
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTunable enzyme responses in amphiphilic nanoassemblies through alterations in the unimer–aggregate equilibrium†
Jingjing
Gao
a,
Hui
Wang
 ab,
Jiaming
Zhuang
a and
S.
Thayumanavan
*acd
ab,
Jiaming
Zhuang
a and
S.
Thayumanavan
*acd
aDepartment of Chemistry, University of Massachusetts Amherst, Amherst, MA 01003, USA. E-mail: thai@umass.edu
bU.S. Army Edgewood Chemical Biological Center, 8198 Blackhawk Road, Aberdeen Proving Ground, MD 21010, USA
cMolecular and Cellular Biology Program, University of Massachusetts Amherst, Amherst, MA 01003, USA
dCenter for Bioactive Delivery, Institute for Applied Life Sciences, University of Massachusetts Amherst, Amherst, MA 01003, USA
First published on 15th January 2019
Abstract
Developing design rules that offer tailorability in materials' response to enzymes is of great importance, as such materials are of interest in a variety of biomedical applications including sensing, diagnostics and drug delivery. Using an amphiphilic oligomeric platform, we show that the degree of polymerization and hydrophilic–lipophilic balance variations can be utilized to alter the unimer–aggregate equilibrium, which in turn offers robust tunability of the host–guest properties of the amphiphilic nanoassemblies. We found that oligomeric assemblies with higher degree of polymerization are less sensitive to enzymatic degradation and release the guest molecules at a slower rate. Similarly, increasing the hydrophilicity makes these assemblies more sensitive to enzymes. These trends can be understood by correlating these changes to predictable modifications in the dynamics of the unimer–aggregate equilibrium, which affects the substrate availability for enzymes. These findings provide insights into rationally tuning the response of enzyme-sensitive supramolecular assemblies.
Introduction
Enzymes, as one of the most essential class of macromolecules in living organisms, are known to catalyse more than 5000 biochemical reactions efficiently and serve a variety of functions in biological processes.1 Therefore, dysregulation of enzymatic activities has been associated with many human pathologies.2–6 In this context, introducing enzymes as stimuli to trigger specific responses in artificial supramolecular assemblies has been of interest, as they have potential application in areas such as activity profile based biological imaging and drug delivery.7–15 A promising design strategy that leads to such materials involves covalent incorporation of substrate functionalities in self-assembling molecules, such as amphiphilic macromolecules, where the specific catalytic action of an enzyme covalently modifies the substrate moiety. If it were to be designed such that the product of this enzymatic reaction exhibits distinctly different self-assembly features, compared to the substrate, then there exists a unique opportunity for programmable changes in the nanostructures and their host–guest properties.Many supramolecular systems including polymeric nanoparticles, hydrogels, silica nanoparticles and gold nanoparticles have displayed adaptive behaviours toward enzymes.16–27 Tunability of the kinetics of the enzymatic response still remains a challenge, as it is mainly influenced by two factors: accessibility of an enzyme to the substrate moiety and the degree of difference in the host–guest properties between the reactant and product assemblies. In the case of amphiphilic assemblies, our group and others have shown that enzymatic activation usually occurs in the unimeric state, where the substrate is more accessible to the enzyme than in the assembled micellar form.28,29 Following these findings, we have been interested in investigating how the reaction kinetics and the ensuing changes in the host–guest characteristics would be affected by tuning the unimer–aggregate equilibrium to alter the assemblies' accessibility to the enzyme. Moreover, we were interested in identifying as to how structural changes in host assemblies, induced by an enzyme, would affect the rate of disassembly and kinetics of guest molecule release. We envisaged that oligomeric amphiphiles would be an ideal choice to address this question, because: (i) these molecules have critical aggregation concentrations (CACs) that are quite low and compare very well with those of amphiphilic polymers; (ii) despite the fact that they do exhibit low CACs, unlike polymers, these are amenable to a well-defined structure–property relationship study as the degree of oligomerization can be precise. Here we report a new modular design of oligomeric amphiphiles with which a precise control over the degree of polymerization (DP) and functional group placement in the scaffolds can be achieved (Fig. 1). These oligomers are expected to self-assemble in the aqueous phase and host hydrophobic guests in their interiors. By varying the DP and hydrophilic moieties of host molecules, we explore the molecular features that underlie the kinetics of enzymatic response in these supramolecular assemblies.
