
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArtificial chimeric exosomes for anti-phagocytosis and targeted cancer therapy†
Kai-Long
Zhang
a,
Ying-Jie
Wang
a,
Jin
Sun
bc,
Jie
Zhou
a,
Chao
Xing
a,
Guoming
Huang
 c,
Juan
Li
*ab and
Huanghao
Yang
*a
c,
Juan
Li
*ab and
Huanghao
Yang
*a
aMOE Key Laboratory for Analytical Science of Food Safety and Biology, Fujian Provincial Key Laboratory of Analysis and Detection Technology for Food Safety, State Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou 350116, P. R. China. E-mail: lijuan@fzu.edu.cn; hhyang@fzu.edu.cn
bInstitute of Molecular Medicine, Renji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai Jiao Tong University, Shanghai, 200240, P. R. China
cCollege of Biological Science and Engineering, Fuzhou University, Fuzhou 350116, P. R. China
First published on 27th November 2018
Abstract
Development of exosome-based delivery systems is still facing some formidable challenges, including the lack of standardized isolation and purification methods, non-large-scale production and low drug-loading efficiency. Inspired by biomimetic technologies, we turned to the design of artificial chimeric exosomes (ACEs) constructed by integrating cell membrane proteins from multiple cell types into synthetic phospholipid bilayers. For benchmarking, hybrid membrane proteins derived from red blood cells (RBCs) and MCF-7 cancer cells were selected as models. The resulting ACEs were engineered much like “Emperor Qin's Terra-Cotta Warriors”, simultaneously equipped with armor (anti-phagocytosis capability from RBCs) and dagger-axes (homologous targeting ability from cancer cells). ACEs demonstrated higher tumor accumulation, lower interception and better antitumor therapeutic effect than plain liposomes in vivo, alongside large-scale standardized preparation, stable structure, high drug-loading capacity and custom-tailored functionality, highlighting the suitability of ACEs as promising alternatives of exosomes in clinical applications.
1. Introduction
Exosomes are nanosized and membrane-enclosed extracellular vesicles derived from most cells, which are found in blood, saliva and other biological fluids.1–3 They carry diverse biomolecules, including proteins, mRNAs, miRNAs, DNA and lipid molecules, carrying information related to their source cells.4–7 Generally, exosomes are regarded as important mediators of intercellular communication and thus play critical roles in regulating many physiological and pathological processes, including immune responses, inflammation, infection and tumor growth.8–11Furthermore, as natural vesicles, exosomes hold great promise to become ideal delivery systems for biomedical application.12 Up to now, several exosome-based vehicles derived from stem cells,13,14 dendritic cells15–17 and cancer cells18–21 have emerged for delivery of therapeutic nucleic acids,22 proteins23 and drugs.24–26 Self-derived exosomes provide multiple advantages over traditional synthetic delivery vehicles,27,28 such as limited immunogenicity, intrinsic cell targeting properties, enhanced stability in circulation and innate biocompatibility.29 In particular, the presence of CD47 on exosomes plays a key role in evading phagocytosis and increasing accumulation in tumor tissue.30 CD47 is a transmembrane protein overexpressed on the surface of red blood cells (RBCs), which is capable of interacting with its receptor signal regulatory protein alpha (SIRPα) present on macrophages.31 The binding interaction between CD47 and SIRPα initiates the “don't eat me” signal to protect RBCs or exosomes from clearance.30,32–34 Additionally, exosome-based vesicles, or membrane-derived vesicles, demonstrated a homologous targeting ability as a result of inheriting homologous surface adhesion proteins from their source cells.35–39 Despite these advantages, some challenges still need to be overcome before exosome-based vesicles can be used for broad applications. First, it is difficult to achieve standardized isolation and purification at a suitable clinical grade.40,41 Another challenge is the lack of approaches for efficient loading of drugs or genes into exosomes.42–44
In consideration of these challenges, the design and fabrication of artificial exosome-mimetic nanovesicles would be of great interest.45 However, studies on artificial exosomes are still very rare. Herein, to address the aforementioned limitations of exosomes, we designed artificial chimeric exosomes (ACEs) using a combined bottom-up and top-down approach (Scheme 1).46,47 It is well known that exosomes have a phospholipid bilayer structure similar to that of nanoliposomes. Due to the special nature (hydrophobicity) in the transmembrane domain of cell membrane proteins, we hypothesize cell membrane proteins would be embedded into phospholipid bilayers during the nanoliposomes assemble. Along this line, we constructed nanometer scale ACEs by integrating cell membrane proteins from multiple cell types into synthetic phospholipid bilayers. In this case, hybrid membrane proteins derived from RBCs and MCF-7 cancer cells were selected as models. Membrane proteins from RBCs contain high CD47 levels, which are capable of inhibiting phagocytosis, while those from MCF-7 cancer cells contain specific adhesion proteins, which can adhere to homologous cancer cells. Meanwhile, the FDA-approved fluorescent dye indocyanine green (ICG) and doxorubicin (DOX) were loaded into ACEs as a traceable fluorescent probe and a model drug, respectively.48–50 The resulting ACEs were engineered like “Emperor Qin's Terra-Cotta Warriors”, the Terra-Cotta Army said to protect the tomb of China's first emperor, which is an apt metaphorical reference. The ACEs were simultaneously equipped with armor (anti-phagocytosis capability from RBCs) and dagger-axes (homologous targeting ability from cancer cells). Importantly, such ACEs can be easily custom-tailored and produced on a large-scale with standardized preparation, stable structure and high drug-loading capacity, making them promising as the alternative of exosomes in clinical medicine.
