
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxygen transfer in electrophilic epoxidation probed by 17O NMR: differentiating between oxidants and role of spectator metal oxo†
Christian
Ehinger‡
 ,
Christopher P.
Gordon‡
and
Christophe
Copéret
*
,
Christopher P.
Gordon‡
and
Christophe
Copéret
*
Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir Prelog Weg 1-5, 8093, Zürich, Switzerland. E-mail: ccoperet@ethz.ch
First published on 3rd December 2018
Abstract
Peroxide compounds are used both in laboratory and industrial processes for the electrophilic epoxidation of olefins. Using NMR-spectroscopy, we investigate why certain peroxides engage in this type of reaction while others require activation by metal catalysts, e.g. methyltrioxorhenium (MTO). More precisely, an analysis of 17O NMR chemical shift and quadrupolar coupling parameters provides insights into the relative energy of specific frontier molecular orbitals relevant for reactivity. For organic peroxides or H2O2 a large deshielding is indicative of an energetically high-lying lone-pair on oxygen in combination with a low-lying σ*(O–O) orbital. This feature is particularly pronounced in species that engage in electrophilic epoxidation, such as peracids or dimethyldioxirane (DMDO), and much less pronounced in unreactive peroxides such as H2O2 and ROOH, which can however be activated by transition-metal catalysts. In fact, for the proposed active peroxo species in MTO-catalyzed electrophilic epoxidation with H2O2 an analysis of the 17O NMR chemical shift highlights specific π- and δ-type orbital interactions between the so-called metal spectator oxo and the peroxo moieties that raise the energy of the high-lying lone-pair on oxygen, thus increasing the reactivity of the peroxo species.
Introduction
Electrophilic epoxidations are at the core of numerous processes, ranging from the industrial synthesis of propylene oxide to enzymatic oxygenase reactions.1–8 In organic synthesis, this ubiquitous transformation is commonly achieved by using stoichiometric epoxidation agents such as meta-chloroperoxybenzoic acid (mCPBA), dimethyldioxirane (DMDO), or oxaziridines.9–14 H2O2 and ROOH can also be used for epoxidation but they require a catalyst. The most commonly used catalysts are (i) organorhenium trioxides (especially methyltrioxorhenium, MTO)15–21 and group 6 metal dioxo compounds22–24 (Fig. 1) or (ii) early transition-metal alkoxides (e.g. Ti and V),25–29 that involve peroxo-species as key reaction intermediates.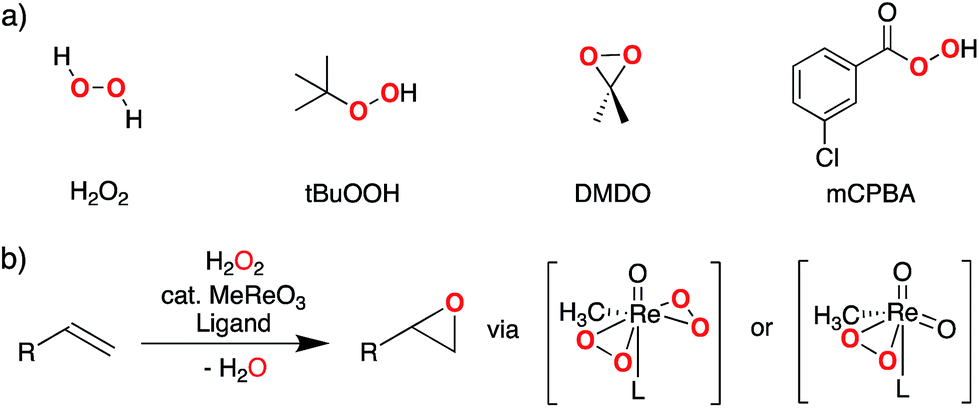 | ||
| Fig. 1 (a) Representative primary peroxide oxidants used for electrophilic epoxidation, (b) MTO-catalyzed olefin epoxidation involving bisperoxo- or monoperoxo-species (L = pyridine or water). | ||
While the reactivity of oxidizing agents such as mCPBA or DMDO towards olefins is well established and exploited synthetically, the origin of their reactivity towards C–C double bonds has not been studied in detail. This question is particularly apparent when considering that other peroxides, such as H2O2 or tBuOOH, are usually not reactive towards olefins, unless combined with metal catalysts.
Recent work has shown that analysis of the 13C NMR chemical shift tensor (CST) of metal alkyl compounds can give valuable insights into the electronic structure and the reactivity of ubiquitous reaction intermediates in organometallic chemistry and homogeneous catalysis.30–37 Considering the large chemical shift window of 17O nuclei (around 1200 ppm), we reasoned that analysis of the 17O NMR chemical shift tensor of oxidants would allow for a detailed understanding of the electronic structure and associated reactivity of these molecules. In addition, the quadrupolar coupling constant of 17O (nuclear spin I = 5/2) can provide valuable information on the charge distribution around the nucleus.38–42 In fact, 17O NMR spectroscopy has been used to identify and study peroxo species as well as related compounds containing O–N bonds.43–46
The isotropic chemical shift δiso (eqn (1)) and the three principal components (δ11 ≥ δ22 ≥ δ33) of the CST contain a considerable amount of information on the electronic structure of NMR active nuclei. The corresponding shielding values (σ, eqn (2)), can be decomposed computationally into diamagnetic (σdia) and paramagnetic contributions, which also include contributions from spin–orbit coupling (σpara+SO, eqn (3)). While the diamagnetic contributions, which arise from a molecule's electronic ground state, lead to shielding and are usually similar for all nuclei of a given kind independent of their chemical environment, the paramagnetic contributions, which give mostly rise to deshielding, originate from magnetically induced coupling of excited states to the ground state, by action of the angular momentum operator ![[L with combining circumflex]](https://www.rsc.org/images/entities/b_i_char_004c_0302.gif) i, as described in a 2nd order perturbation approach in eqn (4).47
i, as described in a 2nd order perturbation approach in eqn (4).47
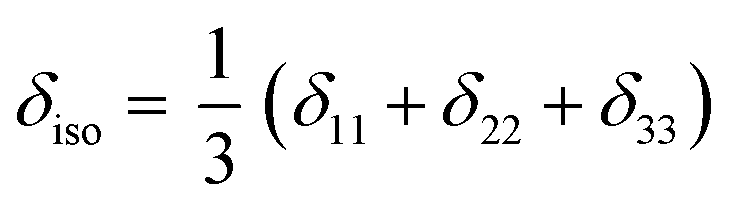 | (1) |
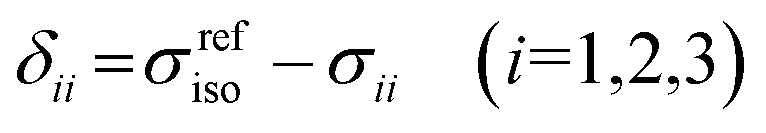 | (2) |
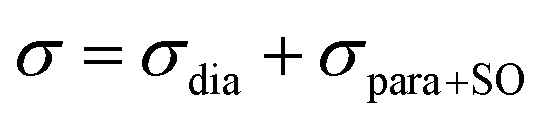 | (3) |
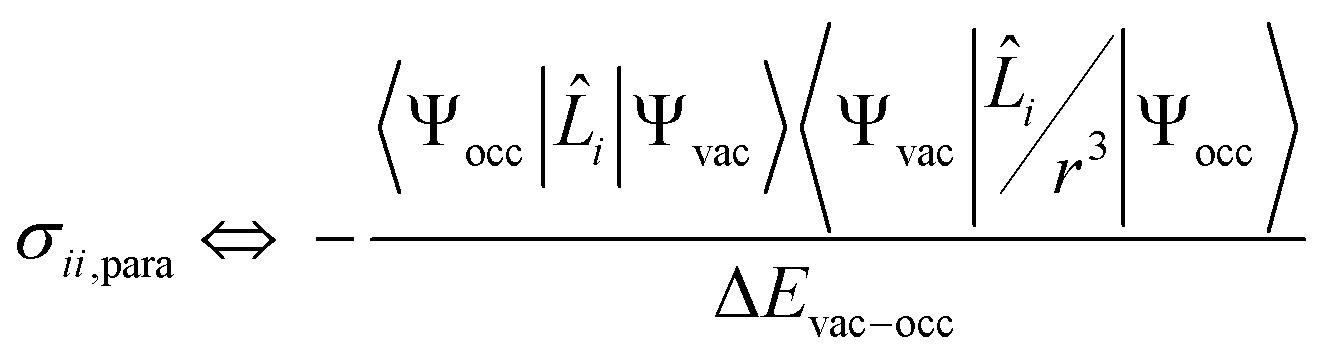 | (4) |
According to eqn (4), deshielding of a nucleus is expected along the direction i, if an occupied orbital on this nucleus can be “superimposed” onto a vacant orbital on the same nucleus rotated by 90° along the axis i (Fig. 2). Since the extent of deshielding increases with a decreasing energy gap between the two orbitals, the paramagnetic contribution to shielding is most strongly affected by frontier molecular orbitals (FMOs) – energetically high-lying occupied and low-lying vacant orbitals.
