
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-mediated Minisci C–H alkylation of heteroarenes with unactivated alkyl halides using O2 as an oxidant†‡
Jianyang
Dong
a,
Xueli
Lyu
a,
Zhen
Wang
a,
Xiaochen
Wang
a,
Hongjian
Song
a,
Yuxiu
Liu
a and
Qingmin
Wang
 *ab
*ab
aState Key Laboratory of Elemento-Organic Chemistry, Research Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, People's Republic of China. E-mail: wangqm@nankai.edu.cn
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300071, People's Republic of China
First published on 27th November 2018
Abstract
Herein, we report a protocol for direct visible-light-mediated Minisci C–H alkylation of heteroarenes with unactivated alkyl halides using molecular oxygen as an oxidant at room temperature. This mild protocol is compatible with a wide array of sensitive functional groups and has a broad substrate scope. Notably, functionalization of (iso)quinolines, pyridines, phenanthrolines, quinazoline, and other heterocyclic compounds with unactivated primary, secondary, and tertiary alkyl halides proceeds smoothly under the standard conditions. The robustness of this protocol is further demonstrated by the late-stage functionalization of complex nitrogen-containing natural products and drugs.
Introduction
Heteroaryl motifs are present in a wide variety of natural products, organic materials, small-molecule drugs, and ligands for metal catalysts.1 Substituted heteroarenes can be readily accessed by direct functionalization of the C–H bonds of unsubstituted heteroarenes.2 A useful tool for this purpose is the Minisci reaction, in which a protonated N-heteroarene is attacked by an alkyl radical under oxidative conditions.3 Classic Minisci reactions, which involve carboxylic acids as the alkyl radical sources, often require the use of excess oxidant (e.g., peroxide), excess acid, and high temperature.3,4 Recently, there have been several reports of visible-light-mediated Minisci C–H alkylation reactions of N-heteroarenes with alkyl peroxides, primary alcohols, aliphatic carboxylic acids, boronic acids, alkyltrifluoroborates, and alkyl iodides as the alkyl radical sources (Scheme 1A).5 In addition, Barriault and co-workers described a protocol for direct alkylation of heteroarenes with alkyl bromides in the presence of a gold photoredox catalyst (Scheme 1B).6 However, the protocol employs a high catalyst loading and high-energy UVA (365 nm) irradiation, which can narrow the functional group tolerance and limit the substrate scope.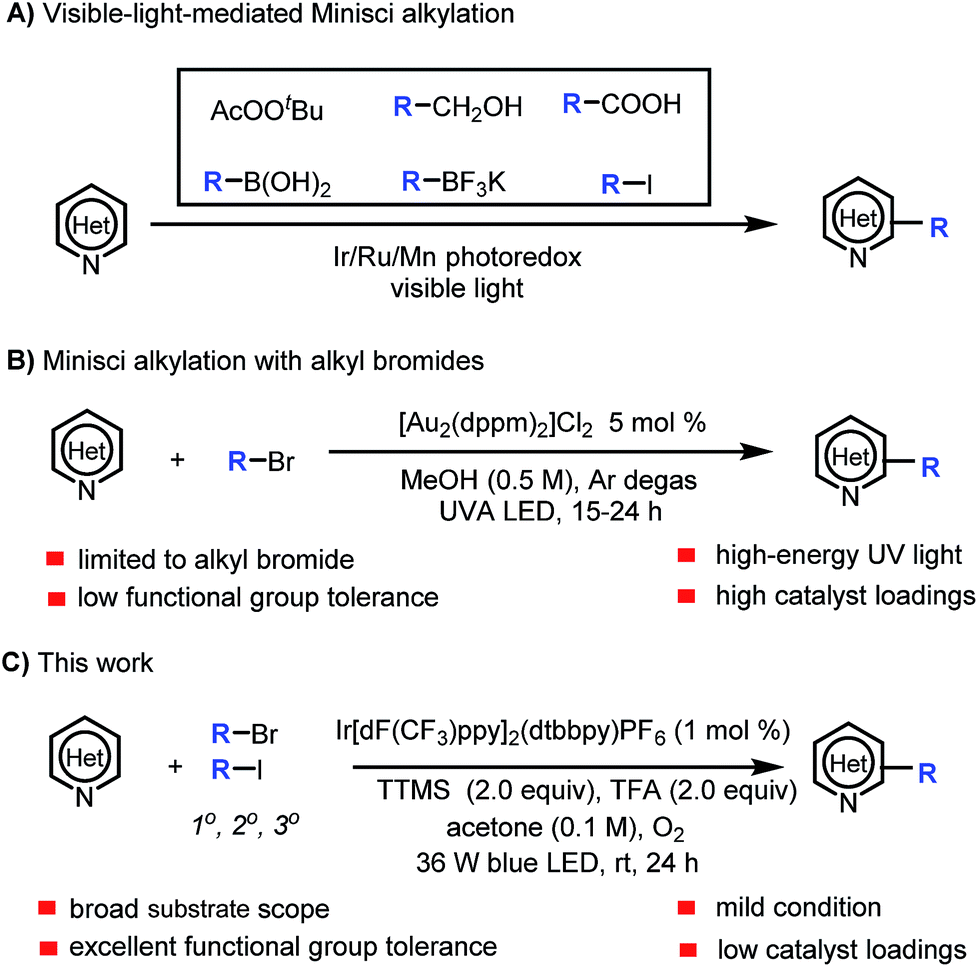 | ||
| Scheme 1 Photoredox-mediated Minisci C–H alkylation of N-heteroarenes. | ||