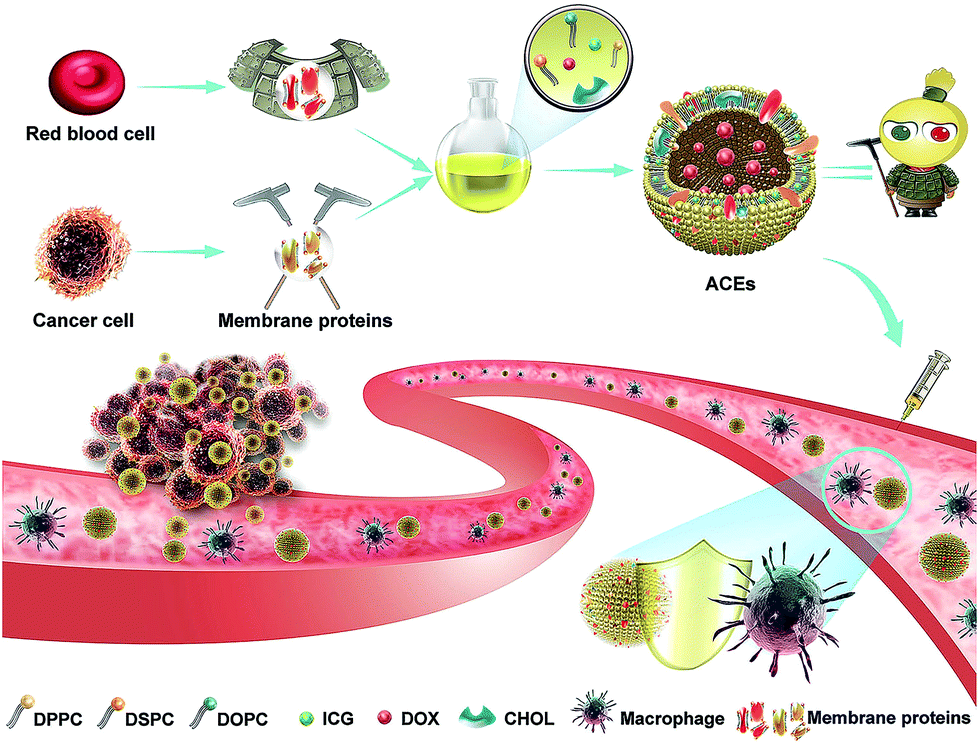 | ||
| Scheme 1 Schematic illustration of the design of biomimetic ACEs for anti-phagocytosis and targeted cancer therapy. ACEs were constructed by integrating cell membrane proteins from multiple cell types into synthetic phospholipid bilayers. | ||
2. Results and discussion
2.1. Preparation and characterization of ACEs
As a proof of concept, biomimetic ACEs were developed by integrating hybrid RBCs and MCF-7 cell membrane proteins into synthetic phospholipid bilayers. To better demonstrate the advantage of hybrid functionalities of ACEs, we fabricated artificial RBC exosomes (AREs) and artificial MCF-7 cell exosomes (AMEs) by integrating single-membrane protein formulations and liposomes without any membrane proteins. As observed by transmission electron microscopy (TEM) and dynamic light scattering (DLS), ACEs were obtained with spherical morphology and homogeneous size (∼100 nm) similar to exosomes (40–150 nm) (Fig. 1a and b). The morphology and size of liposomes, AREs and AMEs were consistent with ACEs (Fig. S1–S3†). In Fig. 1c, the zeta potentials of AREs, AMEs and ACEs were uniform (∼−17 mV) and more positive than that of liposomes (∼−24 mV), possibly a result of the shielding effect of membrane proteins towards the negative charge of the phosphate groups. Topographical analysis by atomic force microscopy (AFM) validated the size of vesicles as measured with DLS and TEM. Also, the AFM images presented increased surface roughness of ACEs compared with liposomes, suggesting the presence of hinged structures in their bilayers (Fig. 1d and e).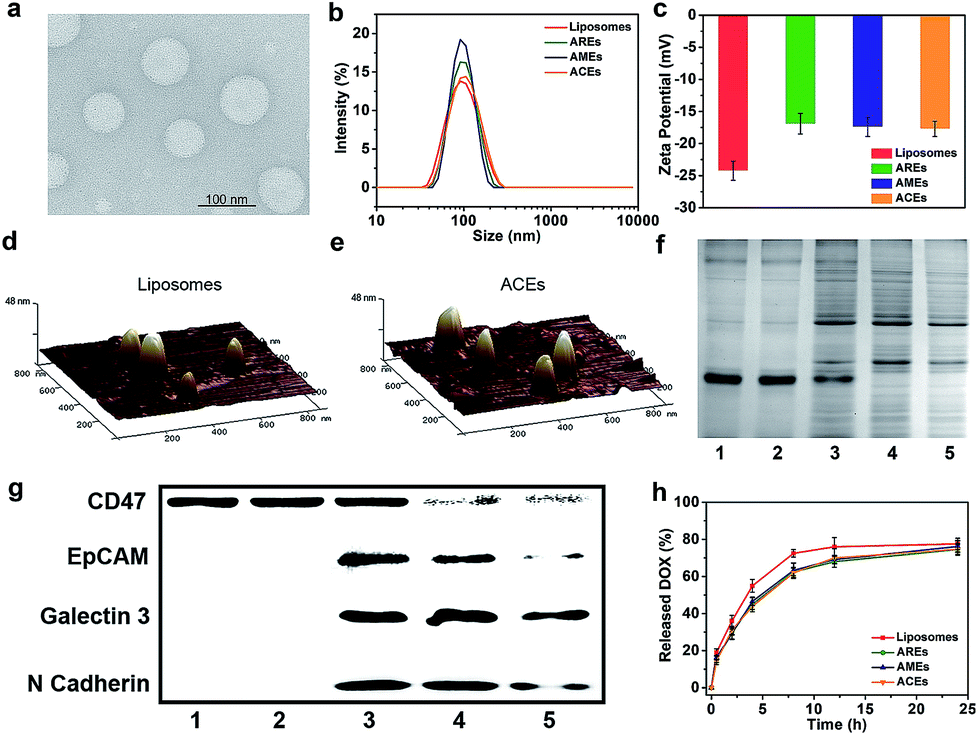 | ||
| Fig. 1 Physicochemical characterization and membrane protein composition analysis of ACEs. (a) TEM images of ACEs, negatively stained with 3% phosphotungstic acid. (b) Hydrodynamic diameter and (c) zeta potential of liposomes, AREs, AMEs and ACEs (means ± SD, n = 3). AFM images of (d) liposomes and (e) ACEs reveal the presence of hinged structures on the surface of ACEs. (f) Protein content visualization of (1) RBCs, (2) AREs, (3) ACEs, (4) AMEs and (5) MCF-7 cells. (g) Membrane protein characterization by Western blotting analysis of (1) RBCs, (2) AREs, (3) ACEs, (4) AMEs and (5) MCF-7 cells. (h) In vitro DOX-release behaviour at 37 °C (means ± SD, n = 3). | ||
Incorporation of the hybrid membrane proteins within the bilayers of ACEs was then examined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting. Compared with AMEs and AREs, ACEs had a profile that represented the union of two single-cell protein formulations. As expected, AMEs and AREs had protein profiles that largely mirrored the corresponding source cell proteins (Fig. 1f). To analyze specific protein markers, Western blotting analysis was carried out (Fig. 1g). CD47,31 a marker of self on RBCs, was present on RBCs, AREs and ACEs, and also, to a lesser degree, on AMEs and MCF-7 cells. EpCAM, Galectin 3 and N Cadherin are important cellular adhesion molecules35–39 on MCF-7 cells, and all were present on MCF-7 cells, ACEs and AMEs, but not on RBCs and AREs. ACEs simultaneously present both RBC-specific and MCF-7 cells-specific markers. Furthermore, the simultaneous presence of RBCs membrane proteins and MCF-7 cell membrane proteins on each individual ACE was examined by high-sensitivity flow cytometer (HSFCM). RBCs membrane proteins and MCF-7 cell membrane proteins were respectively labelled with TAMRA-SE dyes and FITC dyes before the preparation of ACEs. The flow cytometry data showed most of each individual ACE simultaneously contained RBCs membrane proteins and MCF-7 cell membrane proteins (over 98%), suggesting that ACEs was not merely a mixture of AREs and AMEs (Fig. S4†). The incorporation efficiency of membrane protein on AREs, AMEs and