
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIonic polypeptide tags for protein phase separation†
Rachel A.
Kapelner
and
Allie C.
Obermeyer
 *
*
Department of Chemical Engineering, Columbia University, New York, NY 10027, USA. E-mail: aco2134@columbia.edu; Tel: +1-212-853-1315
First published on 17th January 2019
Abstract
Polyelectrolytes of opposite charge in aqueous solution can undergo a liquid–liquid phase separation known as complex coacervation. Complex coacervation of ampholytic proteins with oppositely charged polyelectrolytes is of increasing interest as it results in a protein rich phase that has potential applications in protein therapeutics, protein purification, and biocatalysis. However, many globular proteins do not phase separate when mixed with an oppositely charged polyelectrolyte, and those that do phase separate do so over narrow concentration, pH, and ionic strength ranges. The protein design factors that govern complex coacervation under varying conditions are still relatively unexplored. Recent work indicates that proteins with an intrinsically disordered region, a higher net charge, or a patch of charged residues are more likely to undergo a phase transition. Based on these design parameters, polyionic coacervation tags were designed and assessed for their ability to promote protein complex coacervation with oppositely charged polyelectrolytes. The phase behavior of a panel of engineered proteins was evaluated with the strong polycation poly(4-vinyl N-methyl pyridinium iodide). Proteins containing the ionic tags formed liquid coacervate droplets, while isotropically charged protein variants formed solid precipitates. The ionic tags also promoted phase separation at higher salt concentrations than an isotropic distribution of charge on the protein surface. The salt dependence of the protein complex coacervation could be predicted independently for tagged or isotropic variants by the ratio of negative-to-positive residues on the proteins and universally by calculating the distance between like charges. The addition of just a six residue polyionic tag generated a globular protein capable of liquid–liquid phase separation at physiological pH and ionic strength. This model system has provided the initial demonstration that short, ionic polypeptide sequences (6–18 amino acids) can drive the liquid–liquid phase separation of globular proteins.
Introduction
Complex coacervation, a liquid–liquid phase separation driven by electrostatic interactions between two oppositely charged polyelectrolytes, has the potential to stabilize and deliver proteins for therapeutic or enzymatic applications.1 Complex coacervation can encapsulate high concentrations of protein, up to 200 mg mL−1 of lysozyme has been reported, while still maintaining protein functionality and preventing protein aggregation.1,2 Not all proteins, especially those with a low charge density, phase separate with oppositely charged polyelectrolytes.3 The development of robust design rules and straightforward strategies for globular protein complex coacervation would enable new applications of protein phase separation in biocatalysis and biomedicine.1,4–7Engineered polypeptide sequences that promote complex coacervation are a promising avenue for investigation as polypeptides that promote simple coacervation have been developed successfully. Perhaps the most well studied are elastin-like polypeptides (ELPs), which demonstrate a lower critical solution temperature (LCST). Design rules established over the past several decades enable prediction of the transition temperature for canonical sequences (VPGXG),8,9 synthesis of novel sequences with a LCST or upper critical solution temperature (UCST),10 and creation of multi-domain phase separated structures.11 These 5–40 kDa polypeptide tags and their design rules have enabled applications in protein purification,12,13 therapeutic delivery,14 and hydrogel toughening.15 Analogous sequences and predictive rules for polypeptides that promote complex coacervation have yet to be established. The few reported examples of anionic polypeptide tags have shown the ability of ionic tags to drive the assembly of protein materials and their potential use for encapsulating enzymes and intracellular protein delivery.16,17 The work herein develops design criteria for ionic polypeptide tags that promote protein complex coacervation and characterizes the phase behavior of model protein-tag fusions.
Initial design criteria were based on globular proteins that formed complex coacervates.7,18–21 Previous work has shown that supercharging a globular protein by chemical modification can result in a protein that phase separates even when it would not in its native state.3,21 It was determined that a ratio of negative-to-positive residues (α) greater than 1.1 was sufficient to induce phase separation with a polycation and α > 1.5–2.0 enabled high levels of protein incorporation in the coacervate phase.3 Additionally, efforts focused on using complex coacervation to purify proteins identified charge anisotropy or “patchiness” as a key parameter for phase separation. This was determined because a patch of like charges provided high affinity for proteins with an oppositely charged polyelectrolyte at a specific pH.22 At this pH, the global charge of the protein may be neutral or even the same charge as the polyelectrolyte, but the local charge patch will be opposite to that of the polyelectrolyte.23,24 This has been observed in the coacervation of a hyaluronic acid with bovine serum albumin, which has a positive charge patch at pH 6.9.25 Taken together, these findings indicate that varying the number of ionizable residues in an anionic polypeptide tag should create a supercharged patch that enables a protein to undergo phase separation at a desired pH.
Design parameters for the ionic polypeptide tags were also based on proteins that natively phase separate. Proteins can undergo liquid–liquid phase separation in vivo to form membraneless organelles such as nucleoli, signaling puncta, stress granules, and P granules.26–32 Many of these organelles share the same properties as complex coacervates including rapid exchange of their contents with the bulk and the ability to coalesce. These membraneless organelles frequently consist of proteins and polyelectrolytes such as RNA or DNA, although they can also arise from simple coacervation of a single protein.29 A common feature among proteins found in membraneless organelles is the presence of an intrinsically disordered region (IDR).29 These IDRs are of low complexity and contain repeats of a subset of residues that promote disorder in solution.11 These biological examples suggest that inclusion of a disordered tag containing only a few residues should promote phase separation with an oppositely charged polyelectrolyte.
The final design criteria were based on the phase behavior of synthetic polyelectrolytes. Characterization of the complex coacervation of sequenced polypeptides has indicated a relationship between charge patterning and the entropic gain from counterion release.33 In these polypeptide-based complex coacervates, longer repeats of charged residues demonstrated a higher entropic gain upon coacervation due to expulsion of more condensed counterions and reduction in electrostatic repulsion from neighboring like charges.33–36 These combined observations suggested that the introduction of a charged block of amino acids via an ionic polypeptide tag should promote protein complex coacervation.