Our group is interested in visible-light-mediated Minisci reactions, and we recently achieved α-aminoalkylation of N-heteroarenes by means of an iridium photoredox catalysis approach.7 Because alkyl bromides are readily available and inexpensive, we wished to expand the substrate scope of C–H alkylation of heteroarenes with alkyl bromides by developing a practical, mild protocol with a broader functional group tolerance. In addition, we wanted the new Minisci reaction protocol to employ oxygen, which is environmentally benign, as the only oxidant and to avoid the need for high catalyst loadings and high-energy UV light. However, we needed to overcome the challenge posed by the fact that Ru- and Ir-based photoredox catalysts cannot undergo efficient excited-state quenching with unactivated alkyl bromides (ca. −2.0 V vs. SCE) to generate alkyl radicals under standard Minisci conditions (which are acidic and oxidative).8
Halogen abstraction from alkyl bromides by tris(trimethylsilyl)silyl radical for thermal generation of alkyl radicals has been widely studied, and photomediated processes have also been reported.9 However, halogen abstraction by a silyl radical under Minisci reaction conditions has not been explored. In this study, we focused our attention on developing a practical method for Minisci-type reactions using alkyl bromides as the alkyl radical sources and tris(trimethylsilyl)silane (TTMS) as the halogen-abstraction agent under photoredox conditions. Specifically, we herein report a protocol for visible-light-mediated Minisci C–H alkylation of heteroarenes with unactivated primary, secondary, and tertiary alkyl halides (Scheme 1C). The high efficiency, broad substrate scope, excellent functional group tolerance, and mildness of this protocol make it particularly suitable for late-stage functionalization of complex nitrogen-containing natural products and drugs.
Results and discussion
As a model reaction, we investigated the alkylation of lepidine (1, 1.0 equiv.) with bromocyclohexane (2, 2.0 equiv.) under various conditions (Table 1). First, a number of silanes were screened with 1 mol% of Ir[dF(CF3)ppy]2(dtbbpy)PF6 as the photocatalyst, trifluoroacetic acid as the proton source, molecular oxygen as the oxidant, and acetone as the solvent under irradiation with a 36 W blue LED (see the ESI‡). To our delight, desired product 3 was obtained in excellent yield when TTMS was used as a silyl radical precursor (entry 1). Using the above-described conditions, we then varied the photocatalyst (entries 2–4). However, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 proved to be the most effective catalyst. Other solvents (acetonitrile, methanol, and dimethylformamide) gave lower yields (entries 5–7). Control experiments showed that the reaction failed to proceed in the absence of light, photocatalyst, TTMS, or molecular oxygen (entries 8–11).|
|
|||
|---|---|---|---|
| Entry | Photocatalyst | Solvent | Yieldb (%) |
| a General conditions, unless otherwise noted: 1 (0.2 mmol), 2 (0.4 mmol), photocatalyst (0.002 mmol), TTMS (0.4 mmol), trifluoroacetic acid (TFA, 0.4 mmol), and acetone (2 mL) under O2 atmosphere. NR = no reaction. b Isolated yields are given. c Reaction performed in the absence of light. d Reaction performed in the absence of TTMS. e Reaction performed under argon atmosphere. | |||