ACEs was similar and determined to be about 45% using the BCA assay kit (Table S1†). To further demonstrate that the membrane proteins integrated into the phospholipid bilayers still retained primary biological ability, we used transferrin receptor (TfR) as a model. ACEs and liposomes were respectively incubated with transferrin-conjugated gold nanoparticles (5 nm), followed by TEM imaging (Fig. S5†). Results showed that gold nanoparticles could interact with TfR on the surface of ACEs, suggesting that they retained biofunctionality. Ultimately, the assays carried out here indicated that we had successfully fabricated the biomimetic ACEs that mimicked the morphological and physiological composition of exosomes.
Next, we carried out a series of assays to investigate the ability of ACEs as drug carriers. DOX encapsulation efficiencies of liposomes, AREs, AMEs and ACEs were respectively 93.6 ± 1.2%, 91.2 ± 2.0%, 91.4 ± 2.3% and 90.7 ± 1.8%, and the ICG encapsulation efficiencies of liposomes, AREs, AMEs and ACEs were respectively 90.2 ± 0.8%, 83.4 ± 1.3%, 84.1 ± 0.9% and 83.9 ± 1.1% (Table S1†). These results demonstrated that the incorporation of membrane proteins did not have a significant impact on drug-loading ability. The release profiles of DOX from all groups were investigated in PBS of pH 7.4 at 37 °C. AREs, AMEs and ACEs presented similar release profiles in PBS and a slight delay compared with liposomes (Fig. 1h). Then, we investigated vesicle stability in PBS (pH 7.4) and 20% FBS media. DLS analysis revealed that all vesicles were stable without significant change in vesicle size within 24 hours (Fig. S6†). These results demonstrated that ACEs could effectively achieve drug loading and drug release.
2.2. Intracellular uptake and cytotoxicity of ACEs
In an effort to investigate homologous targeting ability, the intracellular uptake of DOX-loaded ACEs in MCF-7 cells (target cells) and HeLa cells (control cells) was studied (Fig. 2a). In MCF-7 cancer cells, the fluorescence intensity of cells treated with ACEs was much stronger than that of the liposome-treated group, indicating that the hybrid membrane proteins on the surface of ACEs remarkably increased the cellular uptake of vesicles (Fig. S7†). To confirm the crucial role of MCF-7 cancer cell membrane proteins in facilitating internalization with MCF-7 cells, the intracellular uptake of AREs and AMEs in MCF-7 cancer cells was evaluated. The AMEs-treated group exhibited fluorescence intensity similar to that of the ACEs-treated group and obviously enhanced fluorescence intensity compared with that of the AREs-treated group, suggesting that the enhanced internalization of ACEs was caused by membrane proteins of the MCF-7 cells. In HeLa cells, none of the treated groups showed significant increase of red fluorescence (Fig. S8†). These results further demonstrated that membrane proteins of MCF-7 cells could selectively facilitate the uptake of ACEs by the source cancer cells. The intracellular uptake of ACEs was then quantified by the fluorescence intensity of DOX (Fig. 2b). In MCF-7 cells, the fluorescence intensity of the ACEs-treated group was about 3.3-fold higher than the fluorescence intensity of liposomes- or AREs-treated groups after incubation for 2 h. In contrast, neither ACEs nor AMEs exhibited their advantages to HeLa cells compared to liposomes and AREs. Collectively, these results show that ACEs had successfully inherited homologous targeting ability from cancer cells, which was consistent with AMEs, and had much better targeting ability to MCF-7 cells than liposomes and AREs in vitro.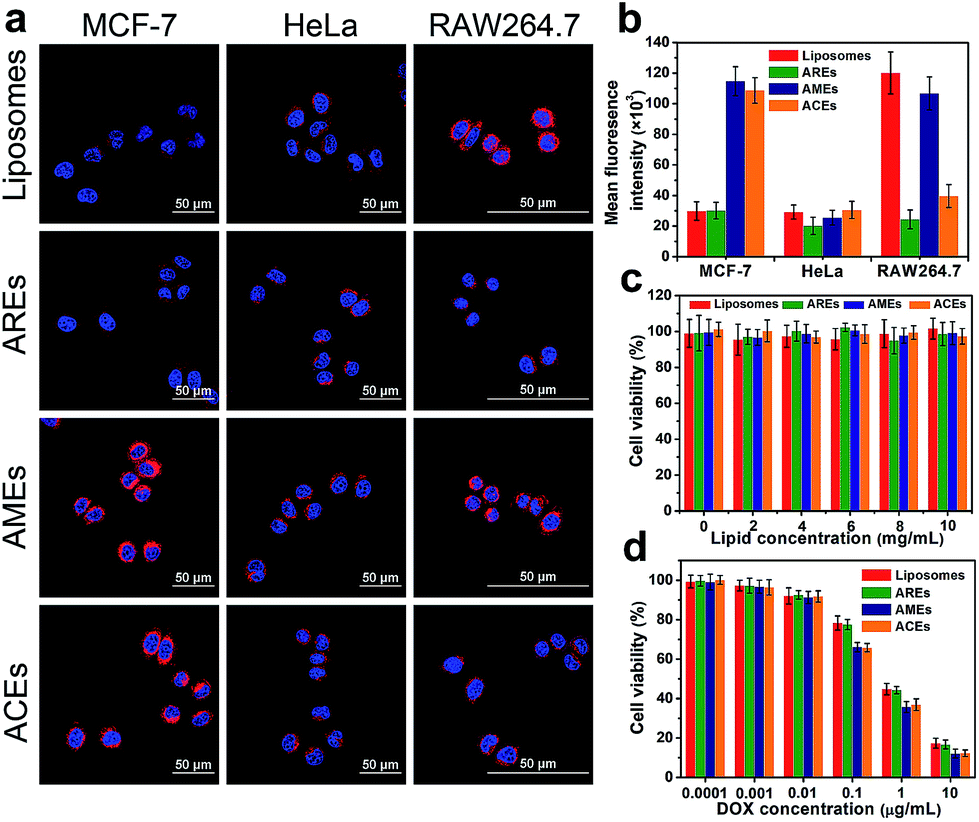 | ||