 | ||
| Fig. 2 Magnetically induced coupling of occupied and vacant orbitals leading to a deshielding along the i-axis. | ||
In this work, we make use of chemical shift to evidence specific high-lying occupied and low-lying vacant orbitals in the aforementioned oxidizing agents, thereby probing their electronic structure and connection to the observed reactivities.
As 17O is a quadrupolar nucleus (I = 5/2), the quadrupolar coupling, which typically complicates the interpretation of spectra by line broadening, holds information about the distribution of charges around the nucleus. The quadrupolar interaction is proportional to the electric field gradient (EFG) tensor ![[V with combining umlaut]](https://www.rsc.org/images/entities/b_i_char_0056_0308.gif) (eqn (5)), where e is the electron charge, Q is the quadrupolar moment of 17O, ħ is the reduced Planck constant (ħ = h/2π), Î is the nuclear spin operator and I the nuclear spin quantum number.
(eqn (5)), where e is the electron charge, Q is the quadrupolar moment of 17O, ħ is the reduced Planck constant (ħ = h/2π), Î is the nuclear spin operator and I the nuclear spin quantum number.
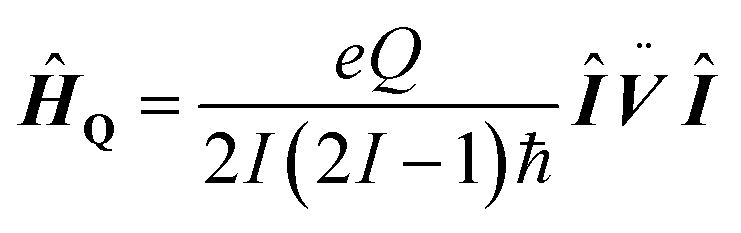 | (5) |
V is a traceless second rank tensor (V11+ V22+ V33 = 0) where we follow the notation |V33| ≥ |V22| ≥ |V11| for the three principal components. The EFG tensor can be described by two independent variables – usually the largest principal component (V33) and the asymmetry parameter ηQ (eqn (6)).
| ηQ = (V11 − V22)/V33 | (6) |
The quadrupolar coupling constant CQ arises from the interaction of the quadrupole moment of 17O (I = 5/2) with the EFG and is proportional to V33 (eqn (7)).38 Since the electric quadrupole moment Q of the 17O nucleus is negative, V33 and CQ have opposing signs.
| CQ = (eQV33/h) | (7) |
V 33 and hence CQ are indicative of how symmetric the EFG and thus the charge distribution around the nucleus is. This has been a valuable tool to assess local symmetry around quadrupolar nuclei (e.g.17O, 27Al, 45Sc).38,41,42,46,48–51
Results & discussion
CSTs of non-metal-based peroxides
We calculated the chemical shift tensors (CSTs) of selected peroxides relevant to epoxidation reactions, as well as associated reduced compounds. We chose to investigate DMDO and mCPBA, two compounds showing activity towards electrophilic epoxidation, as well as H2O2 and tBuOOH that do not participate in this transformation, unless activated by metal catalysts. In order to benchmark our calculations, we also experimentally determined the 17O NMR chemical shift tensors of H2O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 52 and acetone by solid-state 17O NMR spectroscopy53–55 (see ESI† for experimental details).
52 and acetone by solid-state 17O NMR spectroscopy53–55 (see ESI† for experimental details).
The measured and calculated chemical shifts are given in Table 1. Generally, a good agreement between calculated and experimental data (when available) is obtained. The oxygen atoms of the unsymmetric peroxides (tBuOOH and mCPBA) are labelled (O), for the oxygen bound to a carbon atom and (OH) for the oxygen connected to the hydrogen.
| Compound | δ iso | δ 11 | δ 22 | δ 33 | Ω |
|---|---|---|---|---|---|
| a Ω = δ11 – δ33. b Note that the quadrupolar nature of 17O can lead to inaccuracies in the determination of the chemical shift tensors – see ESI for a more detailed discussion of the experimental measurements. Additionally, the presence of solvents can also significantly impact the chemical shift. | |||||
| H2O2 | 195 (195)b | 364 (362) | 227 (232) | −7 (−9) | 371 (370) |
| H2O | 0 (0) | 25 | 8 | −32 | 57 |
| tBuOOH (O) | 273 (246)56 | 413 | 335 | 73 | 340 |
| tBuOOH (OH) | 205 (206)56 | 386 | 228 | 2 | 384 |
| tBuOH | 95 (62)57 | 130 | 104 | 52 | 78 |
| mCPBA (O) | 308 (320)58 | 428 | 261 | 236 | 192 |
| mCPBA (OH) | 278 (275)58 | 540 | 245 | 50 | 490 |
mCBA (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) O) |
398 | 618 | 535 | 42 | 576 |
| mCBA (OH) | 193 | 342 | 161 | 76 | 266 |
| DMDO | 291 (302)59 | 636 | 164 | 73 | 564 |
| Acetone | 667 (625)b | 1239 (1185) | 740 (705) | 23 (−15) | 1216 (1200) |
A comparison of the isotropic chemical shifts given in Table 1 reveals that all peroxide species (H2O2, tBuOOH, mCPBA, and DMDO) show significantly more deshielded δiso values in comparison with H2O, tBuOH and mCBA, albeit less deshielded than carbonyl oxygens (e.g. mCBA (CO) and acetone). While the differences among the various peroxides are less pronounced, the chemical shift (δiso) of the oxygen atom which is transferred during epoxidation reactions is more deshielded for DMDO and mCPBA (OH) as compared to H2O2 and tBuOOH (OH). A closer inspection of the principal components of the chemical shift tensor reveals that this is mostly due to the δ11 component of the CST which is significantly more deshielded in DMDO and the OH–oxygen of mCPBA (636 and 540 ppm) than in H2O2 and tBuOOH (364 and 386 ppm). This highly deshielded component is accompanied by a significantly larger span Ω of the CST in the former compounds.