| 1 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | Acetone | 82 |
| 2 | Ir(ppy)3 | Acetone | Trace |
| 3 | [Ru(bpy)3](PF6)2 | Acetone | NR |
| 4 | Eosin-Y | Acetone | Trace |
| 5 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | CH3CN | 68 |
| 6 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | MeOH | 52 |
| 7 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | DMF | 23 |
| 8c | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | Acetone | NR |
| 9 | — | Acetone | NR |
| 10d | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | Acetone | NR |
| 11e | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | Acetone | 12 |

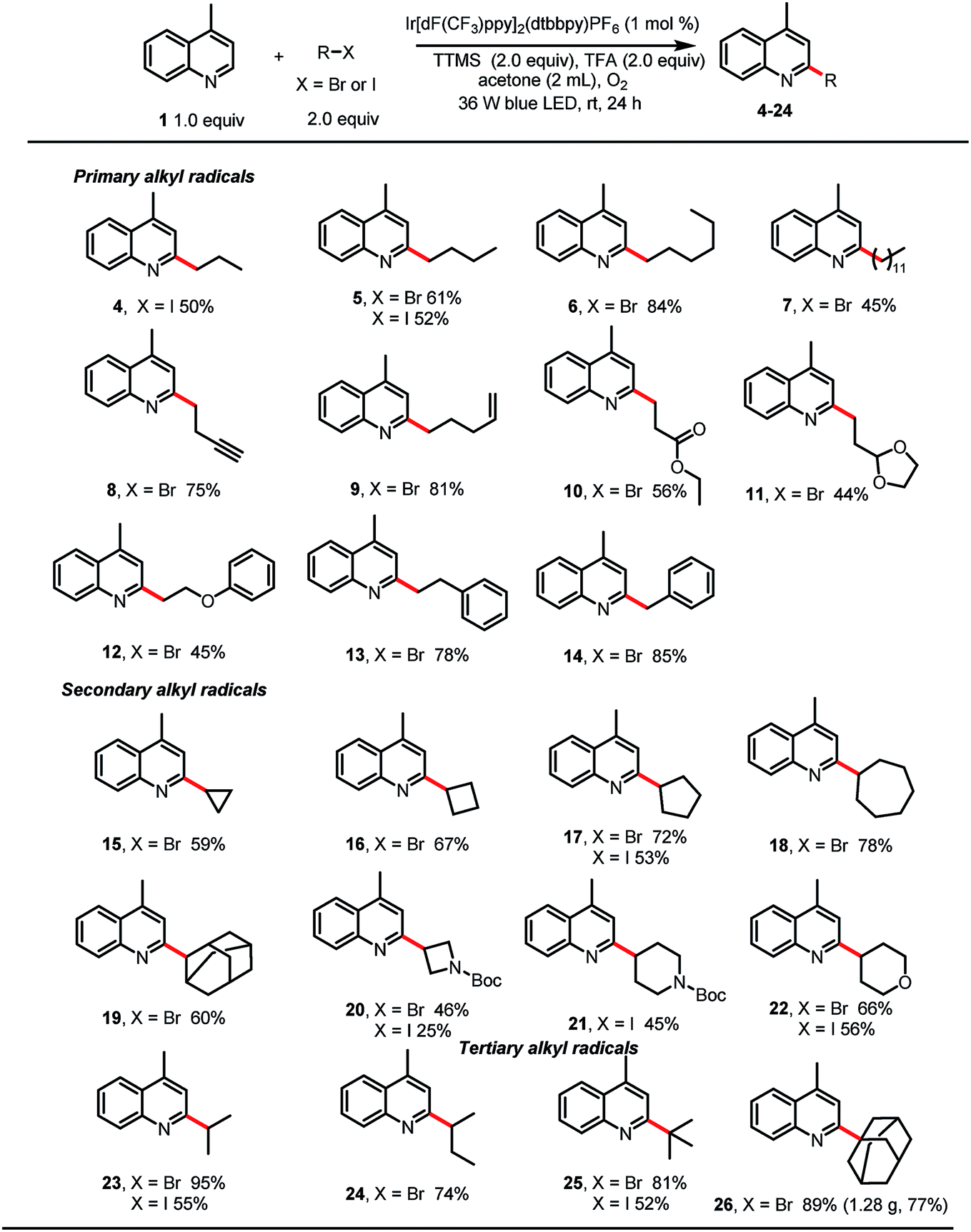
With the optimized reaction conditions in hand, we studied the scope of the reaction with respect to the alkyl bromide (Table 2). The reaction was found to be amenable to a wide range of primary alkyl bromides, which gave the desired products in good to excellent yields. For example, linear alkyl bromides afforded the corresponding alkylated heteroarenes (5–7) in 45–84% yields. Minisci reactions of primary alkyl radicals are more challenging than reactions of secondary alkyl radicals, owing to the lower stability and nucleophilicity of the former.10 However, we were pleased to observe that primary alkyl substituents carrying various functional groups (e.g., a terminal alkyne, a terminal alkene, an ester, an acetal, or a phenoxy group; 8–12, respectively) could be incorporated in good yields. Phenylethyl bromide and benzyl bromide were also suitable substrates, giving 13 and 14 in 78 and 85% yields. Alkylation reactions of secondary bromoalkanes also proceeded under the standard conditions to afford the corresponding alkylated heteroarenes in moderate to excellent yields (15–20, 22–24). Notably, ether, amine, and tert-butyl carbamate groups were also tolerated. Finally, tertiary-bromoalkane-functionalized lepidine derivatives 25 and 26 could be obtained in good to excellent yields, and the protocol was amenable to scale up; 26 was isolated in 77% yield when the reaction was carried out on a 6 mmol scale.
| a Reactions were performed on a 0.3 mmol scale. Isolated yields are given. |
|---|
|
Next, we explored alkylation reactions with alkyl iodides (Table 2). Gratifyingly, we found that unactivated primary, secondary, and tertiary iodoalkanes underwent the desired reaction, and the corresponding products were obtained in moderate to good yields. Although in each case the yield was lower than that obtained with the corresponding alkyl bromide, the ability to use alkyl iodides broadens the scope of the reaction with respect and would be helpful when the alkyl bromide is not available. Unfortunately, unactivated primary, secondary, and tertiary alkyl chlorides showed no reactivity.