| Fig. 2 Intracellular uptake and cytotoxicity of ACEs. (a) In vitro DOX fluorescence imaging of ACEs in MCF-7 cells, HeLa cells, and RAW264.7 cells after 2 h incubation. The nucleus was stained with Hoechst 33342 (blue). The vesicles were loaded with DOX (red). (b) Semiquantitative intracellular uptake of ACEs determined by the averaged DOX fluorescence intensity of each cell (means ± SD, n = 5). In vitro cytotoxicity of different nanovesicles (c) without DOX and (d) with various concentrations of DOX after 24 h incubation with MCF-7 cells (means ± SD, n = 3). | ||
Anti-phagocytosis capability was also investigated via escape from macrophage RAW264.7 cells (Fig. S9†). Compared with liposomes and AMEs, ACEs showed lower cellular uptake, which was similar to that of AREs, possibly owing to the fact that CD47 from RBCs could suppress the uptake by macrophage cells in the mononuclear phagocyte system (MPS). These results proved that ACEs and AREs had successfully inherited anti-phagocytosis capability from RBCs, leading to more effective escape from macrophage phagocytosis than either liposomes or AMEs in vitro.
The biocompatibility and antiproliferative effects of ACEs were investigated in MCF-7 cells. Without DOX, all vesicles exhibited negligible cytotoxicity in the detected concentrations, indicating the good biocompatibility of vesicles (Fig. 2c). After loading with DOX, all vesicles exhibited dose-dependent cytotoxicity and could effectively inhibit the growth of cancer cells (Fig. 2d). It was notable that the viability of MCF-7 cells in the ACEs- and AMEs-treated groups was lower than that in the liposomes- and AREs-treated groups when DOX concentration exceeded 0.1 μg mL−1, which could have resulted from higher cellular uptake by MCF-7 cells.
2.3. In vivo biodistribution and pharmacokinetics of ACEs
After confirming ACEs with enhanced cellular uptake by homologous targeting and reduced macrophage phagocytosis at the cellular level, the biodistribution and pharmacokinetics of ACEs in vivo were evaluated. Fluorescent ICG dyes were loaded into all groups to serve as traceable fluorescent probe for in vivo fluorescence imaging in MCF-7 tumor-bearing nude mice. As shown in Fig. 3a, the liposomes-treated group showed no obvious fluorescence signals in tumor after 8 h. In contrast, the AREs- and AMEs-treated groups exhibited obvious fluorescence signals in tumor after 8 h, indicating that AREs and AMEs had acquired enhanced tumor accumulation. As expected, the fluorescence intensity of tumor in the ACEs-treated group was obviously stronger than that of the other treated groups, indicating that ACEs exhibited higher tumor accumulation. To further investigate the in vivo biodistribution of all vesicles at 24 h post-injection, the tumor and major organs of mice were collected, and DOX concentration in different tissues was examined by HPLC analysis. As shown in Fig. 3b, liposomes achieved a small amount of tumor accumulation, but a large amount of that accumulated in liver and kidney. Compared with liposomes, AREs obtained a 2.1-fold increased tumor accumulation, a 40% reduced liver accumulation and a 31% reduced kidney accumulation. On the other hand, AMEs showed a 2.5-fold increased tumor accumulation, a 15% reduced liver accumulation, and a 12% reduced kidney accumulation compared with liposomes. Importantly, ACEs finally obtained a 3.4-fold increased tumor accumulation, a 37% reduced liver accumulation and a 36% reduced kidney accumulation compared with liposomes. These results proved that ACEs had achieved high tumor accumulation and low interception in MCF-7 breast tumor xenograft models, further demonstrating the advantage of hybrid functionalities in ACEs.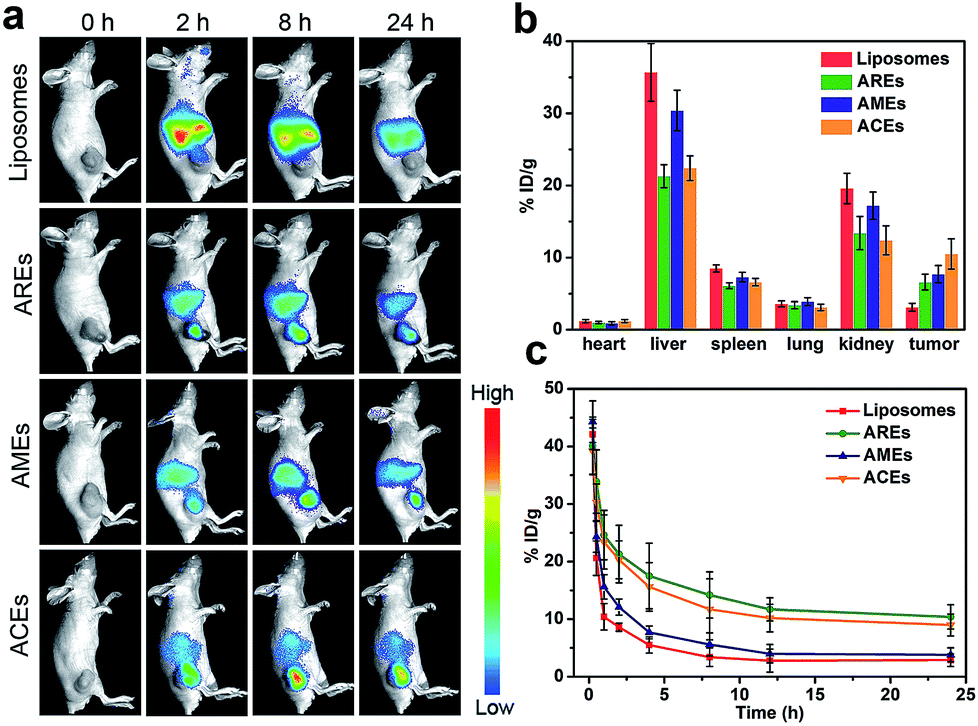 | ||