Orientation of the CSTs
In order to further understand these observed trends, we investigated the orientation of the 17O NMR chemical shielding tensors (CSTs) as obtained by DFT calculations. The calculated CSTs are shown in Fig. 3. Notably, the 17O CST is similarly oriented in all the aforementioned peroxides, with the most deshielded δ11 component being oriented perpendicular to the O–O axis and lying in the O–O–H/R plane. The δ33 component points along the O–O axis, while δ22 is perpendicular to both, δ11 and δ33. Minor deviations to this pattern are found for mCPBA (O) and DMDO, where the orientation of the δ11 and δ33 components is slightly tilted, by comparison with the other compounds (vide infra). | ||
| Fig. 3 Orientation of the CST in (a) H2O2, (b) tBuOOH, (c) DMDO, and (d) mCPBA. The principal components σ11, σ22, and σ33 are indicated by red, green, and blue arrows, respectively. The 17O NMR shielding values (σ) are indicated in ppm next to the direction of each principal component, with the corresponding chemical shift values (δ) given in parenthesis. | ||
Orbital analysis of the CSTs
We decided to further elucidate the origin of deshielding in the individual components of the CST in a Natural Chemical Shielding (NCS) analysis.47,48,60–65 This analysis allows for a decomposition of the σii/δii components into diamagnetic (σdia) and paramagnetic/spin–orbit (σpara+SO) contributions (eqn (3)). The paramagnetic term can then be further decomposed into contributions of the various NLMOs (bonds and lone pairs) surrounding the nucleus of interest. Due to the orbital energy difference in the denominator of eqn (4), the orbitals contributing most strongly to σpara+SO are the frontier molecular orbitals (FMOs) of the molecule with a non-vanishing coefficient on the investigated nucleus.Since the largest differences in the CST of the investigated peroxides originate from the σ11/δ11 component of the CST, its orbital analysis will be further discussed (see Fig. S3 and S4† for other components).
The NCS analysis of the σ11 component of the various compounds (Fig. 4b) reveals, that the diamagnetic contributions to this component are essentially invariant throughout the whole series of compounds. The differences in δ11 result from the paramagnetic contributions, which are mainly affected by four different Natural Localized Molecular Orbitals (NLMOs). These correspond to two lone-pairs on oxygen (denoted as LP ‘p’ and LP ‘s’), as well as the two σ-bonding orbitals (denoted as σ(O–R) and σ(O–O)). These four NLMOs are visualized for the case of hydrogen peroxide in Fig. 4a; for the other compounds they are shown in Fig. S11–S13.†
 | ||
| Fig. 4 (a) Relevant NLMOs contributing to the paramagnetic shielding of the investigated peroxides. (b) Results of the NCS analysis of the most deshielded component of the shielding tensor, σ11, represented in a bar diagram. | ||
Notably, the dominant contribution to the deshielding of σ11/δ11 originates from the LP ‘p’ on oxygen in all cases given in Fig. 4, with the exception of mCPBA (O) (vide infra). The observation of the large deshielding perpendicular to the O–O axis originating from this lone pair indicates the presence of a low-lying vacant orbital, oriented perpendicular to both the lone-pair and the direction of σ11/δ11; i.e. oriented along the O–O bond (eqn (4)). This vacant orbital corresponds to σ*(O–O), the lowest unoccupied molecular orbital with contribution of oxygen in all of the investigated peroxides. As the extent of deshielding along a CST principal axis scales with the inverse of the energy difference between the two coupled orbitals (eqn (4)), a large deshielding of the δ11 component indicates the presence of a high-lying LP ‘p’ on oxygen, derived from the filled bonding and antibonding π(O–O) and π*(O–O) orbitals, and/or a low-lying σ*(O–O) orbital. The comparatively small contribution of the LP ‘p’ to δ11 in the case of the oxygen bound to the carbonyl group in mCPBA, which is associated with a different orientation of this component compared to the other compounds (Fig. 3), can be rationalized by the conjugation to the carbonyl group, which decreases the energy of the lone pair LP ‘p’ and thus renders the coupling to the σ*(O–O) less efficient. Additionally, this conjugation leads to a non-zero coefficient of a vacant orbital with π-symmetry on the oxygen. The C–O σ-bond can couple to this vacant orbital, ultimately leading to the significant contribution of the NLMO σ(O–R) and the tilting of the CST component δ11. In the case of DMDO the slightly tilted orientation of the CST is rationalized by the deviation of the electron density from the internuclear axis in this strained system. This bonding situation can be understood by Walsh-type orbitals (“banana bonds”).66 The tilting of the CST hence evidences the deviation of the σ*(O–O) orbital from the O–O axis. This is consistent with the result of the Natural Hybrid Orbital (NHO) Directionality and Bond Bending Analysis which indicates a deviation of the localized orbital from the internuclear (O–O) axis by 16.8°, as opposed to 2.5° in the case of H2O2 (the values for other investigated compound are given in Table S6†).67
Energetic considerations in oxygen-transfer reactions
The strong deshielding of the δ11 component in the case of DMDO and mCPBA evidences a particularly low-lying σ*(O–O) orbital in combination with a high-lying lone pair LP ‘p’ in these compounds. In order to further understand the impact of this orbital situation on reactivity we investigated the corresponding transition states for the epoxidation of ethylene with DMDO and mCPBA. In both transition states, the olefin is oriented perpendicularly to the plane consisting of the peroxo moiety and the respective substituents (Fig. 5). The free energies of activation for DMDO and mCPBA are found at 25.6 and 25.7 kcal mol−1, respectively. | ||
| Fig. 5 Transition state geometries for the electrophilic epoxidation with (a) DMDO and (b) mCPBA. | ||
The geometry of these transition states can be rationalized by the orbital situation derived from the CST analysis: while a low-lying σ*(O–O) orbital of the peroxide allows for a good interaction with the π(CC) bond of the olefin, the oxygen lone pair LP ‘p’ involved in the filled π*(O–O) orbital can interact with the empty π*(CC) orbital (Fig. 6). Both of these interactions are energetically favorable and are expected to lower the transition state energy for olefin epoxidation. The deshielded δ11 component of the CST is thus indicative of the epoxidation propensity of DMDO and mCPBA.