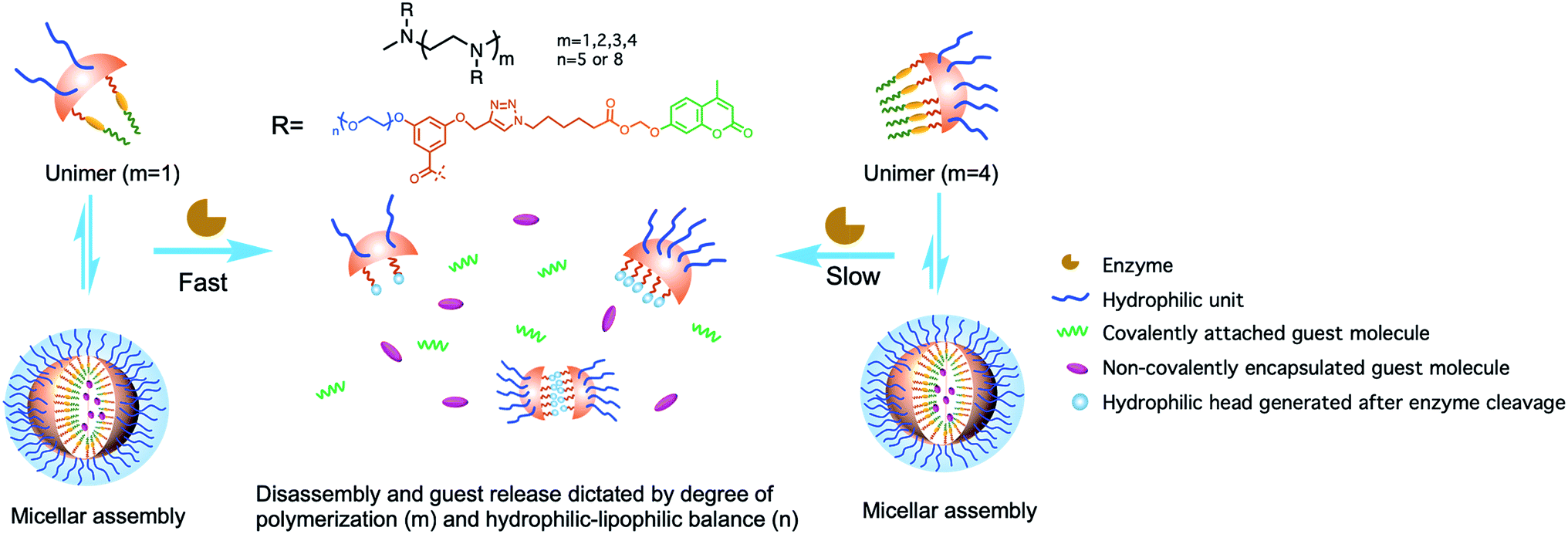 | ||
| Fig. 1 Schematic representation of enzyme-induced disassembly and guest release from oligomeric assemblies. | ||
Results and discussion
Since enzymatic activation usually occurs in the unimeric state, where the substrate is more accessible to the enzyme than in the assembled micellar form, we envisaged that shifting the equilibrium between the unimer and the assembled state would provide an opportunity to alter the enzymatic reaction rate. Degree of polymerization is one of the key factors that can alter this equilibrium30–33 and thus change the accessibility of an enzyme to its substrate. To test this possibility, it is critical that all the designed amphiphiles possess the same hydrophilic–lipophilic balance (HLB). For this purpose, a series of oligomeric amphiphiles from dimer (2-EG5) to pentamer (5-EG5), have been synthesized (Scheme 1). To further evaluate the effects of DP on the enzymatic response, a polymer, P-EG5, with ∼14 repeating units was also synthesized. In these amphiphiles, penta-ethylene glycol (EG5) monomethyl ether moieties are installed as the hydrophilic functionality, while alkylated coumarin moieties are used as the hydrophobic units. Both these units are attached to the meta-positions of a benzoyl building block, which are then attached to well-defined oligoamines to generate amphiphiles with different degrees of oligomerization. In all these systems, the coumarin moiety is chosen as the covalently-appended model guest molecule. In order to release this guest molecule in the presence of an enzyme, we use an acetal–ester linkage to connect the coumarin to the oligomer. The esterase-induced cleavage of the carboxylate moiety would create a hemi-acetal coumarin, which is hydrolytically unstable. This hemi-acetal therefore rapidly hydrolyzes further to generate a highly fluorescent, 4-methylumbelliferone. In addition to releasing this covalently attached molecule, this transformation also replaces the aryl moiety on the hydrophobic side of these amphiphiles with a carboxylic acid moiety. This results in a significant change in the HLB of the amphiphile. Note that this series of amphiphiles share all the common structural features including the backbone, and hydrophobic and hydrophilic functionalities; the only variation within this series of amphiphiles is DP. Therefore, this investigation allows us to inquire about the impact of DP upon self-assembly and enzyme induced disassembly events.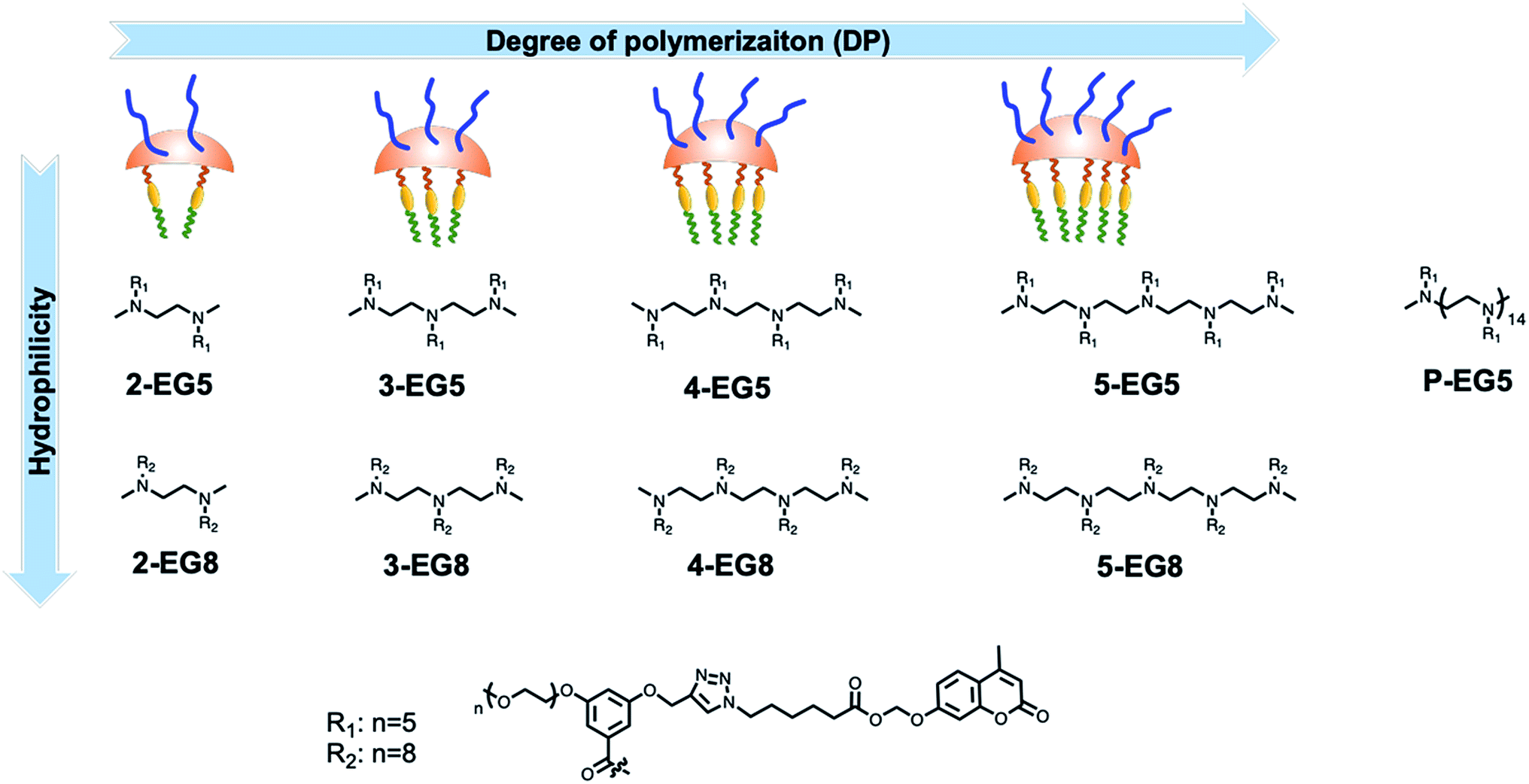 | ||
| Scheme 1 Molecular structures of oligomers: legends of each oligomer indicate increased degree of polymerization from 2-EG5 to P-EG5; EG5 indicates oligomers with five ethyleneglycol units and EG8 indicates oligomers with eight ethyleneglycol units as hydrophilic moieties. | ||
