
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharge transport modulation in pseudorotaxane 1D stacks of acene and azaacene derivatives†
Cristian
Gozalvez
a,
Jose L.
Zafra
b,
Akinori
Saeki
 c,
Manuel
Melle-Franco
*d,
Juan
Casado
*b and
Aurelio
Mateo-Alonso
*ae
c,
Manuel
Melle-Franco
*d,
Juan
Casado
*b and
Aurelio
Mateo-Alonso
*ae
aPOLYMAT, University of the Basque Country UPV/EHU, Avenida de Tolosa 72, E-20018 Donostia-San Sebastian, Spain. E-mail: amateo@polymat.eu
bDepartment of Physical Chemistry, University of Málaga, Campus de Teatinos s/n, 229071 Málaga, Spain. E-mail: casado@uma.es
cDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan
dCICECO—Aveiro Institute of Materials, Department of Chemistry, University of Aveiro, 3810-193, Aveiro, Portugal. E-mail: manuelmelle.research@gmail.com
eIkerbasque, Basque Foundation for Science, E-48011 Bilbao, Spain
First published on 9th January 2019
Abstract
Acenes have received a lot of attention because of their inherent and tunable absorbing, emissive, and charge transport properties for electronic, photovoltaic, and singlet fission applications, among others. Such properties are directly governed by molecular packing, and therefore, controlling their arrangement in the solid state is of utmost importance in order to increase their performance. Herein, we describe a new solid-state ordering strategy that allows obtaining 1D mixed π-stacks of acene and azaacene derivatives. In addition, we illustrate that charge transport can be modulated by the electronic nature of the encapsulated phenazine, opening new perspectives in the design, preparation and development of supramolecular organic semiconductors.
Introduction
The performance of organic field-effect transistors, light-emitting diodes, and solar cells (including perovskite solar cells) critically depends on the charge transport efficiency of organic semiconductors (OSCs).1 This is the efficiency with which electrons or holes move within the organic semiconducting layer(s). The three-dimensional organisation of OSCs in the solid state is one of the key factors that dominate the charge transport efficiency. Among different packing configurations, lamellar (1D or 2D) π-stacking (face-to-face) has been identified as an optimal configuration for electronic transport, as a result of an efficient intermolecular electronic coupling.1 Therefore, improving the ability to organise OSCs into lamellar face-to-face π-stacking configurations in the solid state is crucial in order to increase the performance of electronic devices.Acenes are polycyclic aromatic hydrocarbons constituted of linearly fused aromatic rings that have received a lot of attention as OSCs because their inherent and tunable absorbing, emissive, and charge transport properties have shown a lot of potential for electronic,2 photovoltaic,3 and singlet fission4 applications, among others. However, the most common packing configuration observed in acenes is the herringbone packing without π–π overlap (edge-to-face) rather than the lamellar π-stacking configuration (face-to-face) optimal for charge transport applications.2a,b Several strategies have been successfully developed to enforce such lamellar face-to-face π-stacking configurations in acenes, which include the introduction of bulky substituents,2a–e heteroatoms,5 and hydrogen bonding motifs,5c,6 the polarisation of the aromatic framework,7 and their integration on cyclophane8 or foldamer9 architectures. Yet, predicting the solid-state organization of acenes is a difficult task since small structural modifications in the acene framework can have an unexpected impact on packing,2a,b something that continuously defies our ability to control solid-state organisation.
Herein, we describe a new solid-state ordering approach that allows obtaining lamellar 1D mixed π-stacks of acene and azaacene derivatives (Scheme 1). Through this approach, a set of different phenazine derivatives have been intercalated within a macrocycle with two anthracene moieties into a supramolecular complex with a pseudorotaxane architecture, in which the three components are arranged in a face-to-face configuration. Such pseudorotaxanes pile up, giving rise to lamellar 1D face-to-face π-stacks in the solid state, which are optimal for charge transport applications. This packing configuration is persistent over the set of six phenazine derivatives studied. In addition, we show that charge transport can be modulated by the electronic nature of the encapsulated phenazine, opening new perspectives in the design, preparation and development of supramolecular OSCs.