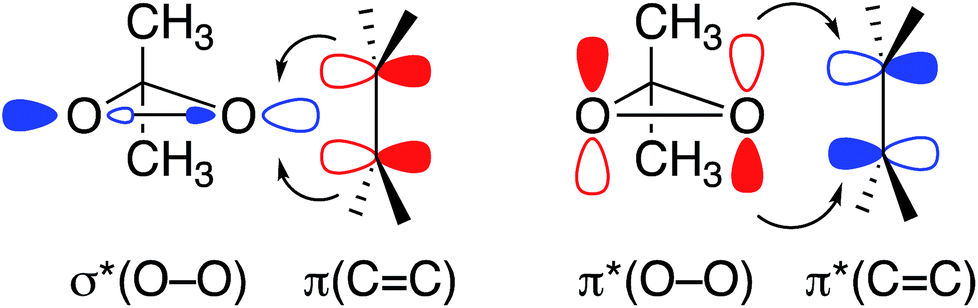 | ||
| Fig. 6 Relevant orbital interactions in epoxidation, shown for the case of DMDO. Electron-donating and -accepting orbitals are colored in red and blue, respectively. | ||
The 17O NMR chemical shift of the investigated peroxides shows that DMDO and mCPBA, the two molecules engaging in electrophilic epoxidation, feature a high-lying lone pair LP ‘p’ and a low-lying σ*(O–O) orbital. This observation can be understood based on the geometry of these compounds: In DMDO and mCPBA the lone pairs LP ‘p’ of the peroxo oxygens are co-planar, resulting in a maximized filled–filled interaction (α-effect), and thus a high-lying filled antibonding π*(O–O) orbital. Hence, in the epoxidation process, the backdonation of the lone pair of the attacked oxygen into the π*(CC) of the olefin is more efficient for these reagents.
The importance of this “backdonation” was further explored by investigating transition state geometries where the olefin is perpendicular to the oxygen lone pair and hence co-planar with the dioxirane moiety in DMDO or the carbonyl group in mCPBA. The corresponding transition state (2nd order saddle point) energies for the epoxidation where the rotation around the reaction coordinate was restricted were found at 32.7 and 29.0 kcal mol−1 for DMDO and mCPBA, respectively. The backdonation of the LP ‘p’ into the π*(CC) of the olefin hence gives rise to a significant stabilization of the transition state energy in both cases (7.1 kcal mol−1 for DMDO and 3.3 kcal mol−1 for mCPBA).
Quadrupolar coupling parameters
To complement the analysis of the 17O NMR parameters we calculated the quadrupolar coupling parameters of the aforementioned peroxides. The respective 17O CQ and ηQ-values are reported in Table 2.| Compound | C Q [MHz] | η Q |
|---|---|---|
| H2O | 10.3 | 0.78 |
| H2O2 | −17.0 (−16) | 0.93 (0.8) |
| tBuOOH (O) | −17.5 | 0.95 |
| tBuOOH (OH) | −16.1 | 0.97 |
| mCPBA (O) | −14.4 | 0.54 |
| mCPBA (OH) | −18.8 | 0.75 |
| DMDO | 18.5 | 0.79 |
| Acetone | 11.8 (12) | 0.52 (0.6) |
For all investigated peroxide oxygens, the absolute magnitude of the quadrupolar coupling constant CQ (and V33 accordingly) is significantly larger than for water. The orientation of the EFG tensor is similar in all of these peroxides, with the most positive component oriented along the O–O bond, and the most negative component oriented in the direction of LP ‘p’ (selected examples in Fig. 7a–c, see Fig. S14† for other compounds). This orientation is hence indicative of a region of high electron density in the direction of LP ‘p’, and a region of depleted electron density in the direction of the O–O bond. The reversed sign of CQ observed for DMDO in contrast to other peroxides is due to the definition of V33, which always corresponds to the EFG tensor component with the largest absolute value (indicated by a red arrow in Fig. 7). While the EFG tensor is similar in all peroxides, the negative EFG tensor component perpendicular to the O–O bond is slightly larger for DMDO by absolute value as compared to the positive EFG tensor component along the O–O bond. For the other investigated peroxides, the situation is reversed, leading to a change in sign of V33 and hence of CQ. The large CQ value for peroxides in combination with the specific orientation of the EFG tensor is consistent with the presence of a high-lying occupied orbital (LP ‘p’) oriented perpendicular to a low-lying vacant σ*(O–O) orbital in these species. This observation parallels what is seen from the NCS analysis of the δ11 component of the CST.
 | ||
| Fig. 7 EFG-tensor orientations of (a) H2O2, (b) mCPBA (OH), and (c) DMDO. Negative regions of the EFG tensor are represented in red, positive regions are depicted in blue. The direction of V33 is indicated with a red arrow. | ||
MTO-catalyzed epoxidation
As both H2O2 and tBuOOH are inactive in electrophilic epoxidation but are rendered active in the presence of transition-metal catalysts, we investigated the process of activation and the properties of the proposed active species. We chose methyltrioxorhenium (MTO) as a prototypical catalyst because of its high efficiency in olefin epoxidation with H2O2 (Fig. 1b), and the existence of detailed mechanistic studies (isolation of reaction intermediates, measurements of 17O NMR parameters and computational studies).15,68–71 We hence calculated the chemical shift tensors of MTO (Table 3 and Fig. 8c) and of the water and pyridine adducts of the corresponding mono- and bisperoxides (Table 3). The bisperoxo pyridine adduct will be discussed in more detail; the tensors are shown in Fig. 8a and b.17–19,72 The CSTs and NCS analyses for other intermediates are provided in the ESI† (Table S1, Fig. S1 and Tables S2–S4, Fig. S8–S10,† respectively) and further commented below for comparison.| Compound | δ iso | δ 11 | δ 22 | δ 33 | Ω | C Q |
|---|---|---|---|---|---|---|
| a Ω = δ11 − δ33. b The monoperoxo species is proposed not to coordinate an additional water-ligand; in fact, coordination of water has little effect on the NMR parameters and the water-ligand dissociated upon optimizing the O-transfer transition state.75 c Same data as shown in Table 1. | ||||||
| MTO | 894 (820) | 1392 (1326) | 718 (629) | 571 (506) | 821 (820) | −4.6 (−4) |
| MeReO(O2)2L cis | 416 | 563 | 358 | 326 | 237 | −15.0 |
| MeReO(O2)2L trans | 378 | 528 | 406 | 201 | 328 | −15.2 |
| MeReO(O2)2(OH2) cis | 422 (422)68 | 572 | 365 | 329 | 243 | −15.0 |
| MeReO(O2)2(OH2) trans | 364 (363)68 | 507 | 394 | 192 | 315 | −15.0 |
| MeReO2(O2)L cis | 419 | 565 | 380 | 312 | 253 | −17.2 |
| MeReO2(O2)L trans | 425 | 695 | 357 | 221 | 474 | −13.0 |
| MeReO2(O2) cisb | 437 | 602 | 374 | 336 | 266 | −16.2 |
| MeReO2(O2) transb | 405 | 646 | 335 | 235 | 411 | −13.3 |
| H2O2c | 195 (195) | 364 (362) | 227 (232) | −7 (−9) | 371 (370) | −17.0 (−16) |
 | ||
| Fig. 8 Chemical shielding tensor orientation in (a) MeReO(O2)2L cis, (b) MeReO(O2)2L trans and (c) MTO. The direction of the principal components σ11, σ22, and σ33 are indicated by red, green, and blue arrows, respectively. The chemical shielding values (σ) are indicated next to each principal component, with the corresponding chemical shift values (δ) given in parenthesis. | ||
CST of peroxo intermediates
