
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functionality in metal–organic framework minerals: proton conductivity, stability and potential for polymorphism†
Igor
Huskić
 a,
Novendra
Novendra
b,
Dae-Woon
Lim
c,
Filip
Topić
a,
Hatem M.
Titi
a,
Igor V.
Pekov
d,
Sergey V.
Krivovichev
e,
Alexandra
Navrotsky
*b,
Hiroshi
Kitagawa
*c and
Tomislav
Friščić
*a
a,
Novendra
Novendra
b,
Dae-Woon
Lim
c,
Filip
Topić
a,
Hatem M.
Titi
a,
Igor V.
Pekov
d,
Sergey V.
Krivovichev
e,
Alexandra
Navrotsky
*b,
Hiroshi
Kitagawa
*c and
Tomislav
Friščić
*a
aDepartment of Chemistry, McGill University, Montreal, Canada. E-mail: tomislav.friscic@mcgill.ca
bPeter A. Rock Thermochemistry Laboratory and NEAT ORU, University of California Davis, Davis, CA, USA. E-mail: anavrotsky@ucdavis.edu
cDivision of Chemistry, Graduate School of Science, Kyoto University, Kitashirakawa-Oiwakecho, Sakyo-ku, Kyoto, 606-8502 Japan. E-mail: kitagawa@kuchem.kyoto-u.ac.jp
dKola Science Centre, Russian Academy of Sciences, Apatity and Department of Crystallography, Saint Petersburg State University, Saint Petersburg, Russia
eFaculty of Geology, Lomonosov Moscow State University, Moscow, Russia
First published on 2nd April 2019
Abstract
Rare metal–organic framework (MOF) minerals stepanovite and zhemchuzhnikovite can exhibit properties comparable to known oxalate MOF proton conductors, including high proton conductivity over a range of relative humidities at 25 °C, and retention of the framework structure upon thermal dehydration. They also have high thermodynamic stability, with a pronounced stabilizing effect of substituting aluminium for iron, illustrating a simple design to access stable, highly proton-conductive MOFs without using complex organic ligands.
Introduction
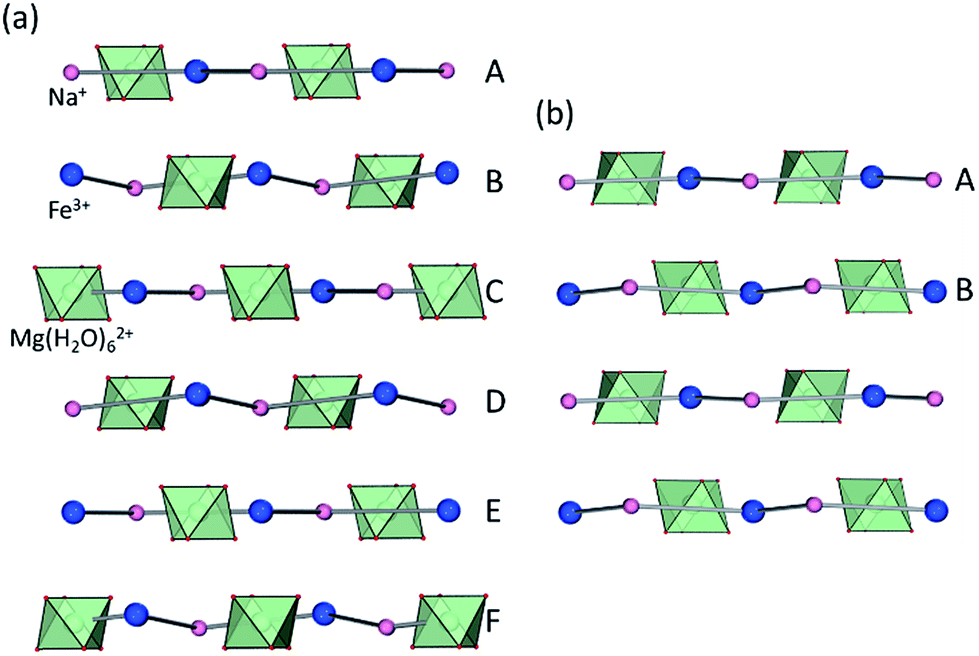
Whereas most minerals are inorganic solids,1 recent work highlighted complex organic or metal–organic structures of minerals found in unusual or extreme environments.2 Examples include mellitic acid on Mars,3 hydrocarbon cocrystals as prospective minerals on Titan,4 metal oxalate coordination polymers (humboldtine, lindbergite),5 or geoporphyrins (abelsonite).6 The search to discover and understand the properties of such non-conventional minerals has been galvanized by the Carbon Mineral Challenge project,7 exploring the role of carbon in mineral and biological evolution on Earth and other planets.8 A recent addition to this set of