefore, it is expected that TFA would first protonate the carboxylate anion and then the fullerenyl anion. When only 1 equiv. of TFA was added, the carboxylate anion of 1a2− would be preferably protonated, and subsequent intramolecular cyclization by the attack of the fullerenyl anion to the formed carboxyl group afforded C60-fused ketone 2a. In comparison, when excess amounts (3 equiv.) of TFA were added, both the carboxylate anion and the fullerenyl anion were protonated to give hydrofullerene 3a as the most stable 1,2-adduct. On the other hand, 1 equiv. of NaH would selectively deprotonate the more acidic fullerenyl proton rather than the carboxyl group of 3a, followed by a cyclization process to provide 2a.
:1) at room temperature under an argon atmosphere provided 2a in 71% yield (Scheme 2b). The reported pKa values of TFA, t-BuC60H, PhCO2H, and AcOH in DMSO were 3.45, 5.7, 11.1, and 12.3, respectively.12 Although their corresponding pKa values in ODCB or a mixture of ODCB and CH3CN are unavailable, it is reasonable to assume that the relative pKa values of the same order are retained in these solvent systems. Therefore, it is expected that TFA would first protonate the carboxylate anion and then the fullerenyl anion. When only 1 equiv. of TFA was added, the carboxylate anion of 1a2− would be preferably protonated, and subsequent intramolecular cyclization by the attack of the fullerenyl anion to the formed carboxyl group afforded C60-fused ketone 2a. In comparison, when excess amounts (3 equiv.) of TFA were added, both the carboxylate anion and the fullerenyl anion were protonated to give hydrofullerene 3a as the most stable 1,2-adduct. On the other hand, 1 equiv. of NaH would selectively deprotonate the more acidic fullerenyl proton rather than the carboxyl group of 3a, followed by a cyclization process to provide 2a.
 | ||
| Scheme 2 Control experiments. | ||
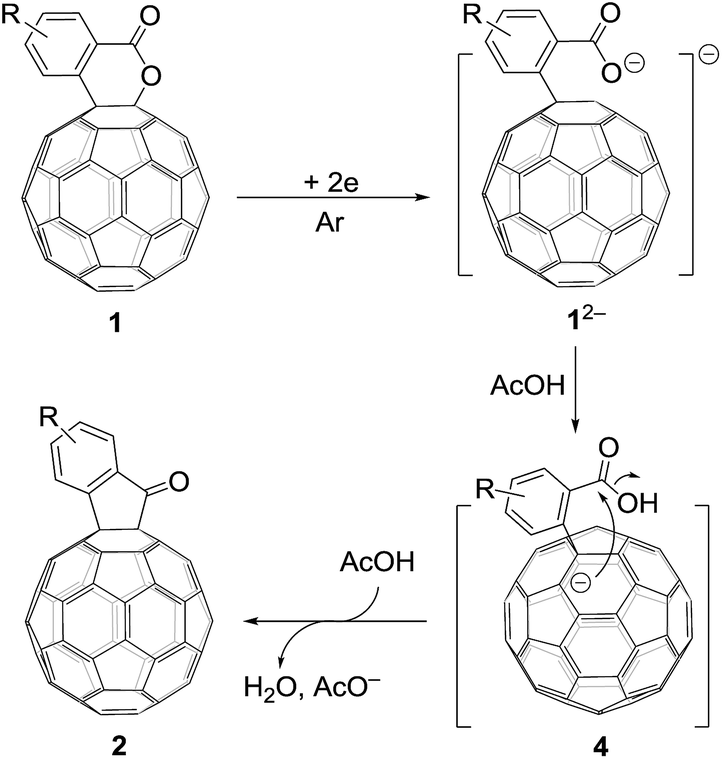
Based on the above results and previous literature,9 a plausible reaction mechanism for the formation of 2 is depicted in Scheme 3. Firstly, C60-fused lactone 1 is electrochemically reduced with a cleavage of the C–O bond to generate ring-opened dianionic 12−. Since AcOH is the weakest acid in the order of the above-mentioned acids (TFA, t-BuC60H, PhCO2H and AcOH), only the carboxylate anion of dianion 12− seems to be protonated even in the presence of excess AcOH to give monoanion 4. Finally, intermediate 4 undergoes intramolecular cyclization accompanied by the removal of the hydroxide ion, which is assisted by the neutralization with another molecule of AcOH, to provide product 2.