In this article, we demonstrate that short ionic polypeptide tags promote the liquid–liquid phase separation of a model globular protein, green fluorescent protein (GFP). Genetic engineering was used to precisely control the number and location of charges on a model protein. The phase behavior of protein-tag fusions was compared to proteins with the same expected charge isotropically distributed on the protein surface. Each comparison demonstrated the benefits of localizing the charged amino acid residues to a short polypeptide tag. As further proof-of-concept, an ionic polypeptide tag was appended to another globular protein (chloramphenicol acetyltransferase, CAT) and was similarly shown to promote phase separation.
Results and discussion
Protein library design
With the goal of developing strategies for the complex coacervation of globular proteins, a panel of 8 proteins of varying charge and charge distribution were designed and biosynthesized (Fig. 1a). Superfolder GFP (sfGFP) was selected as a model protein scaffold as it has previously been supercharged and can be monitored by fluorescence and optical microscopy.37 Four isotropic variants with increasing net negative charge (iso-GFP(-7), (-12), (-18), and (-24)) were prepared in a similar fashion to previously reported supercharged mutants.38–40 These supercharged proteins were designed to establish predictive rules for globular protein complex coacervation. But the global redesign of protein surface charge presents challenges when extended to non-model proteins. For example, even for this well-behaved model protein, isotropic superanionic variants showed decreased fluorescence due to protonation of the GFP chromophore (Fig. 2).41 Therefore, additional design criteria for globular protein complex coacervation were needed. As charge patchiness and intrinsically disordered sequences have previously been demonstrated to promote complex coacervation, these features were incorporated into the design of ionic polypeptide tags. An additional four GFP variants with charged residues localized to a C-terminal tag were prepared. These ionic tag variants were engineered to have the same overall charge as the isotropically charged GFP variants by appending repeats of the sequence [Asp–Glu–Glu–Glu–Asp–Asp] to the C-terminus. This enabled evaluation of the effect of both net charge and charge distribution on protein complex coacervation. An ionic tag was appended to an additional protein (CAT) to probe the generalizability of the results from the GFP model system to other proteins. The engineered proteins were expressed in E. coli and purified by Ni-NTA chromatography. Protein purity was confirmed by gel electrophoresis and MALDI-TOF mass spectrometry (Fig. S1 and S2†). Initial comparison indicated that altered charge distribution had a minimal impact on the measured isoelectric point of each protein (Fig. 1b and S1†). It was also determined that the ionic tags minimally affected GFP fluorescence (Fig. 2). The addition of 18 amino acids as an ionic tag (tag-GFP(-24)) resulted in only a minimal reduction in fluorescence (<10%), whereas mutation of 11 surface residues (iso-GFP(-24)) resulted in a 60% reduction in fluorescence. From the perspective of protein activity, ionic tags demonstrated improved performance relative to isotropic supercharging. The influence of charge distribution on phase behavior was the focus of the remaining studies. | ||
| Fig. 1 (a) Electrostatic surface potential representations of the solvent accessible surface area for GFP variants used in this study as calculated using the linearized Poisson–Boltzmann equation with Adaptive Poisson–Boltzmann Solver (APBS in Pymol; ±5kBT/e; blue for positive and red for negative). (b) Measured isoelectric points of GFP variants determined by isoelectric focusing (raw data, Fig. S1†). | ||
 | ||
| Fig. 2 Relative fluorescence of GFP mutants (λex = 488 nm, λem = 530 nm) normalized to iso-GFP(-7). Samples were prepared in triplicate in 10 mM Tris buffer, pH 7.4 at 1 mg mL−1 as described in the ESI.† | ||
Phase behaviour of engineered proteins
