
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly enantioselective “inherently chiral” electroactive materials based on a 2,2′-biindole atropisomeric scaffold†
Serena
Arnaboldi
 a,
Tiziana
Benincori
*b,
Andrea
Penoni
b,
Luca
Vaghi
c,
Roberto
Cirilli
d,
Sergio
Abbate
e,
Giovanna
Longhi
e,
Giuseppe
Mazzeo
e,
Sara
Grecchi
a,
Monica
Panigati
af and
Patrizia Romana
Mussini
*a
a,
Tiziana
Benincori
*b,
Andrea
Penoni
b,
Luca
Vaghi
c,
Roberto
Cirilli
d,
Sergio
Abbate
e,
Giovanna
Longhi
e,
Giuseppe
Mazzeo
e,
Sara
Grecchi
a,
Monica
Panigati
af and
Patrizia Romana
Mussini
*a
aDipartimento di Chimica, Università degli Studi di Milano, Via Golgi, 19, 20133 Milano, Italy. E-mail: patrizia.mussini@unimi.it
bDipartimento di Scienza e Alta Tecnologia, Università degli Studi dell'Insubria, Via Valleggio 11, 22100 Como, Italy. E-mail: tiziana.benincori@uninsubria.it
cDipartimento di Scienze dei Materiali, Università di Milano-Bicocca, Via R. Cozzi 55, 20125 Milano, Italy
dIstituto Superiore di Sanità, Centro Nazionale per il Controllo e la Valutazione dei Farmaci, Viale Regina Elena 299, 00161 Roma, Italy
eDipartimento di Medicina Molecolare e Traslazionale, Università degli Studi di Brescia, Viale Europa 11, 25123 Brescia, Italy
fIstituto per lo Studio delle Macromolecole, Consiglio Nazionale delle Ricerche (ISMAC-CNR), Via E. Bassini, 15, 20133 Milano, Italy
First published on 5th January 2019
Abstract
Chiral oligothiophene monomers with C2 symmetry, based on 3,3′-bithiophene atropisomeric cores with high racemization barriers, have recently been shown to provide excellent chiral starting materials with high electroactivity for the easy preparation of enantiopure electroactive films endowed with powerful chirality manifestations. We now introduce an inherently chiral monomer based on a 2,2′-biindole core, as the prototype of a new inherently chiral monomer family, whose properties could be modulable through functionalization of the pyrrolic N atoms. By fast, regular electrooligomerization the new monomer yields inherently chiral films with high, reversible electroactivity and, above all, impressive enantioselectivity towards very different chiral probes, some of pharmaceutical interest, as general-scope electrode surfaces. Such results, while opening the way to a new, attractive inherently chiral selector class, nicely confirm the general validity of the inherent chirality strategy for chiral electrochemistry. Furthermore, the enantioselectivity of the new selectors not only holds with electroactive chiral probes, but also with circularly polarized light components as well as electron spins, resulting in good chiroptical and spin filter performances, which suggests fascinating correlations between the three contexts.
1 Introduction
Thiophene-based monomers with C2 symmetry, including either an atropisomeric 3,3′-dibenzothiophene (BT2-T4)1 or a 3,3′-bithiophene core2 with high racemization barriers, have recently been shown to provide excellent chiral starting materials with high electroactivity for the easy chemical or electrochemical preparation of enantiopure electroactive films endowed with powerful chirality manifestations. As chiral electrode surfaces they show outstanding enantioselectivity toward enantiomers of different chiral probes, resulting in very different peak potentials, so that they can be recognized without preliminary separation steps;1,2,4–6 thus, they could provide outstanding tools for chiral voltammetry (and, more generally, for chiral electrochemistry), particularly considering that most of the many previous approaches to this target suffered from one or more drawbacks, resulting e.g. in current rather than in potential differences for the probe enantiomers, being specifically tailored for a single probe, being difficult and/or expensive to prepare, lacking robustness, and so on.7,8 At the same time, the same films exhibited chiroptical properties (circularly polarized luminescence as well as intense circular dichroism, reversibly controlled by electrochemical polarization)1,3,4 and, very recently, unprecedented molecular spin filter performances.9 The search for efficient molecular spin filters as an attractive alternative for inorganic active materials in spintronics is a hot topic, prompted by the inspiring work of Ron Naaman and collaborators describing the “Chiral Induced Spin Selectivity” (CISS) effects of chiral molecular materials. They report e.g. significant spin polarization in photo-ejected electrons transmitted through an enantiopure thin layer on Au as well as small but significant current differences for achiral redox probes in magnetoelectrochemical experiments on ferromagnetic electrodes functionalized with enantiopure chiral molecular layers, inverting the magnetic field orientation.10–13 The authors explain such effects in terms of different transmission probabilities of α and β electrons through the chiral layers. In this context, the above mentioned spin filter experiments performed by some of us appear groundbreaking since potential differences are observed rather than current ones when working with chiral oligothiophene-based oligomer films on ITO electrodes under an applied magnetic field.9 Spin-modulated electrochemical potentials indeed appear to be an attractive tool, giving the convenience of electrode potential for selection, activation, or transduction purposes.The outstanding enantiodiscrimination performances of our electroactive films based on atropisomeric scaffolds were justified by their peculiar design, implying (i) “inherent chirality”, since the stereogenic element coincides with the functional group determining the specific material property (here, electroactivity), i.e. the whole main conjugated backbone featuring a tailored torsion with a racemization barrier too high to be overcome under working conditions, so that the monomer exists as two very stable enantiomers;2 (ii) high regioregularity, since the monomers have equivalent, homotopic terminal positions for oligomerization, so that the monomer torsion cannot propagate into regio- and stereoregular foldamer structures (of course retaining the same configuration of the starting monomer). Such very stable foldamers can lead to powerful and robust chirality manifestations, much higher than those for chiral polymers from achiral monomers grown under asymmetric conditions, and even higher than those for chiral polymers in which the stereogenic element consists in stereocentres external to the main conjugated backbone.8 An additional important feature is that, in the case of the 3,3′-dibenzothiophene-based inherently chiral monomer, the oligomer mixture obtained by (electro)oligomerization was found to include many cyclic terms4,14 that can be regarded as a mixture of attractive chiral cavities of different dimensions with many available heteroatoms, promoting diastereomeric interactions with chiral guests.
To confirm the general validity of the above strategy and related interpretation, as well as to explore the possibility of modulating functional properties, it is particularly important to explore materials derived from inherently chiral starting monomers of different chemical nature. Song and co-authors recently studied a series of monomers with thiophene-based wings and BINOL atropisomeric cores15 by preparing electrode surface films and testing them with aminic probes forming acid/base interactions with the core hydroxy groups. Some enantioselectivity manifestations were observed, e.g. in terms of differences in potential vs. concentration trends for the enantiomers of the chiral probe working in potentiometric mode, although they are less useful for enantiorecognition purposes than the above CV tests. Such interesting observations do not, however, look decisive for the above discussed electroanalytical applications.
In this context, we are considering the 2,2′-biindole scaffold as a very appealing candidate for the atropisomeric core of inherently chiral monomers. In fact, it is electron richer and thus more easily oxidable than the thiophene-based cores; moreover, unlike them, it can be functionalized on nitrogen atoms, a feature that can be exploited to prepare a wide palette of monomers with fine modulation of key properties, e.g. solubility, important for chemical processing, and torsional angle.
In this work we introduce 3,3′-bis(2,2′-bithiophen-5-yl)-1,1′-dimethyl-1H,1′H-2,2′-biindole (1), nicknamed (N-Me-IND)2-T4 (Fig. 1a), as a very convenient starting monomer to prepare inherently chiral films for use as general-scope electrode surfaces with high and reversible electroactivity and impressive enantioselectivity towards very different electroactive chiral probes, also of pharmaceutical interest; at the same time, it also exhibits high chiroptical activity as well as impressive spin filter features. This highlights fascinating correlations between the three contexts, for which we propose a tentative sketch.