 | ||
| Scheme 3 Proposed reaction mechanism for the formation of C60-fused ketones from C60-fused lactones. | ||
We also explored the possibility for the retro Baeyer–Villiger reaction of C60-fused lactones by utilization of their radical monoanions with 1a as an example. The irreversible first redox process in the CV of 1a (Fig. 1a) hinted that its lactone moiety would rupture to provide the ring-opened 1a˙− after receiving one electron. The Vis/NIR spectrum of 1a˙− showed significantly lower intensities at 986 and 652 nm than that of 12− at the same concentration (Fig. 1b), suggesting that only some of 1a˙− had a ring-opened structure with the negative charge distributed on the fullerene skeleton. The synthesis of 2a by the reaction of 1a˙− with 10 equiv. of AcOH was attempted, yet 2a could be obtained in only 54% yield (Scheme 4), much lower than that (91%) from the reaction of 12−. The exact reaction pathway leading to 2a is not clear and currently under investigation. Therefore, the retro Baeyer–Villiger reaction of C60-fused lactones was much more efficiently achieved through their dianionic intermediates rather than with their radical monoanionic species.
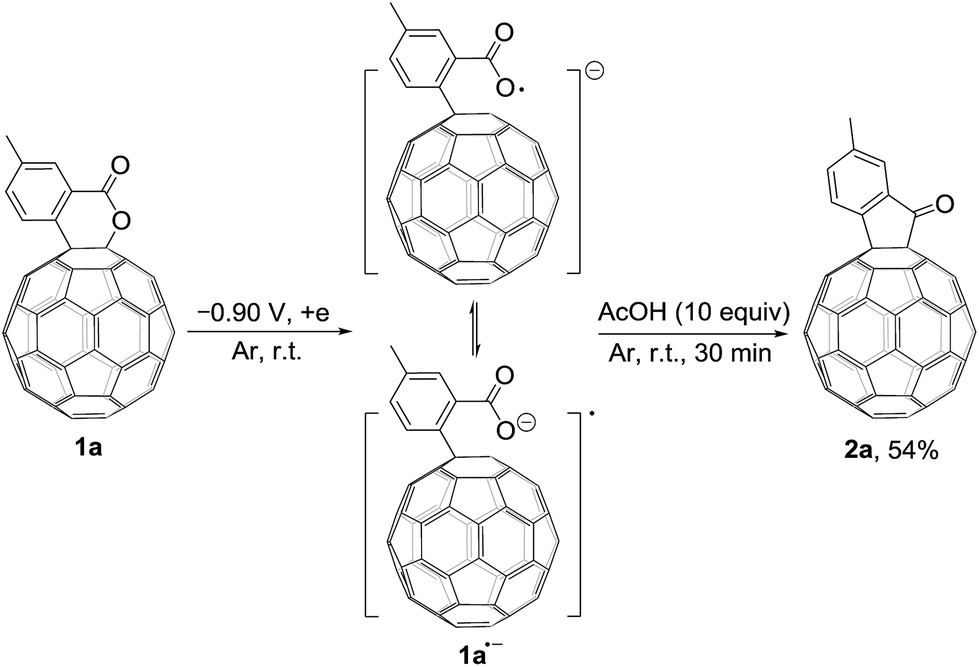 | ||
| Scheme 4 Synthesis of 2a by the reaction of 1a˙− with AcOH. | ||
The half-wave reduction potentials of C60-fused ketones 2a–i and hydrofullerene 3a along with those of C60 were investigated by CV and are summarized in Table 2. All of their electrochemical properties were quite similar and showed two reversible redox processes. As shown in Table 2, the first reduction potentials of products 2a–i and 3a were more negative than that of C60, indicating that they possess higher LUMO energy levels than C60 and may have potential for application in organic photovoltaic devices as acceptors.13
| Compd | E 1 | E 2 |
|---|---|---|
| a Versus ferrocene/ferrocenium. Experimental conditions: 1.0 mM compound and 0.1 M TBAP in anhydrous ODCB; reference electrode: SCE; working electrode: Pt disc; auxiliary electrode: Pt wire; scan rate: 50 mV s−1. | ||
| C60 | –1.076 | –1.460 |
| 2a | –1.121 | –1.503 |
| 2b | –1.127 | –1.522 |
| 2c | –1.125 | –1.520 |
| 2d | –1.128 | –1.522 |
| 2e | –1.106 | –1.495 |
| 2f | –1.104 | –1.500 |
| 2g | –1.103 | –1.490 |
| 2h | –1.117 | –1.499 |
| 2i | –1.111 | –1.483 |
| 3a | –1.130 | –1.518 |
Conclusions
In summary, we have achieved a highly efficient synthesis of various C60-fused ketones from C60-fused lactones for the first time via electrochemical reduction, an unprecedented dehydrative retro Baeyer–Villiger reaction. The present protocol shows advantages of mild reaction conditions, a short reaction time, excellent product yields, and remarkable functional group tolerance. Moreover, control experiments have been performed to elucidate the plausible reaction mechanism for the formation of C60-fused ketones. The electrochemical properties of the synthesized C60-fused ketones have been characterized and may be utilized in solar cell devices.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are thankful for financial support from the National Natural Science Foundation of China (21572211) and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20000000).Notes and references
- For selected reviews, see: (a) G. R. Krow, Org. React., 1993, 43, 251 CAS; (b) G. Strukul, Angew. Chem., Int. Ed., 1998, 37, 1198 CrossRef; (c) G.-J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Chem. Rev., 2004, 104, 4105 CrossRef PubMed.
