DOI:
10.1039/C8SC04115F
(Edge Article)
Chem. Sci., 2019,
10, 1796-1801
A metal-catalyzed new approach for α-alkynylation of cyclic amines†
Received
16th September 2018
, Accepted 24th November 2018
First published on 26th November 2018
Abstract
The first catalytic α-alkynylation of cyclic amines with the help of the N-propargylic group to afford 2-(1-alkynyl) N-allylic cyclic amines with an exclusive E-stereoselectivity for the in situ formed C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond has been realized. Based on mechanistic studies, it is proven that the reaction proceeds through metal-mediated anti-1,5-hydride transfer forming an iminonium intermediate, which accepts the addition of the in situ generated 1-alkynyl metal species. The synthetic application has also been demonstrated.
C bond has been realized. Based on mechanistic studies, it is proven that the reaction proceeds through metal-mediated anti-1,5-hydride transfer forming an iminonium intermediate, which accepts the addition of the in situ generated 1-alkynyl metal species. The synthetic application has also been demonstrated.
Introduction
Due to the synthetic1 and bio-importance2–7 of cyclic amines much attention has been focused on the development of the related methodologies. One straightforward approach is α-functionalization of readily available cyclic amines.8 Oxidative coupling of N-protected cyclic amines with terminal alkynes or 1-alkynyl trifluoroborate in the presence of a stoichiometric amount of an oxidant (eqn (1)) and the three-component reaction of N-non-protected cyclic amines with terminal alkynes and aldehydes have been well established (eqn (2)).9,10 Starting from 2010, we have reported the ZnX2,11,12 CuI,13 or CuBr2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14-mediated allenylation of terminal alkynes (ATA) reaction with aldehydes in the presence of different amines forming allenes. In this reaction, the second step is the metal-mediated 1,5-H transfer reaction of propargylic amines in situ formed in the first step, which was proven to be non-stereoselective by Nakamura et al. affording allylic propargylic amines with an E/Z ratio of 58/42–63/37 with acyclic amine.15 Herein, we wish to report a highly stereoselective N-propargylic cyclic amine-based α-alkynylation providing stereodefined N-(E)-allylic 2-alkynyl cyclic amines by using CdBr2 (or ZnI2) as the catalyst (Scheme 1).
14-mediated allenylation of terminal alkynes (ATA) reaction with aldehydes in the presence of different amines forming allenes. In this reaction, the second step is the metal-mediated 1,5-H transfer reaction of propargylic amines in situ formed in the first step, which was proven to be non-stereoselective by Nakamura et al. affording allylic propargylic amines with an E/Z ratio of 58/42–63/37 with acyclic amine.15 Herein, we wish to report a highly stereoselective N-propargylic cyclic amine-based α-alkynylation providing stereodefined N-(E)-allylic 2-alkynyl cyclic amines by using CdBr2 (or ZnI2) as the catalyst (Scheme 1).
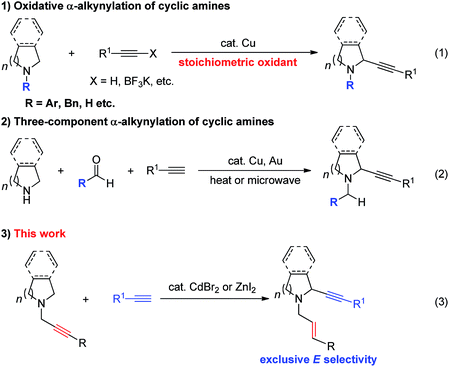 |
| | Scheme 1 Different approaches for α-alkynylation of cyclic amines. | |
Results and discussion
Optimization of the reaction
When we studied the mechanism of the Cu-catalyzed allenylation of terminal alkynes in the presence of an amine,13 it was observed that the reaction between N-alkynylic amine 1a and phenylacetylene 2a under CuBr catalysis provided a new product 3aa in a low yield of 13% with 64% of starting material 1a being recovered as judged by 1H NMR analysis. This new product was identified as α-alkynylated cyclic amine with an N-allylic group bearing an exclusive E CC bond (Table 1, entry 1). Due to the importance of cyclic amines, we further optimized the reaction conditions by screening a variety of metal salts such as CuX2, ZnX2, AgOTf and CdX2, and CdBr216 turned out to be the best providing the product 3aa in 42% yield and 52% recovery of 1a (Table 1, entries 2–7). On increasing the temperature to 120 °C, the yield was improved to 56% with 20% recovery (Table 1, entry 8).