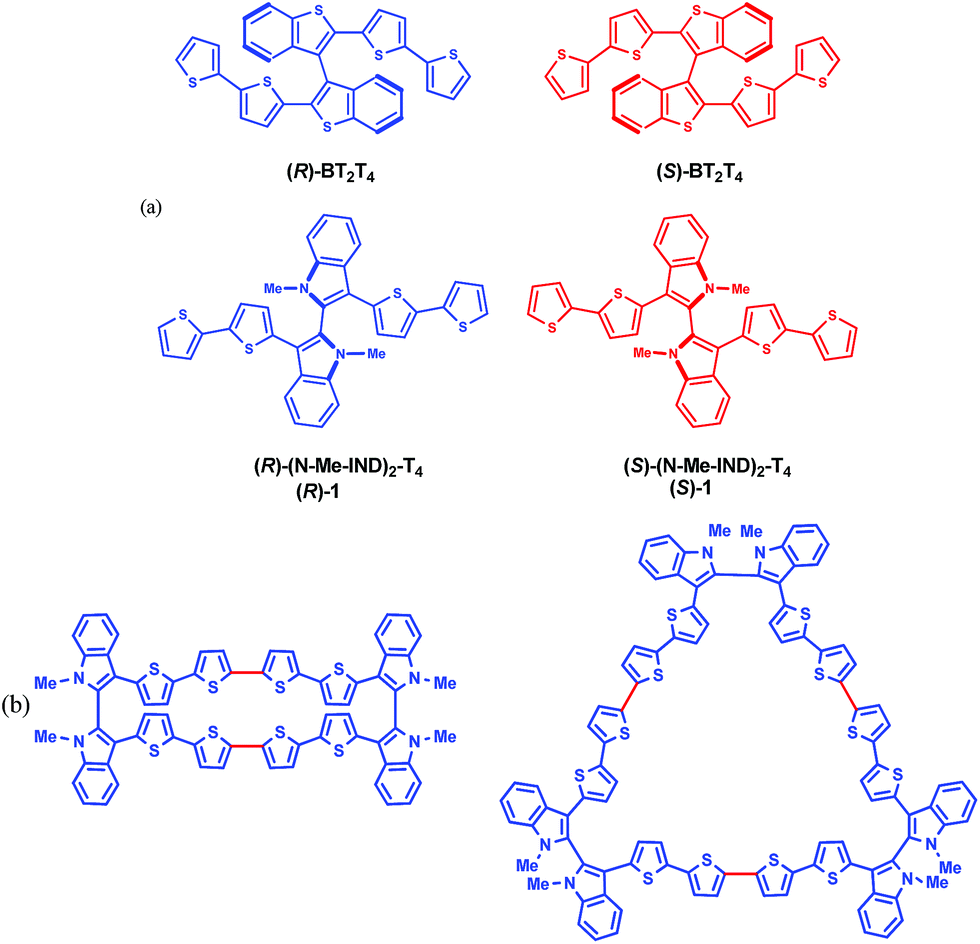 | ||
| Fig. 1 (a) Enantiomers of the inherently chiral monomers discussed; (b) planar molecular formulas of cyclic dimers and trimers of monomer 1 (red bonds indicate the oligomerization sites). | ||
2 Results and discussion
2.1 The starting inherently chiral monomer as a racemate
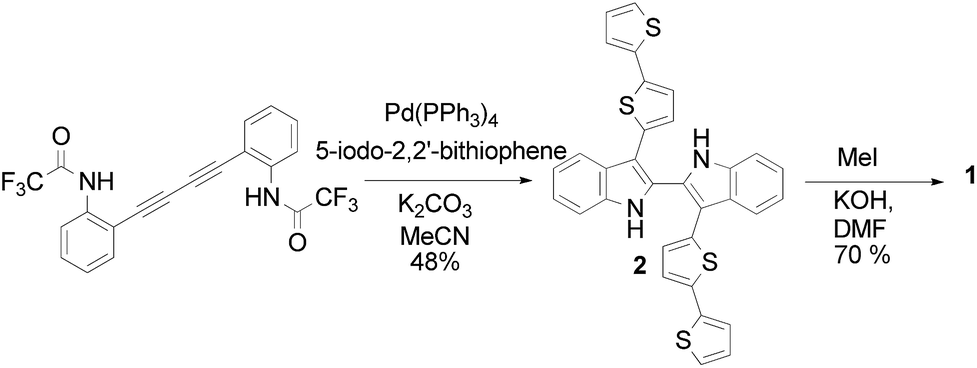 | ||
| Scheme 1 | ||
The reaction, catalyzed by Pd(0), requires a double alkyne derivative, namely 2,2,2-trifluoro-N-(2-(4-[2,2,2-trifluoro-acetylamino-phenyl]-buta-1,3-diynyl)-phenyl)-acetamide and a suitable aromatic haloderivative. We exploited this strategy using 5-iodo-2,2′-bithiophene, obtained in turn by the direct iodination of 2,2′-bithiophene with N-iodosuccinimide in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CHCl3:CH3COOH solution.18 The reaction was carried out in refluxing acetonitrile using palladium(tetrakis) as the catalyst and K2CO3 as the base. This approach allowed the formation of the interannular bond between the two indole units and their functionalization with the bithienyl units in a single step.
:1 CHCl3:CH3COOH solution.18 The reaction was carried out in refluxing acetonitrile using palladium(tetrakis) as the catalyst and K2CO3 as the base. This approach allowed the formation of the interannular bond between the two indole units and their functionalization with the bithienyl units in a single step.
Racemic monomer 1 was obtained in good yields by reaction of the bisanion of 2, generated by employing KOH in N,N-dimethylformamide at 0 °C, with methyl iodide.