unusual natural structures are metal–organic frameworks (MOFs) in the form of rare minerals zhemchuzhnikovite (ZH, [Mg(H2O)6][NaFexAl1−x(C2O4)3]·3H2O, x ∼ 0.6) and stepanovite (ST, [Mg(H2O)6][NaFe(C2O4)3]·3H2O), discovered in the Lower Lena region of Siberia.9,10 Single crystal X-ray diffraction analysis of natural samples reveals that the minerals are based on open two-dimensional (2-D) honeycomb (3,6)-topology (hcb-topology) anionic nets composed of oxalate linkers and a combination of Na+ with either only Fe3+ (in ST) or a mixture of Al3+ and Fe3+ nodes (in ZH) (Fig. 1a).11 The sheets are separated by layers of ordered guest water molecules, and charge-balanced by Mg(H2O)62+ ions located in cavities of each hcb-sheet. These structures are hybrid organic–inorganic materials analogous to oxalate MOFs investigated for optical, magnetic and particularly proton-conductive properties.12,13 The structural similarity raises the exciting possibility that ZH and ST minerals should exhibit functional properties expected for related MOFs, notably proton conductivity and stability upon heating or guest removal.11,14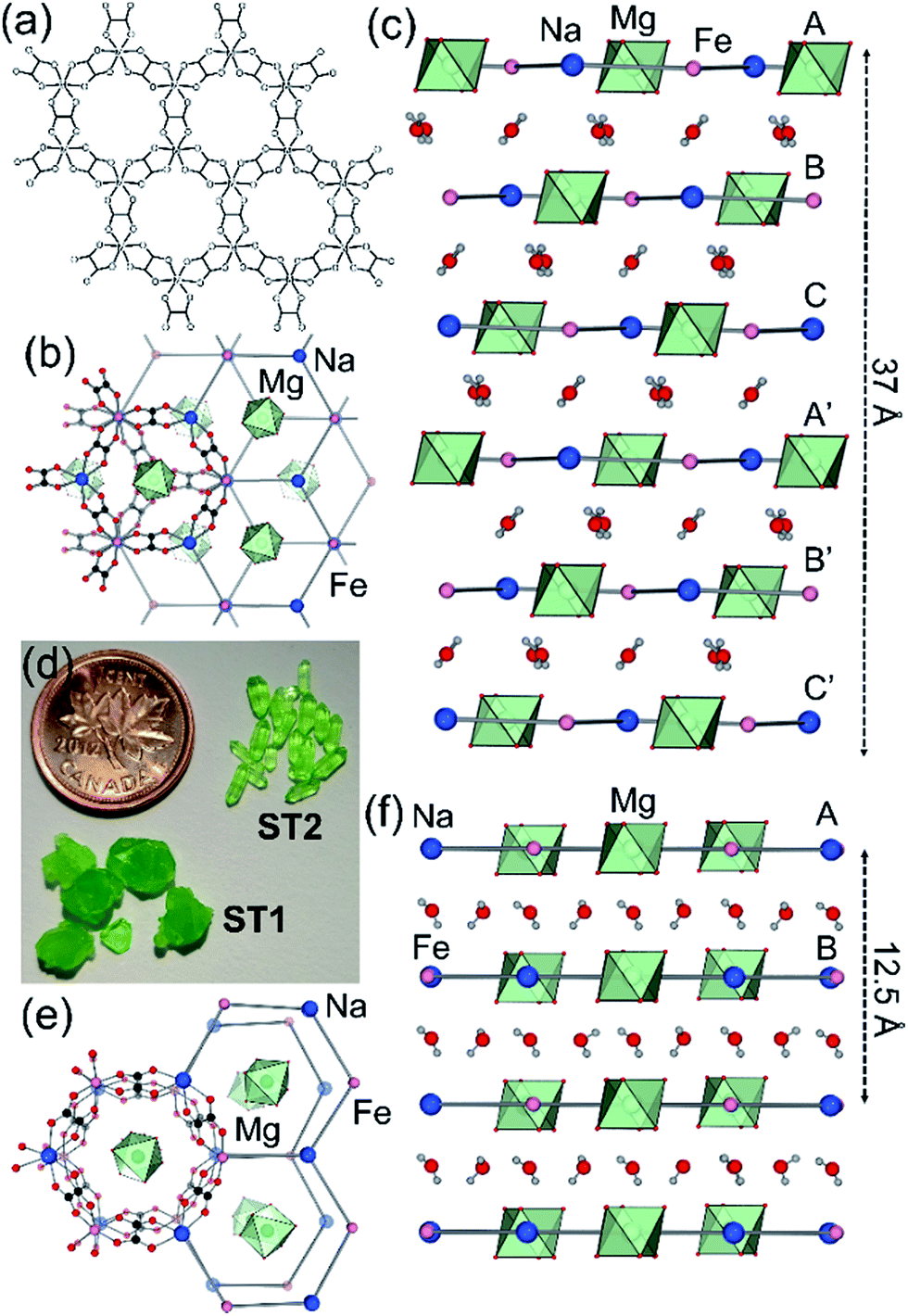 | ||
| Fig. 1 (a) Scheme of hcb-layers in stepanovite and zhemchuzhnikovite; (b) hcb-layers in synthetic stepanovite (ST1) viewed along and (c) perpendicular to the crystallographic c-axis, showing ABCA′B′C′ stacking. (d) Samples of ST1 and ST2 shown next to a Canada penny coin (∼1.9 cm diameter) for size comparison; (e) hcb-layers in ST2 viewed parallel to the c-axis, showing channel formation and (f) perpendicular to the c-axis, showing AB stacking. Green octahedra represent coordination environments of Mg(H2O)62+ ions. | ||
However, scarcity of the minerals has prevented the measurement of their properties. Moreover, while it was reported that ZH and ST undergo reversible loss of guest water molecules,11 there has been no evidence that such dehydration proceeds with retention of the hcb-networks, which would be expected in a functional MOF material.14
We now show that ZH and ST can indeed exhibit functional behavior on par with known oxalate MOF proton conductors. Specifically, synthetic mineral analogues exhibit high proton conductivities at room temperature, undergo thermal removal of included water guests without changes to the underlying hcb-networks and, by using solution calorimetry, we demonstrate they are also of high thermodynamic stability.