In addition to DP, the HLB of oligomers is another factor that impacts the unimer–aggregate equilibrium. To test this possibility, with the same oligomer series as above, we simply increased the length of the oligoethyleneglycol chain length from five to eight units. Thus, we synthesized four more oligomers 2-EG8, 3-EG8, 4-EG8, and 5-EG8 (Scheme 1). We hypothesized that the increase in hydrophilicity upon going from penta-ethylene glycol monomethyl ether (EG5) to octa-ethylene glycol (EG8) monomethyl ether would increase the dynamics of the unimer–aggregate equilibrium, which will then increase the availability of the substrate moiety for the enzymes. In this study, we also test this hypothesis.
The amphiphilic oligomers were designed in such a way that they can be synthesized in a modular fashion, providing a facile way to vary the number of repeating units and functional group placement. The synthetic routes to the target oligomers are exemplified by the synthesis of trimer 3-EG5 in Scheme 2 (see the ESI† for detailed procedures and characterization). The 3,5-disubstituted-benzoyl chloride molecule 1a was reacted with N,N′′-dimethyl diethylenetriamine under basic conditions to generate the substituted oligoamine scaffold 1b. This molecule now contains the pentaethyleneglycol hydrophilic unit and the alkyne moiety to anchor the hydrophobic unit. The hydrophobic and fluorogenic enzyme substrate was then attached to all three repeat units of the oligomer using the Huisgen 1,3-dipolar cycloaddition reaction, the so-called “click” chemistry,16 to yield the desired oligomer 3-EG5 (Scheme 2).
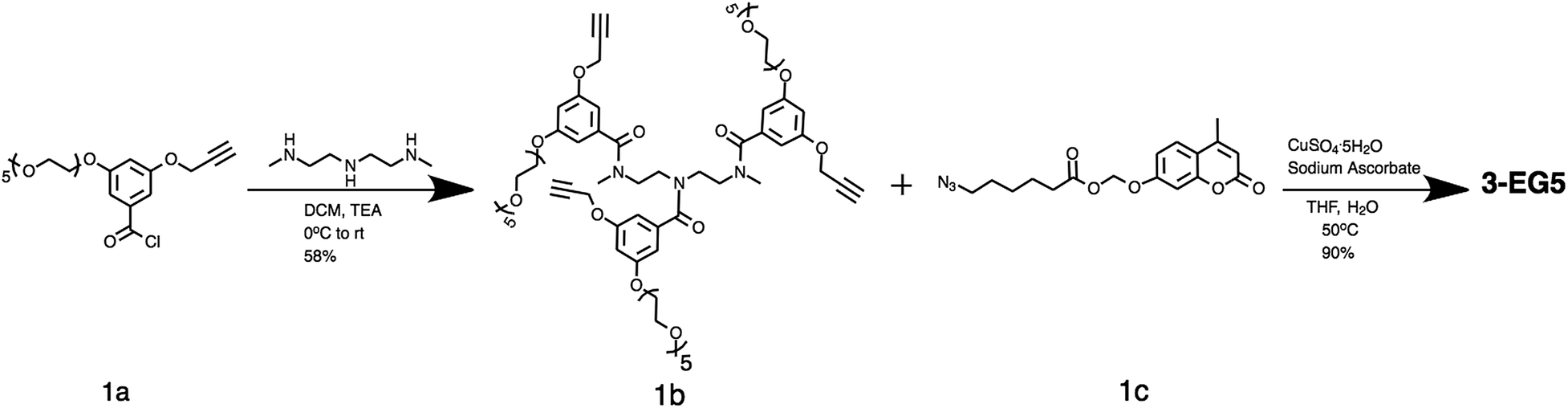 | ||
| Scheme 2 Synthesis route to oligomers exemplified using 3-EG5 scheme. | ||
We first investigated whether these oligomeric amphiphiles would form aggregates in the aqueous phase, since they contain both hydrophobic and hydrophilic moieties. If self-assembly occurs, the interior of these assemblies would have the capability to non-covalently encapsulate hydrophobic molecules. To test this, the oligomers were directly dissolved in phosphate buffer and non-covalent incorporation of a solvatochromic dye, Nile Red, within these assemblies was attempted. We found that at lower concentrations of oligomers, the emission intensity of Nile Red was quite low. However, once the concentration of the oligomers reached a certain point, a rather sharp increase in emission intensity was observed. This onset point is taken to be the onset of hydrophobicity-driven aggregation, which is estimated to be the critical aggregation concentration (CAC) of these oligomers. As shown in Table 1, with DP increasing from 1 to 13, the CAC values of these oligomers vary from 75 μM to 0.58 μM (Fig. S1†). In general, oligomers with higher DP tend to aggregate at lower concentrations, despite the fact that the HLBs of all these oligomers are identical. At the same DP, the systems with longer ethylene glycol chains as the hydrophilic moiety exhibited higher CAC values.