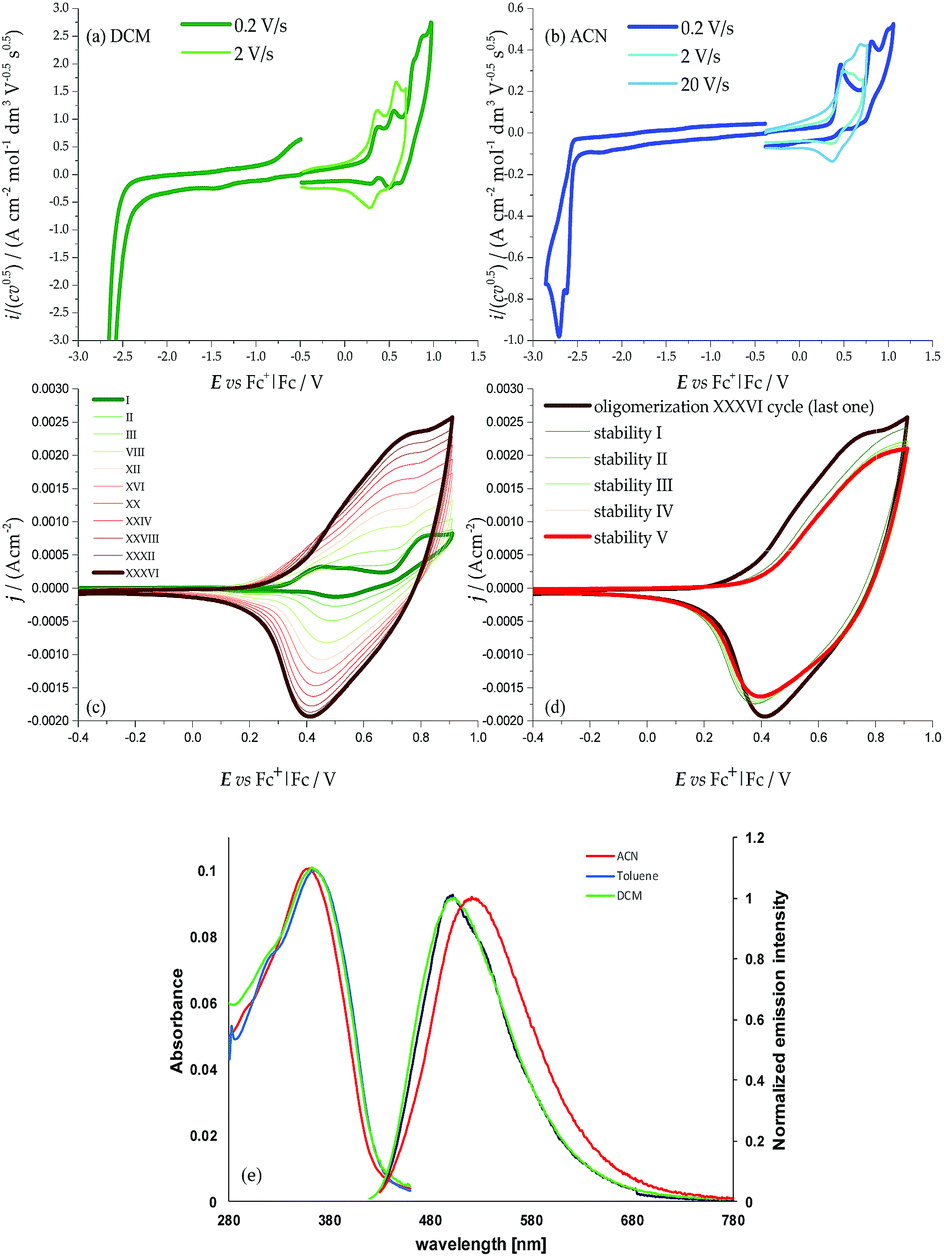 | ||
| Fig. 2 CV patterns of monomer 1 in (a) DCM and (b) ACN, with 0.1 M TBAPF6 supporting electrolyte, on a GC electrode. Oxidative and reductive half cycles are considered separately to avoid electrode conditioning by electron transfer products; (c) electrooligomerization of monomer 1 (0.00093 M in DCM + 0.1 M TBAP) on the GC electrode by oxidative potential cycling at 0.2 V s−1. (d) Stability cycles in monomer-free solution; (e) solvent effect on the absorption and emission spectra of 1. | ||
The monomer is a C2 symmetric atropisomeric system consisting of two equal moieties with a torsional barrier of 27 kcal mol−1 according to computations (ESI 2 with related ESI Table 2.1 and ESI Fig. 2.1–2.4†) and 29.9 kcal mol−1 according to off-column racemization experiments analyzed by enantioselective HPLC (ESI 3†). Such a barrier is high enough to yield stable (R)- and (S)-(N-Me-IND)2-T4 enantiomers at room temperature (ESI Fig. 2.5†), while also allowing some partial residual communication between the two moieties through the interanular bond, besides through-space interactions. Such communication is more efficient than in BT2T4, which has a significantly higher torsional barrier, 43 kcal mol−1, as well as a nearly 90° torsional angle (ESI Fig. 1.1†).1
The two moieties are quite active both electrochemically, as redox sites (ESI 3†) and spectroscopically, as chromophores (ESI 4†), the two features being strictly interconnected. The HOMOs and LUMOs mainly involve the pyrrole–bithiophene PTT conjugated systems (ESI Fig. 2.4†), and are therefore both shifted to much higher energies than those in the BT2T4 case (which involves terthiophene TTT conjugated systems) consistent with the electron-richer nature of the pyrrole ring with respect to the thiophene one. This is evidenced by the significant negative potential shifts of the corresponding CV features (easier oxidation, more difficult reduction; Fig. 2, Table 1). Consistently, the HOMO–LUMO gaps are similar to the BT2T4 ones19,20 (Table 1), but translated to higher energies.
| E p,c vs. Fc+|Fc/V | E p,a vs. Fc+|Fc/V | E LUMO,ECa/eV | E HOMO,ECa/eV | λ abs/nm (104ε/M−1 cm−1) | λ em/nm (Φ) | τ/ns | H–L gap/eV | |
|---|---|---|---|---|---|---|---|---|
| a Estimated as ELUMO(HOMO),EC = −1e(Ep,Ic(Ia)/V vs. Fc+|Fc + 4.8/V Fc+|Fc vs. 0). b Electronic gap. c Optical bandgap. | ||||||||
| (N-Me-IND)2-T4 | ||||||||
| ACN | −2.62, −2.71 | 0.47; 0.82; 0.99 | −2.18 | −5.27 | 364 (3.13) | 503 (0.15) | 1.1 | 3.09b (3.41)c |
| DCM | n.d. | 0.38, 0.56; 0.78; 0.87 | −5.18 | 367 | 500 (0.16) | (3.38)c | ||
| Toluene | 358 | 523 (0.08) | (3.47)c | |||||
|
||||||||
| BT2-T4 | ||||||||
| ACN19 | −2.28 | 0.78 | −2.52 | −5.58 | 372 (4.80) | 507 (0.16) | 1.7 | 3.06b (3.33)c |
| DCM20 | n.d. | 0.67, 0.80 | −5.47 | 378 | 505 (0.18) | (3.28)c | ||
| Toluene | 371 | 507 (0.15) | (3.34)c | |||||
The presence of the pyrrole ring also results in asymmetry in the main conjugated system with respect to the BT2T4 case, with significant localization of the HOMO on the pyrrole side (i.e. on the monomer core) and of the LUMO on the bithiophene side (i.e. on the monomer terminals). This is confirmed by electronic spectroscopy (ESI 5†), in which absorption and emission wavelengths exhibit some solvatochromism, pointing to a charge transfer character of the electronic transition (Fig. 2e), and above all by CV experiments, where, unlike the BT2T4 case, the first oxidation appears to be chemically reversible with no coupling follow-up (Fig. 2a); this is consistent with stable radical cation formation close to the molecule core. Instead to achieve oligomerization it is necessary to activate the thiophene wings by potential cycling around the relevant, much more positive, oxidation peak (Fig. 2c; see also ESI Fig. 2.3† for characterization of charged species via DFT calculations).
Interestingly, the presence of two equivalent, partially interacting PTT moieties/redox sites nicely results in a twin-peak system for the monomer first oxidation in CV, corresponding to the formation of two partially interacting radical cations, each one on a PTT moiety, each of them mostly localized on the pyrrole ring (as discussed above). In other words, partial reciprocal interaction results in loss of degeneracy between the two equivalent redox sites, which are therefore activated at different potentials/energies. Such peak splitting is dependent on solvent polarity; in particular, the higher charge screening ability of acetonitrile with respect to dichloromethane results in twin peak merging; concurrently chemical reversibility appears to decrease too, at least at low scan rates (ESI 4†).
One could draw a parallel between the loss of degeneracy of the two PTT moieties as redox sites and the loss of degeneracy as chromophores in absorption spectroscopy in terms of “exciton coupling” due to interaction between electric dipole transition moments, resulting in a splitting in absorption wavelengths for the two moieties/chromophores. This Davydov splitting21,22 is evidenced in electronic circular dichroism (ECD) spectroscopy (Section 2.4.1.1) by a symmetrical couplet for the monomer enantiomers (Fig. 3a) centered close to the UV-vis absorption maximum wavelength (Fig. 2e). Interaction of neighbouring electric fields in space can justify both phenomena. This analogy, or more appropriately, connection, between redox potential splitting and wavelength splitting in atropisomeric conjugated systems is, to our knowledge, pointed out here for the first time. Careful reexamination of former research19,20 confirms the simultaneous presence of spectroscopic (Davydov) splitting and CV peak splitting in other atropisomeric inherently chiral systems with two equivalent redox sites/chromophore moieties, as discussed in more detail in ESI 6†.