All 8 engineered GFPs phase separated with a polycation as monitored by solution turbidity. Associative phase separation of the engineered proteins was evaluated with several polycations. One strong polycation, poly(4-vinyl N-methyl pyridinium iodide) (qP4VP), and two weak polycations, poly(ethylene imine hydrochloride) (PEI) and poly-L-lysine hydrochloride (PLL) were investigated. qP4VP has previously been demonstrated to form complex coacervates and precipitates with chemically supercharged proteins and was therefore expected to phase separate with this panel of engineered proteins.3 PEI and PLL were selected as they are regularly used to form electrostatic polyplexes for nucleic acid delivery.42,43 The strong polycation was tested with all GFP variants, while the two weak polycations were tested with only one pair of GFP mutants with the same expected charge (-18). GFP and qP4VP, PEI, or PLL were mixed at a total macromolecule concentration of 1 mg mL−1. Phase behavior was measured over a broad range of macromolecule mixing ratios (Fig. 3, S4a and S5†). Phase separation was examined as a function of the positive charge fraction (f+), which is the ratio of positive charge (M+) to total charge (M+ + M−). M+ and M− are the charge per mass of the positive and negative species respectively. The charge per mass for the proteins, M− was calculated using the expected charge determined by the Henderson–Hasselbalch equation and the isolated side chain pKa values (ESI Tables S1 and S2†).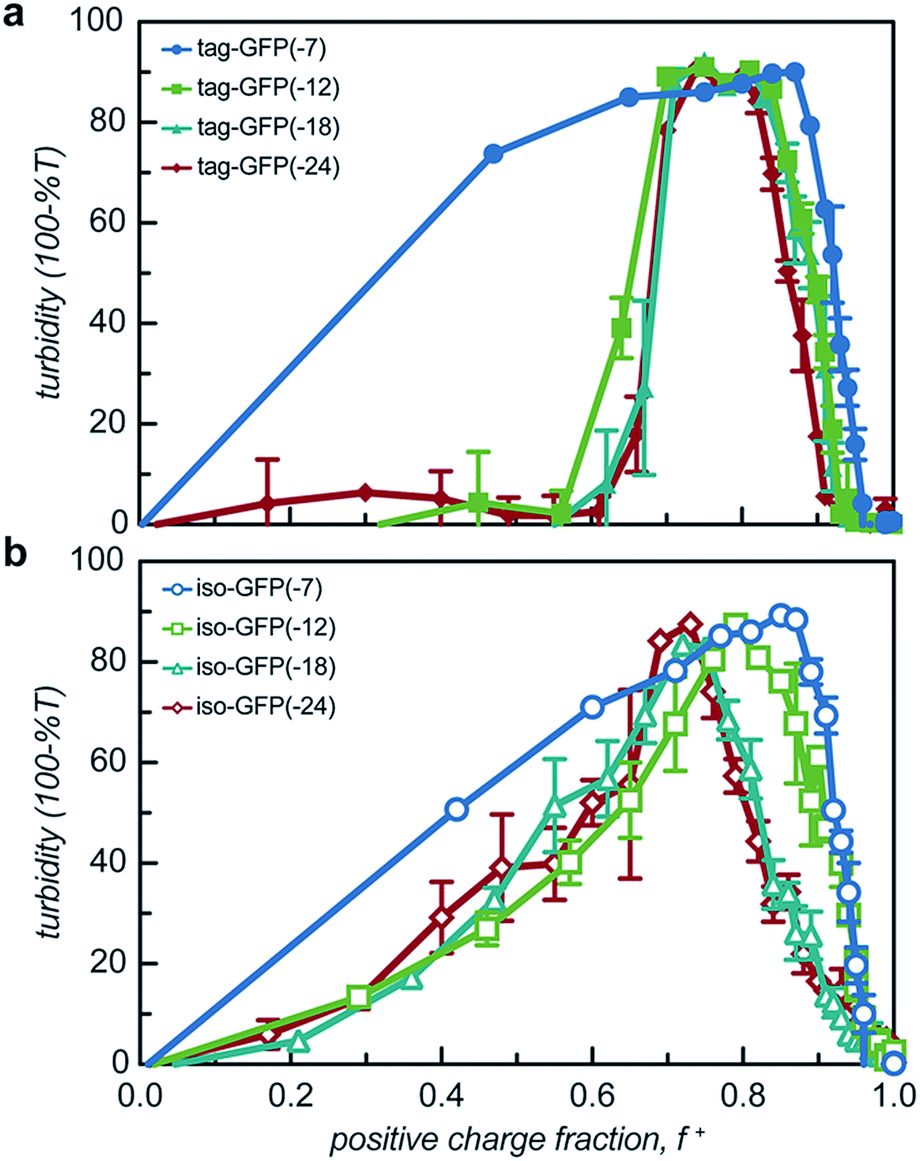 | ||
| Fig. 3 Turbidity studies of GFP and qP4VP solutions. Turbidity profiles as a function of positive charge fraction in 10 mM Tris buffer, pH 7.4 for (a) tagged mutants and (b) isotropically charged mutants. Total macromolecule concentration is 1 mg mL−1. | ||
All the GFP mutants phase separated over a broad range of positive charge fractions (Fig. S4a and S5†). All of the proteins had a negative-to-positive residue ratio, α, greater than 1.1, suggesting these mutants were sufficiently supercharged and should phase separate in the presence of a strong polycation at low ionic strength.3 A maximum in turbidity centered at f+ = 0.5 would indicate that phase separation occurs optimally when the system is charge neutral. The turbidity maxima for both the isotropic and tag mutants were shifted toward higher positive charge fractions with the maxima of the isotropic mutants shifted to f+ ∼ 0.7–0.9 and the tagged mutants shifted to f+ ∼ 0.8. This shift to higher charge fractions indicates that phase separation is favored in the presence of excess cationic charges on the polycation. Several factors are hypothesized to contribute to this shift from the expected charge neutral ratio. This shift from charge neutrality can be explained by the ability of the ionizable groups on the protein to undergo induced charging, charge anisotropy, and the globular protein structure.3,44 First, upon complexation with qP4VP, the ionizable functional groups on the protein can change protonation state, altering the overall charge of the protein, making it more negative. This would require fewer proteins to complex with the polycation to result in charge neutrality, resulting in f+ > 0.5 at the maximum of phase separation.3,44 In support of this hypothesis is the observation that the fluorescence of iso-GFP(-18) and iso-GFP(-24) increased in the presence of qP4VP. This demonstrates that qP4VP influenced the protonation state of the GFP chromophore. This increased fluorescence was eliminated when sufficient NaCl was added to screen interactions with the polycation. Second, the shift of the tagged mutants can also be explained by an anisotropic charge patch that could allow for complexation with qP4VP that results in local, but not overall, charge neutrality.3 Finally, it is possible that the coacervation between a protein colloid and polyelectrolyte does not occur at equal charge stoichiometry. This can be rationalized by the geometric mismatch between the folded globular structure of the protein and the extended polymer.