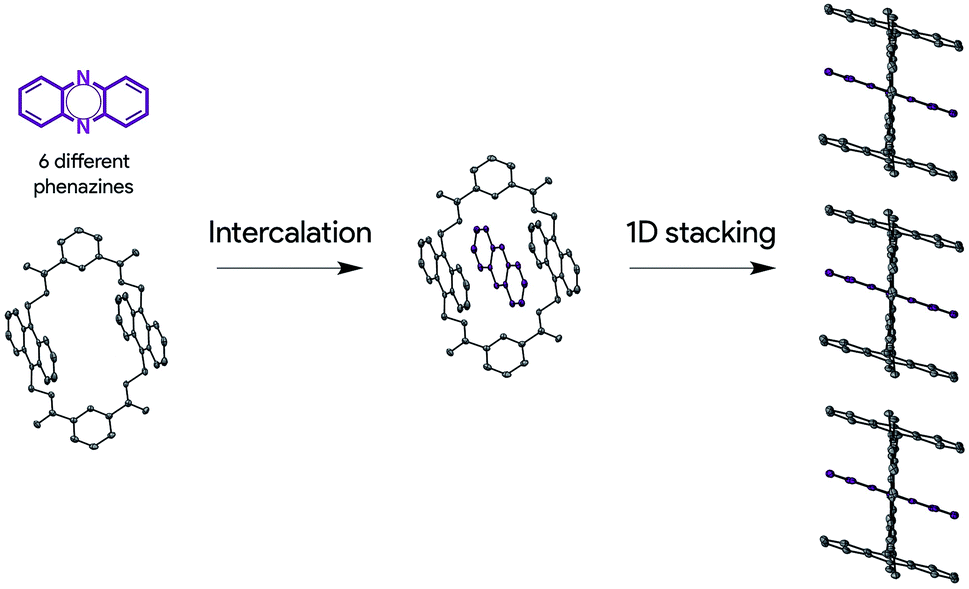 | ||
| Scheme 1 Schematic representation of the solid-state ordering approach. | ||
Results and discussion
Design, synthesis and structural characterisation
Pseudorotaxanes 2@1–7@1 were prepared by combining macrocycle 1 with the individual phenazine derivatives 2–7, respectively (Scheme 1 and Fig. 1). Pseudorotaxane precursors 1–7 were synthesised according to the procedures described in the ESI.† | ||
| Fig. 1 Chemical structures of macrocycle 1, phenazines 2–7 and pseudorotaxanes 2@1–7@1. | ||
We selected macrocycle 110 as a host because of (i) its inner cavity with a face to face distance from anthracene to anthracene of 7.21 Å (Fig. 2) that allows the encapsulation of an aromatic compound; (ii) the presence of the two anthracene units that provides an element for recognition both at the encapsulation level and at the supramolecular organisation level; (iii) the amide hydrogen bond donating binding sites that promote the encapsulation of hydrogen bond accepting guests. Macrocycle 110 and its derivatives11 have proven to be quite promiscuous and a variety of organic dyes, sugars, and even inorganic ions have been found to accommodate within its cavity.
 | ||
| Fig. 2 X-ray structures and packing of 1 and 2@1–7@1. | ||
Phenazine derivatives 2–7 (Fig. 1) were selected as guests because of (i) their structural and electronic complementarity with macrocycle 1, with three linearly fused rings in all cases, and (ii) the presence of hydrogen bond accepting moieties in the appropriate arrangement for binding the macrocycle's amides. Phenazine derivatives 2–7 can be organised in two groups, unsubstituted (2–4) and dimethoxy-substituted (5–7), which allow studying the effect of the different types of hydrogen bond accepting moieties present in the phenazine framework (sp2 N atoms, N-oxide and di-N-oxide) on the overall electronic structure and packing.
First, macrocycle 1 was combined with the individual phenazine derivatives 2–7, in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in chloroform. Then, single crystals suitable for X-ray diffraction studies were obtained from these 1:1 solutions using liquid–liquid diffusion protocols. The crystallization conditions are summarized in the ESI (Table S2†).
:1 ratio in chloroform. Then, single crystals suitable for X-ray diffraction studies were obtained from these 1:1 solutions using liquid–liquid diffusion protocols. The crystallization conditions are summarized in the ESI (Table S2†).