Table 1 Optimization of catalytic α-alkynylation of 1-(2-alkynyl) cyclic amine 1a with 2aa
|
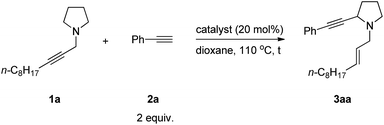
|
| Entry |
Catalyst |
Time (h) |
Yield of 3aab (%) |
Recovery of 1ab (%) |
|
The reaction was conducted using 1a (1.0 mmol) and alkyne 2a (2.0 mmol) in 6 mL of dioxane at 110 °C.
Determined by 1H NMR analysis with CH2Br2 as the internal standard.
The reaction was conducted at 120 °C.
The reaction was conducted at 130 °C.
|
| 1 |
CuBr |
12 |
13 |
64 |
| 2 |
CuBr2 |
10 |
13 |
— |
| 3 |
ZnCl2 |
10 |
24 |
— |
| 4 |
ZnBr2 |
10 |
39 |
— |
| 5 |
AgOTf |
12 |
5 |
95 |
| 6 |
CdI2 |
10 |
40 |
— |
| 7 |
CdBr2 |
24 |
42 |
53 |
| 8c |
CdBr2 |
36 |
56 |
20 |
| 9d |
CdBr2 |
12 |
47 |
26 |
Effect of solvents
Then solvents were screened: when tBuOMe was used as the solvent, the desired product 3aa could be obtained in 63% yield with complete consumption of 1a (Table 2, entries 1–7). In addition, reducing the catalyst loading to 10 mol% improved the yield slightly to 66% (Table 2, entries 8–9). Further reducing the catalyst loading resulted in the recovery of 1a (Table 2, entry 10). Thus, 1a (1 equiv.), 2a (2 equiv.), and CdBr2 (10 mol%) in tBuOMe at 120 °C were defined as the optimized reaction conditions for further study of this reaction.
Table 2 Optimization of reaction conditions for catalytic α-alkynylation of N-internal 2-alkynylic cyclic amine 1a with 2aa
|
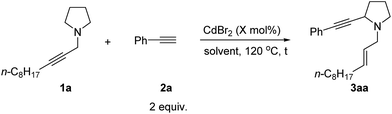
|
| Entry |
X
|
Solvent |
t (h) |
Yield of 3aab (%) |
Recovery of 1bb (%) |
|
The reaction was conducted using 1a (0.5 mmol) and alkyne 2a (1.0 mmol) in 3 mL of solvent.
Determined by 1H NMR analysis with CH2Br2 as the internal standard.
The reaction was conducted using 1a (1.0 mmol) and alkyne 2a (2.0 mmol) in 6 mL of tBuOMe at 120 °C.
|
| 1 |
20 |
DMF |
23 |
43 |
25 |
| 2 |
20 |
DMSO |
23 |
20 |
35 |
| 3 |
20 |
Toluene |
23 |
40 |
— |
| 4 |
20 |
THF |
23 |
48 |
— |
| 5 |
20 |
DCE |
23 |
3 |
— |
| 6 |
20 |
CH3CN |
23 |
39 |
— |
| 7 |
20 |
t
BuOMe |
36 |
63 |
— |
| 8 |
15 |
t
BuOMe |
36 |
64 |
— |
|
9
|
10
|
t
BuOMe
|
36
|
66
|
—
|
| 10 |
5 |
t
BuOMe |
36 |
69 |
10 |
Substrate scope
With the optimal reaction conditions in hand, diversified terminal alkynes were investigated to examine the scope of this α-alkynylation reaction with amine 1a. Terminal aryl acetylenes bearing electron-donating p-Me and p-MeO, and electron-withdrawing and synthetically attractive p-F, p-Cl, m-Cl, p-NO2, p-EtOOC, p-CN and p-Ac groups on the aryl ring could all afford the corresponding product 3 in moderate yields (Table 3, entries 1–10). In addition, alkyl-substituted terminal alkynes, such as 1-decyne (2k) and cyclohexylacetylene (2l), were found to be sluggish affording the corresponding products in 31% and 40% yields, respectively (Table 3, entries 11 and 12). Interestingly, trimethylsilylacetylene could react with 1a to furnish 3am in 76% yield (Table 3, entry 13). Other substituted propargylic amines, such as 1b, 1c, 1d and 1g could also react smoothly to afford the desired products 3ba, 3ca, 3da and 3ga in 40–65% yields (Table 3, entries 14–16, 21). N-Terminal propargylic amine 1e was next exposed to the optimized conditions with arylacetylenes substituted with different functional groups, such as electron-donating p-MeO and electron-withdrawing p-F and p-Cl, affording the corresponding products 3ea–3ee in moderate yields with 30 mol% of ZnI2 (Table 3, entries 17–20).