Turbidity measurements provide an indication of phase separation, but they do not offer insight into the nature of phase separation. Both liquid–liquid and solid–liquid phase separation have been observed in protein–polymer systems.22,40,45,46 Optical microscopy was used to distinguish these two modes of phase separation. Samples were prepared at the optimal charge ratio as determined by the initial turbidity screens with a total macromolecule concentration of 1 mg mL−1 in 10 mM Tris, pH 7.4 (Tables S1 and S2†). All tagged mutants underwent liquid–liquid phase separation with qP4VP, as indicated by the spherical droplets that were observed to coalesce over time (Fig. 4 and S8†). These phase separated droplets demonstrated a viscous liquid-like recovery when portions of the droplets were photobleached (representative data, Fig. S6†). Two of the isotropic mutants also formed a liquid at the midpoint of phase separation. The two most negatively charged isotropic mutants underwent solid–liquid phase separation with qP4VP. Phase separation with the weak polyelectrolytes appeared to result in liquid–liquid phase separation for both the tagged and isotropic mutants. This is consistent with prior findings that indicate that strong polyelectrolytes are more likely to result in solid precipitate formation.22 The solid–liquid phase separation was observed to occur quickly after mixing (<1 min). We hypothesize that this precipitation is due to the system reaching a local, but potentially not global, thermodynamic minimum. The high charge on these proteins might result in kinetic trapping as there are more interaction sites than on a protein with lower charge density. It is hypothesized that the tagged mutants of equivalent charge undergo liquid–liquid phase separation because the tag reduces the ability of the proteins to pack uniformly. Both the isotropic and tagged GFP mutants underwent liquid–liquid phase separation with PLL (Fig. S4c†). It is hypothesized that iso-GFP(-18) undergoes liquid–liquid phase separation with PLL because the weak polyelectrolyte does not cause the system to become kinetically trapped. It is indeterminate whether the tagged and isotropic GFP(-18) mutants underwent solid–liquid or liquid–liquid phase separation with PEI as both liquid and solid phases were observed simultaneously. Therefore, it is possible that these samples indicate either both liquid–liquid and solid–liquid phase separation happening concurrently or that the system is transitioning from liquid–liquid phase separation to solid–liquid phase separation as has been observed for some native intrinsically disordered sequences.47,48
 | ||
| Fig. 4 Optical microscopy images of GFP and qP4VP at the midpoint of phase separation as determined by the initial turbidity screens. Samples were prepared with a total macromolecule concentration of 1 mg mL−1 in 10 mM Tris buffer, pH 7.4. Samples were mixed by pipetting. Many proteins exhibited liquid–liquid (labeled L) as demonstrated by spherical droplets that coalesced over time. Alternately, some isotropic samples displayed solid–liquid (labeled S) phase separation as indicated by irregular aggregates. Scale bar represents 25 μm. | ||
Factors that influence phase behaviour
While two macroions of opposite charge are generally required to induce complex coacervation, there are many system parameters that can, either favorably or not, alter phase behavior. The critical factor that determines if a system phase separates is the charge stoichiometry of the polyelectrolytes. Other system parameters, such as pH and ionic strength, can impact the charge state of the polyelectrolytes and the entropic gain from counterion release, respectively, ultimately impacting the macromolecule ratios where phase separation is observed.1 The effect of these solution parameters on the phase separation of the engineered GFPs with varied charge distribution was determined.Both the tagged and isotropic mutants tested had at least one stoichiometric ratio that incorporated nearly all (>90%) of the initial protein added to the system (Fig. 5 and S7†). The proteins that underwent liquid–liquid phase separation exhibited maxima in the fraction encapsulated at the macromolecule ratio near the midpoint of the observed phase separation regime. A relationship between the nature of phase separation and the efficiency of protein encapsulation at varying macromolecule ratios was also observed. The proteins that underwent solid–liquid phase separation exhibited increasing encapsulation efficiency with increasing positive charge fraction. This is attributed to the nature of phase separation; any additional protein also precipitates out of solution.