In all cases the X-ray structures reveal that the different phenazines intercalate between the two anthracenes of macrocycle 1 in a pseudorotaxane architecture (Fig. 2). Either the sp2 N atoms of the central pyrazine ring or the N-oxides engage in hydrogen bonds with the macrocycle's amides, giving rise to a hydrogen bond network with the additional contribution of the intramacrocycle hydrogen bonds between the amides and the pyridinic N. In addition, all phenazines are sandwiched between anthracene moieties at π-stacking distances between 3.38 and 3.54 Å (Fig. 2).
In terms of packing, macrocycle 1 is organized in chain-like structures in the crystal, where one pyridyl ring stacks in an antiparallel fashion with another pyridyl ring to an adjacent macrocycle (Fig. 2). The packing motif is quite different for the crystal structures of pseudorotaxanes 2@1–7@1. For instance, the inclusion of phenazine derivatives 2–7 in the cavity of macrocycle 1 gives rise to 1D columnar structures of the resulting pseudorotaxanes, in which one pseudorotaxane is stacked on top of the other by the anthracene moieties at π-stacking distances between 3.45 and 3.61 Å (Fig. 2). This packing motif is persistent over the whole pseudorotaxane series, even in the presence of crystallisation solvents in some of the structures (not shown), which tend to accommodate in between the pseudorotaxane columns.
Spectroscopic characterization in the solid state
To gain additional insight on the nature of the intrapseudorotaxane interactions, the optoelectronic and vibrational properties of the pseudorotaxanes in the solid state were investigated. Polycrystalline powder samples obtained by EtOH assisted precipitation of the pseudorotaxanes assembled in CHCl3 solutions were used in these experiments in order to obtain reasonable provisions for spectroscopic characterisation. These powder samples of 2@1–7@1 show the same colour of the corresponding single crystals. Furthermore, the colour of macrocycle 1 