Table 3 The scope of catalytic α-alkynylation of N-internal 2-alkynylic cyclic aminesa
Tetrahydroisoquinoline is the core skeleton of a variety of natural bio-active compounds and drugs.17 We first applied CdBr2 in tBuOMe to N-propargylic tetrahydroisoquinoline derivative 1f and phenylacetylene 2a. The 1-alkynated product was obtained exclusively in 57% isolated yield with 24% 1f recovery. Interestingly, using 10 mol% ZnI2 as the catalyst and dioxane as the solvent, the reaction afforded 3fa in a yield of 78%. Trimethylsilylacetylene (2m) and 1-hexyne (2n) are also compatible (Scheme 2).
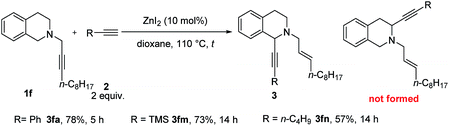 |
| | Scheme 2 The scope of catalytic α-alkynylation of tetrahydroisoquinoline 1f. The reaction was conducted using 1f (1.0 mmol) and alkyne 2 (2.0 mmol) in 6 mL of dioxane at 110 °C. | |
For piperidine derivative 1h, a larger catalyst-loading is required and toluene was also necessary since the reaction in MTBE resulted in 13% yield of the target product with 89% recovery of 1h. Unfortunately, morpholine did not work (Scheme 3).
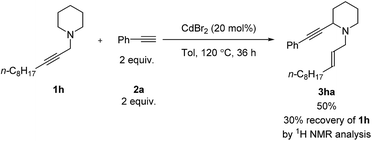 |
| | Scheme 3 Catalytic α-alkynylation of piperidine 1h. The reaction was conducted using 1h (1.0 mmol) and phenylacetylene 2a (2.0 mmol) in 6 mL of toluene at 120 °C. | |
Furthermore, several non-cyclic amines were investigated. The reaction of diisopropylamine 1i with phenylacetylene 2a generated 55% yield of 1,2-undecadiene11–14 (Scheme 4). When we applied diisobutylamine 1j and diallylamine 1k under the standard reaction conditions, such reactions were not observed.
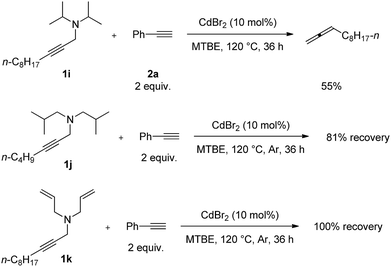 |
| | Scheme 4 The reaction of non-cyclic amine 1i–1k. The reaction was conducted using 1i–1k (1.0 mmol) and phenylacetylene 2a (2.0 mmol) in 6 mL of MTBE at 120 °C. | |
Deuterium experiments
To gain insight into the mechanism of this reaction, deuterium-labeled d4-1a was treated with 2a under standard conditions to give d4-3aa with 95% D incorporation, which reveals that the hydrogen at the γ-position of the allylic group comes from the α-position of the amine unit (Scheme 5a). In addition, 24% of deuterium incorporation was observed in the 2-position of the N-allylic group in product d2-3aa of the reaction between deuterium-labeled d-2a and 1a (Scheme 5b). The control experiment of treating 3aa with d-2a led to no deuterium incorporation (Scheme 5c).