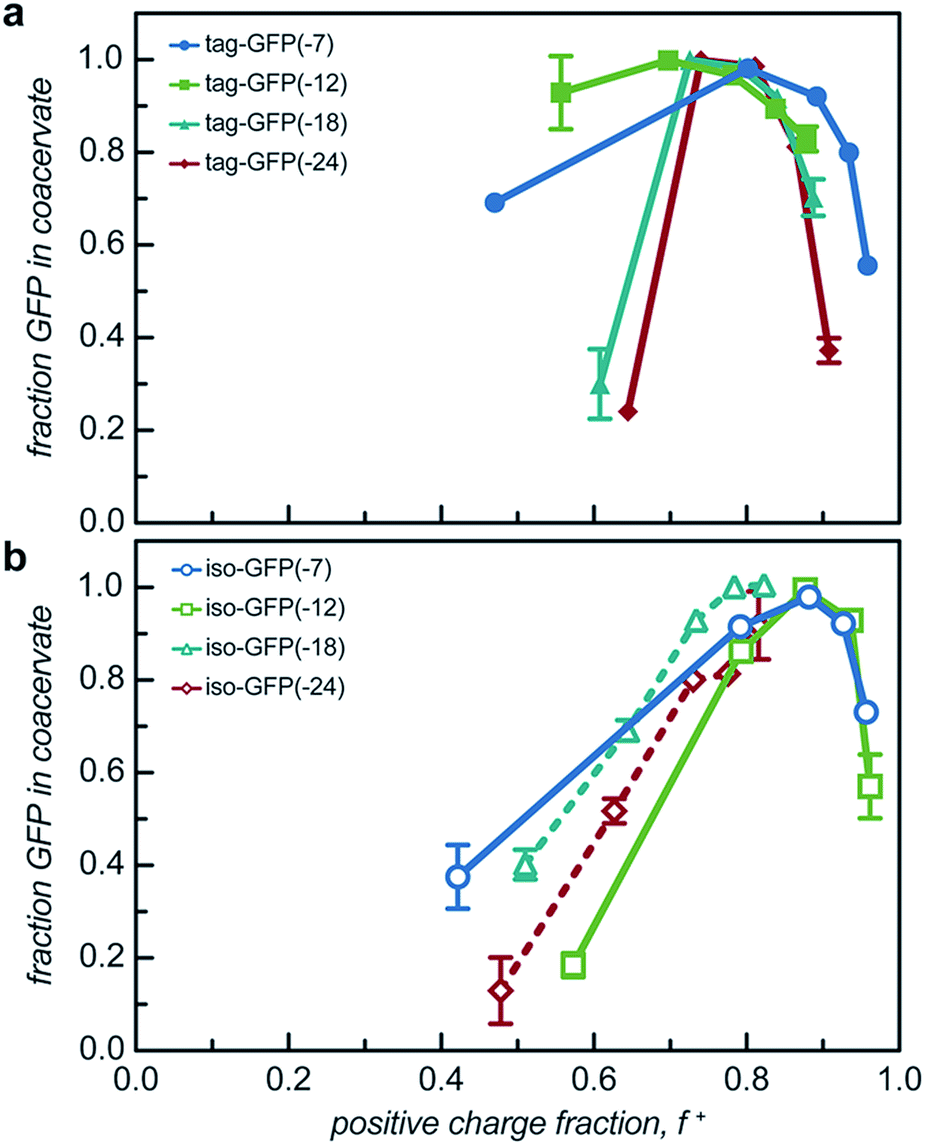 | ||
| Fig. 5 Encapsulation efficiency of (a) tagged GFP and (b) isotropic GFP with qP4VP in the coacervate (solid line) or precipitate (dashed line) phase at 5 macromolecule ratios determined by initial turbidity studies. | ||
The engineered proteins are weak polyelectrolytes with many ionizable side chains, which means that the phase separation will be dependent on the solution pH. It should be noted that these model proteins also contained an N-terminal His-tag, which contains several ionizable residues. The pH dependence of the phase separation for all the model protein mutants was determined by titrating the pH of protein and qP4VP solutions. The weak polycations were not tested as it had previously been demonstrated that the polyelectrolyte component had a minimal influence on the critical pH of protein complex coacervation.41 Protein and polymer were initially mixed at pH 3.0 and the critical pH for phase separation was then determined by increasing the pH of the solution and monitoring the change in turbidity. Each protein exhibited a critical pH of phase separation between 4.5 and 6.2, indicating that for pH responsive materials, the protein charge can be tuned to control the pH of the phase transition (Fig. S9†). All the isotropic proteins and tag-GFP(-7) and tag-GFP(-12) phase separated below the experimentally determined isoelectric points (Fig. 6). Previous studies have indicated the ampholytic nature of proteins can lead to formation of a charge patch with sufficient charge for phase separation below the pI of the protein.1,22 For these proteins, it is proposed that the distance between negative residues is sufficient to produce a localized charge patch that promotes phase separation in the presence of a polycation at a pH below the pI of the protein. This is supported by the fact that the pH of phase separation decreased with increasing negative charge for all mutants. In contrast, GFPs with long ionic tags, tag-GFP(-18) and (-24) had a critical pH for complex coacervation above the pI of the protein. This is proposed to result from a higher configurational entropy penalty upon complexation for the long, flexible polypeptide tags that must be counterbalanced by additional proton losses.49 Another possible explanation for the apparent differences in behaviour observed for tag-GFP(-18) and (-24) is that at the same pH the longer tags do not produce a charge patch as the ionized residues are distributed over an increased number of residues thus increasing the distance between like charges.
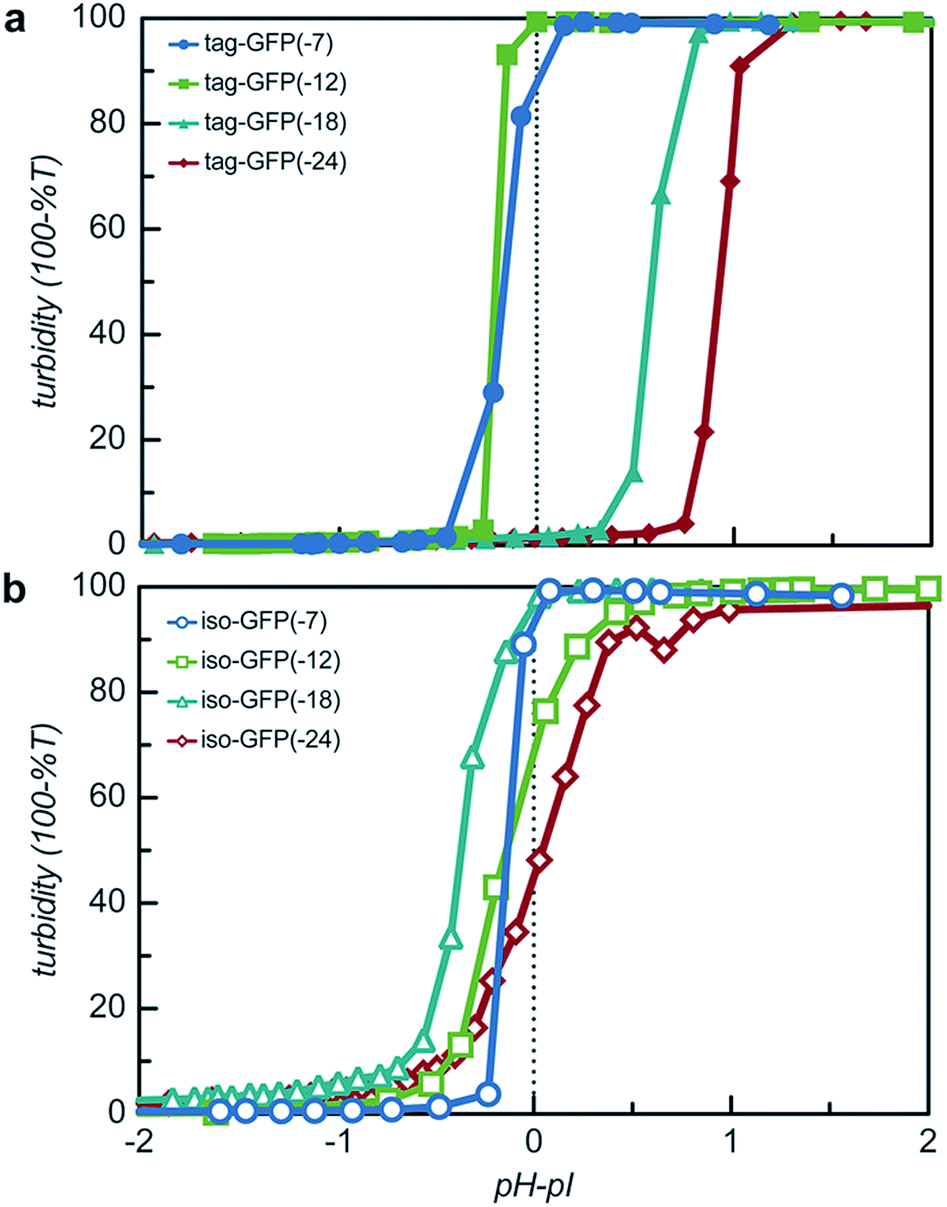 | ||
| Fig. 6 The pH dependence of protein phase separation was determined via pH titration studies. Turbidity as a function of the difference between the solution pH and the isoelectric point (pI) for (a) tagged mutants and (b) isotropically charged mutants. Samples were prepared in 10 mM Tris buffer (pH 3). Total macromolecule concentration was 1 mg mL−1. Absorbance and pH were measured after the addition of a NaOH solution in 1 μL increments. | ||
Both the overall charge and the distribution of charge were expected to significantly impact the critical salt concentration for protein coacervation, as charge patterning had recently been demonstrated to affect the charge–charge interactions that drive this associative phase separation.34 The effect of increased salt concentration on phase separation was explored by titrating 5 M NaCl into phase separated solutions of protein and qP4VP and monitoring the change in turbidity. It was determined that increasing charge on the protein resulted in a system that was resistant to dissolution by salt (Fig. 7a). Increasing the expected charge of the protein increased the salt stability of the coacervate from 30–40% over a less negatively charged mutant. We hypothesize that this could be attributed to the fact that increasing the number of negative residues on the protein increases the number of interaction sites for the polycation to initially bind to the protein as well as an increased number of charged residues to release counterions.50
 | ||