| Oligomer | m | n | M w (kDa) | CAC (μM) | Size (nm) |
|---|---|---|---|---|---|
| 2-EG5 | 1 | 5 | 1594 | 66 | 240 |
| 3-EG5 | 2 | 5 | 2392 | 7.8 | 192 |
| 4-EG5 | 3 | 5 | 3189 | 2 | 310 |
| 5-EG5 | 4 | 5 | 3986 | 0.74 | 184 |
| P-EG5 | 13 | 5 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 162 162 |
0.58 | 97 |
| 2-EG8 | 1 | 8 | 1858 | 75 | 208 |
| 3-EG8 | 2 | 8 | 2789 | 8.3 | 127 |
| 4-EG8 | 3 | 8 | 3716 | 3.8 | 202 |
| 5-EG8 | 4 | 8 | 4647 | 0.95 | 125 |
The solution phase sizes of these nanoassemblies were then measured by dynamic light scattering (DLS) at a concentration above their CACs. We observed an average hydrodynamic diameter ranging from ∼100 to 300 nm for these assemblies (Table 1). The spherical morphology and size of these assemblies were further ascertained using transmission electron microscopy (TEM), as shown in Fig. S2.†
Note that we hypothesized that if the HLB of the oligomers was kept constant, then oligomeric amphiphiles with higher DP would be hydrolyzed by the enzyme at a slower rate than their counterparts with lower DP. To test this, we first measured the enzymatic cleavage rates of all oligomers. Since the enzymatic reaction releases the fluorescent byproduct 4-methylumbelliferone, we were able to monitor the cleavage rates spectroscopically. For an accurate comparison, it is necessary that all these oligomer solutions are not only prepared at concentrations above their respective CACs but also contain the same concentration of the substrate functionalities, regardless of their DP. To meet these two criteria, we prepared oligomer solutions that contain 200 μM enzyme substrates (based on coumarin), i.e. 100 μM dimer, 66.7 μM trimer, 50 μM tetramer, and 40 μM pentamer, and then treated with 60 nM esterase. As shown in Fig. 2, a clear trend of the enzymatic reaction rate was observed for these oligomers with EG5 as the hydrophilic moiety; amphiphile 2-EG5 exhibited the fastest enzymatic rate over 48 hours, systematically followed by 3-EG5, 4-EG5 and 5-EG5. Moreover, when the same concentrations of the enzyme and the substrate were used in the case of the 14-mer P-EG5, the molecular weight of which is comparable to that of polymers, little hydrolysis was observed from the emission spectra. These results are consistent with our hypothesis that a higher DP would result in a slower enzymatic reaction rate, which in turn provides a convenient handle to tune reaction rates of enzymes and the resultant release of the covalently bound molecules.