and phenazines 2–7 in the powder form is completely different from the colour of the pseudorotaxanes 2@1–7@1 (Fig. 4), as previously observed in solution. In particular, macrocycle 1 is white-yellowish and, in general, phenazines are yellow with the exception of di-N-oxy phenazines 4 and 7, which are red, whereas pseudorotaxanes show much darker colours ranging from orange to garnet, depending on the encapsulated phenazine.| Compound | CT peak (onset)/nm | CT peak/nm, M06-2X 6-31+g(d,p) | PL/nm |
|---|---|---|---|
| 1 | — | — | — |
| 2@1 | 412 (455) | 459 | 588 |
| 3@1 | 519 (691) | 500 | 650 |
| 4@1 | 535 (651) | 567 | — |
| 5@1 | 476 (587) | 447 | 566 |
| 6@1 | 551 (665) | 512 | 616 |
| 7@1 | 537 (649) | 530 | — |
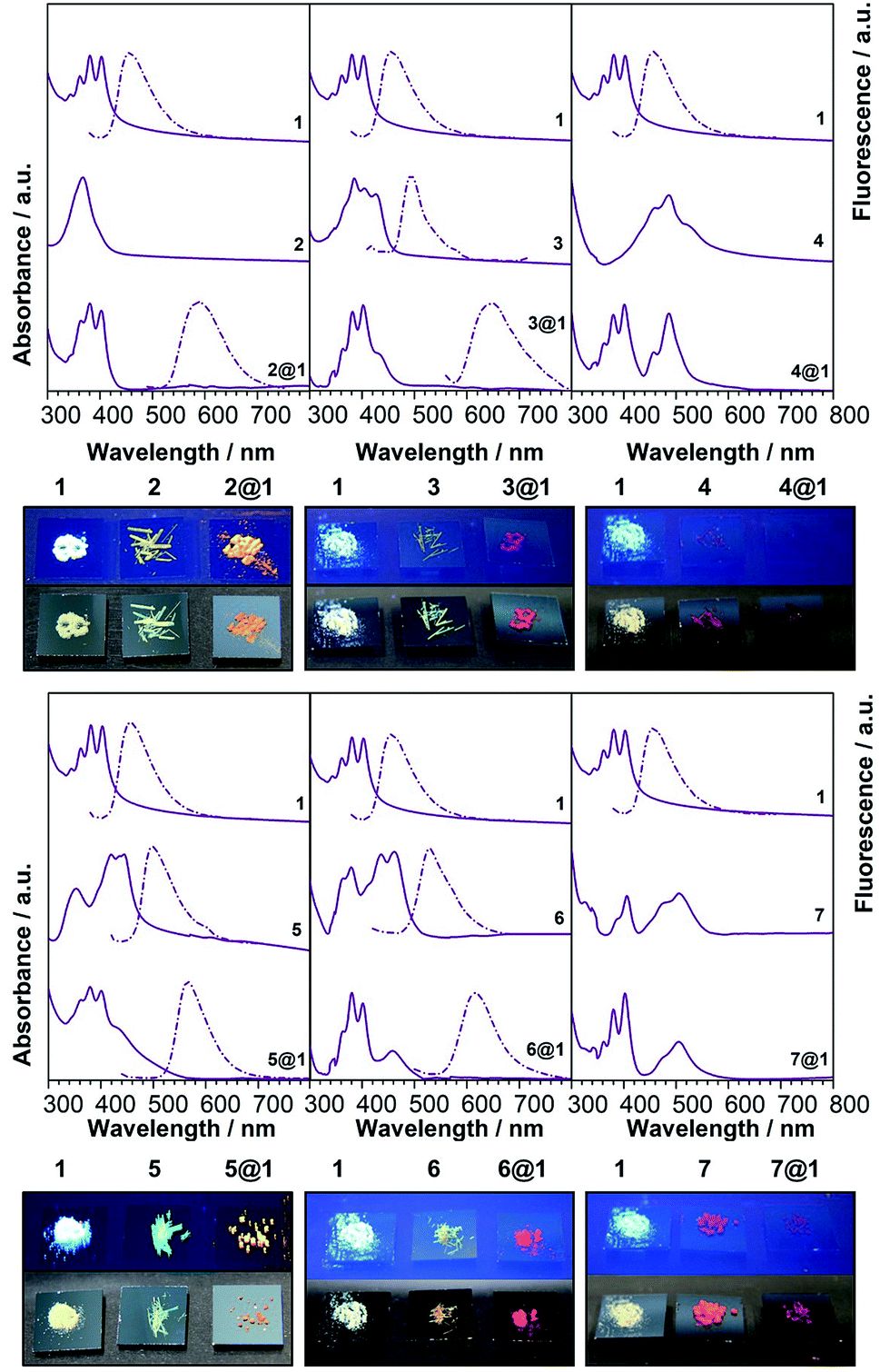 | ||
| Fig. 3 Electronic absorption, photoluminescence and colour of the solids (under natural (bottom) and UV (top) light. | ||
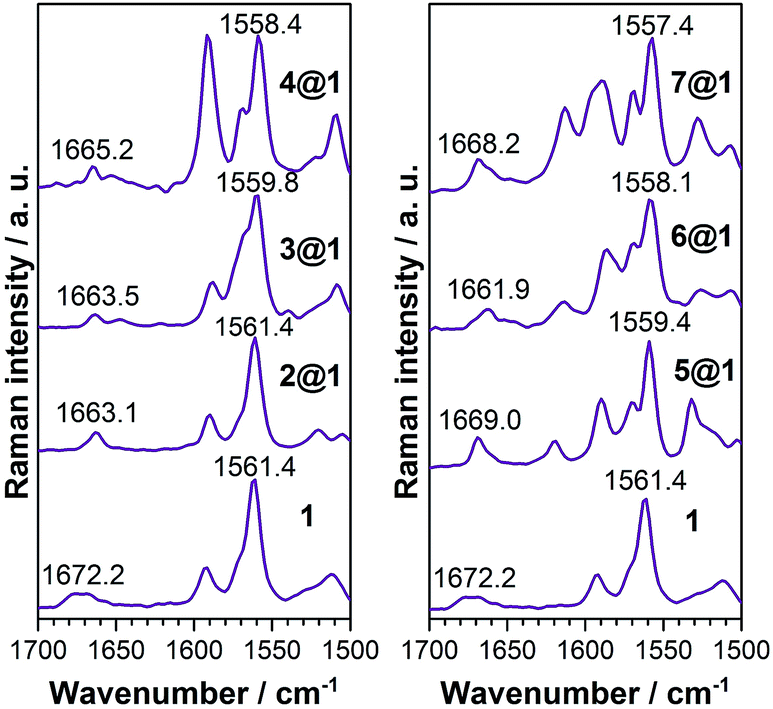 | ||
| Fig. 4 Solid-state Raman characterisation. | ||
The calculated electronic spectra (M062X/6-31+g(d,p)) (Tables 1 and S3†) show a good match with the experimental energies of the CT bands, which are the result of HOMO→LUMO contributions larger than 90%. The existing CT interactions can be also illustrated by calculating the orbital energies and the charge densities (B3LYP/6-31+g(d,p)) (Fig. S5†). In all cases, the largest densities for the HOMO of the complex are on the anthracenes, while the LUMO of all complexes shows larger densities on the encapsulated phenazines.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) modes as they display a frequency downshift (Δν) upon ground electronic state charge transfer (electrons released from the anthracenes) or ground electronic state polarization (between the donor and acceptor groups of the same molecule, i.e., by methoxy and N-oxide functionalizations), and this will be the spectroscopic Raman fingerprint of the electronic interaction mode between macrocycle 1 and the intercalated phenazines.