| Fig. 7 Effects of salt on GFP and qP4VP complexes. (a) Titration studies of GFP and qP4VP complex dissolution. Measurements were performed by mixing GFP and qP4VP at a total macromolecule concentration of 1 mg mL−1 in 10 mM Tris buffer, pH 7.4 at the midpoint of phase separation as determined by the initial turbidity studies. Absorbance was measured after 1 μL additions of 5 M NaCl in 10 mM Tris buffer, pH 7.4. (b) Critical salt concentration as a function of charge patch size as approximated by the mean distance between each negatively charged residue and the nearest negatively charged neighbor. Purple star represents tag-CAT(-18) and was not included in the initial regression. Black dashed line represents the 95% confidence band; grey dashed line represents the 95% prediction band. (c) Effect of salt on complex formation. GFP and qP4VP were mixed at a total macromolecule concentration of 0.97 mg mL−1 in 150 mM NaCl in 10 mM Tris buffer, pH 7.4 at the midpoint of phase separation as determined by initial turbidity screens. | ||
It was also observed that the tagged mutants remained phase separated at higher salt concentrations than their isotropic counterparts. In fact, the least negatively charged tagged mutants had a critical salt concentration (CSC) that was 25% higher than the isotropic equivalent and the most negatively charged mutant saw a nearly 50% increase in the CSC as compared to its isotropic counterpart (Fig. 7a). This can be attributed to the fact that by adding the tag the number of closely neighboring negatively charged residues increases. We hypothesize that this would result in the polycation interacting with a larger number of negative residues on the tag in order to reduce repulsion between residues of like charge. This increase in the number of interactions between the two polyelectrolytes would increase the salt concentration where counterion release is no longer entropically favorable, resulting in the transition from a two-phase system to a single-phase system at a higher salt concentration.50 The CSC for the tagged and isotropic variants was correlated with the ratio of negative-to-positive residues (α), but the different charge patterns demonstrated distinctly different correlations with α (Fig. S10†). By quantifying the average minimum distance between negatively charged residues, the CSC for all 8 engineered proteins could be predicted from a single master curve (Fig. 7b). To test the applicability of these findings to other proteins, an ionic tag was appended to a different globular protein, CAT. The salt dependence of the complex coacervation of CAT and qP4VP was determined in the same manner as for GFP (Fig. 7b). The slight increase in the CSC relative to the prediction is attributed to the assembly state of CAT. sfGFP forms a weak dimer that is likely enhanced in the coacervate phase, while CAT forms a stable trimer in solution.51 Based on this finding, it is hypothesized that truly monomeric proteins may likely exhibit a slightly lower CSC than is predicted. However, this simple prediction is still able to provide a reasonable estimation of the CSC for the complex coacervation of the strong polycation, qP4VP, with proteins with an acidic pI.
The polyion chemistry was also expected to influence the CSC, as has been observed in both polymer/polymer and protein/polymer systems.34,41,52 The salt dependence of phase separation for tag- and iso-GFP(-18) was also determined with PEI and PLL (Fig. S4b†). These two weak polycations behaved similarly to each other, but quite differently than qP4VP. The benefits of having the charge localized to the C-terminal tag were eliminated when complexing with a weak polyelectrolyte. We hypothesize that when both components are ionizable, both the protein and polymer can charge regulate to minimize excess charge and repulsion between like-charges. This minimizes the importance of a charge patch and can explain why the C-terminal tags do not enhance the CSC relative to isotropic controls when complexing with weak polyelectrolytes.
From these initial titration experiments, it was also predicted that tag-GFP(-12) remains phase separated at 150 mM NaCl. This was confirmed by mixing polymer and protein (0.97 mg mL−1, 150 mM NaCl, 10 mM Tris pH 7.4) at the optimal stoichiometric ratio and measuring the turbidity and observing the sample by optical microscopy (Fig. 7c and S11†). The combination of these results indicates that the addition of a single six amino acid tag is sufficient to promote the phase separation of a globular protein under physiological salt and pH conditions.