The measured and calculated chemical shifts of bis- and mono-peroxo intermediates of the MTO-catalyzed olefin epoxidation are given in Table 3. Both the isotropic chemical shift (δiso) and the three principal components of the CST are significantly more deshielded in the metal-peroxide compound by comparison with H2O2 (Table 3, where cis/trans denotes the peroxo oxygen pseudo-cis/trans with respect to the methyl substituent). This observation suggests a change in the electronic structure of the peroxide oxygen atoms, which is likely connected to their increased reactivity towards olefins. Note the significantly larger deshielding found for the oxo-ligands as typically observed for metal-oxo compounds.73,74 The CSTs of the peroxo oxygen atoms have similar orientations as shown in Fig. 8 (and Fig. S1† for the water adducts and the monoperoxo species). For both peroxo oxygens, σ11/δ11 and σ22/δ22 are in the O–Re–O plane (defined by Re and the two peroxo O-atoms) whereas σ33/δ33 is perpendicular to it. In contrast to H2O2, the most deshielded component of the metal peroxo species, δ11, is no longer oriented perpendicularly to the O–O axis but is significantly tilted, while remaining in the peroxo O–Re–O plane. One can also note differences of chemical shifts observed for the cis and trans peroxo oxygens in the bisperoxo compounds, the former being slightly more deshielded than the latter for both δiso and δ11. These values are not strongly affected by the apical ligand (pyridine vs. water), suggesting a similar electronic structure in all bisperoxo intermediates. For the monoperoxo species, the δiso of both peroxo oxygens are more similar, albeit slightly more deshielded than in the bisperoxo intermediates.Orbital analysis of the CSTs
An orbital analysis reveals that the largest contribution to the paramagnetic deshielding of the σ11/δ11 component arises from the LP ‘p’, as was also observed for non-metal-based peroxides (shown in Fig. 9 for the bisperoxo pyridine adduct). Note however, that a significant contribution of the σ(O–Re) bond is also observed for the metal-peroxo species. The other components and peroxo species are given in Tables S2–S4 and Fig. S6–S10.† | ||
| Fig. 9 (a) Orbitals with a significant contribution to the paramagnetic shielding shown for the case of the oxygen pseudo-cis to the methyl group. (b) NCS analysis of the most deshielded component of the CST, σ11, for both peroxo oxygens. | ||
While the contributions of the LP ‘p’ is again indicative of a low-lying σ*(O–O) orbital in combination with high-lying lone-pairs LP ‘p’ on oxygen, the significant contribution of the σ(O–Re) bond to σ11/δ11 evidences the presence of a low-lying vacant orbital perpendicular to the O–Re–O plane with oxygen-contribution. A closer inspection reveals that the bonding combination of the oxygen lone pairs LP ‘p’ perpendicular to the O–Re–O plane – π(O–O) – can interact with an empty metal d-orbital (i.e. dyz) forming a π-bond while the anti-bonding combination of the LP ‘p’ – π*(O–O) – can interact with another empty d-orbital (i.e. dxz) in a δ type fashion (Fig. 10). In fact, the calculated molecular orbitals suggest that the antibonding combinations of the above mentioned δ- and π-bonds are the LUMO and LUMO+1 of the MTO bisperoxide, respectively.
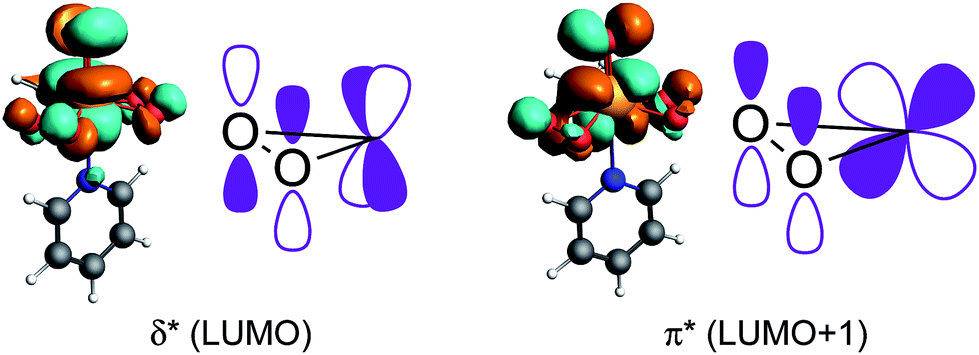 | ||
| Fig. 10 δ*- and π*-orbitals, which were found to be the LUMO and LUMO+1 of the MTO bisperoxide. | ||
As observed for non-metal-based peroxides, the MTO-derived bisperoxide shows rather large CQ values (−15.0 MHz and −15.2 MHz for the cis- and trans-oxygen, respectively, see Table S5 and Fig. S15†) by comparison with MTO (−4.6 MHz). This again indicates a large electric field gradient around the peroxide O-atoms, consistent with a high-lying lone pair oriented perpendicularly to a low-lying σ*(O–O) orbital. As observed for the CST, also the CQ values of the peroxo oxygen atoms are rather insensitive towards replacement of the pyridine ligand by water; the corresponding water adduct shows a CQ of −15.0 MHz on both oxygen atoms. These values differ more in the monoperoxo species, where CQ's are equal to −17.2 and −13.0 MHz for the cis- and trans-oxygen in the pyridine adduct, respectively, consistent with a more dissymmetric structure (Table 3).
Energetic considerations in oxygen-transfer reactions
Transition state energy calculations were performed both for the attack of ethylene at the oxygen pseudo-cis to the methyl group and the oxygen pseudo-trans to it. The obtained geometries for the bis-peroxo pyridine adducts are shown in Fig. 11 (others are given as coordinate files in the ESI†). | ||
| Fig. 11 Optimized transition state geometries for the epoxidation of ethylene with the bisperoxide of MTO, for the attack at the oxygen (a) pseudo-cis and (b) pseudo-trans to the methyl group. | ||
The free energy barriers from the bis-peroxo pyridine adduct are 34.2 kcal mol−1 and 27.2 kcal mol−1 for transfer of the cis and trans oxygens, respectively. Notably, the more deshielded oxygen atom (pseudo-cis to the methyl ligand) is associated with a less favorable oxygen transfer step, consistent with a CST already close to what is observed in metal-oxo species, showing the connection between reactant and product. While in H2O2 the dihedral angle (H–O–O–H) is calculated to be 114° with the two LP ‘p’ on the oxygen pointing away from each other, the lone pairs are coplanar in a peroxo metal complex, introducing again a maximized α-effect (similar to DMDO and mCPBA). In order to understand and quantify the effect of the LP ‘p’ backdonation into the olefin π*-orbital to the transition state energy, and hence probe the importance of the α-effect, a transition state (2nd order saddle point), where the attacking olefin is coplanar with the peroxo O–Re–O plane was calculated for the case of the oxygen pseudo-trans to the methyl group. The obtained free energy of activation was found to be at 31.6 kcal mol−1 and thus 4.4 kcal mol−1 higher in energy than for the perpendicular transition state. This is consistent with what has been found for the epoxidation with non-metal-based peroxides, again evidencing the importance of the backdonation of a high-lying LP ‘p’ on oxygen – the filled π*(O–O) orbital – into the olefin π*(CC) orbital.