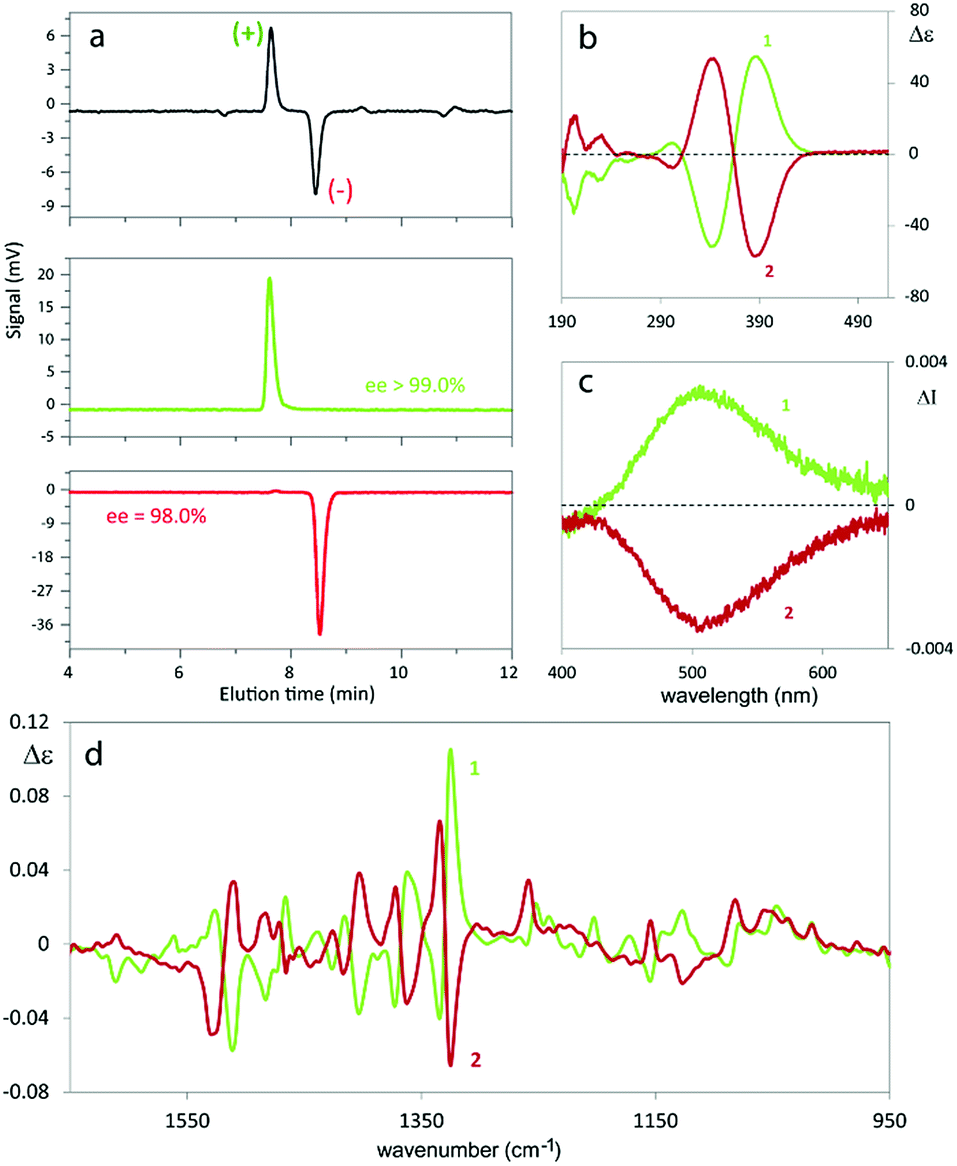 | ||
| Fig. 3 (a) Analytical HPLC resolution of 1 and determination of the enantiomeric purity of the fractions collected on a semipreparative scale. Column: Chiralpak IB (250 mm × 4.6 mm I.D.), eluent: n-hexane–CH2Cl2–ethanol 100:5:1, flow rate: 1 cm3 min−1, temperature: 25 °C, detector: CD at 380 nm; (b) experimental Electronic Circular Dichroism (ECD) spectra of both enantiomers of (N-Me-IND)2-T4; I and II designate the elution order. (c) Experimental Circularly Polarized Luminescence (CPL) spectra. (d) Vibrational Circular Dichroism (VCD). | ||
2.2 Conversion of the monomer into oligomer films
The monomer features can be propagated and amplified due to regioregularity, converting the monomer into an oligomer film, which can be done either chemically or electrochemically. Chemical oxidation of racemic monomer 1 can be performed with an excess of FeCl3 in CHCl3 at room temperature, as previously described for BT2-T4, and it is processed as described in the Experimental section. High Resolution Laser Desorption Ionization (HR-LDI) experiments reveal that the oligomer mixture contains dimers and trimers (height peak ratio 40), their isotopic patterns unequivocally demonstrating that all of them are fully conjugated macrocycles (ESI Fig. 7.1†). Concurrently, HPLC analysis on the Chiral Stationary Phase (CSP) (CD detector) of a pure sample of cyclic dimers, isolated by column chromatography, demonstrates the presence of enantiomers and meso-compounds in quite similar amounts (ESI Fig. 7.2†).Oligomer films can also be directly electrodeposited on a suitable electrode support by repeated potential cycling around the potential corresponding to the third oxidation peak (the first one involving the bithiophene wings with free terminals); fast formation of an electroactive film on the electrode surface is observed in DCM (Fig. 2c); the growth is even faster upon further widening of the oxidative cycle.
The films show good stability upon subsequent potential cycling in monomer-free solution, as required for use as an enantioselective electrode surface (Fig. 2d). The onset potential for the film is only slightly more negative than the starting monomer one, consistent with the oxidation potentials of the bisindole core and of linear tetrathiophene units (which are formed upon monomer coupling).
2.3 Preparation of enantiopure monomer and oligomer selectors
The antipodes of monomer 1 can be successfully separated on a semi-preparative scale by HPLC on the polysaccharide-based Chiralpak IB CSP under chlorinated normal phase conditions. Fig. 3 shows the analytical HPLC chromatogram of 1 obtained by on-line CD detection and the determination of the enantiomeric purity of the isolated antipodes. The [α]D20 values are +808° and −818° (0.1 M in CHCl3) for the first and the second eluted enantiomers, respectively.From each enantiopure monomer antipode the corresponding enantiopure oligomer film can then be easily obtained. In fact both enantiomers undergo very fast and regular electrooligomerization in DCM + TBAP 0.1 M on GC as the working electrode at a 0.2 V s−1 potential scan rate, cycling 36 times around the fourth oxidation peak (ESI Fig. 8†).
2.4 Enantiodiscrimination performances by enantiopure selectors
2.4.1.1. Enantiopure monomers. The monomer antipodes show, as expected, enantioselectivity towards the left-handed L and right-handed D circularly polarized light components, with clear-cut differences in (i) L vs. D absorption, resulting in intense circular dichroism23–26 spectra, both electronic (ECD, Fig. 3b) and vibrational (VCD, Fig. 3d), as well as in (ii) L vs. D emission, resulting in circularly polarized luminescence23–26 (CPL, Fig. 3c).
ECD and VCD experimental spectra are reported in Fig. 3b and d. As already mentioned, they are characterized by a strong “exciton coupling” between the two equal chromophores corresponding to the monomer moieties, which results in loss of degeneracy in the absorption wavelength λabs (“Davydov splitting”). This splitting, unperceivable in the UV-vis absorption spectrum, is evidenced in the ECD one by the dominating couple of bands with maxima/minima at 347 and 390 nm, specular for the two enantiomers and intersecting each other on the λ axis at 364 nm,27 close to the UV-vis absorption maximum. Also VCD spectra, while displaying few very weak features below 1300 cm−1, contain a good number of (+, −) couplets above 1300 cm−1, again as a consequence of the molecule's C2-symmetry.