C) modes as they display a frequency downshift (Δν) upon ground electronic state charge transfer (electrons released from the anthracenes) or ground electronic state polarization (between the donor and acceptor groups of the same molecule, i.e., by methoxy and N-oxide functionalizations), and this will be the spectroscopic Raman fingerprint of the electronic interaction mode between macrocycle 1 and the intercalated phenazines.
Macrocycle 1 is the common component of all complexes and thus serves as the reference. Intercalation with phenazine 2 produces moderate alteration of the ground electronic state structure due to wavefunction overlap between the components and this increases proportionally to the number of N-oxides included (Δν1→2@1 = 0 cm−1, Δν1→3@1 = 1.6 cm−1, and Δν1→4@1 = 3 cm−1). These changes are typical of long distance π–π stacking. The inclusion of electron-donating dimethoxy groups in the phenazine significantly reinforces the coupling in the ground electronic state between the three stacked acenes in comparison to the situation without substitution (Δν1→5@1 = 2.0 cm−1, Δν1→6@1 = 3.3 cm−1, and Δν1→7@1 = 4.0 cm−1). This assignment is also consistent with the analysis of amide I stretching modes (see ESI†) and with the electronic absorption spectra (see above).
 | ||
| Fig. 5 Correlation between electronic absorption and Raman spectroscopy. Top (red): representation of the variation (Δν) of the wavenumber value of the main ν(CC) Raman band of the complexes regarding the same Raman band of the individual macrocycle. Bottom (green): normalized variation (ΔE) of the energy of the CT electronic absorption band of the complexes regarding the absorption band of the macrocycle. | ||
Charge transport properties in the solid state
To understand how charge transport is affected by the nature of the encapsulated phenazine, the charge transport properties of macrocycle 1, phenazines 2–7 and pseudorotaxanes 2@1–7@1 were probed at the intrapseudorotaxane level by time-resolved microwave conductivity (TRMC)12 of polycrystalline powder samples. This method makes possible the evaluation of the intrinsic and anisotropic charge-transport properties of materials without introducing electrodes on the charge separation/recombination length-scale, which fall within the intrapseudorotaxane regime. Here, the transient conductivity is defined as ϕ∑μ, where ϕ and ∑μ are the photocarrier-generation yield and the sum of the mobilities of the generated charge carriers, respectively. The values were extracted from the resultant rise and decay profiles of transient conductivities (Fig. S7 and S8†). The ϕ∑μ maxima can be considered the minimum mobility of the material.The ϕ∑μ values observed for pseudorotaxanes 2@1–7@1 were in the order of 10−4 cm2 V−1 s−1, while a ϕ∑μ = 6.9 × 10−5 cm2 V−1 s−1 was observed for empty macrocycle 1 which was used as a reference (Fig. 6). This clearly evidences that the intercalation of the phenazine within the macrocycle has an enhancing effect on the transient conductivities in all cases, which is consistent with the structure, as the phenazine interconnects the external macrocycle's anthracenes by π-stacking, opening intrapseudorotaxane channels optimal for charge transport. In addition, these studies reveal the interdependence of the electronic properties of the encapsulated phenazine with intrapseudorotaxane charge transport.