Conclusions
A panel of genetically engineered proteins was prepared in order to develop a simple strategy for the complex coacervation of globular proteins. The introduction of 6–18 anionic amino acids (Asp, Glu) was sufficient to promote liquid–liquid phase separation of GFP with polycations at physiologically relevant conditions (∼25 μM GFP, Tris buffered saline, pH 7.4). When compared to proteins with the same expected net charge, the addition of ionic polypeptide tags demonstrated several key advantages. First, the tags minimally perturbed GFP fluorescence. Engineered GFPs with the same expected charge isotropically distributed on the protein surface showed significantly reduced fluorescence, despite having fewer introduced mutations. Second, the ionic polypeptide tags promoted liquid–liquid phase separation even at high net charge. Conversely, isotropically supercharged GFP was prone to precipitation instead of complex coacervation. Finally, proteins with the ionic polypeptide tags demonstrated higher (25–50%) critical salt concentrations (CSC) than proteins with isotropic charge when mixed with a strong polycation. Differences in the CSC based on charge distribution could be eliminated by correlating the CSC to the calculated mean minimum distance between negatively charged residues. These combined findings point to the utility of short, ionic polypeptides to promote phase transitions of globular proteins. These polypeptide tags can easily be appended to any protein with an acidic pI to artificially promote complex coacervation with polycations. Based on prior work with supercationic GFP, we believe that a similar strategy can be adopted for proteins with a basic pI.41 A tag of cationic residues (Lys, Arg, His) is expected to provide the same advantages for basic proteins that the anionic tag provides for acidic proteins. Additionally, if desired, a protease site could be introduced to remove the polypeptide tag after complex coacervation to furnish the soluble, native protein of interest.53 This approach provides biological responsivity and minimal protein modification when compared to chemical or genetic supercharging. The ionic polypeptide tags characterized here provide insight into how protein charge distribution influences the complex coacervation of globular proteins. This initial report demonstrates the utility of these tags for the complex coacervation of globular proteins and characterizes the influence of several additional parameters on protein phase behavior. However, as has been demonstrated in polymer/polymer systems, even relatively subtle changes to the polymer, buffer, or salt can significantly alter the complex coacervation process. The impact of these additional parameters will be the focus of future investigations. These tags also provide a method to promote the liquid–liquid phase separation of proteins that is complementary to current methods that rely on simple coacervation of temperature-responsive polypeptides. The design parameters established here for small (∼750 Da) ionic tags should enable the investigation of protein complex coacervation for applications including protein stabilization, delivery, and purification.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Fu School of Engineering and Applied Sciences at Columbia University.Notes and references
- W. C. Blocher and S. L. Perry, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9(4), e1442 Search PubMed.
- S. Lindhoud and M. M. A. E. Claessens, Soft Matter, 2016, 12, 408–413 RSC.
- A. C. Obermeyer, C. E. Mills, X.-H. Dong, R. J. Flores and B. D. Olsen, Soft Matter, 2016, 12, 3570–3581 RSC.
- M. Jaturanpinyo, A. Harada, X. Yuan and K. Kataoka, Bioconjugate Chem., 2004, 15, 344–348 CrossRef CAS PubMed.
- S. R. MacEwan and A. Chilkoti, Biopolymers, 2010, 94, 60–77 CrossRef CAS PubMed.
- N. A. Chaniotakis, Anal. Bioanal. Chem., 2004, 378, 89–95 CrossRef CAS PubMed.
- C. L. Cooper, P. L. Dubin, A. B. Kayitmazer and S. Turksen, Curr. Opin. Colloid Interface Sci., 2005, 10, 52–78 CrossRef CAS.
- D. W. Urry, D. C. Gowda, T. M. Parker, C.-H. Luan, M. C. Reid, C. M. Harris, A. Pattanaik and R. D. Harris, Biopolymers, 1992, 32, 1243–1250 CrossRef CAS PubMed.
- T. Christensen, W. Hassouneh, K. Trabbic-Carlson and A. Chilkoti, Biomacromolecules, 2013, 14, 1514–1519 CrossRef CAS PubMed.
- F. G. Quiroz and A. Chilkoti, Nat. Mater., 2015, 14, 1164–1171 CrossRef CAS PubMed.
- J. R. Simon, N. J. Carroll, M. Rubinstein, A. Chilkoti and G. P. López, Nat. Chem., 2017, 9, 509–515 CrossRef CAS PubMed.
- M. R. Banki, L. Feng and D. W. Wood, Nat. Methods, 2005, 2, 659–662 CrossRef CAS PubMed.
- W. Hassouneh, T. Christensen and A. Chilkoti, Curr. Protoc. Protein Sci., 2010, 6.11 Search PubMed.
- W. Hassouneh, S. R. MacEwan and A. Chilkoti, Methods Enzymol., 2012, 502, 215–237 CAS.
- M. J. Glassman, R. K. Avery, A. Khademhosseini and B. D. Olsen, Biomacromolecules, 2016, 17, 415–426 CrossRef CAS PubMed.
- J. E. Glasgow, S. L. Capehart, M. B. Francis and D. Tullman-Ercek, ACS Nano, 2012, 6, 8658–8664 CrossRef CAS PubMed.
- R. Mout and V. M. Rotello, Bio-Protoc., 2017, 7(24), e2661 Search PubMed.
- M. Nigen, T. Croguennec, D. Renard and S. Bouhallab, Biochemistry, 2007, 46, 1248–1255 CrossRef CAS PubMed.
- T. Croguennec, G. M. Tavares and S. Bouhallab, Adv. Colloid Interface Sci., 2017, 239, 115–126 CrossRef CAS PubMed.
- A. B. Kayitmazer, D. Seeman, B. B. Minsky, P. L. Dubin and Y. Xu, Soft Matter, 2013, 9, 2553–2583 RSC.
- Y. Lee, T. Ishii, H. Cabral, H. J. Kim, J.-H. Seo, N. Nishiyama, H. Oshima, K. Osada and K. Kataoka, Angew. Chem., Int. Ed. Engl., 2009, 48, 5309–5312 CrossRef CAS PubMed.
- F. Comert and P. L. Dubin, Adv. Colloid Interface Sci., 2017, 239, 213–217 CrossRef CAS PubMed.
- E. Kizilay, A. B. Kayitmazer and P. L. Dubin, Adv. Colloid Interface Sci., 2011, 167, 24–37 CrossRef CAS PubMed.
- Y. Xu, M. Mazzawi, K. Chen, L. Sun and P. L. Dubin, Biomacromolecules, 2011, 12, 1512–1522 CrossRef CAS PubMed.
- X. Du, P. L. Dubin, D. A. Hoagland and L. Sun, Biomacromolecules, 2014, 15, 726–734 CrossRef CAS PubMed.
- A. Molliex, J. Temirov, J. Lee, M. Coughlin, A. P. Kanagaraj, H. J. Kim, T. Mittag and J. P. Taylor, Cell, 2015, 163, 123–133 CrossRef CAS PubMed.
- S. Elbaum-Garfinkle, Y. Kim, K. Szczepaniak, C. C.-H. Chen, C. R. Eckmann, S. Myong and C. P. Brangwynne, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7189–7194 CrossRef CAS PubMed.
- M. Feric, N. Vaidya, T. S. Harmon, D. M. Mitrea, L. Zhu, T. M. Richardson, R. W. Kriwacki, R. V. Pappu and C. P. Brangwynne, Cell, 2016, 165, 1686–1697 CrossRef CAS PubMed.
- M.-T. Wei, S. Elbaum-Garfinkle, A. S. Holehouse, C. C.-H. Chen, M. Feric, C. B. Arnold, R. D. Priestley, R. V. Pappu and C. P. Brangwynne, Nat. Chem., 2017, 9, 1118–1125 CrossRef CAS PubMed.
- C. P. Brangwynne, P. Tompa and R. V. Pappu, Nat. Phys., 2015, 11, 899–904 Search PubMed.
- Y. Shin and C. P. Brangwynne, Science, 2017, 357, eaaf4382 CrossRef PubMed.
- S. F. Banani, H. O. Lee, A. A. Hyman and M. K. Rosen, Nat. Rev. Mol. Cell Biol., 2017, 18, 285–298 CrossRef CAS PubMed.
- L.-W. Chang, T. K. Lytle, M. Radhakrishna, J. J. Madinya, J. Vélez, C. E. Sing and S. L. Perry, Nat. Commun., 2017, 8, 1273 CrossRef PubMed.
- G. S. Manning, J. Chem. Phys., 1969, 51, 924–933 CrossRef CAS.
- S. Dou and R. H. Colby, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 2001–2013 CrossRef CAS.
- Z. Ou and M. Muthukumar, J. Chem. Phys., 2006, 124, 154902 CrossRef PubMed.
- J.-D. Pédelacq, S. Cabantous, T. Tran, T. C. Terwilliger and G. S. Waldo, Nat. Biotechnol., 2006, 24, 79–88 CrossRef PubMed.
- M. S. Lawrence, K. J. Phillips and D. R. Liu, J. Am. Chem. Soc., 2007, 129, 10110 CrossRef CAS PubMed.
- C. N. Lam, H. Yao and B. D. Olsen, Biomacromolecules, 2016, 17, 2820–2829 CrossRef CAS PubMed.
- C. S. Cummings and A. C. Obermeyer, Biochemistry, 2018, 57, 314–323 CrossRef CAS PubMed.
- G. J. Bartlett, C. T. Porter, N. Borkakoti and J. M. Thornton, J. Mol. Biol., 2002, 324, 105–121 CrossRef CAS PubMed.
- K. Taira, K. Kataoka and T. Niidome, Non-viral Gene Therapy: Gene Design and Delivery, Springer Science & Business Media, 2006 Search PubMed.
- R. J. Christie, N. Nishiyama and K. Kataoka, Endocrinology, 2010, 151, 466–473 CrossRef CAS PubMed.
- W. M. de Vos, F. A. M. Leermakers, A. de Keizer, M. A. Cohen Stuart and J. M. Kleijn, Langmuir, 2010, 26, 249–259 CrossRef CAS PubMed.
- C. G. de Kruif, F. Weinbreck and R. de Vries, Curr. Opin. Colloid Interface Sci., 2004, 9, 340–349 CrossRef CAS.
- F. Comert, A. J. Malanowski, F. Azarikia and P. L. Dubin, Soft Matter, 2016, 12, 4154–4161 RSC.
- A. Patel, H. O. Lee, L. Jawerth, S. Maharana, M. Jahnel, M. Y. Hein, S. Stoynov, J. Mahamid, S. Saha, T. M. Franzmann, A. Pozniakovski, I. Poser, N. Maghelli, L. A. Royer, M. Weigert, E. W. Myers, S. Grill, D. Drechsel, A. A. Hyman and S. Alberti, Cell, 2015, 162, 1066–1077 CrossRef CAS PubMed.
- S. Alberti, S. Saha, J. B. Woodruff, T. M. Franzmann, J. Wang and A. A. Hyman, J. Mol. Biol., 2018, 430, 4806–4820 CrossRef CAS PubMed.
- A. B. Kayitmazer, A. F. Koksal and E. K. Iyilik, Soft Matter, 2015, 11, 8605–8612 RSC.
- D. Priftis and M. Tirrell, Soft Matter, 2012, 8, 9396–9405 RSC.
- A. G. W. Leslie, J. Mol. Biol., 1990, 213, 167–186 CrossRef CAS PubMed.
- J. Fu, H. M. Fares and J. B. Schlenoff, Macromolecules, 2017, 50, 1066–1074 CrossRef CAS.
- B. S. Schuster, E. H. Reed, R. Parthasarathy, C. N. Jahnke, R. M. Caldwell, J. G. Bermudez, H. Ramage, M. C. Good and D. A. Hammer, Nat. Commun., 2018, 9, 2985 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, alternate plots, and supplementary data. See DOI: 10.1039/c8sc04253e |
| This journal is © The Royal Society of Chemistry 2019 |