- The terminology of “retro Baeyer–Villiger reaction” in retrosynthetic analyses of complex molecules appeared in the literature. See: (a) L. A. Paquette, M. J. Wyvratt, O. Schallner, D. F. Schneider, W. J. Begley and R. M. Blankenship, J. Am. Chem. Soc., 1976, 98, 6744 CrossRef CAS; (b) L. A. Paquette, M. J. Wyvratt, O. Schallner, J. L. Muthard, W. J. Begley, R. M. Blankenship and D. Balogh, J. Org. Chem., 1979, 44, 3616 CrossRef CAS; (c) D. Enders and M. Knopp, Tetrahedron, 1996, 52, 5805 CrossRef CAS.
- The indirect retro Baeyer–Villiger reaction via multiple steps has been reported. See: J.-M. Dollat, J.-L. Luche and P. Crabbé, J. Chem. Soc., Chem. Commun., 1997, 761 Search PubMed.
- For selected reviews, see: (a) E. Nakamura and H. Isobe, Acc. Chem. Res., 2003, 36, 807 CrossRef CAS PubMed; (b) L. Sánchez, N. Martín and D. M. Guldi, Angew. Chem., Int. Ed., 2005, 44, 5374 CrossRef PubMed; (c) C. Thilgen and F. Diederich, Chem. Rev., 2006, 106, 5049 CrossRef CAS PubMed; (d) F. Giacalone and N. Martín, Chem. Rev., 2006, 106, 5136 CrossRef CAS PubMed; (e) Y. Matsuo and E. Nakamura, Chem. Rev., 2008, 108, 3016 CrossRef CAS PubMed; (f) B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58 CrossRef CAS PubMed; (g) D. M. Guldi, B. M. Illescas, C. M. Atienza, M. Wielopolski and N. Martín, Chem. Soc. Rev., 2009, 38, 1587 RSC; (h) J. L. Delgado, P.-A. Bouit, S. Filippone, M. Á. Herranz and N. Martín, Chem. Commun., 2010, 46, 4853 RSC; (i) M. D. Tzirakis and M. Orfanopoulos, Chem. Rev., 2013, 113, 5262 CrossRef CAS PubMed; (j) S.-E. Zhu, F. Li and G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7535 RSC; (k) B. M. Illescas, J. Rojo, R. Delgado and N. Martín, J. Am. Chem. Soc., 2017, 139, 6018 CrossRef CAS PubMed. For a recent example of biological applications, see: (l) J. Luczkowiak, A. Muñoz, M. Sánchez-Navarro, R. Ribeiro-Viana, A. Ginieis, B. M. Illescas, N. Martín, R. Delgado and J. Rojo, Biomacromolecules, 2013, 14, 431 CrossRef CAS PubMed.
- For selected reviews, see: (a) M. Murata, Y. Murata and K. Komatsu, Chem. Commun., 2008, 6083 RSC; (b) G. C. Vougioukalakis, M. M. Roubelakis and M. Orfanopoulos, Chem. Soc. Rev., 2010, 39, 817 RSC; (c) K. Itami, Chem. Rec., 2011, 11, 226 CrossRef CAS PubMed; (d) G.-W. Wang and F.-B. Li, Curr. Org. Chem., 2012, 16, 1109 CrossRef CAS; (e) E. E. Maroto, M. Izquierdo, S. Reboredo, J. Marco-Martínez, S. Filippone and N. Martín, Acc. Chem. Res., 2014, 47, 2660 CrossRef CAS PubMed; (f) L. Gan, Chem. Rec., 2015, 15, 189 CrossRef CAS PubMed.