Similar trends are observed for the bis-peroxo water adduct with free energy barriers of 34.3 and 27.8 kcal mol−1, for the transfer of the cis- and trans-oxygen, respectively. For the mono-peroxo intermediate, the free energy barriers for O-transfer are typically slightly higher (>29.0 kcal mol−1) for both oxygen atoms, at the exception of the cis-peroxo oxygen of the pyridine adduct (26.8 kcal mol−1). These results suggest that the bisperoxo complex is possibly the more reactive species in water, and that in the presence of pyridine both mono- and bis-peroxo complexes are reactive.72,75 Thus pyridine may have a dual role, i.e. as a phase transfer agent and as a ligand to accelerate catalysis.17,76 Notably, for all metal-peroxo compounds, the easier oxygen transfer is associated with the peroxo oxygen with smaller δiso and δ11, while the oxygen atom which is not transferred displays a larger oxo-character, evidenced by the larger δiso and δ11.
As for DMDO and mCPBA, the coplanarity of the two oxygen lone pairs LP ‘p’ in the MTO peroxo species maximizes the α-effect, raising the energy of the lone pairs thereby increasing their reactivity. In addition, the π-interaction of the peroxo moiety with the metal (Fig. 10) assists the oxygen transfer process in olefin epoxidation, during which a fully developed π(ReO) bond is formed.
Notably, the spectator oxo-ligand in the apical position in the bisperoxo Re complex is not innocent: this oxo-ligand interacts with the peroxo moiety via the metal d-orbitals involved in the π- and δ-interactions (Fig. 10). This interaction minimizes the stabilization of the peroxo LP ‘p’ by weakening the (stabilizing) δ- and π-bonds. In addition, the presence of the spectator oxo-ligand also provides a driving force for the formation of the metal-peroxo species in the catalytic cycle: formation of the peroxo species from MTO strengthens the bond of the apical “spectator” oxo-ligand as evidenced by a slight decrease of the ReO bond length on going from MTO (1.69 Å) to the monoperoxo (1.68 Å) and then the bisperoxo (1.67 Å) pyridine adducts. This effect is reminiscent of the spectator oxo effect discussed for metallacyclobutane formation from metal alkylidenes during the olefin metathesis reaction, as well as for the formation of metallacycle oxetane intermediates in the reaction of alkenes with metal oxo compounds.77–79
Conclusions
Overall, peroxide compounds are associated with significantly deshielded 17O chemical shifts that indicate the presence of low-lying vacant and high-lying occupied orbitals, corresponding to the σ*(O–O) and the lone pairs on oxygens, associated with π(O–O) and π*(O–O), for both metal-based and non-metal-based peroxides. These specific electronic features are particularly pronounced in peroxide species that engage in electrophilic epoxidation reactions (DMDO, mCPBA, and MTO bisperoxo), as evidenced by their remarkably large deshielding. This is due to the coplanarity of the oxygen lone-pairs in these peroxides which is induced by their strained cyclic structure or by H-bonding in the case of mCPBA. Both maximize overlaps and in fine raises the HOMO (α-effect) and increases reactivity towards electrophilic epoxidation. In metal peroxo species, this HOMO is further raised in energy by the presence of a spectator oxo-ligand in the apical position. In fact, this “spectator” oxo species participates in modulating the reactivity of peroxo intermediates in transition-metal-catalyzed oxidation processes; it is thus not surprising that such a moiety is ubiquitous in efficient epoxidation catalysts that use H2O2 as a primary oxidant. The α-effect and the presence of a strained cyclic structure goes hand in hand with a weakening of the O–O bond. Both the high-lying lone pair of the filled π*(O–O) and the low-lying σ*(O–O) orbital drive the observed reactivity in electrophilic epoxidation, in which these two orbitals interact with the π*(CC) and π(CC) orbitals of the olefin, respectively. Thus, 17O NMR chemical shift provides a powerful descriptor to pinpoint key electronic features that are decisive for reactivity in oxidation chemistry (Fig. 12).
 | ||
| Fig. 12 NMR chemical shift probes specific electronic features with relevance for reactivity in electrophilic epoxidation. | ||
Epilogue
The frontier orbital interactions shown in Fig. 6 highlight the following observation: while an epoxidation with DMDO or mCPBA is typically thought of as an electrophilic epoxidation, with the oxidant acting as electrophile and the olefin acting as nucleophile, these molecular orbital interactions indicate that both substrates act as nucleophiles and electrophiles by exploiting the low-lying σ*(O–O) orbital and the high-lying lone pair LP ‘p’ (O) induced by the α-effect. This is reminiscent of synergistic effects observed in transition metal chemistry, for example in olefin complexes or in oxidative addition processes (Fig. 13a and b). Considering that olefin epoxidation with DMDO or mCPBA is isolobal to epoxidations with oxaziridines and halogenation reactions by X2 or NXS (X = Cl, Br, I), a similar orbital picture can be anticipated for these “electrophilic” additions (Fig. 13c).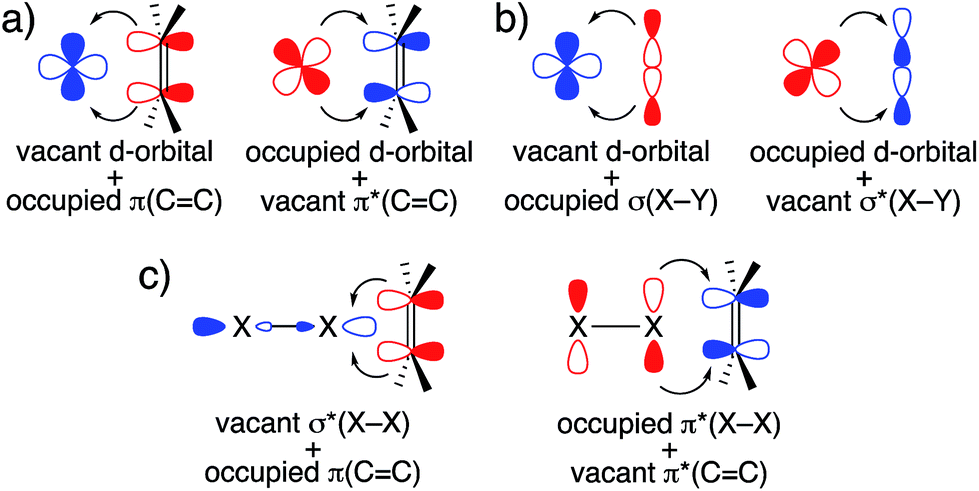 | ||
| Fig. 13 Relevant synergistic orbital interactions in (a) metal-olefin complexes, (b) oxidative addition processes, and (c) olefin halogenation (X = Cl, Br, I). Electron-donating and -accepting orbitals are colored in red and blue, respectively. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. P. G is a recipient of the Scholarship Fund of the Swiss Chemical Industry. E. Lam is acknowledged for helpful discussions. K. Yamamoto and W.-C. Liao are acknowledged for the synthesis and the NMR characterization of 17O-labelled H2O2, respectively.Notes and references
- R. A. Sheldon, J. Mol. Catal., 1980, 7, 107–126 CrossRef CAS
.