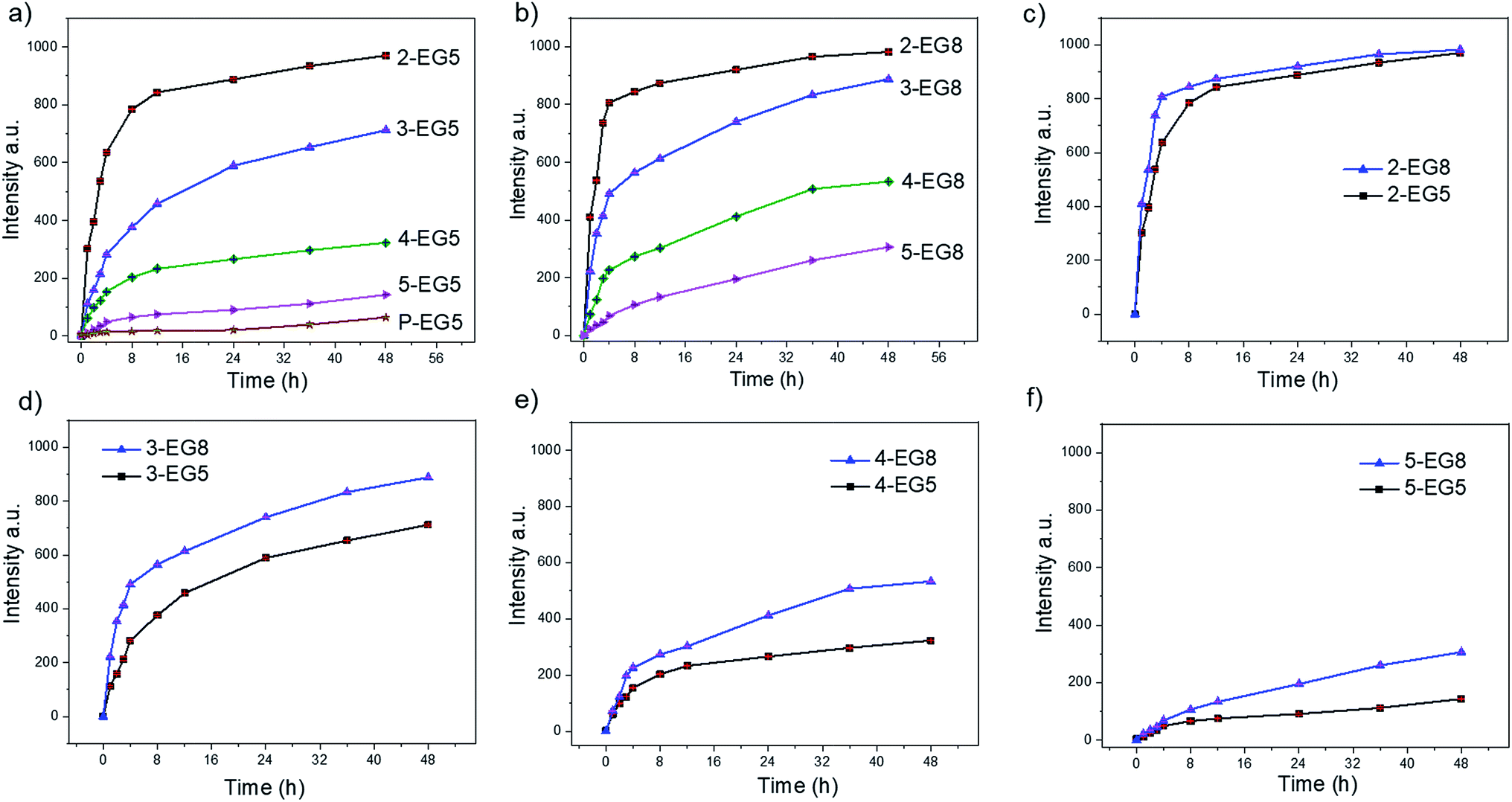 | ||
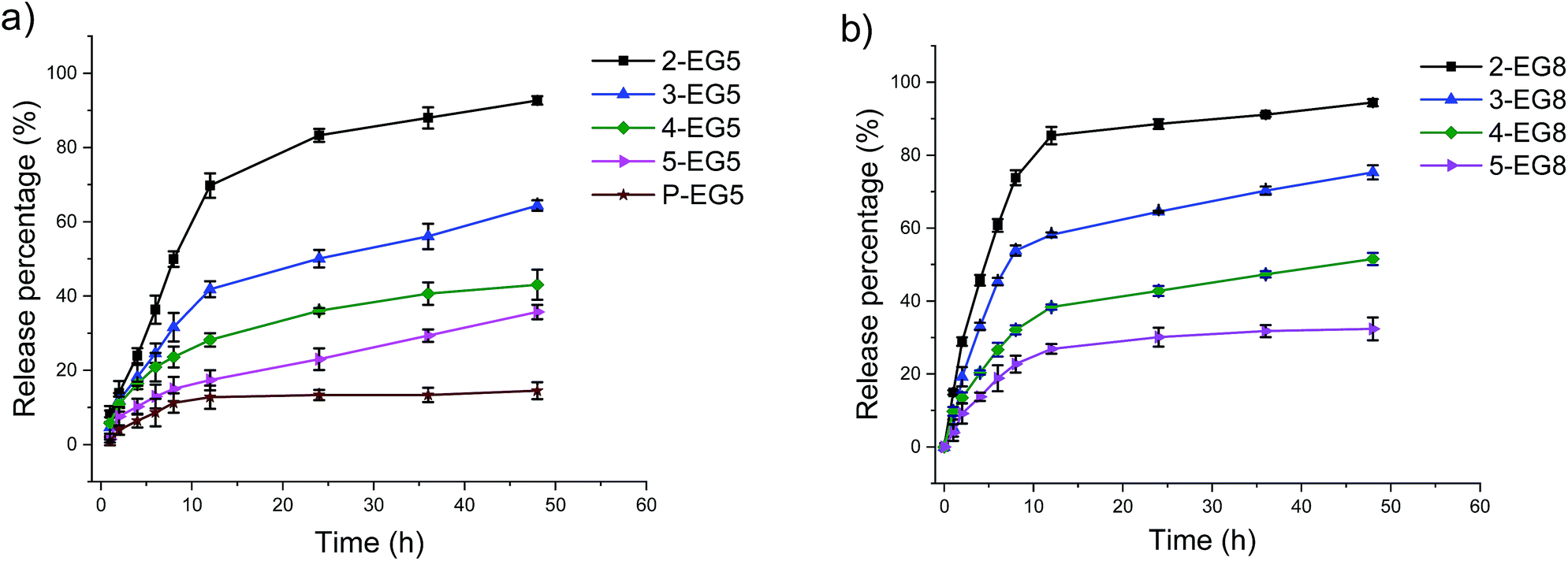
| Fig. 2 Enzymatic hydrolysis of oligomeric assemblies based on coumarin release. (a) Oligomer assemblies with EG5 as the hydrophilic moiety, (b) oligomer assemblies with EG8 as the hydrophilic moiety, and enzymatic hydrolysis comparison between oligomer-EG5 and the corresponding oligomer-EG8 (c–f). | ||
When the same experiments were performed with the second series of oligomers (the EG8 series) that contain longer ethylene glycol chains as the hydrophilic group, a similar trend was indeed observed, i.e. the hydrolysis rate decreases for oligomers with higher DP. These results again confirmed our hypothesis that amphiphiles with higher DP are less accessible to enzymes and thus more stable compared with oligomers with lower DP. Meanwhile, comparison of the two series of oligomers also allows us to evaluate the HLB effects on the enzymatic hydrolysis rates of these oligomers. Note that the basis for our hypothesis that a higher degree of oligomerization would cause a slower reaction rate is that the dynamics of the unimer–aggregate equilibrium would be slower at higher DP. The results above support this hypothesis. If this were true, then it should also follow that if the hydrophilicity of these oligomers changes, the dynamics of the unimer–aggregate equilibrium would also be affected, which would in turn alter the sensitivity of these oligomers to enzymes. To test this idea, we compared the hydrolysis rates of EG5 oligomers and EG8 oligomers under the same experimental conditions. Interestingly, we observed that the cleavage rates of the covalently attached molecules from 2-EG5 and 2-EG8 were very similar (Fig. 2c). However, when the DP increases to trimeric or higher, n-EG8 oligomers with longer ethylene glycol chains indeed consistently exhibited faster cleavage, compared to their corresponding n-EG5 oligomers with shorter ethyleneglycol chains (Fig. 2d–f). These results suggest that lowering the hydrophilicity of oligomers will make them more stable in the presence of esterase. This is reasonably expected, because an increase in hydrophilicity is expected to increase the dynamics in the unimer–aggregate equilibrium, which facilitates the enzyme's access to its substrate functionalities. We attribute the lack of significant difference between 2-EG5 and 2-EG8 assemblies to the fact that these low order oligomers are already sufficiently dynamic, such that there is no significant advantage to increasing the hydrophilicity of the oligomeric amphiphiles from EG5 to EG8.
Next, we were interested in evaluating the effect of the enzyme-induced changes in the HLB of the amphiphiles upon their host characteristics for hydrophobic guest molecules. We were especially interested in identifying whether this anticipated molecule release event will follow a DP- and hydrophilicity-dependent trend observed in the covalent modification of the amphiphile. To test this, we encapsulated a hydrophobic fluorophore, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine (DiI), into these assemblies. The DiI-encapsulated oligomeric assemblies were treated with esterase and the molecule release was assessed by fluorescence change. A change in fluorescence is anticipated in this case because the DiI molecule is insoluble in aqueous solutions and therefore precipitates out of solution, upon release from the hydrophobic pockets of these amphiphilic assemblies. As with the experiments above, the concentrations of esterase and the substrate functionalities in the oligomers were maintained for appropriate comparison of the relative rates of molecule release.