Experimental
All reagents were available commercially and were used without additional purification. Oxalic acid, NaOH, NaHCO3, MgO, MgCl2·6H2O and FeCl3·6H2O were obtained from Sigma Aldrich.Synthesis of ST1 and ST2
Synthesis of ST1 and ST2 was performed by dissolving FeO(OH) (first obtained by mixing of aqueous solutions of FeCl3·6H2O and NaHCO3, followed by filtration) in an aqueous solution of oxalic acid. After addition of NaOH and MgCl2·6H2O, the solution was filtered and placed in a refrigerator at 4 °C for crystallization and slow evaporation. The resulting ST1 and ST2 crystals were separated by hand, based on crystal morphology. Slow evaporation led to mixtures containing ST1 as the major product, while faster evaporation, e.g. in a rotary evaporator, gave ST2 as the major product.Synthesis of ZH
Synthesis of ZH was performed in two steps. In the first step, NaMgAl(C2O4)3·9H2O was obtained by dissolving aluminum metal in an aqueous solution of oxalic acid with sonication, followed by addition of NaOH, MgCl2·6H2O and filtration. Evaporation of the liquid led to crystallization of NaMgAl(C2O4)3·9H2O, isolated by filtration. In the second step, equivalent amounts of NaMgAl(C2O4)3·9H2O and NaMgFe(C2O4)3·9H2O (synthesized as described for ST1 and ST2 above) were dissolved in a small amount of water. After several days at 4 °C, yellow-green needles of ZH were isolated. Bulk ZH sample was obtained by milling equimolar amounts of NaMgAl(C2O4)3·9H2O and NaMgFe(C2O4)3·9H2O with 40 μL H2O for 30 min in 10 mL Teflon® jars with one zirconia ball (10 mm diameter) at 25 Hz.Dehydration of ST1 and ST2 single crystals
Dehydration of ST1 and ST2 single crystals was performed with gentle heating at 50 °C under vacuum over a period of 12 hours. The compounds are further denoted as ST1d and ST2d denoting dehydrated ST1 and ST2 respectively.Powder X-ray diffraction (PXRD)
Powder X-ray diffraction (PXRD) data for rehydration experiments was collected on a PROTO AXRD benchtop instrument equipped with a DECTRIS MYTHEN2R 1D detector, using nickel-filtered CuKα (λ = 0.154056 Å) radiation. Rehydration was performed in a custom-made sealed sample holder15 kept at 98% relative humidity with the aid of a saturated aqueous solution of K2SO4.Variable temperature (VT)
Variable temperature (VT) dehydration experiments and verification after impedance measurements were performed on a Bruker D8 Advance instrument equipped with a LYNXEYE XE-T detector using nickel-filtered CuKα (λ = 0.154056 Å) radiation. The setup was equipped with Anton Paar CHC plus+ chamber. Diffractograms were collected in a stepwise fashion in a dry environment. After each collection (ca. 15 min), temperature in the chamber was raised by 1 °C at a rate of 1 °C min−1 and a new collection started.Crystal structures of ST2, ST1d and ST2d
Crystal structures of ST2, ST1d and ST2d were determined by single crystal X-ray diffraction using a Bruker D8 APEX2 X-ray diffractometer and graphite-monochromated MoKα (λ = 0.71073 Å) radiation. Structures were solved by intrinsic phasing in SHELXT16 and refined on F2 using SHELXL.17 Wherever possible, hydrogen atoms participating in hydrogen bonds were located from the electron density map. Crystallographic data in CIF format has been deposited with the Cambridge Structural Database (CSD), CCDC 1868681–1868685.Fourier-transform infrared attenuated total reflectance (FTIR-ATR)
Fourier-transform infrared attenuated total reflectance (FTIR-ATR) studies were done on a Bruker VERTEX 70 instrument with a PLATINUM diamond crystal ATR unit.Thermogravimetric analysis and differential scanning calorimetry (TGA and DSC) analysis
Thermogravimetric analysis and differential scanning calorimetry (TGA and DSC) analysis was done on a Mettler-Toledo TGA DSC 1 Star system thermobalance using alumina crucibles under a stream of nitrogen (50 mL min−1) and a heating rate of 5 °C min−1 from 25 °C until 200 °C, and in a stream of air (50 mL min−1) at a heating rate of 10 °C min−1 from 200 °C to 700 °C. Sample size was between 2 mg and 10 mg.Deuterated ST1 and ZH
Deuterated ST1 and ZH were prepared by first fully dehydrating the parent phases NaMgFe(C2O4)3·9H2O (ST1) and NaMgAl(C2O4)3·9H2O under vacuum at 70 °C, after which the materials were dissolved in D2O and recrystallized in a dry N2 atmosphere. Deuteration was confirmed by FTIR-ATR, and the identity of samples was verified by PXRD.Alternative current (AC) impedance measurement
Alternative current (AC) impedance measurement was performed by the conventional two-probe method. Samples (ST1, ST2, and ZH1) were pelletized about ∼1.2 mm in thickness and 2.5 mm in diameter. The pellet was connected to electrode with gold paste and gold wires (50 μm in diameter). The test is performed in a temperature- and humidity-controlled chamber, which was connected to a Solartron SI 1260 Impedance/Gain-Phase Analyzer and 1296 Dielectric Interface. The impedance measurement was executed in the frequency range from 1 Hz to 10 MHz at 298 K. Proton conductivity and activation energy barrier were estimated by eqn (1) and (2), where σ is the conductivity (S cm−1), L is the measured sample thickness (cm), S is the electrode area (cm2) and Z is the impedance value. In eqn (2), A is a pre-exponential factor, k is the Boltzmann constant, and Ea is the activation energy of ionic conduction.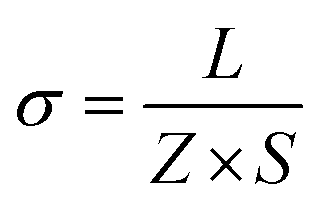 | (1) |
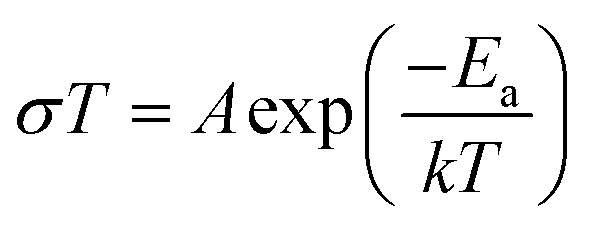 | (2) |
Water vapor sorption measurements
Water vapor sorption measurements were carried out on ZH, ST1, and ST2 using an automated micropore gas analyser BELSORP-max (MicrotracBEL Corp.). Before measurement, the water for adsorbate was degassed through a freeze-thaw cycle, and all samples were activated at 80 °C under vacuum (10−3 Pa) overnight. The water isotherms were measured at each equilibrium pressure by the static volumetric method at 298 K under P/P0 = 0.95.Calorimetric measurements