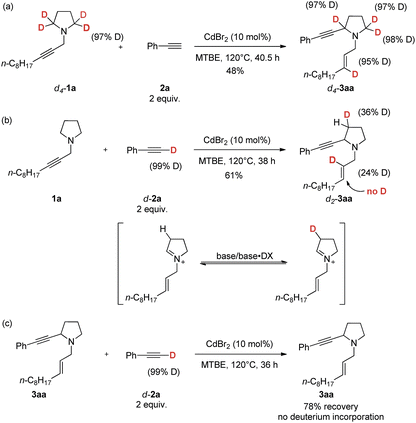 |
| | Scheme 5 Deuterium labeling experiments. | |
Based on the above deuterium labeling experiments and the products in the E configuration, a plausible mechanism proposed is shown in Scheme 6. The propargylic amine 1 coordinates to MX2 to form Int 1, which would undergo anti-1,5-hydride transfer to form cationic Int 2 in the E configuration.15 Subsequently, 1-alkynyl cadmium species Int 3, in situ generated from terminal alkyne, CdBr2, and amine, would react with the iminium ion Int 2 to afford the corresponding α-substituted cyclic amine 3 (Scheme 6). In addition, the possibility of forming the product from allenyl amine 1′ is excluded since there is no D-incorporation at the 3-position of the E-allylic unit in the product of eqn (b) of Scheme 5. It is believed that CdBr2 may coordinate better with the C–C triple bond to trigger the 1,5-H transfer reaction.
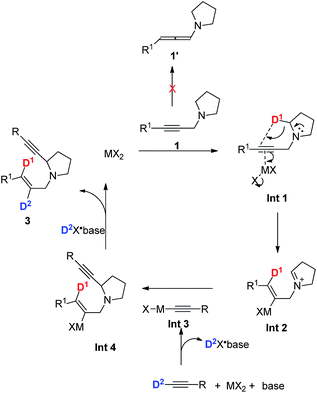 |
| | Scheme 6 A plausible mechanism for the formation of 3. | |
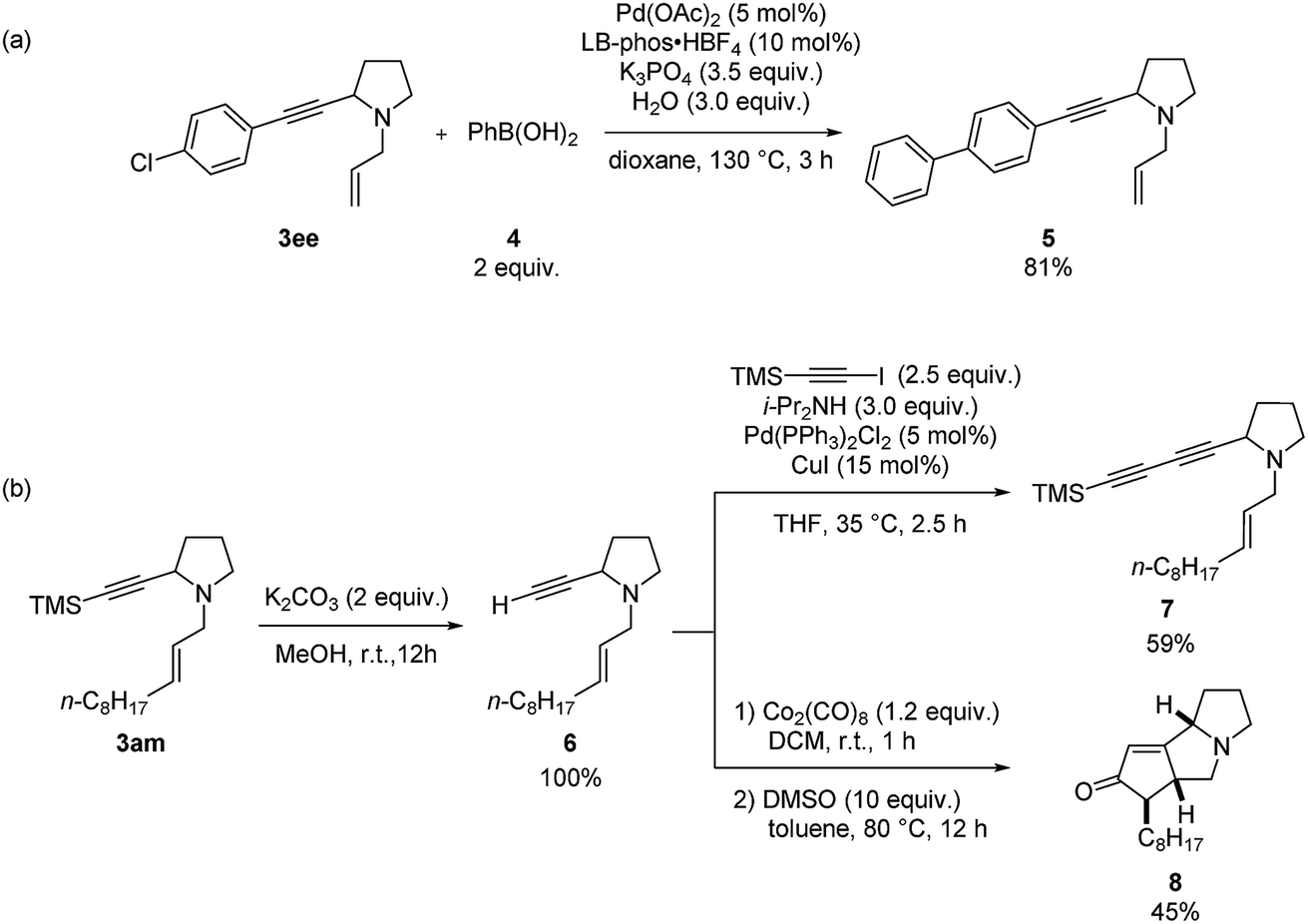
Finally, we conducted a gram-scale synthesis of both 3ee and 3am (Scheme 7).
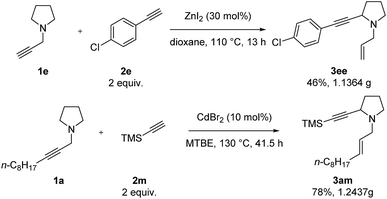 |
| | Scheme 7 Gram-scale synthesis. | |
Synthetic applications
Furthermore, diversified synthetic utilities of these two products were demonstrated. Suzuki coupling between 3ee and phenyl boronic acid using LB-Phos·HBF418 affords 5 in 81% yield (Scheme 8a). Deprotection of the TMS group in 3am with K2CO3 in MeOH afforded enyne 6, which may react with 1-trimethylsilylethynyl iodide to afford conjugated diyne 7 (Scheme 8b). Sequential treatment of 6 with 1.2 equiv. of Co2(CO)8 and 10 equiv. of DMSO afforded the Pauson–Khand reaction product 8 in 45% yield.19
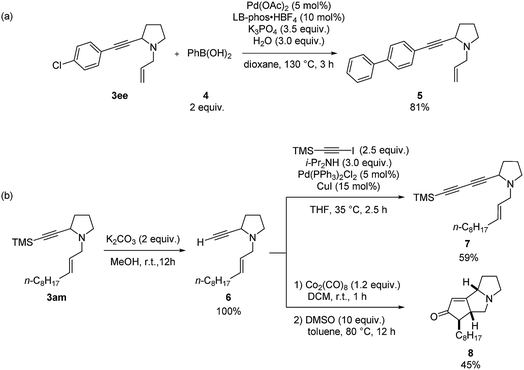 |
| | Scheme 8 Synthetic applications. | |
Conclusions
In conclusion, we have succeeded in developing a catalytic α-alkynylation of N-propargylic cyclic amines, providing 1-(2(E)-alkenyl) 2-(1-alkynyl) cyclic amines highly stereoselectively. Further studies on identifying the chiral catalyst, the scope of nucleophiles, and their applications to natural products are being actively pursued in the laboratory.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the National Basic Research Program of China (2015CB856600) and the National Natural Science Foundation of China (21772172) is greatly appreciated. We thank Di Zhai in this group for reproducing the results for 3ac, 3am, and 3ec presented in Table 3.
Notes and references
- For reviews:
(a) H.-P. Husson and J. Royer, Chem. Soc. Rev., 1999, 28, 383 RSC
 ;
(b) J. Cossy, Chem. Rec., 2005, 5, 70 CrossRef CAS PubMed ;
(c) C. Escolano, M. Amat and J. Bosch, Chem.–Eur. J., 2006, 12, 8198 CrossRef CAS PubMed .
;
(b) J. Cossy, Chem. Rec., 2005, 5, 70 CrossRef CAS PubMed ;
(c) C. Escolano, M. Amat and J. Bosch, Chem.–Eur. J., 2006, 12, 8198 CrossRef CAS PubMed .
- J. E. Henningfield and M. Zeller, Psychopharmacology, 2006, 184, 286 CrossRef CAS PubMed .
- S. T. Lee, B. T. Green, K. D. Welch, J. A. Pfister and K. E. Panter, Chem. Res. Toxicol., 2008, 21, 2061 Search PubMed .
- R. K. Hill, T. H. Chan and J. A. Joule, Tetrahedron, 1965, 21, 147 CrossRef CAS PubMed .
- P. Mungkornasawakul, S. Chaiyong, T. Sastraruji, A. Jatisatienr, C. Jatisatienr, S. G. Pyne, A. T. Ung, J. Korth and W. Lie, J. Nat. Prod., 2009, 72, 848 CrossRef CAS PubMed .
- J. P. Michael, Nat. Prod. Rep., 2008, 25, 139 RSC .
- T. Tokuyama, N. Nishimori, A. Shimada, M. W. Edwards and J. W. Daly, Tetrahedron, 1987, 43, 643 CrossRef CAS .
- For reviews on α-functionalization of cyclic amines:
(a) S.-I. Murahashi, Angew. Chem., Int. Ed., 1995, 34, 2443 CrossRef CAS ;
(b) S. Doye, Angew. Chem., Int. Ed., 2001, 40, 3351 CrossRef CAS ;
(c) K. R. Campos, Chem. Soc. Rev., 2007, 36, 1069 RSC ;
(d) S.-I. Murahashi and D. Zhang, Chem. Soc. Rev., 2008, 37, 1490 RSC ;
(e) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed ;
(f) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16, 2654 CrossRef CAS PubMed ;
(g) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed ;
(h) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed ;
(i) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed ;
(j) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC ;
(k) E. A. Mitchell, A. Peschiulli, N. Lefevre, L. Meerpoel and B. U. W. Maes, Chem.–Eur. J., 2012, 18, 10092 CrossRef CAS PubMed ;
(l) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687 RSC ;
(m) D. Hoppe and T. Hense, Angew. Chem., Int. Ed., 1997, 36, 2282 CrossRef CAS ;
(n) P. Beak, A. Basu, D. J. Gallagher, Y. S. Park and S. Thayumanavan, Acc. Chem. Res., 1996, 29, 552 CrossRef CAS ;
(o) M. C. Haibach and D. Seidel, Angew. Chem., Int. Ed., 2014, 53, 5010 CrossRef CAS PubMed ;
(p) K. Lauder, A. Toscani, N. Scalacci and D. Castagnolo, Chem. Rev., 2017, 117, 14091 CrossRef CAS PubMed ;
(q) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587 CrossRef CAS PubMed .
- Examples on oxidative α-alkynylation of cyclic amines:
(a) Z. Li and C.-J. Li, J. Am. Chem. Soc., 2004, 126, 11810 CrossRef CAS PubMed ;
(b) Z. Li, D. S. Bohle and C.-J. Li, Proc. Natl. Acad. Sci. U. S. A., 2006, 8928 CrossRef CAS PubMed ;
(c) I. D. Jurberg, B. Peng, E. Wçstefeld, M. Wasserloos and N. Maulide, Angew. Chem., Int. Ed., 2012, 51, 1950 CrossRef CAS PubMed ;
(d) R.-Y. Zhang, L.-Y. Xi, L. Zhang, S. Liang, S.-Y. Chen and X.-Q. Yu, RSC Adv., 2014, 4, 54349 RSC ;
(e) W. Chen, L. Ma, A. Paul and D. Seidel, Nat. Chem., 2018, 10, 165 CrossRef CAS PubMed .