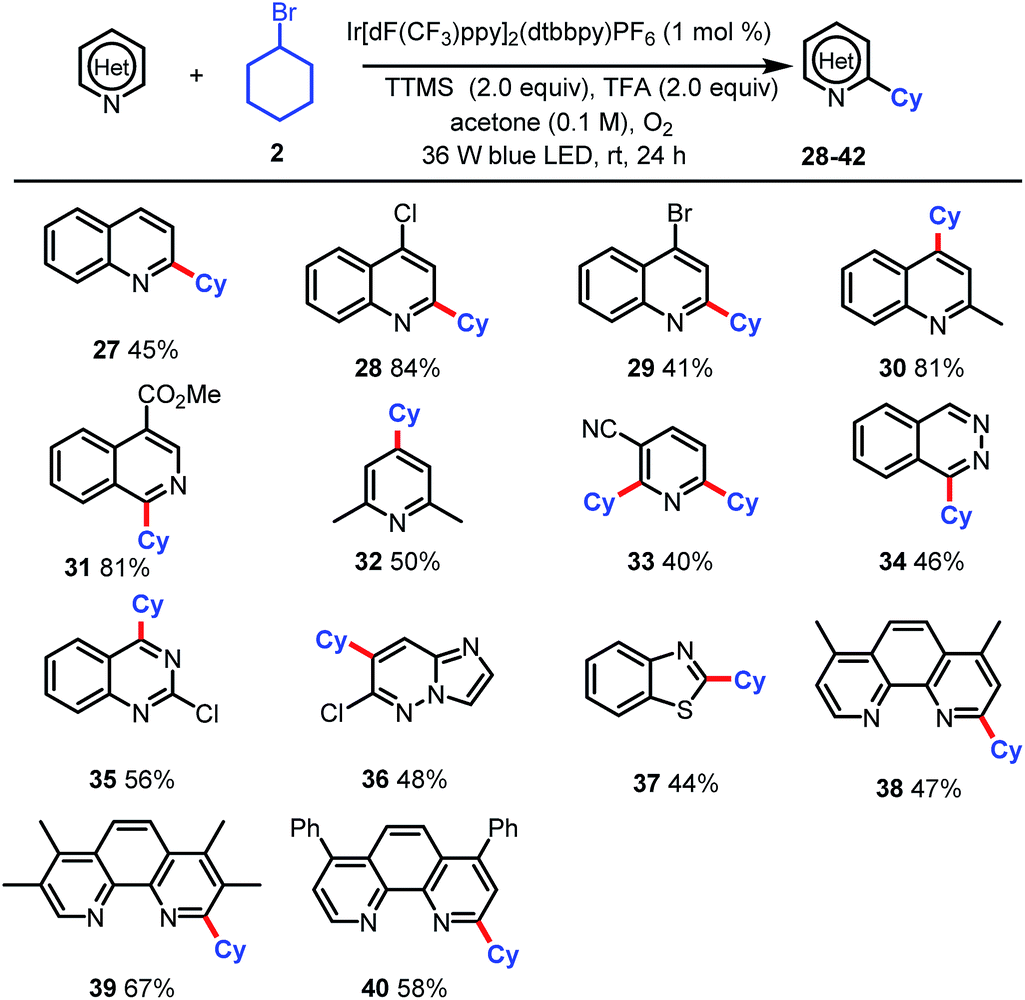
Finally, we tested this new alkylation protocol with various N-heteroarenes (Table 3). Electron-deficient heteroarenes were readily alkylated at the most electrophilic position with bromocyclohexane (2) in fair to excellent yields. Specifically, quinoline was selectively alkylated with 2 at the C2 position to afford 27 in 45% yield. The use of 4-chloro- and 4-bromo-quinoline and 2-methyl-quinoline resulted in selective alkylation with 2 at C2 for the halogenated substrates and at C4 for the methylated substrate (28–30, 41–84% yields). Reactions of 2 with 4-methoxycarbonyl-substituted isoquinolines afforded products of selective α-aminoalkylation at C2 position (31, 81% yield). Alkylation of 2,6-dimethylpyridine afforded a fair yield (50%) of C4-functionalized product 32. When nicotinonitrile was used, dialkylated product 33 (40%) was the major product. The scope of the reaction was further extended to phthalazine (34, 46%), 2-chloroquinazoline (35, 56%), 6-chloroimidazo[1,2-b]pyridazine (36, 48%), and benzothiazole (37, 44%). Notably, the reaction could be used to modify commercially available phenanthroline ligands, as demonstrated by the selective monoalkylation with 2 at the C2 position to afford fair to good yields of 38–40. The selective monoalkylation of these phenanthroline ligands suggests that this protocol may find applications in the synthesis of ligands for catalysis.