 | ||
| Fig. 6 ϕ∑μ maxima of macrocycle 1 and pseudorotaxanes 2@1–7@1 in the solid state. | ||
In particular, in the group of unsubstituted phenazines, pseudorotaxane 2@1 shows a ϕ∑μ value (2.1 × 10−4 cm2 V−1 s−1) that is three times higher than that of macrocycle 1. The ϕ∑μ value of 3@1 (2.1 × 10−4 cm2 V−1 s−1) is similar to that of 2@1. In contrast, the ϕ∑μ value of 4@1 (2.7 × 10−4 cm2 V−1 s−1) shows a 4-fold increase in comparison to that of 1. In the group of dimethoxyphenazines, pseudorotaxanes 5@1–7@1 also display higher ϕ∑μ values in comparison to bare macrocycle 1. Pseudorotaxane 5@1 exhibits a ϕ∑μ value that shows a 4-fold increase (2.8 × 10−4 cm2 V−1 s−1) compared to that of macrocycle 1 and is the highest of the whole pseudorotaxane series. Although the overall ϕ∑μ is higher than in the unsubstituted phenazine series, the same trend was observed in 5@1–7@1, as the ϕ∑μ decreases for mono-N-oxidized 6@1 (2.2 × 10−4 cm2 V−1 s−1) and remains slightly lower for di-N-oxidized 7@1 (2.7 × 10−4 cm2 V−1 s−1).
Conclusions
We have reported an ordering approach that allows effective control over the molecular orientation of anthracene and phenazine derivatives in the solid state. Through this approach, a phenazine derivative is encapsulated by macrocycle 1 with two anthracene moieties into a supramolecular complex with a pseudorotaxane architecture held together by hydrogen bonds and charge transfer interactions, which upon crystallisation provides lamellar 1D π-stack architectures in the solid state. The electronic nature of the intercalated phenazine directly affects the several optoelectronic properties of intrapseudorotaxanes, such as electronic absorption, photoluminescence and Raman frequencies that shine light on the nature of the intrapseudorotaxane interactions. Remarkably, the intercalation of phenazines in the cavity of the macrocycle improves the ϕ∑μ values of intrapseudorotaxanes up to four times in comparison to the empty macrocycle, which evidences that the intercalation of phenazines enables a contact between the macrocycle's anthracenes. This contact opens up an optimal intrapseudorotaxane charge transport pathway that can be electronically tuned with the nature of the intercalated phenazine. We foresee that this approach will pave the way for the preparation of supramolecular and mechanically interlocked semiconductors based on higher acenes and azaacenes, which may combine the properties of acenes with those of molecular devices and machines.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Basque Science Foundation for Science (Ikerbasque), POLYMAT, the University of the Basque Country (Grupo de Investigación GIU17/054 and SGIker), Gobierno de España (Ministerio de Economía y Competitividad and FEDER CTQ2016-77970-R), Gobierno Vasco (BERC programme), the Portuguese Foundation for Science and Technology (IF/00894/2015) and CICECO — Aveiro Institute of Materials, POCI-01-0145-FEDER-007679 (FCT ref. UID/CTM/50011/2013). J. C. thanks support from MINECO/FEDER of Spain Government (CTQ2015-69391-P) and J. L. Z. thanks the Research Central Services (SCAI) of the University of Málaga. This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 664878. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no 722951).Notes and references
- (a) M. Mas-Torrent and C. Rovira, Chem. Rev., 2011, 111, 4833–4856 CrossRef CAS PubMed; (b) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed; (c) I. Yavuz, B. N. Martin, J. Park and K. N. Houk, J. Am. Chem. Soc., 2015, 137, 2856–2866 CrossRef CAS PubMed; (d) R. Pfattner, S. T. Bromley, C. Rovira and M. Mas-Torrent, Adv. Funct. Mater., 2016, 26, 2256–2275 CrossRef CAS; (e) C. Sutton, C. Risko and J.-L. Brédas, Chem. Mater., 2016, 28, 3–16 CrossRef CAS.
- (a) J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed; (b) J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS; (c) U. H. F. Bunz, Chem.–Eur. J., 2009, 15, 6780–6789 CrossRef CAS; (d) U. H. F. Bunz, Pure Appl. Chem., 2010, 82, 953–968 CAS; (e) U. H. F. Bunz, Acc. Chem. Res., 2015, 48, 1676–1686 CrossRef CAS PubMed; (f) Q. Miao, Adv. Mater., 2014, 26, 5541–5549 CrossRef CAS PubMed.