- Recent examples on three-component α-alkynylation of cyclic amines:
(a) D. Das, A. X. Sun and D. Seidel, Angew. Chem., Int. Ed., 2013, 52, 3765 CrossRef CAS PubMed ;
(b) J. Li, H. Wang, J. Sun, Y. Yang and L. Liu, Org. Biomol. Chem., 2014, 12, 2523 RSC ;
(c) W. Lin, T. Cao, W. Fan, Y. Han, J. Kuang, H. Luo, B. Miao, X. Tang, Q. Yu, W. Yuan, J. Zhang, C. Zhu and S. Ma, Angew. Chem., Int. Ed., 2014, 53, 277 CrossRef CAS PubMed ;
(d) C. Zhao and D. Seidel, J. Am. Chem. Soc., 2015, 137, 4650 CrossRef CAS ;
(e) H. Cheng, J. Wen and C. Bolm, Chem.–Eur. J., 2017, 23, 12100 CrossRef CAS ;
(f) D. Seidel, Org. Chem. Front., 2014, 1, 426 RSC .
-
(a) J. Ye, S. Li, B. Chen, W. Fan, J. Kuang, J. Liu, Y. Liu, B. Miao, B. Wan, Y. Wang, X. Xie, Q. Yu, W. Yuan and S. Ma, Org. Lett., 2012, 14, 1346 CrossRef CAS PubMed ;
(b) J. Ye, W. Fan and S. Ma, Chem.–Eur. J., 2013, 19, 716 CrossRef CAS PubMed ;
(c) J. Ye, R. Lü, W. Fan and S. Ma, Tetrahedron, 2013, 69, 8959 CrossRef CAS ;
(d) R. Lü, J. Ye, T. Cao, B. Chen, W. Fan, W. Lin, J. Liu, H. Luo, B. Miao, S. Ni, X. Tang, N. Wang, Y. Wang, X. Xie, Q. Yu, W. Yuan, W. Zhang, C. Zhu and S. Ma, Org. Lett., 2013, 15, 2254 CrossRef .
-
(a) J. Kuang and S. Ma, J. Am. Chem. Soc., 2010, 132, 1786 CrossRef CAS PubMed ;
(b) J. Kuang, X. Tang and S. Ma, Org. Chem. Front., 2015, 2, 470 RSC .
- J. Kuang, H. Luo and S. Ma, Adv. Synth. Catal., 2012, 354, 933 CrossRef CAS .
-
(a) X. Huang, T. Cao, Y. Han, X. Jiang, W. Lin, J. Zhang and S. Ma, Chem. Commun., 2015, 51, 6956 RSC ;
(b) X. Tang, X. Huang, T. Cao, Y. Han, X. Jiang, W. Lin, Y. Tang, J. Zhang, Q. Yu, C. Fu and S. Ma, Org. Chem. Front., 2015, 2, 688 RSC .
- T. Sugiishi and H. Nakamura, J. Am. Chem. Soc., 2012, 134, 2504 CrossRef CAS PubMed .
- For limited reports on CdBr2-mediated reactions, see:
(a) X. Tang, C. Zhu, T. Cao, J. Kuang, W. Lin, S. Ni, J. Zhang and S. Ma, Nat. Commun., 2013, 4, 2450 CrossRef PubMed ;
(b) X. Tang, Y. Han and S. Ma, Org. Lett., 2015, 14, 1176 CrossRef PubMed .
- J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669 CrossRef CAS .
- B. Lü, C. Fu and S. Ma, Tetrahedron Lett., 2010, 51, 1284 CrossRef .
- The relative configurations of product 8 were tentatively assigned, A. Bandaru and K. P. Kaliappan, Synlett, 2012, 23, 1473 CrossRef CAS .
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04115f |
| ‡ These two authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ac
*ac