| a Reactions were performed on a 0.3 mmol scale. Isolated yields are given. |
|---|
|
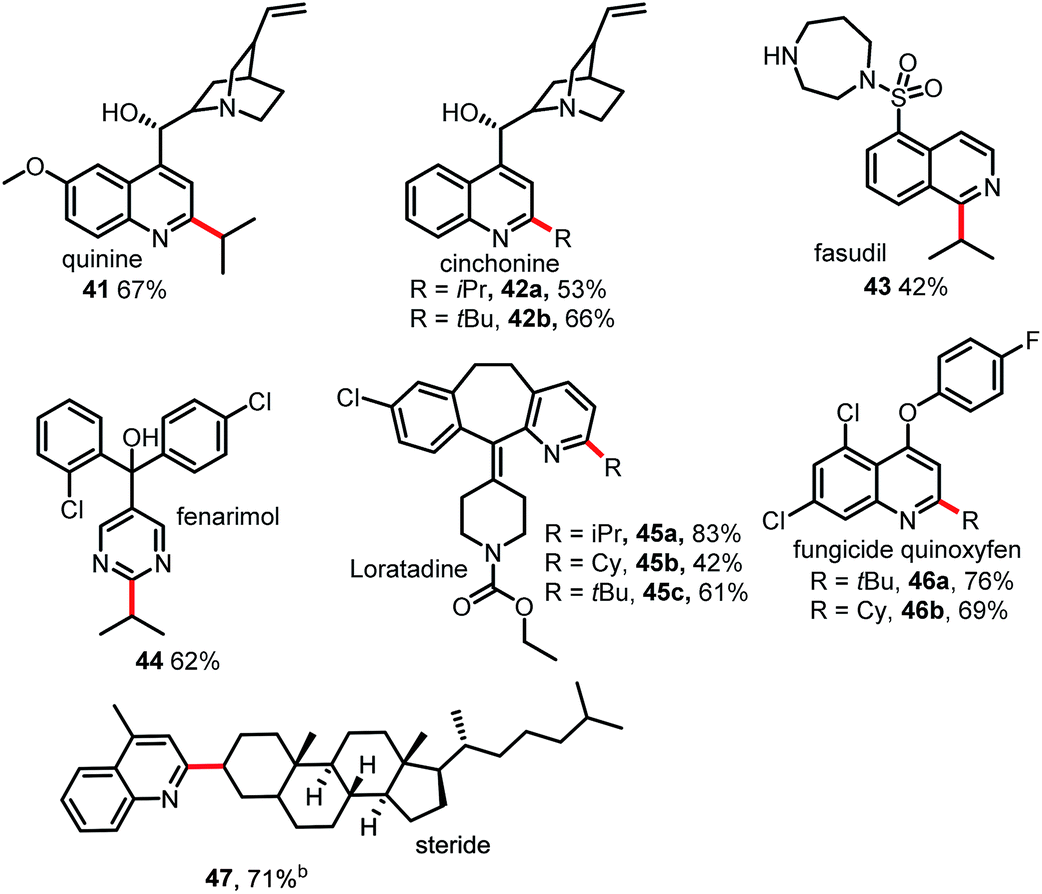
As shown in Table 4, this Minisci C–H alkylation protocol could be readily used to functionalize complex natural products and drug molecules.5,11 For instance, quinine and cinchonine, both of which have a free OH group as well as amine and vinyl groups, could be selectively alkylated at the C2 position with either an isopropyl group or a tert-butyl group to afford 41 and 42 in good yields. Fasudil, which bears a free secondary NH group, was selectively alkylated at C1 to give a fair yield of 43. Fenarimol, which has a tertiary OH group, could also tolerate the reaction conditions and was selectively monoalkylated to afford 44 in 62% yield. Loratadine, which has amide and vinyl groups, could be alkylated selectively at the C2 position of the pyridine ring to give 45. The fungicide quinoxyfen was alkylated at C2 of the quinoline ring to give 46 in good yield. Remarkably, the alkylation of steride also proceeded smoothly, affording a 71% yield of 47. Considering that alkyl halides are readily available and inexpensive, this new Minisci protocol may prove highly useful in drug discovery research.12
Having explored the substrate scope and utility of the reaction, we turned our attention to the mechanism (Scheme 2). When a radical scavenger, 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or 1,1-diphenylethylene, was present in a reaction mixture containing 1 and 2, the formation of 3 was completely inhibited. When benzyl acrylate (48) was used as a radical scavenger, benzyl 3-(1,1,1,3,3,3-hexamethyl-2-(trimethylsilyl)trisilan-2-yl)propanoate (49) was isolated in 5% yield, which suggests that a silyl radical was generated.9f In addition, the product of cyclohexyl radical trapping, benzyl 3-cyclohexylpropanoate (50), was observed by mass spectrometry.9f To lend additional support to our proposed mechanism, we carried out a radical clock experiment.6,13 Alkylation of 1 with 6-bromohex-1-ene (51) resulted in 5-exo trig cyclization prior to heteroarene addition and afforded a 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 52a and 52b. Alkylation of 1 with (bromomethyl)cyclopropane (53) under the standard conditions gave ring-opened product 54 in 45% yield. (Bromomethyl)cyclobutane 55 remained mostly unopened, affording 56a and 56b in a 14:1 ratio. These experiments clearly point to a radical pathway. Moreover, we conducted a light/dark experiment, which showed that coupling product 3 formed only under continuous irradiation (see ESI‡). This result suggests that radical chain propagation was not involved in the reaction.
:1 mixture of 52a and 52b. Alkylation of 1 with (bromomethyl)cyclopropane (53) under the standard conditions gave ring-opened product 54 in 45% yield. (Bromomethyl)cyclobutane 55 remained mostly unopened, affording 56a and 56b in a 14:1 ratio. These experiments clearly point to a radical pathway. Moreover, we conducted a light/dark experiment, which showed that coupling product 3 formed only under continuous irradiation (see ESI‡). This result suggests that radical chain propagation was not involved in the reaction.