The exciton coupling in the ECD spectra also provides a clue concerning the enantiomer absolute configuration; in particular, the couplet is positive (i.e. with the positive component at lower energy/longer wavelength) for the first eluted enantiomer (I) and negative for the second eluted one (II). According to the rules of Harada, Nakanishi and Berova,27 this points to P and M helicity, respectively, corresponding to S and R axial stereogenicity for the present atropisomeric I and II monomer antipodes. Confirmation comes from the comparison of ECD experimental spectra with those calculated according to DFT and TD-DFT. As described in detail in ESI 2† with related ESI Table S2.1,† the number of conformers participating in shaping the spectra is pretty large, the maximum population factor being less than 15%. All conformers are characterized by a positive value of the central δ dihedral angle, with values between 115° and 80°; the dihedral angles φ1 and φ2, next to δ, are either ca. ±35° or ca. ±145°, while ϑ1 and ϑ2 are either ca. ±160° (s-trans bi-thiophenes) or ca. ±30° (s-cis bi-thiophenes). Importantly, TDDFT calculations on all conformers give the positive exciton couplet observed in the ECD spectrum for the 1st eluted enantiomer, with maxima/minima at 347 and 390 nm (while the minor features between 290 and 190 nm are not as exactly reproduced).
Fig. 4 shows the ECD spectrum computed as the weighted average of the calculated spectra of all the conformers.
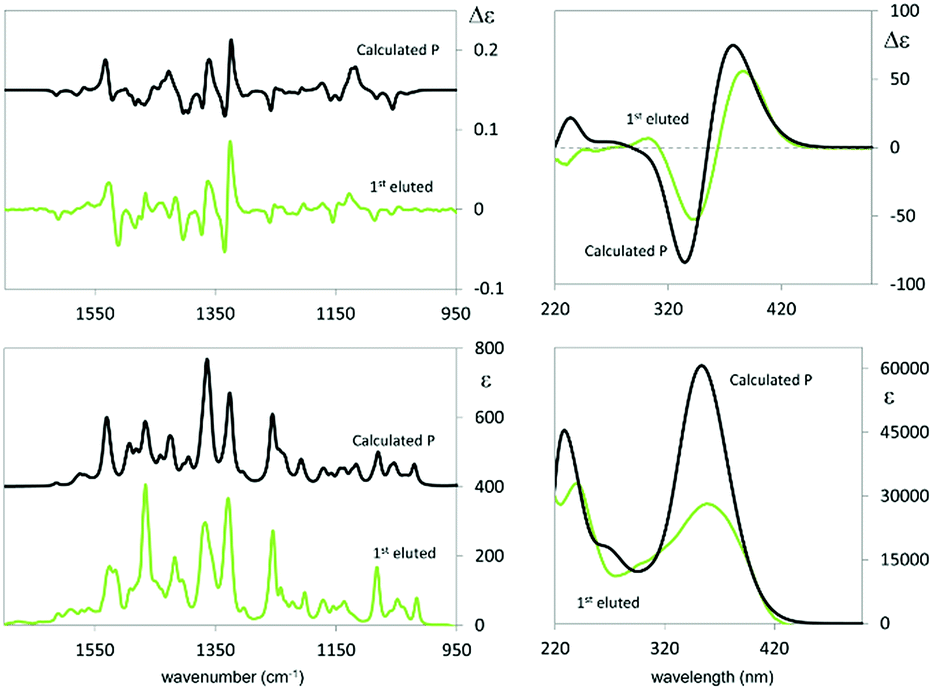 | ||
| Fig. 4 Comparison of experimental and calculated VCD and IR spectra (left) and comparison of experimental and calculated ECD and UV absorption spectra (right): VCD experimental data are for the semi-difference of I and II eluted enantiomers; calculated data are for the (S) enantiomer of (N-Me-IND)2-T4. Calculated wavenumbers in the IR range have been scaled by 0.98. Calculated wavelengths in the UV range have been shifted by 10 nm. | ||
The calculated wavelength of the first electronic transition is similar for all conformers and is in the 320–360 nm range (i.e. 3.45–3.87 eV), a little higher than the first observed maximum/minimum, but it should be considered that a blue shift is expected when employing the CAM-B3LYP functional. By inspecting the molecular orbitals involved in the couplet (see ESI Fig. 2.5†), one may see that the transitions giving rise to the two features at 390 and 347 nm are originated by transitions from HOMO and HOMO−1 orbitals localized on the pyrrole rings and the two nearby thiophene rings, to LUMO and LUMO+1 orbitals localized on the two bithiophene rings; this is consistent with the above discussion on the racemate monomer features in Section 2.1.2.
In the case of the VCD spectra, two couplets between 1320 and 1360 cm−1 are clearly positive for antipode I and negative for II, and one couplet at ca. 1500 cm−1 is negative for I and positive for II.21–26 Also in this case the spectra have been calculated for each conformer (ESI Fig. 2.6†), and, again, the weighted averages of the IR and VCD calculated spectra are in good correspondence with the experimental ones (Fig. 4).
Not only is the absolute configuration unambiguously assigned, but sensitivity to the conformational structure can also be recognized.
The features corresponding to the VCD couplets at 1320 and 1360 cm−1 can be associated with vibrational exciton coupling28–33 mainly involving indolic in-plane bendings (see ESI Fig. 2.7†); in particular, the first one is common to all conformers while the second one is sharper for conformer 1t (φ1 and φ2 values of about ±150°). Instead the doublets at 1250 cm−1 (scaled wavenumber) and at 1500 cm−1 can be assigned to CH in plane bendings delocalized on thiophenes and indoles, and may be assumed to be a signature of conformers with φ1 and φ2 values of about ±35°, e.g. conformer 2t: the observed weak intensity is due to the presence of the other conformers. Instead the negative feature at 1400 cm−1 is common to all structures and can be assigned to the umbrella mode of the indole methyl groups.
As mentioned above, the high enantiodiscrimination ability of monomer 1 antipodes also concerns polarized light emission in addition to absorption. Neat CPL spectra have been recorded with our home-built apparatus and are shown in Fig. 3.34 The single CPL band is positive for the first eluted enantiomer I and bears the same sign as the longest wavelength feature of the ECD couplet, as expected when the first excited state has similar geometrical and electronic structures as the ground state.35–37 The value for the luminescence dissymmetry factor glum = 2(IL − IR)/(IL + IR) (with IL and IR denoting the intensities of the left-hand and right-hand circularly polarized emitted light components) is about 0.3 × 10−2, which can be considered quite large for non-metal complex organic compounds in solution.24–26 Actually the situation met in the present case, as well as for the previously studied BT2-T44 monomer, looks more like the one met for the thia-bridged triarylamine heterohelicenes,38 defined “helical-responsive”, rather than that for simpler helicene cases,37 where it was defined “substituent-sensitive”. In fact, large ECD features correspond to large CPL features and bear the same sign, irrespective of small perturbation from substituents.
Such intense CPL makes (N-Me-IND)2-T4 an attractive candidate as a material for optoelectronic devices.
2.4.1.2. Enantiopure films. Electrodeposited films have been characterized by ECD spectroscopy, too (Fig. 5). A couplet is observed again, but at higher wavelengths, as expected, due to the increase in conjugation upon oligomerization. The couplet is not completely symmetric, as also observed in the case of BT2-T4,3 as a result of contributions from dimers (cyclic or non-cyclic) and trimers. Besides, the intensities of the two ECD components of the exciton couplet are different, unlike for the monomer. This may be related to the presence of oligomers of different sizes and shapes, even though other explanations cannot be excluded.