Indeed, we found that the guest release depends on the DP of oligomers at constant HLB of the molecule, i.e. within the same oligomer series (EG5 or EG8 series). That is, assemblies from higher order oligomers exhibit the ability to more stably encapsulate the guest molecules and respond to the enzyme much more slowly compared to the lower order oligomers (Fig. 3). Also, assemblies with longer ethylene glycol chains can release guest molecules much faster in the same time range (Fig. 3 and S3†). These results show that a precise control over the release kinetics of non-covalently encapsulated guest molecules can also be achieved by tuning the molecular structures.
 | ||
| Fig. 3 Non-covalent guest (DiI) release from nanoassemblies. | ||
Comparison of data for the covalent molecule release based on the enzymatic cleavage of the substrate functionalities and the release of the non-covalently bound guest molecules revealed that the latter process lags behind the former process. The potential reason behind this difference is that the enzymatic cleavage of the covalently attached guest molecules happens first, which is followed by the loss in capability of the amphiphilic assemblies to hold the guest molecules to cause molecule release. In this scenario, the intermediate states of the aggregated assemblies generated by the enzymatic reaction (e.g. only one of the coumarin moieties cleaved in a pentameric amphiphile) also can bind to guest molecules, but their relative ability to act as a host might be lower. This process in conjunction with the need for a critical concentration of DiI to cause its precipitation likely manifests itself as the lag in the non-covalent guest release, relative to the covalent modification of the oligomers by the enzyme.
Since the enzymatic cleavage of hydrophobic groups seems to be the primary reason for assemblies to lose their stability and capability to hold guest molecules, it is likely that this enzyme reaction induces morphological changes of the aggregated assemblies. To test this possibility, we monitored the temporal evolution of the size of these assemblies by DLS. We found that the size of assemblies changes immediately after esterase was introduced into these systems. As shown in Fig. 4, both 2-EG5 and 3-EG5 completely disassembled in the presence of the enzyme: the size of assembly 2-EG5 sharply decreased from ∼240 nm to ∼20 nm, while assembly 3-EG5 formed an ∼35 nm assembly from an initial size of ∼220 nm in 48 hours; this size change was also confirmed by TEM images which showed clear spherical structures initially but few visible aggregates after 48 hours of enzymatic reactions (as shown in Fig. S4†). However, the size of oligomers 4-EG5 and 5-EG5 remained relatively unchanged over the same timeframe. Furthermore, a similar trend of assembly size change was observed for n-EG8 oligomers with longer EG chains under the same experimental conditions. While 4-EG8 and 5-EG8 were rather more stable in the presence of the enzyme, both 2-EG8 and 3-EG8 completely disassembled in the presence of the enzyme at a faster rate compared with the corresponding n-EG5 oligomers, respectively. These results suggested that enzymatic cleavage can induce the disassembly process. Also, both DP and HLB variations of oligomeric amphiphiles can alter the disassembly kinetics, which correlate well with the guest release profiles of both covalently bound and non-covalently bound hydrophobic molecules.