- Y.-F. Lim, Y. Shu, S. R. Parkin, J. E. Anthony and G. G. Malliaras, J. Mater. Chem., 2009, 19, 3049–3056 RSC.
- (a) D. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, Science, 2013, 340, 334–337 CrossRef CAS PubMed; (b) J. Lee, P. Jadhav, P. D. Reusswig, S. R. Yost, N. J. Thompson, D. N. Congreve, E. Hontz, T. Van Voorhis and M. A. Baldo, Acc. Chem. Res., 2013, 46, 1300–1311 CrossRef CAS PubMed.
- (a) X. Chi, D. Li, H. Zhang, Y. Chen, V. Garcia, C. Garcia and T. Siegrist, Org. Electron., 2008, 9, 234–240 CrossRef CAS; (b) Z. Liang, Q. Tang, R. Mao, D. Liu, J. Xu and Q. Miao, Adv. Mater., 2011, 23, 5514–5518 CrossRef CAS PubMed; (c) Z. He, D. Liu, R. Mao, Q. Tang and Q. Miao, Org. Lett., 2012, 14, 1050–1053 CrossRef CAS PubMed; (d) T. He, M. Stolte and F. Würthner, Adv. Mater., 2013, 25, 6951–6955 CrossRef CAS PubMed.
- A. N. Sokolov, T. Friščić and L. R. MacGillivray, J. Am. Chem. Soc., 2006, 128, 2806–2807 CrossRef CAS PubMed.
- Q. Miao, M. Lefenfeld, T.-Q. Nguyen, T. Siegrist, C. Kloc and C. Nuckolls, Adv. Mater., 2005, 17, 407–412 CrossRef CAS.
- Y. Wu, M. Frasconi, D. M. Gardner, P. R. McGonigal, S. T. Schneebeli, M. R. Wasielewski and J. F. Stoddart, Angew. Chem., Int. Ed., 2014, 53, 9476–9481 CrossRef CAS PubMed.
- M. Carini, M. P. Ruiz, I. Usabiaga, J. A. Fernández, E. J. Cocinero, M. Melle-Franco, I. Diez-Perez and A. Mateo-Alonso, Nat. Commun., 2017, 8, 15195 CrossRef CAS PubMed.
- (a) J. J. Gassensmith, E. Arunkumar, L. Barr, J. M. Baumes, K. M. DiVittorio, J. R. Johnson, B. C. Noll and B. D. Smith, J. Am. Chem. Soc., 2007, 129, 15054–15059 CrossRef CAS PubMed; (b) J. J. Gassensmith, J. M. Baumes, J. Eberhard and B. D. Smith, Chem. Commun., 2009, 2517–2519 RSC; (c) W. Liu, A. G. Oliver and B. D. Smith, J. Am. Chem. Soc., 2018, 140, 6810–6813 CrossRef CAS PubMed.
- (a) J. M. Baumes, J. J. Gassensmith, J. Giblin, J.-J. Lee, A. G. White, W. J. Culligan, W. M. Leevy, M. Kuno and B. D. Smith, Nat. Chem., 2010, 2, 1025 CrossRef CAS PubMed; (b) C. Ke, H. Destecroix, M. P. Crump and A. P. Davis, Nat. Chem., 2012, 4, 718 CrossRef CAS PubMed; (c) H. Destecroix, C. M. Renney, T. J. Mooibroek, T. S. Carter, P. F. N. Stewart, M. P. Crump and A. P. Davis, Angew. Chem., Int. Ed., 2015, 54, 2057–2061 CrossRef CAS PubMed.
- (a) A. Saeki, S. Yoshikawa, M. Tsuji, Y. Koizumi, M. Ide, C. Vijayakumar and S. Seki, J. Am. Chem. Soc., 2012, 134, 19035–19042 CrossRef CAS PubMed; (b) A. Saeki, Y. Koizumi, T. Aida and S. Seki, Acc. Chem. Res., 2012, 45, 1193–1202 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the synthesis, characterization and calculations. CCDC 1850666–1850672. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04845b |
| This journal is © The Royal Society of Chemistry 2019 |