 | ||
| Scheme 2 Mechanistic experiments. | ||
On the basis of our experimental observations and literature reports, we propose the mechanism depicted in Scheme 3. The mechanism begins with generation of carbon-based radical A from an alkyl bromide under photoredox conditions, which has been explained previously by MacMillan and co-workers in the context of their cross-electrophile coupling.9e Briefly, alkyl bromide may produce tiny amounts of bromide (Br−) under irradiation of light, oxidation of bromide (Br−) by the photocatalyst generates an electrophilic bromine radical,14 which can abstract the hydrogen from (Me3Si)3SiH, generating a silyl radical species [(Me3Si)3Si˙].9e Subsequent halogen abstraction from alkyl bromide 2 provides nucleophilic radical species A and the bromosilane byproduct. This abstraction step is effectively irreversible owing to the difference in bond dissociation energy between the Si–Br bond of Me3Si–Br (96 kcal mol−1) and the Csp3–Br bond of bromoethane (69 kcal mol−1).15 Radical A then adds to the protonated electron-deficient heteroarene via a Minisci-type pathway to afford radical cation B. Single-electron oxidation of Ir2+ by O2 forms the superoxide radical anion (O2−˙) and completes the photoredox cycle.16 O2−˙ can abstract a hydrogen atom from B to form the final alkylated product, along with HO2−.17
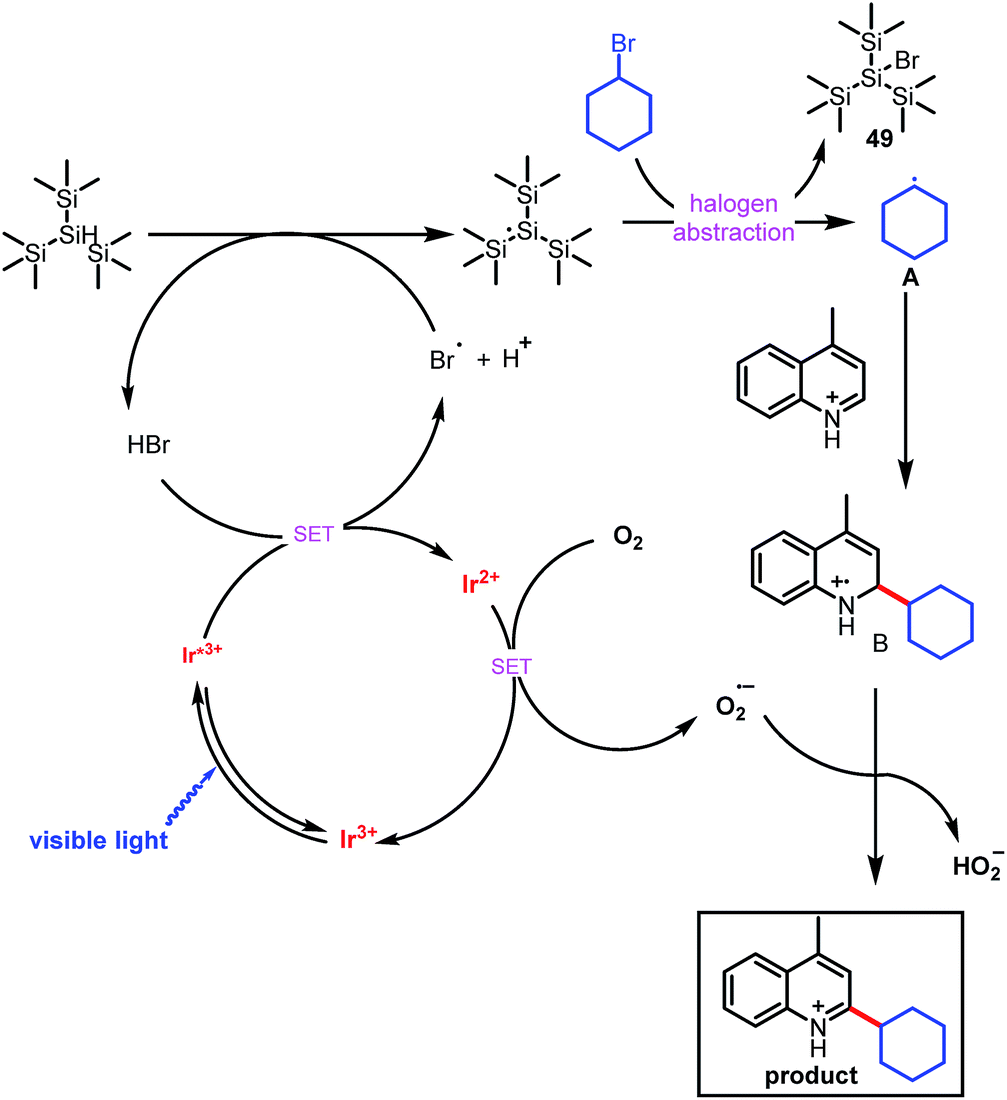 | ||
| Scheme 3 Proposed mechanism for direct C–H alkylation of heteroarenes. | ||
Conclusions
In conclusion, we have achieved visible-light-mediated Minisci C–H alkylation of heteroarenes by using readily available, inexpensive alkyl halides as the alkyl radical sources. A broad range of cyclic and acyclic unactivated primary, secondary, and tertiary alkyl groups can be efficiently incorporated into N-heteroarenes under mild conditions, and the protocol is scalable to the gram level. Its high efficiency, broad substrate scope, excellent functional group tolerance, and mild operation conditions make it particularly suitable for late-stage functionalization of complex nitrogen-containing natural products and drugs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (21732002, 21672117) and the Tianjin Natural Science Foundation (16JCZDJC32400) for generous financial support for our programs.Notes and references
- (a) R. R. Gupta, Bioactive heterocycles V, Topics in Heterocyclic Chemistry V, ed. R. R. Gupta, Wiley-VCH, Springer, Heidelberg, Hoboken, vol. 11, 2008 Search PubMed; (b) M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347 CrossRef CAS PubMed; (c) M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2013, 9, 2265 CrossRef PubMed; (d) J. A. Builla, J. J. Vaquero and J. Barluenga, Cabal in Modern Heterocyclic Chemistry, ed. J. A. Builla, J. J. Vaquero and J. Barluenga, Wiley-VCH, Weinheim, vol. 1, 2011 CrossRef; (e) S. W. Thomas III, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef PubMed.
- (a) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC; (b) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 ( Angew. Chem. , 2009 , 121 , 9976 ) CrossRef CAS PubMed; (c) T. Breckl, R. D. Baxter, R. Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2012, 45, 826 CrossRef PubMed; (d) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (e) H. Bonin, M. Sauthier and F. X. Felpin, Adv. Synth. Catal., 2014, 356, 645 CrossRef CAS; (f) G.-Y.-S. Qiu, Y.-W. Li and J. Wu, Org. Chem. Front., 2016, 3, 1011 RSC; (g) J. Hofmann and M. R. Heinrich, Tetrahedron Lett., 2016, 57, 4334 CrossRef CAS; (h) I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. Kçnig, Acc. Chem. Res., 2016, 49, 1566 CrossRef CAS PubMed; (i) J.-R. Zhang, L. Xu, Y.-Y. Liao, J.-C. Deng and R.-Y. Tang, Chin. J. Chem., 2017, 35, 271 CrossRef CAS; (j) O. Boubertakh and J. P. Goddard, Eur. J. Org. Chem., 2017, 2072 CrossRef CAS; (k) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018 ( Angew. Chem. , 2011 , 123 , 5122 ) CrossRef CAS PubMed; (l) T.-L. Chan, Y. Wu, P.-Y. Choy and F.-Y. Kwong, Chem.–Eur. J., 2013, 19, 15802 CrossRef CAS PubMed; (m) C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219 CrossRef CAS PubMed; (n) D. Mirizzi, S. T. Hilton and K. Jones, Adv. Heterocycl. Chem., 2010, 100, 101 CrossRef CAS; (o) R. A. Rossi, A. B. Pierini and A. B. Penenory, Chem. Rev., 2003, 103, 71 CrossRef CAS PubMed; (p) C. Galli and Z. Rappoport, Acc. Chem. Res., 2003, 36, 580 CrossRef CAS PubMed; (q) N. Zhang, S. R. Samanta, B. M. Rosen and V. Percec, Chem. Rev., 2014, 114, 5848 CrossRef CAS PubMed.
- For reviews on the Minisci reaction, see: (a) J. Tauber, D. Imbr and T. Opatz, Molecules, 2014, 19, 16190 CrossRef PubMed; (b) C. Punta and F. Minisci, Trends Heterocycl. Chem., 2008, 13, 1 CAS; (c) F. Minisci, E. Vismara and F. Fontana, Heterocycles, 1989, 28, 489 CrossRef CAS; (d) F. Minisci, F. Fontana and E. Vismara, J. Heterocycl. Chem., 1990, 27, 79 CrossRef CAS; (e) M. A. Duncton, MedChemComm, 2011, 2, 1135 RSC.
- (a) G. A. Molander, V. Colombel and V. A. Braz, Org. Lett., 2011, 13, 1852 CrossRef CAS PubMed; (b) R. Gianatassio, S. Kawamura, C. L. Eprile, K. Foo, J. Ge, A. C. Burns, M. R. Collins and P. S. Baran, Angew. Chem., Int. Ed., 2014, 53, 9851 ( Angew. Chem. , 2014 , 126 , 10009 ) CrossRef CAS PubMed; (c) F. Minisci, E. Vismara and F. Fontana, J. Org. Chem., 1989, 54, 5224 CrossRef CAS; (d) H. Togo, K. Hayashi and M. Yokoyama, Chem. Lett., 1991, 20, 2063 CrossRef; (e) M. A. J. Duncton, M. A. Estiarte, R. J. Johnson, M. Cox, D. J. R. Mahony, W. T. Edwards and M. G. Kelly, J. Org. Chem., 2009, 74, 6354 CrossRef CAS PubMed; (f) F. Fontana, F. Minisci and E. Vismara, Tetrahedron Lett., 1988, 29, 1975 CrossRef CAS; (g) M. Presset, N. Fleury-Bregeot, D. Oehlrich, F. Rombouts and G. A. Molander, J. Org. Chem., 2013, 78, 4615 CrossRef CAS PubMed; (h) H. Togo, K. Hayashi and M. Yokoyama, Chem. Lett., 1993, 22, 641 CrossRef; (i) H. Togo, K. Hayashi and M. Yokoyama, Bull. Chem. Soc. Jpn., 1994, 67, 2522 CrossRef CAS; (j) L. Zhang and Z.-Q. Liu, Org. Lett., 2017, 19, 6594 CrossRef CAS PubMed; (k) B. Liang, Q. Wang and Z.-Q. Liu, Org. Lett., 2017, 19, 6463 CrossRef CAS PubMed; (l) Z. Liu and Z.-Q. Liu, Org. Lett., 2017, 19, 5649 CrossRef CAS PubMed.
- (a) D. A. DiRocco, K. Dykstra, S. Krska, P. Vachal, D. V. Conway and M. Tudge, Angew. Chem., Int. Ed., 2014, 53, 4802 ( Angew. Chem. , 2014 , 126 , 4902 ) CrossRef CAS PubMed; (b) J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87 CrossRef CAS PubMed; (c) R. A. Garza-Sanchez, A. Tlahuext-Aca, G. Tavakoli and F. Glorius, ACS Catal., 2017, 7, 4057 CrossRef CAS; (d) G.-X. Li, C. A. Morales-Rivera, Y. Wang, F. Gao, G. He, P. Liu and G. Chen, Chem. Sci., 2016, 7, 6407 RSC; (e) J. K. Matsui, D. N. Primer and G. A. Molander, Chem. Sci., 2017, 8, 3512 RSC; (f) P. Nuhant, M. S. Oderinde, J. Genovino, A. Juneau, Y. Gagne, C. Allais, G. M. Chinigo, C. Choi, N. W. Sach, L. Bernier, Y. M. Fobian, M. W. Bundesmann, B. Khunte, M. Frenette and O. O. Fadeyi, Angew. Chem., Int. Ed., 2017, 56, 15309 CrossRef CAS PubMed; (g) N. B. Bissonnette, M. J. Boyd, G. D. May, S. Giroux and P. Nuhant, J. Org. Chem., 2018, 83, 10933 CrossRef CAS PubMed; (h) J. Wang, G.-X. Li, G. He and G. Chen, Asian J. Org. Chem., 2018, 7, 1307 CrossRef CAS.
- (a) T. McCallum and L. Barriault, Chem. Sci., 2016, 7, 4754 RSC; (b) C. D. McTiernan, M. Morin, T. McCallum, J. C. Scaiano and L. Barriault, Catal. Sci. Technol., 2016, 6, 201 RSC.
- J. Dong, Q. Xia, X. Luy, C. Yan, H. Song, Y. Liu and Q. Wang, Org. Lett., 2018, 20, 5661 CrossRef CAS PubMed.
- (a) A. J. Fry and R. L. Krieger, J. Org. Chem., 1976, 41, 54 CrossRef CAS; (b) T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562 RSC; (c) J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854 CrossRef CAS PubMed; (d) H. Kim and C. Lee, Angew. Chem., Int. Ed., 2012, 51, 12303 CrossRef CAS PubMed; (e) S. Y. Chow, M. Y. Stevens, L. Akerbladh, S. Bergman and L. R. Odell, Chem.–Eur. J., 2016, 22, 9155 CrossRef CAS PubMed; (f) M. Jiang, H. Yang and H. Fu, Org. Lett., 2016, 18, 5248 CrossRef CAS PubMed.
- (a) C. Chatgilialoglu and J. Lalevée, Molecules, 2012, 17, 527 CrossRef CAS PubMed; (b) C. Chatgilialoglu, Organosilanes in Radical Chemistry, Wiley, Chichester, U.K., 2004 CrossRef; (c) C. Chatgilialoglu, Acc. Chem. Res., 1992, 25, 188 CrossRef CAS; (d) M. Ballestri, C. Chatgilialoglu, K. B. Clark, D. Griller, B. Giese and B. Kopping, J. Org. Chem., 1991, 56, 678 CrossRef CAS; (e) P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084 CrossRef CAS PubMed; (f) A. ElMarrouni, C. B. Rittsb and J. Balsells, Chem. Sci., 2018, 9, 6639 RSC; (g) V. Bacauanu, S. Cardinal, M. Yamauchi, M. Kondo, D. F. Fernández, R. Remy and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 12543 CrossRef CAS PubMed; (h) O. Yamazakl, H. Togo, S. Matsubayashi and M. Yokoyama, Tetrahedron, 1999, 55, 3735 CrossRef.
- H. Togo, Advanced Free Radical Reactions for Organic Synthesis, Elsevier, 2004 Search PubMed.
- (a) T. Bruckl, R. D. Baxter, R. Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2012, 45, 826 CrossRef CAS PubMed; (b) C. S. Leung, S. S. F. Leung, J. Tirado-Rives and W. L. Jorgensen, J. Med. Chem., 2012, 55, 4489 CrossRef CAS PubMed.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546 RSC.
- C. Le, T. Q. Chen, T. Liang, P. Zhang and D. W. C. MacMillan, Science, 2018, 360, 1010 CrossRef CAS PubMed.
- (a) M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. J. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712 CrossRef CAS; (b) S. J. Hwang, D. C. Powers, A. G. Maher, B. L. Anderson, R. G. Hadt, S.-L. Zheng, Y.-S. Chen and D. G. Nocera, J. Am. Chem. Soc., 2015, 137, 6472 CrossRef CAS PubMed.
- For the Si–Br bond dissociation energy, see: R. Walsh, Acc. Chem. Res., 1981, 14, 246 CrossRef CAS; For the C–Br bond dissociation energy, see: A. J. Gordon and R. A. Ford, The Chemist's Companion: A Handbook of Practical Data, Techniques, and References, Wiley, New York, 1972 Search PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC.
- (a) Z. Q. Wang, M. Hu, X. C. Huang, L. B. Gong, Y. X. Xie and J. H. Li, J. Org. Chem., 2012, 77, 8705 CrossRef CAS PubMed; (b) S. Chen, Q. Wan and A. K. Badu-Tawiah, Angew. Chem., Int. Ed., 2016, 55, 4345 CrossRef PubMed.
Footnotes |
| † Dedicated to the 100th anniversary of Nankai University. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04892d |
| This journal is © The Royal Society of Chemistry 2019 |