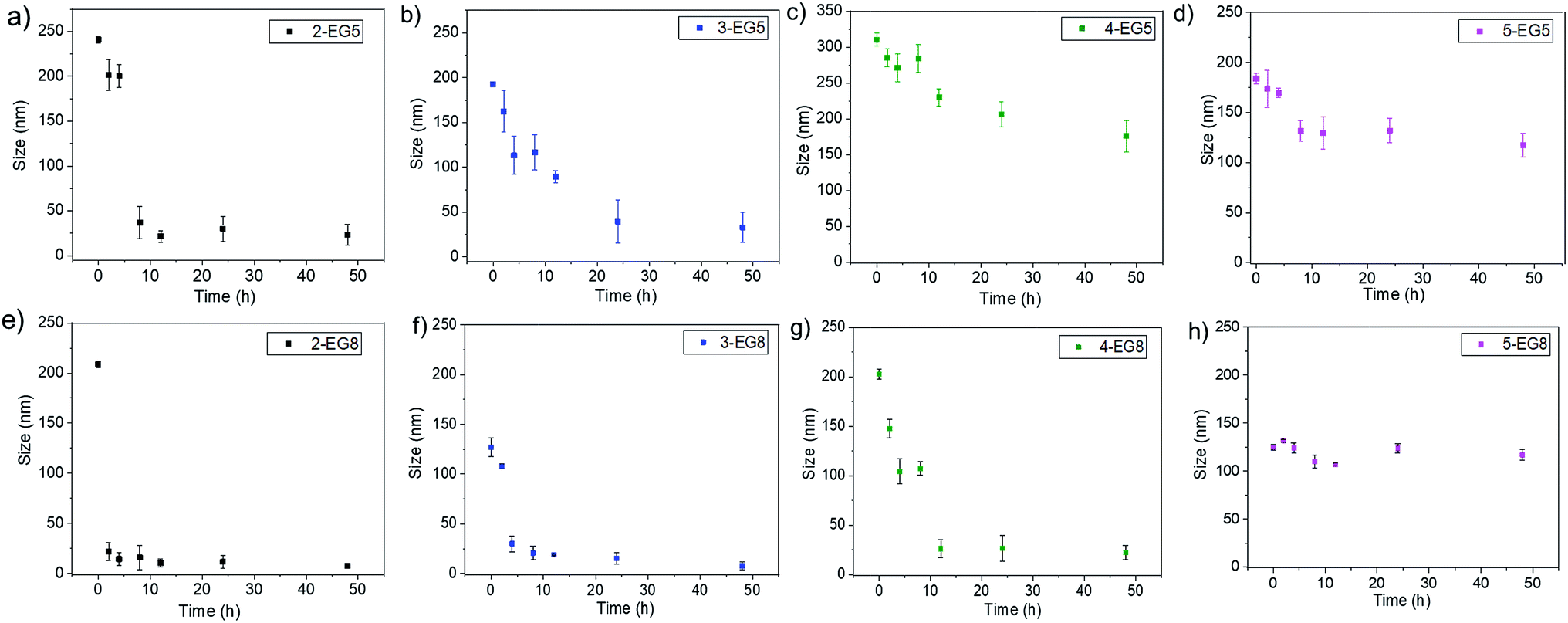 | ||
| Fig. 4 Size evolution of assemblies in the presence of esterase in 48 hours. | ||
Conclusions
In summary, two series of oligomeric amphiphiles were prepared to evaluate the possibility of tuning enzyme-induced changes in their self-assembly properties and host–guest characteristics. We have shown that: (i) when the degree of oligomerization increases in the amphiphiles, the enzymatic reaction rate decreases. This offers a straightforward opportunity to tune the release kinetics of covalently-appended guest molecules. (ii) This reaction kinetics can also be tuned by varying the hydrophilic–lipophilic balance of the self-assembling substrate molecule itself, where an increase in hydrophilicity accelerates the molecule release rates. (iii) In the assemblies where the enzyme-induced alteration in the HLB occurs at a reasonable rate, i.e. in lower order oligomers, a significant change in size and morphology of the assemblies was also observed. (iv) Non-covalently bound guest molecules can also be released from these amphiphilic assemblies in response to the enzyme-induced alteration in the HLB, the trends of which closely follow those observed in the release of the covalently bound guest molecules. The trends in the enzymatic reaction rates and the change in the host–guest characteristics can be understood by correlating structural variations to the change in the dynamics of the unimer–aggregate equilibrium. Factors that lead to faster unimer–aggregate equilibrium dynamics lead to faster enzymatic response. Overall, this study provides two simple and straightforward approaches for altering enzyme-induced changes in amphiphilic assemblies, which in turn offer tunability of the release kinetics of covalently and non-covalently bound guest molecules from these assemblies. The findings presented here could provide a basis for designing enzyme responsive materials with controlled release capabilities for biomedical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank NIGMS of the NIH (GM-065255) for support.Notes and references
- I. Schomburg, A. Chang, S. Placzek, C. Söhngen, M. Rother, M. Lang, C. Munaretto, S. Ulas, M. Stelzer, A. Grote, M. Scheer and D. Schomburg, Nucleic Acids Res., 2013, 41, D764–D772 CrossRef CAS PubMed.
- S. M. Dhanasekaran, T. R. Barrette, D. Ghosh, R. Shah, S. Varambally, K. Kurachi, K. J. Pienta, M. A. Rubin and A. M. Chinnaiyan, Nature, 2001, 412, 822 CrossRef CAS PubMed.