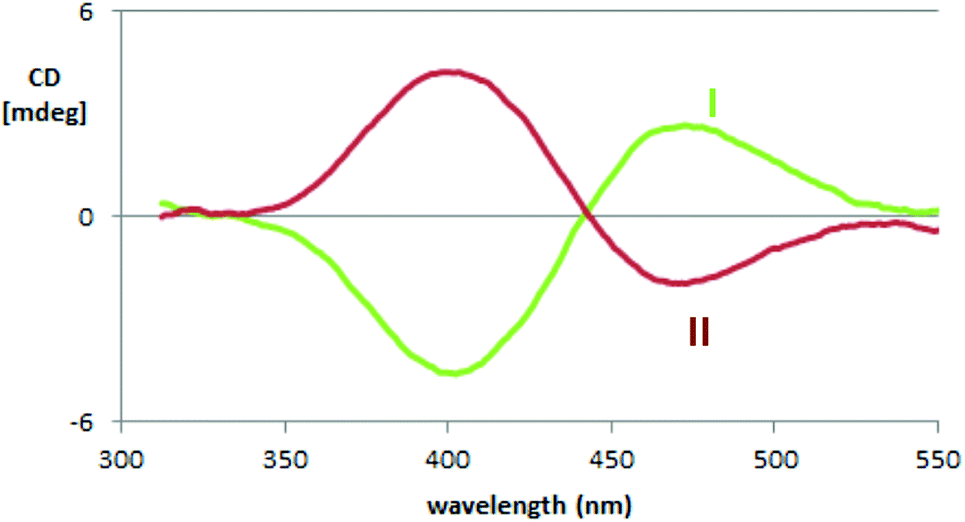 | ||
| Fig. 5 Experimental Electronic Circular Dichroism (ECD) spectra of both enantiomers of (N-Me-IND)2-T4 films; I and II designate the elution order. | ||
Very successful CV tests were carried out with three chiral electroactive molecular probes with different bulkiness, chemical nature and electrochemical activity (Fig. 6). The model chiral probe N,N-dimethyl-1-ferrocenylethylamine can be regarded as a benchmark given that it displays an electrochemically and chemically reversible oxidation process in a region where the film is uncharged (Fig. 6a), the half-wave potential being 0.35 V vs. SCE on a bare GC electrode. Enantioselectivity tests, performed in DCM + 0.1 M TBAP, revealed a spectacular CV peak potential separation of 270 mV between the (S)- and (R)-antipode of N,N-dimethyl-1-ferrocenylethylamine, evaluated as the average of three independent repetitions. This outstanding separation is the highest observed so far in our performed tests with this model probe on inherently chiral thiophene-based films.1,2,4–6
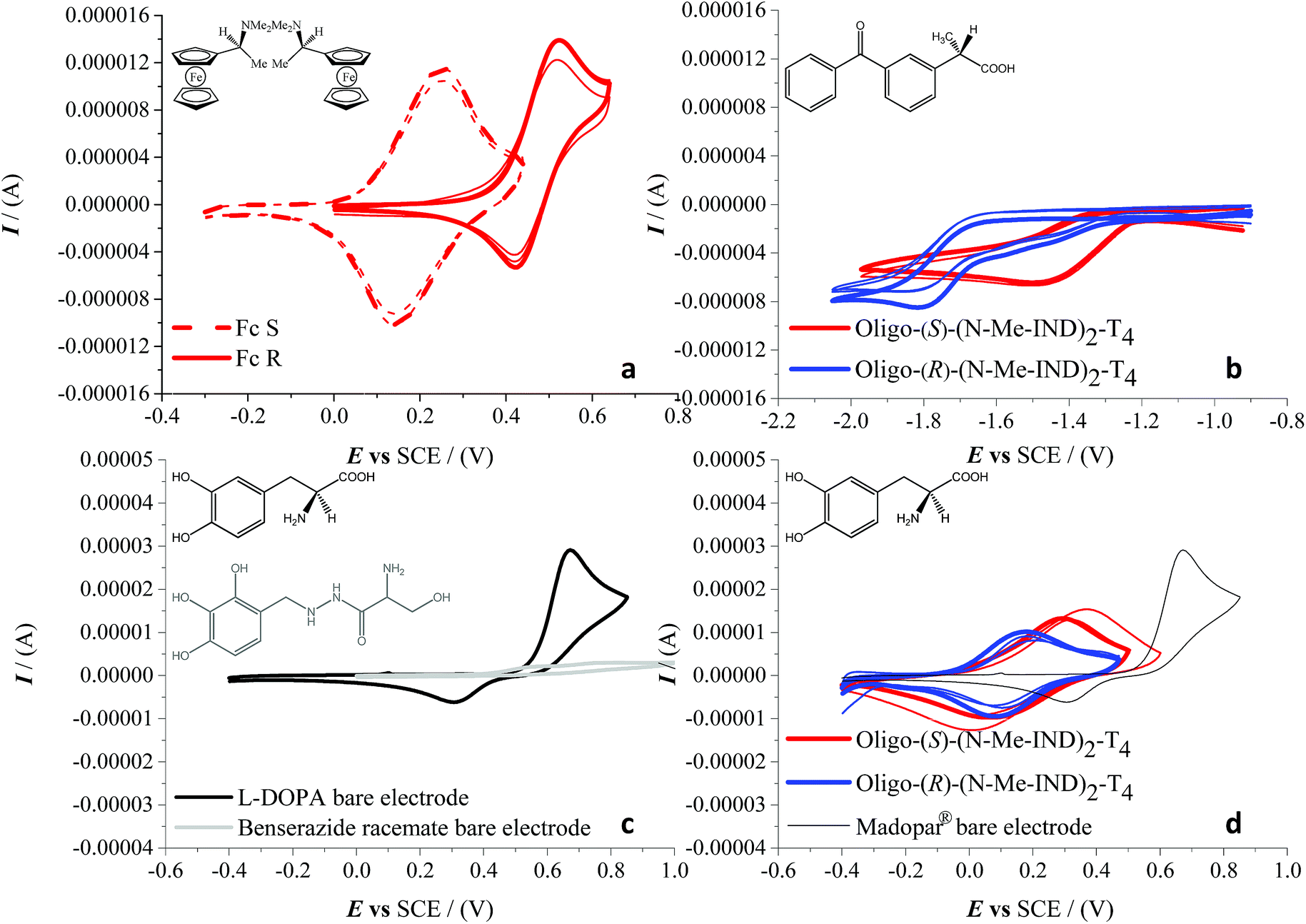 | ||
| Fig. 6 (a) Enantioselection on the enantiopure film oligo-(S)-(N-Me-IND)2T4 towards (S)- and (R)-N,N-dimethyl-1-ferrocenylethylamine (Fc S and Fc R, dashed and solid lines, respectively). (b) Enantioselection on the enantiopure films oligo-(S)-(N-Me-IND)2T4 and oligo-(R)-(N-Me-IND)2T4 (red and blue lines, respectively) towards (S)-(+)-ketoprofen. (c) CVs of L-DOPA (black line) and benserazide (grey line) with a 1:4 weight ratio, recorded on a bare GC electrode. (d) Enantioselection on the enantiopure films oligo-(S)-(N-Me-IND)2T4 (red) and oligo-(R)-(N-Me-IND)2T4 (blue), towards (L)-DOPA (1.9 mg; in tablet form) together with the Madopar® signal (black) on a bare GC electrode. In (a), (b) and (d) experiments, repetition tests have been performed for each probe + selector combination. | ||
We have also verified that the probe/surface interactions underlying the enantiodiscrimination process are reversible and non-destructive towards the chiral film, probably as a consequence of the full chemical reversibility of the probe electron transfer. In fact, the probe-free film can be easily recovered by performing a few CV cycles around the first oxidation potential in a blank solution. This enabled us to perform multiple subsequent enantiorecognition tests, alternating the (S) and (R) probes, on both the enantiomorphic surfaces, starting from either enantiomer. The second chosen analyte was (S)-(+)-ketoprofen, a nonsteroidal anti-inflammatory drug with analgesic and antipyretic effects. Enantiorecognition tests were performed, as in the previous experiments, in dichloromethane with TBAP as the supporting electrolyte. This second case study is very interesting, because it provides an example of enantiodiscrimination based on a reduction process, with respect to the formerly reported oxidation cases. In particular a large, reproducible peak potential difference (about 330 mV) is observed for the carbonyl reduction in the benzophenone system adjacent to the stereogenic center, working on either enantiomer of the indole-based film (Fig. 6b). It should be noted that in this case, since only one antipode of the pharmaceutical probe was available, the discrimination was verified only by inverting the configuration of the inherently chiral selector (i.e. the film). The last tested molecule was L-3,4-dihydroxyphenylalanine (L-DOPA), the active substance for the control of akinesia commonly used in the treatment of Parkinson's disease, in a real matrix, i.e. as the main component of the Madopar® drug in combination with benserazide. The latter acts as an L-DOPA decarboxylation inhibitor, preventing its bioconversion into inactive dopamine by the peripheral decarboxylases before reaching the brain. The weight ratio of L-DOPA and benserazide in Madopar® is 4:1 (even higher, 84%, if we consider the mole percentage). Again the enantiopure inherently chiral surface was prepared by monomer electrooligomerization in DCM, but in this case enantiodiscrimination tests were carried out in an aqueous HCl solution at pH 2, due to the insolubility of the analyte in the organic medium. The CV pattern of Madopar® on a bare electrode (black line, Fig. 6d), with a single oxidation peak at 0.67 V (SCE), is comparable in form and consistent in position (considering the pH effect) with that of pure L-DOPA (reported in a previous study at 0.35 V (SCE) in a pH 7 buffer solution and at 0.53 V (SCE) in a pH 4 one5,6). Preliminary tests were carried out by recording signals of L-DOPA and benserazide separately but using the same 1:4 weight ratio of the formulation of Madopar®; under such conditions the benserazide signals, as shown in Fig. 6c, are practically overlapped by the much higher L-DOPA peak on the bare GC electrode. Enantiodiscrimination (here verified, like in the aforementioned ketoprofen case, by testing the drug on either inherently chiral film enantiomer) is very good, reaching a difference of 140 mV evaluated as the average of three independent measurements (Fig. 6d). While the drug is available with L-DOPA only, an experiment with L- and D-DOPA enantiomers in aqueous solution resulting in a specular response on R- and S-films is reported in ESI 9.†