| Fig. 3 In vivo biodistribution and pharmacokinetics of ACEs after intravenous injection. (a) Time-lapse fluorescence imaging of ACEs in breast tumor-bearing nude mice. (b) Biodistribution of DOX at 24 h after intravenous administration of ACEs to breast tumor-bearing nude mice at the dose of 6 mg kg−1 (means ± SD, n = 3). (c) Blood retention profiles of DOX after intravenous administration of ACEs in mice at the dose of 6 mg kg−1 (means ± SD, n = 3). | ||
Subsequently, we performed pharmacokinetics studies to compare the blood retention time among all vesicles. Blood samples were collected at different intervals after a single intravenous injection of liposomes, ACEs, AREs and AMEs (DOX dose of 6 mg kg−1, n = 3), respectively. Plasma concentrations of DOX were determined using HPLC. As illustrated in Fig. 3c, ACEs and AREs displayed similar blood circulation time curves and significantly enhanced blood retention compared with liposomes and AMEs. AREs and ACEs at 24 h post-injection, respectively, exhibited blood retentions of 10.4% and 9.1% ID g−1, which were much higher than those of liposomes and AMEs (2.9% and 3.8% ID g−1, respectively). We next calculated the pharmacokinetic parameters by Drug and Statistics software (DAS, version 3.2.2) using a two-compartment model to fit. The pharmacokinetic profile of AREs and ACEs showed significantly longer elimination half-life (t1/2β), slower clearance rate (CL), smaller elimination rate constant (K10), and higher area under the curve (AUC) compared with those values of liposomes and AMEs (Table S2†).
2.4. In vivo antitumor efficacy of ACEs
To verify the in vivo therapeutic effect of ACEs as drug delivery nanovehicles, different DOX-loaded nanovesicles (liposomes, AREs, AMEs and ACEs) and PBS were intravenously injected every 3 days for a total of five treatments into MCF-7 tumor xenograft models with initial tumor volume of 80 mm3. In vivo therapeutic data showed that tumor growth inhibition was observed in all drug-treated groups, but that the tumor volume of mice treated with ACEs increased more slowly than that of other control groups (Fig. 4a and c). At 18 d after treatment with AREs and AMEs, the tumor volume increased to ∼965 and ∼721 mm3, respectively, values smaller than that of the liposomes-treated group (∼1260 mm3). In marked contrast, the smallest tumor volume was observed in the ACEs-treated group (∼307 mm3), demonstrating that enhanced DOX accumulation in tumor had substantially improved the antitumor efficacy resulting from ACEs targeted delivery. To further confirm the antitumor effect of all groups, we conducted hematoxylin and eosin (H&E) staining of tumor tissues at day 18 after treatment. The results showed that more obvious damage of tumor was seen in the ACEs-treated group compared with that seen in the other treated groups, suggesting excellent anticancer efficacy of ACEs at the animal level (Fig. 4d). Furthermore, the body weights of mice in all treated groups showed no significant loss (Fig. 4b), and the H&E stained histological images of heart, liver, spleen, lung and kidney exhibited no noticeable organ damage or inflammatory lesion (Fig. S10†), suggesting the low toxicity of all treatments.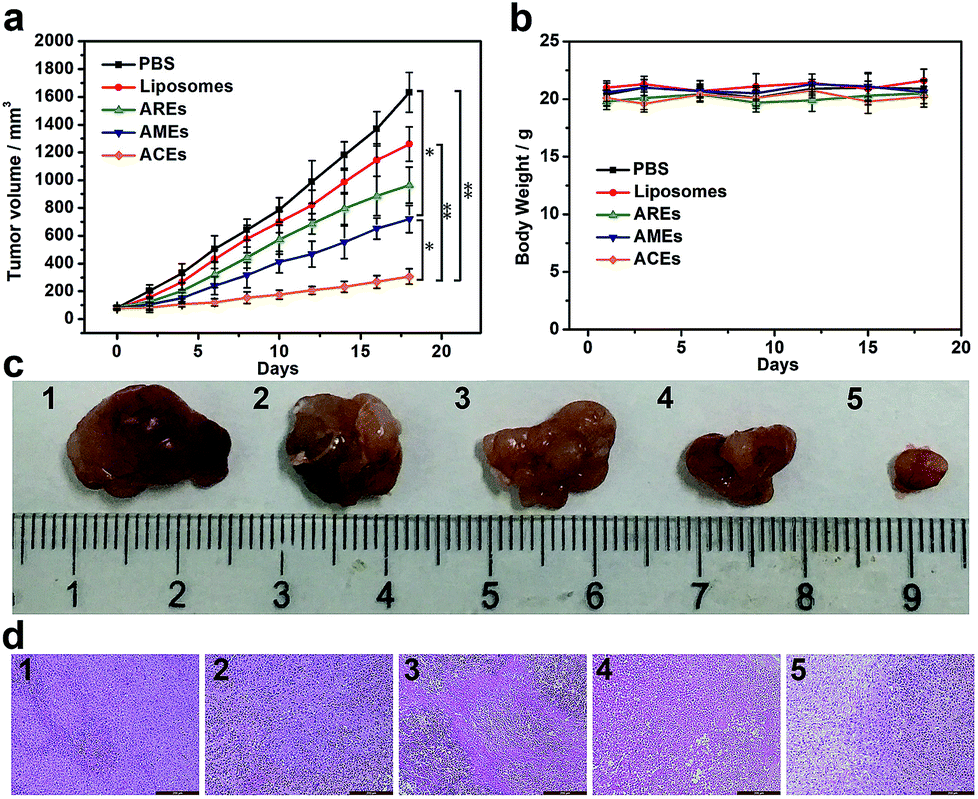 | ||
| Fig. 4 In vivo antitumor efficacy of ACEs to tumor-bearing nude mice. (a) Tumor growth curves of different groups after treatments (means ± SD, n = 5; t-test, *p < 0.05, **p < 0.01). (b) Changes of body weight with increasing time (means ± SD, n = 5). (c) Representative tumor photos and (d) H&E stained tumor sections from tumor-bearing mice after treatment with (1) PBS, (2) liposomes, (3) AREs, (4) AMEs and (5) ACEs (scale bar: 200 μm). | ||
3. Conclusions
In summary, we developed the first example of biomimetic artificial chimeric exosomes (ACEs) for anti-phagocytosis and targeted cancer therapy. ACEs were constructed by integrating cell membrane proteins from multiple cell types into synthetic phospholipid bilayers, which mimicked the morphological and physiological composition of exosomes. In this work, ACEs represented combined anti-phagocytosis capability from RBCs and homologous targeting ability from cancer cells, resulting in increased tumor accumulation, decreased interception by MPS and enhanced antitumor therapeutic effect in an animal model. Furthermore, to meet the demand of individualized treatment, this facile and simple construction strategy allows the integration of multiple functionalities into ACEs as needed. On the other hand, artificial chimeric exosomes can be easily customized, providing a controlled system with which to study protein functions and interactions within cells. Ultimately, with their large-scale standardized preparation, stable structure, high drug-loading capacity and custom-tailored functionality, ACEs will provide a suitable and promising alternative of exosomes in clinical applications.Ethical statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Fujian Medical University and Experiments were approved by the Animal Ethics Committee of Fujian Medical University. BALB/c nude mice and ICR mice were obtained from Shanghai Experimental Animal Center (Shanghai). Animal models were prepared by subcutaneously injecting of 1 × 107 MCF-7 cells into the back of the hind leg of nude mice.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 21635002, U1505221, 21622502), Natural Science Foundation of Fujian Province of China (No. 2016J05035, 2017J06004), the Program for Changjiang Scholars and Innovative Research Team in University (No. IRT15R11), the outstanding youth scientific research personnel training plan of colleges and universities in Fujian Province, and the Health-Education joint research project of Fujian Province (No. WKJ2016-2-23).References
- C. Thery, L. Zitvogel and S. Amigorena, Nat. Rev. Immunol., 2002, 2, 569–579 CrossRef CAS PubMed.
- L. Barile and G. Vassalli, Pharmacol. Ther., 2017, 174, 63–78 CrossRef CAS PubMed.
- S. Wan, L. Q. Zhang, S. Wang, Y. Liu, C. C. Wu, C. Cui, H. Sun, M. L. Shi, Y. Jiang, L. Li, L. P. Qiu and W. H. Tan, J. Am. Chem. Soc., 2017, 139, 5289–5292 CrossRef CAS PubMed.
- M. Kanada, M. H. Bachmann, J. W. Hardy, D. O. Frimannson, L. Bronsart, A. Wang, M. D. Sylvester, T. L. Schmidt, R. L. Kaspar, M. J. Butte, A. C. Matin and C. H. Contag, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E1433–E1442 CAS.
- M. K. McDonald, Y. Z. Tian, R. A. Qureshi, M. Gormley, A. Ertel, R. Gao, E. A. Lopez, G. M. Alexander, A. Sacan, P. Fortina and S. K. Ajit, Pain, 2014, 155, 1527–1539 CrossRef CAS PubMed.
- A. Hoshino, B. Costa-Silva, T. L. Shen, G. Rodrigues, A. Hashimoto, M. T. Mark, H. Molina, S. Kohsaka, A. Di Giannatale, S. Ceder, S. Singh, C. Williams, N. Soplop, K. Uryu, L. Pharmer, T. King, L. Bojmar, A. E. Davies, Y. Ararso, T. Zhang, H. Zhang, J. Hernandez, J. M. Weiss, V. D. Dumont-Cole, K. Kramer, L. H. Wexler, A. Narendran, G. K. Schwartz, J. H. Healey, P. Sandstrom, K. J. Labori, E. H. Kure, P. M. Grandgenett, M. A. Hollingsworth, M. de Sousa, S. Kaur, M. Jain, K. Mallya, S. K. Batra, W. R. Jarnagin, M. S. Brady, O. Fodstad, V. Muller, K. Pantel, A. J. Minn, M. J. Bissell, B. A. Garcia, Y. Kang, V. K. Rajasekhar, C. M. Ghajar, I. Matei, H. Peinado, J. Bromberg and D. Lyden, Nature, 2015, 527, 329 CrossRef CAS PubMed.
- C. Williams, R. Rodriguez-Barrueco, J. M. Silva, W. J. Zhang, S. Hearn, O. Elemento, N. Paknejad, K. Manova-Todorova, K. Welte, J. Bromberg, H. Peinado and D. Lyden, Cell Res., 2014, 24, 766–769 CrossRef PubMed.
- S. A. A. Kooijmans, P. Vader, S. M. van Dommelen, W. W. van Solinge and R. M. Schiffelers, Int. J. Nanomed., 2012, 7, 1525–1541 CAS.
- C. J. He, S. Zheng, Y. Luo and B. Wang, Theranostics, 2018, 8, 237–255 CrossRef CAS PubMed.
- H. Valadi, K. Ekstrom, A. Bossios, M. Sjostrand, J. J. Lee and J. O. Lotvall, Nat. Cell Biol., 2007, 9, 654 CrossRef CAS PubMed.
- A. Kalani, A. Tyagi and N. Tyagi, Mol. Neurobiol., 2014, 49, 590–600 CrossRef CAS PubMed.
- S. El-Andaloussi, Y. Lee, S. Lakhal-Littleton, J. H. Li, Y. Seow, C. Gardiner, L. Alvarez-Erviti, I. L. Sargent and M. J. A. Wood, Nat. Protoc., 2012, 7, 2112–2126 CrossRef CAS PubMed.
- R. C. Lai, R. W. Y. Yeo, K. H. Tan and S. K. Lim, Biotechnol. Adv., 2013, 31, 543–551 CrossRef CAS PubMed.
- J. K. Lee, S. R. Park, B. K. Jung, Y. K. Jeon, Y. S. Lee, M. K. Kim, Y. G. Kim, J. Y. Jang and C. W. Kim, PLoS One, 2013, 8, e84256 CrossRef PubMed.
- M. Alexander, R. Z. Hu, M. C. Runtsch, D. A. Kagele, T. L. Mosbruger, T. Tolmachova, M. C. Seabra, J. L. Round, D. M. Ward and R. M. O'Connell, Nat. Commun., 2015, 6, 7321 CrossRef CAS PubMed.
- L. Alvarez-Erviti, Y. Q. Seow, H. F. Yin, C. Betts, S. Lakhal and M. J. A. Wood, Nat. Biotechnol., 2011, 29, 341 CrossRef CAS PubMed.
- B. Escudier, T. Dorval, N. Chaput, F. Andre, M. P. Caby, S. Novault, C. Flament, C. Leboulaire, C. Borg, S. Amigorena, C. Boccaccio, C. Bonnerot, O. Dhellin, M. Movassagh, S. Piperno, C. Robert, V. Serra, N. Valente, J. B. Le Pecq, A. Spatz, O. Lantz, T. Tursz, E. Angevin and L. Zitvogel, J. Transl. Med., 2005, 3, 10 CrossRef PubMed.
- M. P. Plebanek, N. L. Angeloni, E. Vinokour, J. Li, A. Henkin, D. Martinez-Marin, S. Filleur, R. Bhowmick, J. Henkin, S. D. Miller, I. Ifergan, Y. Lee, I. Osman, C. S. Thaxton and O. V. Volpert, Nat. Commun., 2017, 8, 1913 CrossRef PubMed.
- X. Y. Zhuang, X. Y. Xiang, W. Grizzle, D. M. Sun, S. Q. Zhang, R. C. Axtell, S. W. Ju, J. Y. Mu, L. F. Zhang, L. Steinman, D. Miller and H. G. Zhang, Mol. Ther., 2011, 19, 1769–1779 CrossRef CAS PubMed.
- R. B. Rountree, S. J. Mandl, J. M. Nachtwey, K. Dalpozzo, L. Do, J. R. Lombardo, P. L. Schoonmaker, K. Brinkmann, U. Dirmeier, R. Laus and A. Delcayre, Cancer Res., 2011, 71, 5235–5244 CrossRef CAS PubMed.
- T. Z. Yang, P. Martin, B. Fogarty, A. Brown, K. Schurman, R. Phipps, V. P. Yin, P. Lockman and S. H. Bai, Pharm. Res., 2015, 32, 2003–2014 CrossRef CAS PubMed.
- J. Wahlgren, T. D. Karlson, M. Brisslert, F. V. Sani, E. Telemo, P. Sunnerhagen and H. Valadi, Nucleic Acids Res., 2012, 40, e130 CrossRef CAS PubMed.
- P. F. Zhang, L. Zhang, Z. E. Qin, S. H. Hua, Z. D. Guo, C. C. Chu, H. R. Lin, Y. Zhang, W. G. Li, X. Z. Zhang, X. Y. Chen and G. Liu, Adv. Mater., 2018, 30, 1705350 CrossRef PubMed.
- D. M. Sun, X. Y. Zhuang, X. Y. Xiang, Y. L. Liu, S. Y. Zhang, C. R. Liu, S. Barnes, W. Grizzle, D. Miller and H. G. Zhang, Mol. Ther., 2010, 18, 1606–1614 CrossRef CAS PubMed.
- X. D. Zhang, C. Wang, J. Q. Wang, Q. Y. Hu, B. Langworthy, Y. Q. Ye, W. J. Sun, J. Lin, T. F. Wang, J. Fine, H. Cheng, G. Dotti, P. Huang and Z. Gu, Adv. Mater., 2018, 30, 1707112 CrossRef PubMed.
- X. D. Zhang, J. Q. Wang, Z. W. Chen, Q. Y. Hu, C. Wang, J. J. Yan, G. Dotti, P. Huang and Z. Gu, Nano Lett., 2018, 18, 5716–5725 CrossRef CAS PubMed.
- J. Li, S. X. Wu, C. C. Wu, L. P. Qiu, G. Z. Zhu, C. Cui, Y. Liu, W. J. Hou, Y. Y. Wang, L. Q. Zhang, I. T. Teng, H. H. Yang and W. H. Tan, Nanoscale, 2016, 8, 8600–8606 RSC.
- S. X. Wu, J. Li, H. Liang, L. P. Wang, X. Chen, G. X. Jin, X. P. Xu and H. H. Yang, Sci. China: Chem., 2017, 60, 628–634 CrossRef CAS.
- A. Clayton, C. L. Harris, J. Court, M. D. Mason and B. P. Morgan, Eur. J. Immunol., 2003, 33, 522–531 CrossRef CAS PubMed.
- S. Kamerkar, V. S. LeBleu, H. Sugimoto, S. J. Yang, C. F. Ruivo, S. A. Melo, J. J. Lee and R. Kalluri, Nature, 2017, 546, 498 CAS.
- E. Koh, E. J. Lee, G. H. Nam, Y. Hong, E. Cho, Y. Yang and I. S. Kim, Biomaterials, 2017, 121, 121–129 CrossRef CAS PubMed.
- L. Blanc, A. De Gassart, C. Geminard, P. Bette-Bobillo and M. Vidal, Blood Cells, Mol., Dis., 2005, 35, 21–26 CrossRef CAS PubMed.
- K. L. Zhang, J. Zhou, H. Zhou, Y. Wu, R. Liu, L. L. Wang, W. W. Lin, G. Huang and H. H. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 30502–30509 CrossRef CAS PubMed.
- M. Gao, C. Liang, X. J. Song, Q. Chen, Q. T. Jin, C. Wang and Z. Liu, Adv. Mater., 2017, 29, 1701429 CrossRef PubMed.
- H. P. Sun, J. H. Su, Q. S. Meng, Q. Yin, L. L. Chen, W. W. Gu, P. C. Zhang, Z. W. Zhang, H. J. Yu, S. L. Wang and Y. P. Li, Adv. Mater., 2016, 28, 9581 CrossRef CAS PubMed.
- Z. Chen, P. F. Zhao, Z. Y. Luo, M. B. Zheng, H. Tian, P. Gong, G. H. Gao, H. Pan, L. L. Liu, A. Q. Ma, H. D. Cui, Y. F. Ma and L. T. Cai, ACS Nano, 2016, 10, 10049–10057 CrossRef CAS PubMed.
- Q. Y. Hu, C. G. Qian, W. J. Sun, J. Q. Wang, Z. W. Chen, H. N. Bomba, H. L. Xin, Q. D. Shen and Z. Gu, Adv. Mater., 2016, 28, 9573 CrossRef CAS PubMed.
- X. Ai, M. Hu, Z. Wang, W. Zhang, J. Li, H. Yang, J. Lin and B. Xing, Bioconjugate Chem., 2018, 29, 838–851 CrossRef CAS PubMed.
- K. Xiong, W. Wei, Y. J. Jin, S. M. Wang, D. X. Zhao, S. Wang, X. Y. Gao, C. M. Qiao, H. Yue, G. H. Ma and H. Y. Xie, Adv. Mater., 2016, 28, 7929–7935 CrossRef CAS PubMed.
- S. Mathivanan, H. Ji and R. J. Simpson, J. Proteomics, 2010, 73, 1907–1920 CrossRef CAS PubMed.
- C. Liu, J. Y. Guo, F. Tian, N. Yang, F. S. Yan, Y. P. Ding, J. Y. Wei, G. Q. Hu, G. J. Nie and J. S. Sun, ACS Nano, 2017, 11, 6968–6976 CrossRef CAS PubMed.
- S. A. A. Kooijmans, P. Vader and R. M. Schiffelers, Nat. Biomed. Eng., 2017, 1, 634–636 CrossRef.
- T. S. Chen, F. Arslan, Y. Yin, S. S. Tan, R. C. Lai, A. B. H. Choo, J. Padmanabhan, C. N. Lee, D. P. V. de Kleijn and S. K. Lim, J. Transl. Med., 2011, 9, 47 CrossRef CAS PubMed.
- T. Lener, M. Gimona, L. Aigner, V. Borger, E. Buzas, G. Camussi, N. Chaput, D. Chatterjee, F. A. Court, H. A. del Portillo, L. O'Driscoll, S. Fais, J. M. Falcon-Perez, U. Felderhoff-Mueser, L. Fraile, Y. S. Gho, A. Gorgens, R. C. Gupta, A. Hendrix, D. M. Hermann, A. F. Hill, F. Hochberg, P. A. Horn, D. de Kleijn, L. Kordelas, B. W. Kramer, E. M. Kramer-Albers, S. Laner-Plamberger, S. Laitinen, T. Leonardi, M. J. Lorenowicz, S. K. Lim, J. Lotvall, C. A. Maguire, A. Marcilla, I. Nazarenko, T. Ochiya, T. Patel, S. Pedersen, G. Pocsfalvi, S. Pluchino, P. Quesenberry, I. G. Reischl, F. J. Rivera, R. Sanzenbacher, K. Schallmoser, I. Slaper-Cortenbach, D. Strunk, T. Tonn, P. Vader, B. W. M. van Balkom, M. Wauben, S. El Andaloussi, C. Thery, E. Rohde and B. Giebel, J. Extracell. Vesicles, 2015, 4, 30087 CrossRef PubMed.
- H. De la Pena, J. A. Madrigal, S. Rusakiewicz, M. Bencsik, G. W. V. Cave, A. Selman, R. C. Rees, P. J. Travers and I. A. Dodi, J. Immunol. Methods, 2009, 344, 121–132 CrossRef CAS PubMed.
- R. Molinaro, C. Corbo, J. O. Martinez, F. Taraballi, M. Evangelopoulos, S. Minardi, I. K. Yazdi, P. Zhao, E. De Rosa, M. B. Sherman, A. De Vita, N. E. T. Furman, X. Wang, A. Parodi and E. Tasciotti, Nat. Mater., 2016, 15, 1037–1046 CrossRef CAS PubMed.
- R. Molinaro, M. Evangelopoulos, J. R. Hoffman, C. Corbo, F. Taraballi, J. O. Martinez, K. A. Hartman, D. Cosco, G. Costa, I. Romeo, M. Sherman, D. Paolino, S. Alcaro and E. Tasciotti, Adv. Mater., 2018, 1702749 CrossRef PubMed.
- L. S. Lin, X. Y. Yang, Z. J. Zhou, Z. Yang, O. Jacobson, Y. J. Liu, A. Yang, G. Niu, J. B. Song, H. H. Yang and X. Y. Chen, Adv. Mater., 2017, 29, 1606681 CrossRef PubMed.
- X. R. Song, S. H. Li, J. Y. Dai, L. Song, G. M. Huang, R. H. Lin, J. Li, G. Liu and H. H. Yang, Small, 2017, 13, 1603997 CrossRef PubMed.
- D. Zhang, M. Wu, Z. Cai, N. Liao, K. Ke, H. Liu, M. Li, G. Liu, H. Yang, X. Liu and J. Liu, Adv. Sci., 2018, 5, 1700648 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Comprehensive experimental details and additional details on all experiments. See DOI: 10.1039/c8sc03224f |
| This journal is © The Royal Society of Chemistry 2019 |