- P. Imming, C. Sinning and A. Meyer, Nat. Rev. Drug Discovery, 2006, 5, 821 CrossRef CAS PubMed.
- H. Sato, T. Takino, Y. Okada, J. Cao, A. Shinagawa, E. Yamamoto and M. Seiki, Nature, 1994, 370, 61 CrossRef CAS PubMed.
- L. Dang, D. W. White, S. Gross, B. D. Bennett, M. A. Bittinger, E. M. Driggers, V. R. Fantin, H. G. Jang, S. Jin and M. C. Keenan, Nature, 2009, 462, 739 CrossRef CAS PubMed.
- G. I. Murray, M. E. Duncan, P. O'Neil, W. T. Melvin and J. E. Fothergill, Nat. Med., 1996, 2, 461–462 CrossRef CAS PubMed.
- J. Zhou, G. Yu and F. Huang, Chem. Soc. Rev., 2017, 46, 7021–7053 RSC.
- X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed.
- L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840–7892 CrossRef CAS PubMed.
- Y. Wang, K. Zhou, G. Huang, C. Hensley, X. Huang, X. Ma, T. Zhao, B. D. Sumer, R. J. DeBerardinis and J. Gao, Nat. Mater., 2013, 13, 204 CrossRef PubMed.
- D.-E. Lee, H. Koo, I.-C. Sun, J. H. Ryu, K. Kim and I. C. Kwon, Chem. Soc. Rev., 2012, 41, 2656–2672 RSC.
- A. P. Blum, J. K. Kammeyer, A. M. Rush, C. E. Callmann, M. E. Hahn and N. C. Gianneschi, J. Am. Chem. Soc., 2015, 137, 2140–2154 CrossRef CAS PubMed.
- M. Molina, M. Asadian-Birjand, J. Balach, J. Bergueiro, E. Miceli and M. Calderón, Chem. Soc. Rev., 2015, 44, 6161–6186 RSC.
- M. Zelzer, S. J. Todd, A. R. Hirst, T. O. McDonald and R. V. Ulijn, Biomater. Sci., 2013, 1, 11–39 RSC.
- M. Karimi, A. Ghasemi, P. S. Zangabad, R. Rahighi, S. M. M. Basri, H. Mirshekari, M. Amiri, Z. S. Pishabad, A. Aslani and M. Bozorgomid, Chem. Soc. Rev., 2016, 45, 1457–1501 RSC.
- A. B. Lowe, Polymer, 2014, 55, 5517–5549 CrossRef CAS.
- L. M. Randolph, M.-P. Chien and N. C. Gianneschi, Chem. Sci., 2012, 3, 1363–1380 RSC.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991 CrossRef CAS PubMed.
- J. Gao, B. Zhao, M. Wang, M. A. C. Serrano, J. Zhuang, M. Ray, V. M. Rotello, R. W. Vachet and S. Thayumanavan, J. Am. Chem. Soc., 2018, 140, 2421–2425 CrossRef CAS PubMed.
- S. Goggins, B. J. Marsh, A. T. Lubben and C. G. Frost, Chem. Sci., 2015, 6, 4978–4985 RSC.
- A. Bernardos, E. Aznar, M. D. Marcos, R. Mart'inez-Máñez, F. Sancenón, J. Soto, J. M. Barat and P. Amorós, Angew. Chem., 2009, 121, 5998–6001 CrossRef.
- X. Xu, M. S. Han and C. A. Mirkin, Angew. Chem., 2007, 119, 3538–3540 CrossRef.
- Y. Xiao, F. Patolsky, E. Katz, J. F. Hainfeld and I. Willner, Science, 2003, 299, 1877–1881 CrossRef CAS PubMed.
- J.-H. Kang, D. Asai, J.-H. Kim, T. Mori, R. Toita, T. Tomiyama, Y. Asami, J. Oishi, Y. T. Sato and T. Niidome, J. Am. Chem. Soc., 2008, 130, 14906–14907 CrossRef CAS PubMed.
- R. V. Ulijn, J. Mater. Chem., 2006, 16, 2217–2225 RSC.
- Y. Li, G. Liu, X. Wang, J. Hu and S. Liu, Angew. Chem., Int. Ed., 2016, 55, 1760–1764 CrossRef CAS PubMed.
- J. Gao, X. Liu, H. Secinti, Z. Jiang, O. Munkhbat, Y. Xu, X. Guo and S. Thayumanavan, Chem. - Eur. J., 2018, 24, 1789–1794 CrossRef CAS PubMed.
- A. J. Harnoy, I. Rosenbaum, E. Tirosh, Y. Ebenstein, R. Shaharabani, R. Beck and R. J. Amir, J. Am. Chem. Soc., 2014, 136, 7531–7534 CrossRef CAS PubMed.
- M. A. Azagarsamy, P. Sokkalingam and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 14184–14185 CrossRef CAS PubMed.
- C. J. Hawker, K. L. Wooley and J. M. J. Frechet, J. Chem. Soc., Perkin Trans. 1, 1993, 1287–1297 RSC.
- G. Fleischer, J. Phys. Chem., 1993, 97, 517–521 CrossRef CAS.
- G. R. Newkome, C. N. Moorefield, G. R. Baker, A. L. Johnson and R. K. Behera, Angew. Chem., Int. Ed. Engl., 1991, 30, 1176–1178 CrossRef.
- E. N. Savariar, S. V. Aathimanikandan and S. Thayumanavan, J. Am. Chem. Soc., 2006, 128, 16224–16230 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04744h |
| This journal is © The Royal Society of Chemistry 2019 |