Work is in progress to develop a mechanistic interpretation of such wide-scope enantiorecognition performances in terms of potential differences. Since CV peaks, including chemically and electrochemically reversible ones, appear to undergo rigid shifts rather than morphology changes, the effect should be of thermodynamic rather than kinetic character. In particular, it could involve diastereomeric and therefore energetically different intermolecular interactions between the enantiopure selector and enantiopure probe. Such interactions could particularly rely on the available aromatic rings, heteroatoms and oligomer ringlets. A further possible alternative or synergistic explanation, based on the internal magnetic field of the chiral film, will be outlined in the next section.
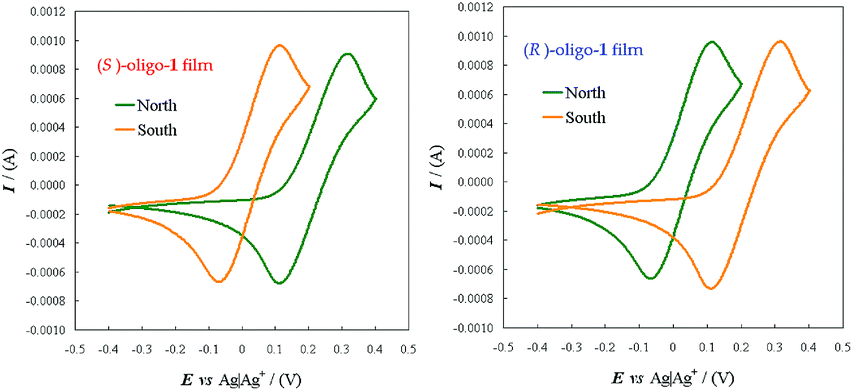 | ||
| Fig. 7 CV patterns for the achiral Fe(CN)63−|Fe(CN)64− redox couple, recorded at 0.05 V s−1 in aqueous solution on ITO electrodes modified by a thin layer of a (S)- or (R)-enantiopure oligo-1 thin film (right and left box, respectively), applying an external magnetic field with NS or SN orientation (green or orange lines respectively). | ||
Achieving spin-modulated electrochemical potentials looks to be a very attractive tool for possible advanced applications, considering the intrinsic convenience of electrode potential for selection, activation, or transduction purposes. Moreover it also suggests an additional interpretative scheme for the potential differences observed when applying enantiopure inherently chiral film electrodes to the discrimination of enantiopure molecular probes (Section 2.5). In fact, diastereomeric changes in the α and β electron energy level splittings, with related differences in redox potentials, could be induced not only by the combination of the (S)- or (R)-internal magnetic field of the chiral enantiopure film with a NS or SN external one, but also by the modulation of the (S)- or (R)-internal magnetic field by the presence of either a (S)- or a (R)-chiral probe. We look forward to performing further investigations concerning this important feature.
3 Conclusions
The striking performances of biindole-based inherently chiral molecular materials confirm the general validity of the “inherent chirality” strategy. The family provides a new attractive tool with multifaceted enantiodiscrimination ability, to be possibly exploited for applications in chiral electrochemistry, chiroptics and spintronics, three fields which appear to share fascinating fundamental correlations to be further explored.We particularly look forward to extending the study to other inherently chiral selectors, including ionic liquid/additive ones, and to ternary combinations (for instance, circular dichroism of the inherently chiral film in the presence of a chiral molecular probe, either chromophore or non-chromophore).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors' inherent chirality work is financially supported by Fondazione Cariplo and Regione Lombardia (2016-0923 RST—Avviso congiunto FC-RL Sottomisura B) rafforzamento (Enhancing VINCE (Versatile INherently Chiral Electrochemistry)). The authors also gratefully acknowledge the use of the advanced facilities at SmartMatLab (cofunded by Regione Lombardia, Fondazione Cariplo and Università degli Studi di Milano (2013-1766)) and the use of the resources of the Big & Open Data Innovation Laboratory (BODaI-Lab), University of Brescia, granted by Fondazione Cariplo and Regione Lombardia. The authors thank Professor Francesco Sannicolò (University of Milano and Laboratori Alchemia S.r.l.) for fruitful discussion and suggestions.References
- F. Sannicolò, S. Arnaboldi, T. Benincori, V. Bonometti, R. Cirilli, L. Dunsch, W. Kutner, G. Longhi, P. R. Mussini, M. Panigati, M. Pierini and S. Rizzo, Angew. Chem., Int. Ed., 2014, 53, 2623 CrossRef PubMed.
- F. Sannicolò, P. R. Mussini, T. Benincori, R. Martinazzo, S. Arnaboldi, G. Appoloni, M. Panigati, E. Quartapelle Procopio, V. Marino, R. Cirilli, S. Casolo, W. Kutner, K. Noworyta, A. Pietrzyk-Le, Z. Iskierko and K. Bartold, Chem.–Eur. J., 2016, 22, 10839 CrossRef PubMed.
- G. Longhi, S. Abbate, G. Mazzeo, E. Castiglioni, P. Mussini, T. Benincori, R. Martinazzo and F. Sannicolò, J. Phys. Chem. C, 2014, 118, 16019 CrossRef CAS.
- F. Sannicolò, P. R. Mussini, T. Benincori, R. Cirilli, S. Abbate, S. Arnaboldi, S. Casolo, E. Castiglioni, G. Longhi, R. Martinazzo, M. Panigati, M. Pappini, E. Quartapelle Procopio and S. Rizzo, Chem.–Eur. J., 2014, 20, 15261 CrossRef.
- S. Arnaboldi, T. Benincori, R. Cirilli, W. Kutner, M. Magni, P. R. Mussini, K. Noworyta and F. Sannicolò, Chem. Sci., 2015, 6, 1706 RSC.
- S. Arnaboldi, T. Benincori, R. Cirilli, S. Grecchi, L. Santagostini, F. Sannicolò and P. R. Mussini, Anal. Bioanal. Chem., 2016, 408, 7243 CrossRef CAS PubMed.
- S. Arnaboldi, M. Magni and P. R. Mussini, Curr. Opin. Electrochem., 2018, 8, 60 CrossRef CAS.
- S. Arnaboldi, S. Grecchi, M. Magni and P. R. Mussini, Curr. Opin. Electrochem., 2018, 7, 188 CrossRef CAS.
- T. Benincori, S. Arnaboldi, M. Magni, S. Grecchi, C. Fontanesi and P. R. Mussini, Chem. Sci., 2018 10.1039/C8SC04126A.
- K. Ray, S. P. Ananathevel, D. H. Waldeck and R. Naaman, Science, 1999, 283, 814 CrossRef CAS PubMed.
- S. G. Ray, S. S. Daube, G. LEitus, Z. VAger and R. Naaman, Phys. Rev. Lett., 2006, 96, 036101 CrossRef CAS PubMed.
- B. Göhler, V. Hamelbeck, T. Z. MArkus, M. Kettner, G. F. Hanne, Z. Vager, R. Naaman and H. Zacharias, Science, 2011, 331, 894 CrossRef PubMed.
- P. C. Mondal, C. Fontanesi, D. H. Waldeck and R. Naaman, Acc. Chem. Res., 2016, 49, 2560 CrossRef CAS PubMed.
- E. Quartapelle Procopio, T. Benincori, G. Appoloni, P. R. Mussini, S. Arnaboldi, C. Carbonera, R. Cirilli, A. Cominetti, L. Longo, R. Martinazzo, M. Panigati and R. Po', New J. Chem., 2017, 41, 10009 RSC.
- S. Kang, I. Cha, J. G. Han and C. Song, Mater. Express, 2013, 3, 119 CrossRef CAS.
- R. C. Larock and E. K. Yum, J. Am. Chem. Soc., 1991, 113, 6689 CrossRef CAS.
- G. Abbiati, A. Arcadi, E. Beccalli, G. Bianchi, F. Marinelli and E. Rossi, Tetrahedron, 2006, 62, 3033 CrossRef CAS.
- U. Boas, A. Dhanabalan, D. R. Greve and E. W. Meijer, Synlett, 2001, 5, 634 CrossRef.
- F. Sannicolò, S. Rizzo, T. Benincori, W. Kutner, K. Noworyta, J. W. Sobczak, V. Bonometti, L. Falciola, P. R. Mussini and M. Pierini, Electrochim. Acta, 2010, 55, 8352 CrossRef.
- T. Benincori, G. Appoloni, P. R. Mussini, S. Arnaboldi, R. Cirilli, E. Quartapelle Procopio, M. Panigati, S. Abbate, G. Mazzeo and G. Longhi, Chem.–Eur. J., 2018, 284, 513 CAS.
- N. Berova, L. Di Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914 RSC.
- J. T. Vázquez, Tetrahedron: Asymmetry, 2017, 28, 1199 CrossRef.
- J. P. Riehl and G. Muller, in Comprehensive Chiroptical Spectroscopy, ed. N. Berova, P. L. Polavarapu, K. Nakanishi and R. W. Woody, Wiley, Hoboken, NJ, 2012, vol. 3, p. 65 Search PubMed.
- E. Sànchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. De la Moya, Chem.–Eur. J., 2015, 21, 13488 CrossRef PubMed.
- G. Longhi, E. Castiglioni, J. Koshoubu, G. Mazzeo and S. Abbate, Chirality, 2016, 28, 696 CrossRef CAS PubMed.
- H. Tanaka, Y. Inoue and T. Mori, ChemPhotoChem, 2018, 2, 1 CrossRef.
- N. Harada, K. Nakanishi and N. Berova, in Comprehensive Chiroptical Spectroscopy, ed. N. Berova, P. L. Polavarapu, K. Nakanishi and R. W. Woody, Wiley, Hoboken, NJ, 2012, vol. 4, p. 115 Search PubMed.
- U. Narayanan and T. A. Keiderling, J. Am. Chem. Soc., 1983, 105, 6406 CrossRef CAS.
- T. Taniguchi and K. Monde, J. Am. Chem. Soc., 2012, 134, 3695 CrossRef CAS PubMed.
- S. Abbate, G. Mazzeo, S. Meneghini, G. Longhi, S. E. Boiadjiev and D. A. Lightner, J. Phys. Chem. A, 2015, 119, 4261 CrossRef CAS PubMed.
- C. L. Covington, V. P. Nicu and P. L. Polavarapu, J. Phys. Chem. A, 2015, 119, 10589 CrossRef CAS PubMed.
- G. Mazzeo, S. Abbate, G. Longhi, E. Castiglioni, S. E. Boiadjiev and D. A. Lightner, J. Phys. Chem. B, 2016, 120, 2380 CrossRef CAS PubMed.
- E. Castiglioni, S. Abbate and G. Longhi, Appl. Spectrosc., 2010, 64, 1416 CrossRef CAS PubMed.
- S. Abbate, T. Bruhn, G. Pescitelli and G. Longhi, J. Phys. Chem. A, 2017, 121, 394 CrossRef CAS PubMed.
- H. P. J. M. Dekkers, in Circular Dichroism: Principles, Applications, ed. N. Berova, K. Nakanishi and R. W. Woody, John Wiley & Sons, NY, 2000, vol. 7, p. 185 Search PubMed.
- G. Longhi, E. Castiglioni, S. Abbate, F. Lebon and D. A. Lightner, Chirality, 2013, 25, 589 CrossRef CAS PubMed.
- S. Abbate, G. Longhi, F. Lebon, E. Castiglioni, S. Superchi, L. Pisani, F. Fontana, F. Torricelli, T. Caronna, C. Villani, R. Sabia, M. Tommasini, A. Lucotti, D. Mendola, A. Mele and D. A. Lightner, J. Phys. Chem. C, 2014, 118, 1682 CrossRef CAS.
- G. Longhi, E. Castiglioni, C. Villani, R. Sabia, S. Menichetti, C. Viglianisi, F. Devlin, S. Abbate, J. Photobiol and A. Photochem, Chemistry, 2016, 331, 138 CAS.
- B. M. W. Langeveld-Voss, R. A. J. Janssen and E. W. Meijer, J. Mol. Struct., 2000, 521, 285 CrossRef CAS.
- G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2011, 40, 4603 RSC.
- G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2014, 43, 5211 RSC.
- P. J. Stephens, Annu. Rev. Phys. Chem., 1974, 25, 201 CrossRef CAS.
- P. J. Stephens, Adv. Chem. Phys., 1976, 35, 197 CAS.
- W. R. Mason, A Practical Guide to Magnetic Circular Dichroism Spectroscopy, Wiley & Sons, Inc., 2007 Search PubMed.
- J.-L. Bredas, Mater. Horiz., 2014, 1, 17 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04862b |
| This journal is © The Royal Society of Chemistry 2019 |