- For selected examples of electrosyntheses, see: (a) C. Caron, R. Subramanian, F. D'Souza, J. Kim, W. Kutner, M. T. Jones and K. M. Kadish, J. Am. Chem. Soc., 1993, 115, 8505 CrossRef CAS; (b) P. L. Boulas, Y. Zuo and L. Echegoyen, Chem. Commun., 1996, 1547 RSC; (c) K. M. Kadish, X. Gao, E. Van Caemelbecke, T. Suenobu and S. Fukuzumi, J. Phys. Chem. A, 2000, 104, 3878 CrossRef CAS; (d) O. Lukoyanova, C. M. Cardona, M. Altable, S. Filippone, Á. Martín-Domenech, N. Martín and L. Echegoyen, Angew. Chem., Int. Ed., 2006, 45, 7430 CrossRef CAS PubMed; (e) M. Zheng, F.-F. Li, L. Ni, W.-W. Yang and X. Gao, J. Org. Chem., 2008, 73, 3159 CrossRef CAS PubMed; (f) W.-W. Yang, Z.-J. Li and X. Gao, J. Org. Chem., 2010, 75, 4086 CrossRef CAS PubMed; (g) F.-F. Li, A. Rodríguez-Fortea, P. Peng, G. A. Campos Chavez, J. M. Poblet and L. Echegoyen, J. Am. Chem. Soc., 2012, 134, 7480 CrossRef CAS PubMed; (h) H.-L. Hou, Z.-J. Li and X. Gao, Org. Lett., 2014, 16, 712 CrossRef CAS PubMed; (i) Y. Xiao and G. Wang, Chin. J. Chem., 2014, 32, 699 CrossRef CAS; (j) H.-S. Lin, Y. Matsuo, J.-J. Wang and G.-W. Wang, Org. Chem. Front., 2017, 4, 603 RSC.
- For the synthesis of other C60-fused ketones, see: (a) S.-C. Chuang, A. Islam, C.-W. Huang, H.-T. Shih and C.-H. Cheng, J. Org. Chem., 2003, 68, 3811 CrossRef CAS PubMed; (b) W. Zhang, J. K. Sprafke, M. Ma, E. Y. Tsui, S. A. Sydlik, G. C. Rutledge and T. M. Swager, J. Am. Chem. Soc., 2009, 131, 8446 CrossRef CAS PubMed; (c) B. Hu, T.-X. Liu, P. Zhang, Q. Liu, J. Bi, L. Shi, Z. Zhang and G. Zhang, Org. Lett., 2018, 20, 4801 CrossRef CAS PubMed.
- G.-W. Wang and B. Zhu, Chem. Commun., 2009, 1769 RSC.
- (a) G.-W. Wang, F.-B. Li and T.-H. Zhang, Org. Lett., 2006, 8, 1355 CrossRef CAS PubMed; (b) W.-W. Yang, Z.-J. Li, F.-F. Li and X. Gao, J. Org. Chem., 2011, 76, 1384 CrossRef CAS PubMed; (c) W.-W. Chang, Z.-J. Li, W.-W. Yang and X. Gao, Org. Lett., 2012, 14, 2386 CrossRef CAS PubMed; (d) R. Liu, F. Li, Y. Xiao, D.-D. Li, C.-L. He, W.-W. Yang, X. Gao and G.-W. Wang, J. Org. Chem., 2013, 78, 7093 CrossRef CAS PubMed; (e) Y. Xiao, S.-E. Zhu, D.-J. Liu, M. Suzuki, X. Lu and G.-W. Wang, Angew. Chem., Int. Ed., 2014, 53, 3006 CrossRef CAS PubMed; (f) J.-J. Wang, H.-S. Lin, C. Niu and G.-W. Wang, Org. Biomol. Chem., 2017, 15, 3248 RSC.
- The CCDC number of compound 2f is 1856172. For more details, see the ESI.†.
- For other selected hydrofullerenes, see: (a) M. Izquierdo, S. Osuna, S. Filippone, A. Martín-Domenech, M. Solà and N. Martín, J. Org. Chem., 2009, 74, 1480 CrossRef CAS PubMed; (b) M. Izquierdo, S. Osuna, S. Filippone, A. Martín-Domenech, M. Solà and N. Martín, J. Org. Chem., 2009, 74, 6253 CrossRef CAS PubMed.
- For selected examples, see: (a) F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS; (b) P. J. Fagan, P. J. Krusic, D. H. Evans, S. A. Lerke and E. Johnston, J. Am. Chem. Soc., 1992, 114, 9697 CrossRef CAS.
- For selected examples, see: (a) Y. Matsuo, A. Iwashita, Y. Abe, C.-Z. Li, K. Matsuo, M. Hashiguchi and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 15429 CrossRef CAS PubMed; (b) C.-P. Chen, C. Luo, C. Ting and S.-C. Chuang, Chem. Commun., 2011, 47, 1845 RSC; (c) M. Hashiguchi, N. Obata, M. Maruyama, K. S. Yeo, T. Ueno, T. Ikebe, I. Takahashi and Y. Matsuo, Org. Lett., 2012, 14, 3276 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterization data, the NMR spectra, and CVs of 2a–i and 3a (PDF). X-ray crystallographic data for 2f (CIF). CCDC 1856172. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05089a |
| This journal is © The Royal Society of Chemistry 2019 |