- B. Notari, Adv. Catal., 1996, 41, 253–334 CAS
-
R. Jira, R. A. Sheldon, P. Lappe, E. Schulz, R. W. Fischer and F. Röhrscheid, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, 2008 Search PubMed
- T. A. Nijhuis, M. Makkee, J. A. Moulijn and B. M. Weckhuysen, Ind. Eng. Chem. Res., 2006, 45, 3447–3459 CrossRef CAS
- T. Zhang, L. Mazaud, L.-M. Chamoreau, C. Paris, A. Proust and G. Guillemot, ACS Catal., 2018, 8, 2330–2342 CrossRef CAS
-
B. Bühler, K. Bühler and F. Hollman, in Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H.Gröger and O.May, Wiley-VCH, Weinheim, 2012 Search PubMed
- Y. Liang, J. Wei, X. Qiu and N. Jiao, Chem. Rev., 2018, 118, 4912–4945 CrossRef CAS PubMed
- J. Dong, E. Fernández-Fueyo, F. Hollmann, C. E. Paul, M. Pesic, S. Schmidt, Y. Wang, S. Younes and W. Zhang, Angew. Chem., Int. Ed., 2018, 57, 9238–9261 CrossRef CAS PubMed
- N. Prileschajew, Ber. Dtsch. Chem. Ges., 1909, 42, 4811–4815 CrossRef CAS
- H. Hussain, A. Al-Harrasi, I. R. Green, I. Ahmed, G. Abbas and N. U. Rehman, RSC Adv., 2014, 4, 12882–12917 RSC
- R. W. Murray and R. Jeyaraman, J. Org. Chem., 1985, 50, 2847–2853 CrossRef CAS
- Y. Zhu, Q. Wang, R. G. Cornwall and Y. Shi, Chem. Rev., 2014, 114, 8199–8256 CrossRef CAS PubMed
- F. A. Davis and A. C. Sheppard, Tetrahedron, 1989, 45, 5703–5742 CrossRef CAS
- K. S. Williamson, D. J. Michaelis and T. P. Yoon, Chem. Rev., 2014, 114, 8016–8036 CrossRef CAS PubMed
- W. A. Herrmann, R. W. Fischer, W. Scherer and M. U. Rauch, Angew. Chem., Int. Ed. Engl., 1993, 32, 1157–1160 CrossRef
- W. A. Herrmann, R. W. Fischer, M. U. Rauch and W. Scherer, J. Mol. Catal., 1994, 86, 243–266 CrossRef CAS
- J. Rudolph, K. L. Reddy, J. P. Chiang and K. B. Sharpless, J. Am. Chem. Soc., 1997, 119, 6189–6190 CrossRef CAS
- C. Copéret, H. Adolfsson and K. B. Sharpless, Chem. Commun., 1997, 0, 1565–1566 RSC
- H. Adolfsson, C. Copéret, J. P. Chiang and A. K. Yudin, J. Org. Chem., 2000, 65, 8651–8658 CrossRef CAS PubMed
- S. Huber, M. Cokoja, M. Drees, J. Mínk and F. E. Kühn, Catal. Sci. Technol., 2013, 3, 388–393 RSC
- F. Dyckhoff, S. Li, R. M. Reich, B. J. Hofmann, E. Herdtweck and F. E. Kühn, Dalton Trans., 2018, 47, 9755–9764 RSC
- G. B. Payne and P. H. Williams, J. Org. Chem., 1959, 24, 54–55 CrossRef CAS
- C. Venturello and R. D'Aloisio, J. Org. Chem., 1988, 53, 1553–1557 CrossRef CAS
- J. Y. Piquemal, S. Halut and J. M. Brégeault, Angew. Chem., Int. Ed., 1998, 37, 1146–1149 CrossRef CAS PubMed
- T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974–5976 CrossRef CAS
- T. Itoh, K. Jitsukawa, K. Kaneda and S. Teranishi, J. Am. Chem. Soc., 1979, 101, 159–169 CrossRef CAS
- J. Jarupatrakorn and T. D. Tilley, J. Am. Chem. Soc., 2002, 124, 8380–8388 CrossRef CAS
- Y. Sawada, K. Matsumoto, S. Kondo, H. Watanabe, T. Ozawa, K. Suzuki, B. Saito and T. Katsuki, Angew. Chem., Int. Ed., 2006, 45, 3478–3480 CrossRef CAS PubMed
- A. Berkessel, T. Günther, Q. Wang and J.-M. Neudörfl, Angew. Chem., Int. Ed., 2013, 52, 8467–8471 CrossRef CAS PubMed
- M. Kaupp, V. G. Malkin, O. L. Malkina and D. R. Salahub, Chem.–Eur. J., 1996, 2, 24–30 CrossRef CAS
- V. H. Gessner, F. Meier, D. Uhrich and M. Kaupp, Chem.–Eur. J., 2013, 19, 16729–16739 CrossRef CAS PubMed
- S. Halbert, C. Copéret, C. Raynaud and O. Eisenstein, J. Am. Chem. Soc., 2016, 138, 2261–2272 CrossRef CAS PubMed
- D. P. Estes, C. P. Gordon, A. Fedorov, W.-C. Liao, H. Ehrhorn, C. Bittner, M. L. Zier, D. Bockfeld, K. W. Chan, O. Eisenstein, C. Raynaud, M. Tamm and C. Copéret, J. Am. Chem. Soc., 2017, 139, 17597–17607 CrossRef CAS PubMed
- C. P. Gordon, K. Yamamoto, W.-C. Liao, F. Allouche, R. A. Andersen, C. Copéret, C. Raynaud and O. Eisenstein, ACS Cent. Sci., 2017, 3, 759–768 CrossRef CAS PubMed
- K. Yamamoto, C. P. Gordon, W. C. Liao, C. Copéret, C. Raynaud and O. Eisenstein, Angew. Chem., Int. Ed., 2017, 56, 10127–10131 CrossRef CAS PubMed
- C. P. Gordon, K. Yamamoto, K. Searles, S. Shirase, R. A. Andersen, O. Eisenstein and C. Copéret, Chem. Sci., 2018, 9, 1912–1918 RSC
- L. Foppa, K. Yamamoto, W.-C. Liao, A. Comas-Vives and C. Copéret, J. Phys. Chem. Lett., 2018, 9, 3348–3353 CrossRef CAS PubMed
- J. Autschbach, S. Zheng and R. W. Schurko, Concepts Magn. Reson., Part A, 2010, 36A, 84–126 CrossRef CAS
- L. L. G. Justino, M. L. Ramos, F. Nogueira, A. J. F. N. Sobral, C. F. G. C. Geraldes, M. Kaupp, H. D. Burrows, C. Fiolhais and V. M. S. Gil, Inorg. Chem., 2008, 47, 7317–7326 CrossRef CAS PubMed
- L. L. G. Justino, M. L. Ramos, M. Kaupp, H. D. Burrows, C. Fiolhais and V. M. S. Gil, Dalton Trans., 2009, 0, 9735–9745 RSC