Calorimetric measurements to determine the enthalpies of dissolution (ΔHds) of MOF samples (ST1, ST2 and ZH) and starting materials were performed using a CSC (Calorimetry Sciences Corporation) 4400 isothermal microcalorimeter at 25 °C. Around 5 mg of the sample was hand-pressed to form a pellet and dropped to a Teflon cell in the calorimeter, filled with 25 g of 5 molar aqueous HCl solution. The solvent was isothermally equilibrated for at least 3 h under mechanical stirring before the introduction of the sample, and the sample was allowed to dissolve in the cell for at least 2 hours, ensuring the return of baseline back to its initial position. In each experiment, the solvent in the cell was replaced with new, fresh solvent.The calibration of the instrument was performed using a NIST standard reference material KCl. The calibration was done by dissolving 15 mg of the KCl pellet into 25 g of H2O (type-1, resistivity = 18.2 MΩ cm), which corresponds to a reference concentration of 0.008 mol kg−1 at 25 °C. The calibration factor was obtained by correlating the integrated data with a known enthalpy of dissolution and dilution of 0.008 mol kg−1 KCl. For each sample, at least 4 measurements were performed. The uncertainties given in the result represents 95% confidence interval.18 Due to incomplete dissolution of γ-FeOOH in 5 molar HCl, its exact concentration was determined by inductively coupled plasma mass spectrometry (ICP-MS) using an Agilent 8900 ICP-MS. The supernatant after calorimetry measurement was diluted 10X and 100X with 3% HNO3 (by volume) prepared from concentrated Trace Metal Grade HNO3 (Fisher Scientific) and deionized water (resistivity value of 18.2 MΩ cm).
Results and discussion
Synthetic ZH was prepared following our previous procedure (see ESI†).11 Analysis of synthetic ZH by PXRD was consistent with the reported mineral structure, based on hcb-layers stacked along the 12.6 Å crystallographic c-axis to form channels filled with Mg(H2O)62+ ions. The mineral ST exhibits a different structure,11 in which hcb-layers stack in ABCA′B′C′ fashion, resulting in a longer crystallographic c-axis of ∼37 Å (Fig. 1b and c, where the A and A′ notations indicate layers with the same overall positions but rotated around the c-axis by 180° relative to each other).9–11 However, a crystallographic study of synthetic samples by Piro et al.19 suggested that ST is isostructural to ZH, i.e. exhibiting AB layer stacking along a 12.6 Å c-axis. To resolve this discrepancy, we have now systematically investigated crystallization of ST under a variety of conditions, unexpectedly revealing that slow evaporation from water produces large hexagonal plate-like crystals, whereas rapid evaporation produces elongated crystals with a hexagonal cross-section. Single crystal X-ray diffraction revealed that slowly grown plates (termed stepanovite 1, ST1) are isostructural to the mineral, with ABCA′B′C′ stacking of hcb-sheets and a ∼37 Å c-axis perpendicular to large hexagonal crystal faces (Table 1, entries 1, 2). In contrast, elongated crystals (termed stepanovite 2, ST2) exhibit structural features and crystallographic parameters (Table 1, entries 3, 4) similar to ZH, including AB-stacking of hcb-sheets and guest-filled channels parallel to a 12.4 Å c-axis (Fig. 1e and f). Identical results were observed using X-ray diffraction data collected at room temperature and at 100 K, confirming that the difference between structures of ST1 and ST2 is not related to temperature. The existence of ST2, a polymorph of synthetic stepanovite that is isostructural to zhemchuzhnikovite, offers an explanation of discrepancy between mineral structure and the synthetic sample structure reported by Piro et al.19Stabilities of synthetic ST1, ST2, and ZH were investigated by room temperature acid dissolution calorimetry.18,20 The enthalpies of formation of synthetic minerals were calculated from herein measured dissolution enthalpies (ΔHds) of the minerals and relevant starting materials (oxalic acid, binary oxides, hydroxides, and oxyhydroxides), and a properly designed thermodynamic cycle (see ESI† for details and equations). This enabled the evaluation of relative energetic stabilities (Table 2) of both ST polymorphs and ZH, as well as their enthalpies of formation from oxides (ΔHf,ox) and from the elements (ΔHf,el).
| Sample | ΔHs | ΔHf,ox | ΔHf,el |
|---|---|---|---|
| ST1 | 91.38 ± 0.66 | −422.31 ± 2.29 | −5847.41 ± 2.59 |
| ST2 | 92.15 ± 0.47 | −423.07 ± 2.24 | −5848.19 ± 2.55 |
| ZH | 64.81 ± 1.06 | −434.03 ± 3.20 | −6113.98 ± 3.43 |
The very exothermic ΔHf,ox indicates that the formation of ST1, ST2 and ZH from binary oxides is thermodynamically driven. The results also show that the difference in energetic stability of ST1 and ST2 is within the experimental error of calorimetric measurements. However, ZH has a noticeably more exothermic enthalpy of formation from oxides compared to ST polymorphs, indicating that structure stabilization by substitution of iron by aluminum is far larger than the effect of polymorphism. This trend of greater stability relative to binary oxide components for the aluminum compounds relative to the iron ones has been seen in many inorganic systems containing both Al3+ and Fe3+, such as in spinels,21 jarosite–alunite and natrojarosite–natroalunite solid solutions22 and zoisite, clinozoisite solid solutions.23
Although ST1 and ST2 are very similar in enthalpy, there is some indication from different growth conditions that ST2 is a kinetic polymorph, while ST1 is thermodynamically preferred. This is supported by the observation that ST2 converts to ST1 by shaking or brief milling with small amounts of water. The metastable character of ST2 compared to ST1 would agree with Goldsmith's simplexity principle,24 stating that kinetically-favored phases are often structurally simpler than their thermodynamically stable counterparts.25 Our calculations of information-based structural complexity parameters26 show that ST1 has a very complex structure (IG = 5.618 bits/atom; IG,total = 1617.979 bits/cell), whereas the structure of ST2 is intermediate in complexity (IG = 4.099 bits/atom; IG,total = 393.510 bits/cell).
Analysis by TGA (see ESI†) and variable-temperature powder X-ray diffraction (VT-PXRD, Fig. 2a and b) show that ST1 and ST2 lose three equivalents of guest water molecules upon mild heating, forming new crystalline phases ST1d and ST2d, respectively. Furthermore, crystals of ST1 and ST2 retained their integrity upon dehydration, providing diffraction-quality crystals of ST1d and ST2d. Structure analysis shows that dehydration leads to partial (ST1) or complete (ST2) corrugation of hcb-layers (Fig. 3), which lowers the space group symmetry of ST1d and ST2d compared to the parent phases (Table 1, entries 5, 6). In ST1d the hcb-layers adopt a more complex ABCDEF repeat pattern as a result of the original ABCA′B′C′-stacking now being combined with alternation of flat and corrugated sheets. In ST2d, each hcb-sheet is corrugated, preserving the AB-stacking of ST2.
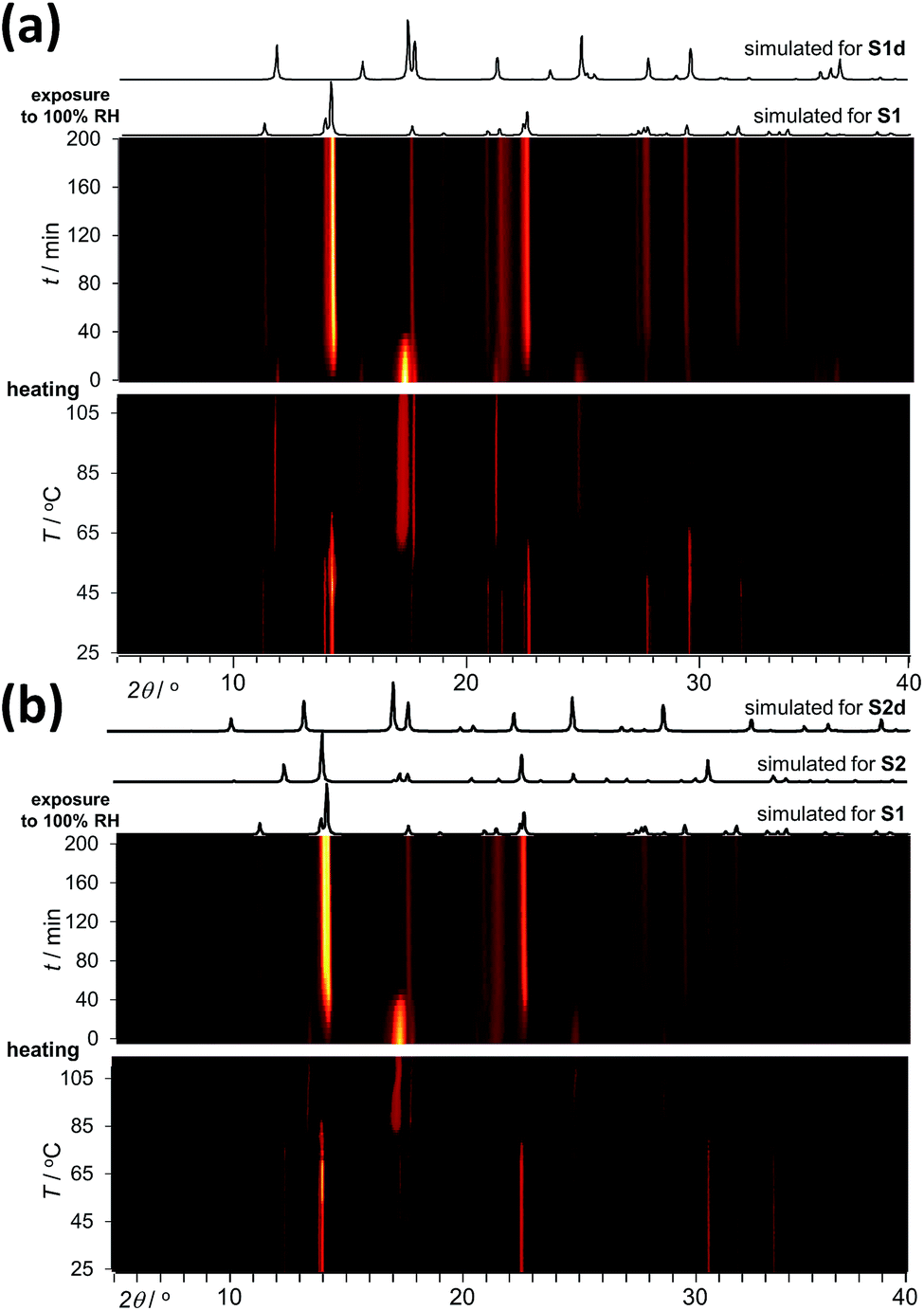 | ||
| Fig. 2 PXRD data for a powdered sample of (a) ST1 and (b) ST2 being (bottom) first dehydrated by heating (2 °C per hour, from 25 °C to 110 °C) and subsequently (top) exposed to 100% RH at room temperature. Simulated PXRD patterns for ST1, ST2, ST1d and ST2d are shown above the in situ data plots. | ||
 | ||
| Fig. 3 Arrangement of hcb-layers in (a) ST1d and (b) ST2d, viewed perpendicular to the crystallographic c-direction. | ||
The accessibility of bulk synthetic samples for ST1, ST2 and ZH enabled the evaluation of suspected proton conductivity by AC impedance measurement on compacted powder pellets. For proton conductivity, all Nyquist plots were obtained under controlled relative humidity (RH) and temperature (see ESI†). Impedance measurements revealed significant enhancement of proton conductivity (σ) in ST1, ST2 and ZH with relative humidity (Fig. 4a). At 90% RH synthetic ZH exhibits a high σ of almost 3 × 10−3 S cm−1, on par with some of the highest room temperature proton conductive MOFs.27–29 Proton conductivity of synthetic ST1 is also high, but almost an order of magnitude lower than that of ZH. Both ST1 and ZH remained crystalline upon conductivity evaluation, as verified by PXRD patterns recorded after the measurements. Proton conductivity for ST2 could not be measured above 70% RH, due to deliquescence at higher RH values. However, at all explored RH levels below 70% the conductivity of ST2 was similar to that of ZH and consistently higher than for ST1. Indeed, at 70% RH and 25 °C the proton conductivity of ST2 is among the top values reported for MOFs at modest humidity and room temperature.30
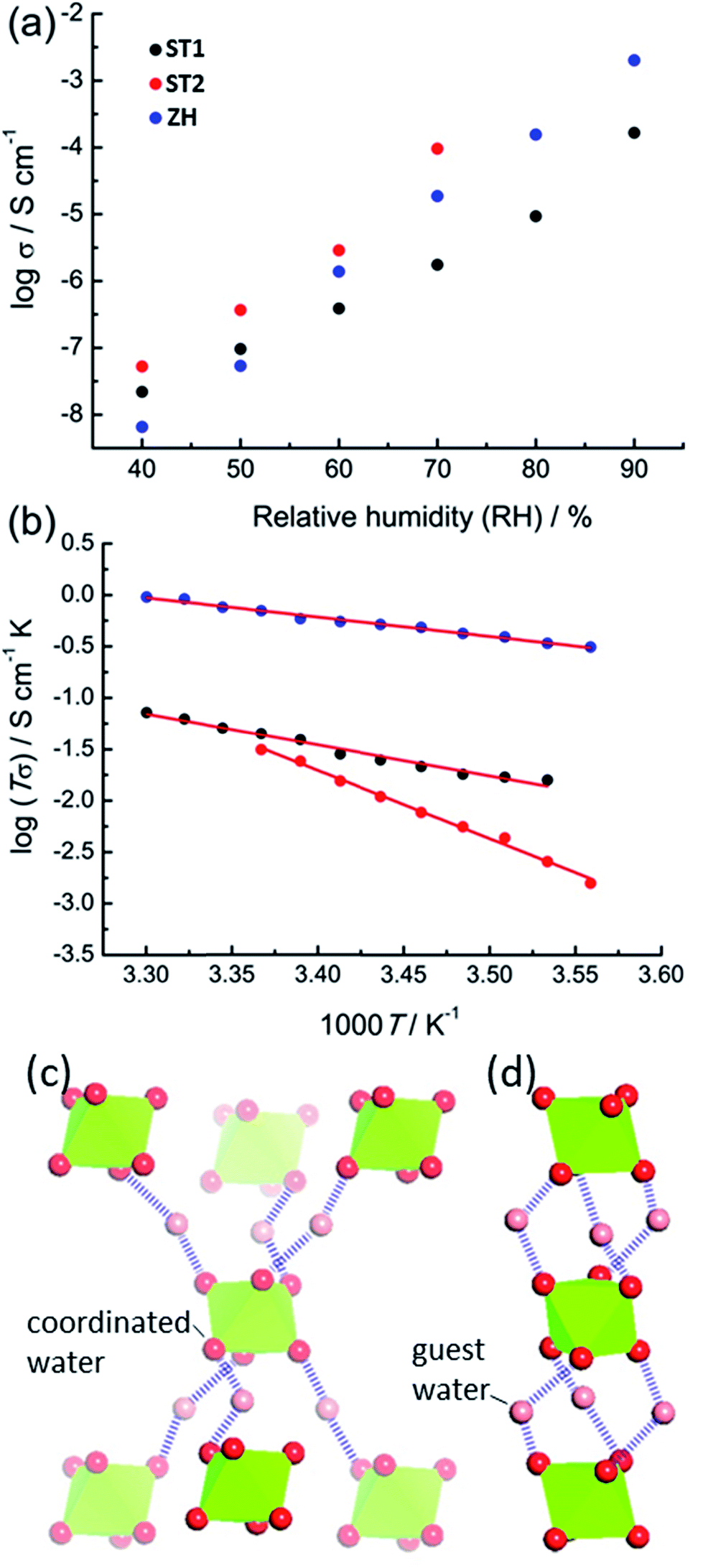 | ||
| Fig. 4 (a) Proton conductivity (σ) at 25 °C for synthetic ST1 (black), ST2 (red) and ZH (blue) at different RH. (b) Arrhenius plots of the proton conductivities for ST1(black, at 90% RH), ST2 (red, at 70% RH), and ZH (blue, at 90% RH), with least-squares fits shown as solid lines. Fragments of hydrogen-bonded networks involving guest water molecules and Mg(H2O)62+ ions in: (c) ST1 and (d) ST2. For clarity, hcb-layers are omitted and Mg(H2O)62+ ions are shown as green octahedra. | ||
The two main mechanisms for proton diffusion are classfied by activation energy (Ea): the vehicle mechanism (Ea > 0.4 eV) and the Grotthuss mechanism (Ea < 0.4 eV).31,32 The activation energies for proton conduction in ST1 (0.59 eV) and ZH (0.37 eV) at 90% RH were established using the Arrhenius method (Fig. 4b). The values are consistent with a mixed Grotthuss–Vehicle proton diffusion mechanism, which puts these materials in the group of superionic conductors, as described by Colomban et al.33 The Ea for ST2 is considerably larger (1.31 eV) compared to ZH and ST1, due to measurements being performed at 70% RH. In addition, PXRD pattern of ST2 after conductivity studies at different temperatures revealed partial structural transformation to ST1, which could also have affected the high Ea (see ESI†).
It was reported that high proton conductivity in oxalate MOFs (≥10−3 S cm−1 at 25 °C, 98% RH) requires the inclusion of additional proton-carrying guests, such as weakly acidic NH4+ or adipic acid.14,34,35 In such systems, conductivity is strongly influenced by the type and organization of proton-carrying guests.36 As the conductivities and activation energies in ST1, ST2 and ZH are comparable to such systems, it is likely that Mg(H2O)62+ ions, in which the proton-donating ability of water molecules is enhanced by coordination to Mg2+, act as such protic guests. Differences in σ between ST1, ST2 and ZH can tentatively be related to different topologies of hydrogen-bonded frameworks formed by Mg(H2O)62+ ions and water guests (Fig. 4c). As ST1 and ST2 have an identical composition, and ST1 and ZH share the same crystallographic structure, it is reasonable to assume that the difference in conduction properties of ST1 and ST2 are associated with ST2 adopting a ZH-like hydrogen-bonded guest structure. To verify whether conductivity is due to proton conduction or contributions of other ionic species, additional impedance measurements were performed on deuterated ST1 and ZH at 25 °C and ca. 70% saturated D2O vapor (see ESI†).37 Measurements on both samples revealed a reduction in conductivity upon deuteration, which is consistent with proton conduction. Specifically, measurements on deuterated ST1 revealed a ca. 1.3-fold reduction in conductivity (from 1.76 × 10−6 S cm−1 to 1.40 × 10−6 S cm−1) along with a comparable activation energy (0.51 eV). For deuterated ZH a more significant 5.8-fold reduction in conductivity was observed (from 1.87 × 10−5 S cm−1 to 3.22 × 10−6 S cm−1), along with a small increase in activation energy (0.51 eV, see ESI†).
Conclusions
In summary, we have shown that MOF minerals zhemchuzhnikovite and stepanovite can exhibit high proton conductivity, enabled by hydrogen-bonding networks involving interstitial water molecules, hydrated metal cation guests and the oxalate based-framework. At the same time, we report the possibility of polymorphism in stepanovite, which enabled observing the effect of hydrogen-bonded framework topology on proton conductivity of an oxalate-based MOF. All of the MOF minerals studied were found to be very stable with respect to the binary oxides, and substitution of iron by aluminum was found to improve the stability of zhemchuzhnikovite. Upon heating, stepanovite polymorphs lose included water guests in a crystal-to-crystal fashion, i.e. without disrupting the 2-dimensional open framework layers.Overall, these results are highly significant in the contexts of geology and materials chemistry, as they demonstrate that naturally occurring MOFs containing small oxalate ligands are thermodynamically stable phases that exhibit functional properties comparable to previously reported advanced materials, including high proton conductivity and framework retention at elevated temperature. We note that this work also presents a simple design for synthesizing high-performing proton conductive materials without sacrificing thermodynamic stability or requiring complex organic components. We are currently investigating the properties of other MOFs based on this design.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by NSERC Discovery Grant (RGPIN-2017-06467) and NSERC E. W. R. Steacie Memorial Fellowship (SMFSU 507347-17). Calorimetry at UC Davis was supported by the U.S. Department of Energy Office of Science, grant DE-SC0016573. D.-W. Lim, and H. Kitagawa acknowledge the support from the ACCEL program, Japan Science and Technology Agency (JST), JPMJAC1501.Notes and references
- T. Echigo and M. Kimata, Can. Mineral., 2010, 48, 1329–1357 CrossRef CAS.
- I. Huskić and T. Friščić, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2018, 74, 539–559 CrossRef.
- S. A. Benner, K. G. Devine, L. N. Matveeva and D. H. Powell, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2425–2430 CrossRef CAS PubMed.
- H. E. Maynard-Casely, M. L. Cable, M. J. Malaska, T. H. Vu, M. Choukroun and R. Hodyss, Am. Mineral., 2018, 103, 343–349 CrossRef.
- (a) T. Echigo and M. Kimata, Phys. Chem. Miner., 2008, 35, 467–475 CrossRef CAS; (b) T. Yamada, M. Sadakkiyo and H. Kitagawa, J. Am. Chem. Soc., 2009, 131, 3144–3145 CrossRef CAS PubMed.
- (a) D. R. Hummer, B. C. Noll, R. M. Hazen and R. T. Downs, Am. Mineral., 2017, 102, 1129–1132 Search PubMed; (b) C. B. Storm, J. Krane, T. Skjetne, N. Telnaes, J. F. Branthaver and E. W. Baker, Science, 1984, 223, 1075–1076 CrossRef CAS PubMed.
- https://mineralchallenge.net/ .
- R. M. Hazen, D. R. Hummer, G. Hystad, R. T. Downs and J. J. Golden, Am. Mineral., 2016, 101, 889–906 CrossRef.
- Y. N. Knipovich, A. I. Komkov and E. I. Nefedov, Tr. Vses. Neft. Nauchno-Issled. Geologorazved. Inst., 1963, 96, 131–135 CAS.
- M. Fleischer, Am. Mineral., 1964, 49, 439–448 Search PubMed.
- I. Huskić, I. V. Pekov, S. V. Krivovichev and T. Friščić, Sci. Adv., 2016, 2, e1600621 CrossRef PubMed.
- (a) M. Sadakiyo, T. Yamada and H. Kitagawa, ChemPlusChem, 2016, 81, 691–701 CrossRef CAS; (b) M. Dan and C. N. R. Rao, Angew. Chem., Int. Ed., 2006, 45, 281–285 CrossRef CAS PubMed; (c) C. N. R. Rao, S. Natarajan and R. Vaidhyanathan, Angew. Chem., Int. Ed., 2004, 43, 1466–1496 CrossRef CAS PubMed; (d) R. Vaidhyanathan, S. Natarajan, A. K. Cheetham and C. N. R. Rao, Chem. Mater., 1999, 11, 3636–3642 CrossRef CAS.
- (a) H. Ōkawa, A. Shigematsu, M. Sadakiyo, T. Miyagawa, K. Yoneda, M. Ohba and H. Kitagawa, J. Am. Chem. Soc., 2009, 131, 13516–13522 CrossRef PubMed; (b) T. Yamada, M. Sadakiyo, A. Shigematsu and H. Kitagawa, Bull. Chem. Soc. Jpn., 2016, 89, 1–10 CrossRef CAS; (c) S. Descurtins, H. W. Schmalle, P. Schneuwly, J. Ensling and P. A. Gütlich, J. Am. Chem. Soc., 1994, 116, 9521–9528 CrossRef.
- (a) D.-W. Lim, M. Sadakiyo and H. Kitagawa, Chem. Sci., 2019, 10, 16–33 RSC; (b) M. Sadakiyo, T. Yamada, K. Honda, H. Matsui and H. Kitagawa, J. Am. Chem. Soc., 2014, 136, 7701–7707 CrossRef CAS PubMed.
- I. Huskić, J.-C. Christopherson, K. Užarević and T. Friščić, Chem. Commun., 2016, 52, 5120–5123 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- J. T. Hughes, T. D. Bennett, A. K. Cheetham and A. Navrotsky, J. Am. Chem. Soc., 2013, 135, 598–601 CrossRef CAS PubMed.
- O. E. Piro, G. A. Echeverría, A. C. González-Baró and E. J. Baran, Phys. Chem. Miner., 2016, 43, 287–300 CrossRef CAS.
- Z. Akimbekov, A. D. Katsenis, G. P. Nagabushana, G. Ayoub, M. Arhangelskis, A. J. Morris, T. Friščić and A. Navrotsky, J. Am. Chem. Soc., 2017, 139, 7952–7957 CrossRef CAS PubMed.
- A. Navrotsky and O. J. Kleppa, J. Inorg. Nucl. Chem., 1968, 30, 479–498 CrossRef CAS.
- C. Drouet, K. L. Pass, D. Baron, S. Draucker and A. Navrotsky, Geochim. Cosmochim. Acta, 2004, 68, 2197–2205 CrossRef CAS.
- E. A. Smelik, G. Franz and A. Navrotsky, Am. Mineral., 2001, 86, 80–91 CrossRef CAS.
- J. R. Goldsmith, J. Geol., 1953, 61, 439–451 CrossRef CAS.
- (a) S. V. Krivovichev, Mineral. Mag., 2013, 77, 275–326 CrossRef CAS; (b) J. Cempirek, E. S. Grew, A. R. Kampf, C. Ma, M. Novak, P. Gadas, R. Skoda, M. Vasinova-Galiova, F. Pezzotta, L. A. Groat and S. V. Krivovichev, Am. Mineral., 2016, 101, 2108–2117 CrossRef; (c) J. Plášil, V. Petříček and J. Majzlan, Acta Crystallogr., Sect. A: Found. Adv., 2017, B73, 856–862 CrossRef PubMed; (d) J. Majzlan, E. Dachs, A. Benisek, J. Plášil and J. Sejkora, Eur. J. Mineral., 2018, 30, 259–275 CrossRef CAS.
- S. V. Krivovichev, Angew. Chem., Int. Ed., 2014, 53, 654–661 CrossRef CAS PubMed.
- X. Meng, H.-N. Wang, S.-Y. Song and H.-J. Zhang, Chem. Soc. Rev., 2017, 46, 464–480 RSC.
- M. R. Karim, K. Hatakeyama, M. Koinuma and S. Hayami, J. Mater. Chem. A, 2017, 5, 7243–7256 RSC.
- P. Ramaswamy, N. E. Wong and G. K. H. Shimizu, Chem. Soc. Rev., 2014, 43, 5913–5932 RSC.
- B. Joarder, J.-B. Lin, Z. H. Romero and G. K. H. Shimizu, J. Am. Chem. Soc., 2017, 139, 7176–7179 CrossRef CAS PubMed.
- K.-D. Kreuer, A. Rabenau and W. Weppner, Angew. Chem., Int. Ed., 1982, 21, 208–209 CrossRef.
- (a) C. J. T. de Grotthuss, Ann. Chim., 1806, 58, 54–73 Search PubMed; (b) N. Agmon, Chem. Phys. Lett., 1995, 244, 456–462 CrossRef CAS.
- P. Colomban and A. Novak, J. Mol. Struct., 1988, 177, 277–308 CrossRef.
- M. Sadakiyo, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2009, 131, 9906–9907 CrossRef CAS PubMed.
- E. Pardo, C. Train, G. Gontard, K. Boubekeur, O. Fabelo, H. Liu, B. Dkhil, F. Lloret, K. Nakagawa, H. Tokoro, S. Ohkoshi and M. Verdaguer, J. Am. Chem. Soc., 2011, 133, 15328–15331 CrossRef CAS PubMed.
- (a) M. Sadakiyo, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2014, 136, 13166–13169 CrossRef CAS PubMed; (b) S. Miyatsu, M. Kofu, A. Nagoe, T. Yamada, M. Sadakiyo, T. Yamada, H. Kitagawa, M. Tyagi, V. García Sakai and O. Yamamuro, Phys. Chem. Chem. Phys., 2014, 16, 17295–17304 RSC.
- (a) S. C. Sahoo, T. Kundu and R. Banerjee, J. Am. Chem. Soc., 2011, 133, 17950–17958 CrossRef CAS PubMed; (b) B. W. Kang, J. H. Song, K. J. Lee, H. G. Lee, J. E. Kim, H. Y. Lee, J. Y. Kim and C. S. Hong, J. Mater. Chem. A, 2017, 5, 17492–17498 RSC; (c) Q. Tang, Y. Liu, S. Liu, D. He, J. Miao, X. Wang, G. Yang, Z. Shi and Z. Zheng, J. Am. Chem. Soc., 2014, 136, 12444–12449 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1868681–1868685. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05088k |
| This journal is © The Royal Society of Chemistry 2019 |