- E. Lam, A. Comas-Vives and C. Copéret, J. Phys. Chem. C, 2017, 121, 19946–19957 CrossRef CAS
- E. Lam and C. Copéret, Helv. Chim. Acta, 2018, 101, e1800120 CrossRef
- M. Leskes, N. E. Drewett, L. J. Hardwick, P. G. Bruce, G. R. Goward and C. P. Grey, Angew. Chem., Int. Ed., 2012, 51, 8560–8563 CrossRef CAS PubMed
- J. Lu, X. Kong, V. Terskikh and G. Wu, J. Phys. Chem. A, 2015, 119, 8133–8138 CrossRef CAS PubMed
- S. Zhang, M. J. Nava, G. K. Chow, N. Lopez, G. Wu, D. R. Britt, D. G. Nocera and C. C. Cummins, Chem. Sci., 2017, 8, 6117–6122 RSC
- R. Gupta, J. Stringer, J. Struppe, D. Rehder and T. Polenova, Solid State Nucl. Magn. Reson., 2018, 91, 15–20 CrossRef CAS PubMed
- C. M. Widdifield and R. W. Schurko, Concepts Magn. Reson., Part A, 2009, 34A, 91–123 CrossRef CAS
- J. Zhu, T. Kurahashi, H. Fujii and G. Wu, Chem. Sci., 2012, 3, 391–397 RSC
- R. N. Kerber, A. Kermagoret, E. Callens, P. Florian, D. Massiot, A. Lesage, C. Copéret, F. Delbecq, X. Rozanska and P. Sautet, J. Am. Chem. Soc., 2012, 134, 6767–6775 CrossRef CAS PubMed
- R. N. Kerber, T. Kerber, X. Rozanska, F. Delbecq and P. Sautet, Phys. Chem. Chem. Phys., 2015, 17, 26937–26945 RSC
- D. Culver, W. Huynh, H. Tafazolian, T.-C. Ong and M. Conley, Angew. Chem., Int. Ed., 2018, 57, 9520–9523 CrossRef CAS PubMed
- K. Yamamoto, S. Tanaka, H. Hosoya, H. Tsurugi, K. Mashima and C. Copéret, Helv. Chim. Acta DOI:10.1002/hlca.201800156
- S. E. Ashbrook and M. E. Smith, Chem. Soc. Rev., 2006, 35, 718–735 RSC
- S. E. Ashbrook, Phys. Chem. Chem. Phys., 2009, 11, 6892–6905 RSC
-
K. Yamada, in Annual Reports on NMR Spectroscopy, ed. G. Webb, Academic Press, 2010, vol. 70, pp. 115–158 Search PubMed
- J. J. Barieux and J. P. Schirmann, Tetrahedron Lett., 1987, 28, 6443–6446 CrossRef CAS
- J. K. Crandall and M. A. Centeno, J. Org. Chem., 1979, 44, 1183–1184 CrossRef CAS
- L. Antolini, R. Benassi, S. Ghelli, U. Folli, S. Sbardellati and F. Taddei, J. Chem. Soc., Perkin Trans. 1, 1992, 0, 1907–1913 RSC
- L. Cassidei, M. Fiorentino, R. Mello, O. Sciacovelli and R. Curci, J. Org. Chem., 1987, 52, 699–700 CrossRef CAS
- G. Schreckenbach and T. Ziegler, J. Phys. Chem., 1995, 99, 606–611 CrossRef CAS
- J. A. Bohmann, F. Weinhold and T. C. Farrar, J. Chem. Phys., 1997, 107, 1173–1184 CrossRef CAS
- E. L. Sceats, J. S. Figueroa, C. C. Cummins, N. M. Loening, P. Van der Wel and R. G. Griffin, Polyhedron, 2004, 23, 2751–2768 CrossRef CAS
- J. Autschbach, J. Chem. Phys., 2008, 128, 164112 CrossRef PubMed
- M. Pascual-Borras, X. Lopez, A. Rodriguez-Fortea, R. J. Errington and J. M. Poblet, Chem. Sci., 2014, 5, 2031–2042 RSC
- I. B. Moroz, K. Larmier, W.-C. Liao and C. Copéret, J. Phys. Chem. C, 2018, 122, 10871–10882 CrossRef CAS
- A. D. Walsh, Trans. Faraday Soc., 1949, 45, 179–190 RSC
-
E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, University of Wisconsin, Madison, 2013 Search PubMed
- M. S. Reynolds and A. Butler, Inorg. Chem., 1996, 35, 2378–2383 CrossRef CAS PubMed
- A. M. Al-Ajlouni and J. H. Espenson, J. Org. Chem., 1996, 61, 3969–3976 CrossRef CAS PubMed
- S. A. Hauser, V. Korinth, E. Herdtweck, M. Cokoja, W. A. Herrmann and F. E. Kühn, Eur. J. Inorg. Chem., 2010, 4083–4090 CrossRef CAS
- P. Gisdakis, S. Antonczak, S. Küstlmeier, W. A. Herrmann and N. Rösch, Angew. Chem., Int. Ed., 1998, 37, 2211–2214 CrossRef CAS PubMed
- W.-D. Wang and J. H. Espenson, J. Am. Chem. Soc., 1998, 120, 11335–11341 CrossRef CAS
- M. Kaupp, O. L. Malkina and V. G. Malkin, J. Chem. Phys., 1997, 106, 9201–9212 CrossRef CAS
- W. A. Herrmann, H. Ding, F. E. Kühn and W. Scherer, Organometallics, 1998, 17, 2751–2757 CrossRef CAS
- A. M. Al-Ajlouni and J. H. Espenson, J. Am. Chem. Soc., 1995, 117, 9243–9250 CrossRef CAS
- D. J. Berrisford, C. Bolm and K. B. Sharpless, Angew. Chem., Int. Ed. Engl., 1995, 34, 1059–1070 CrossRef CAS
- A. K. Rappé and W. A. Goddard III, Nature, 1980, 285, 311 CrossRef
- A. K. Rappé and W. A. Goddard III, J. Am. Chem. Soc., 1982, 104, 448–456 CrossRef
- J. M. Gonzales, R. Distasio, R. A. Periana, W. A. Goddard and J. Oxgaard, J. Am. Chem. Soc., 2007, 129, 15794–15804 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Computational details, calculated chemical shift tensors and quadrupolar coupling parameters, complete results of NCS analysis, visualizations of relevant NLMOs, graphical representations of chemical shift and EFG tensors, experimentally measured NMR spectra and corresponding simulations, coordinates of all computed structures. See DOI: 10.1039/c8sc04868a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |