
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of halogens in the catalyst transfer polycondensation for π-conjugated polymers†
Shuyang
Ye
a,
Scott M.
Foster
a,
Adam A.
Pollit
a,
Susan
Cheng
a and
Dwight S.
Seferos
 *ab
*ab
aDepartment of Chemistry, University of Toronto, 80 St. George St., Toronto, Ontario M5S 3H6, Canada. E-mail: dwight.seferos@utoronto.ca
bDepartment of Chemical Engineering and Applied Chemistry, University of Toronto, 200 College Street, Toronto, Ontario M5S 3E5, Canada
First published on 19th December 2018
Abstract
Catalyst transfer polycondensation is the only method to prepare π-conjugated polymers in a chain-growth manner, yet several aspects that underlie this polymerization are not fully understood. Here, we investigate the nickel-catalyzed polymerization mechanisms of a series of thiophene monomers bearing different halogen functionalities (Cl, Br, I). We have discovered the significant role that halogens and magnesium salts play in this polymerization. More specifically, the catalyst resting state changes depending on the type of halogenated monomer. For chlorinated monomers a mixture of Ni(II)-dithienyl and dissociated Ni(phosphine) complexes are the resting states, which results in uncontrolled polymerization. For brominated monomers, a Ni(II)-dithienyl complex is the resting state, which leads to controlled polymerization. For iodinated monomers, a Ni(II)-thienyl iodide complex is the resting state, and notable inhibition by magnesium salt by-products is observed. The catalyst resting state changes to a Ni(II)-dithienyl complex when a turbo Grignard reagent (i-PrMgCl·LiCl) is used. These findings are used to guide the design of a new monomer, 2-bromo-3-(2-ethylhexyl)-5-iodotellurophene, which enables the first controlled polymerization of a tellurophene monomer containing a sterically encumbered 2-ethylhexyl side chain. These insights are crucial for deepening the mechanistic understanding of Kumada cross coupling reactions and the controlled synthesis of π-conjugated polymers.
Introduction
Transition metal catalyzed cross coupling is one of the most powerful and versatile tools for C–C bond formation.1 In general, cross coupling occurs between two complementary functional groups: an organometallic group (Mg, Zn, Sn or B) and an organic halide (Cl, Br, I). In polymer chemistry, cross coupling reactions are particularly important for the synthesis of π-conjugated polymers.2 Kumada catalyst transfer polycondensation (KCTP) was first developed based on Kumada-type cross coupling.3 Other examples of catalyst transfer polycondensation utilize Negishi, Suzuki, Stille and Murahashi-type couplings.3b,4 These represent the best routes to well-defined, high molecular weight π-conjugated polymers.5 Important considerations for these polymerizations include catalyst design, monomer structure, and the utilization of additives.5b,6 Since the discovery of the quasi-living synthesis of polythiophenes, considerable effort has been devoted to understanding the underlying reaction mechanisms and expanding the scope of applicable monomers. Nickel(II)diphosphine catalysts, such as 1,2-bis-(diphenylphosphino)propane nickel(II) chloride (Ni(dppp)Cl2) and 1,2-bis-(diphenylphosphino)ethane nickel(II) chloride (Ni(dppe)Cl2), are most frequently used for the controlled synthesis of electron-rich conjugated polymers, and their polymerizations are often used as model systems for studying the mechanism of KCTP.7While considerable progress has been made in identifying new functional groups for cross coupling polymerizations, aromatic halides continue to play an irreplaceable role.2a When selecting the halogen, one must typically consider synthetic accessibility, monomer stability, and relative bond strengths (Ar–Cl > Ar–Br > Ar–I). This has led to the predominance of brominated monomers today due to their ease of synthesis, relatively good stability, and intermediate bond strength of brominated species. Aryl chlorides and aryl iodides also afford their own advantages and see significant use in a few select cases, but their polymerizations are less studied. For example, 2-chloro-5-chloromagnesio-3-hexylthiophene can be polymerized with an N-heterocyclic carbene nickel catalyst, but not with the more commonly used nickel(II)diphosphine catalysts.8 In the early stages of polythiophene development, iodine was the halogen of choice. Wudl and coworkers reported that Kumada coupling polymerization of 2,5-diiodothiophene affords high-quality unsubstituted polythiophenes.9 The first synthesis of stable and soluble poly(3-alkylthiophenes) (P3ATs) was demonstrated by Elsenbaumer and coworkers using 3-alkyl-2,5-diiodothiophene as a monomer precursor.10 However, a comprehensive understanding of the role of the halogens in KCTP remains elusive.
Herein we report the first systematic comparison of the polymerizations of thiophenes bearing different halogens (Cl, Br and I). We find that reaction kinetics and chain-growth behaviour vary significantly for different halogens with the polymerization influenced in previously unreported and unexpected ways. In situ31P NMR shows that the catalyst resting state is dependent on the halogen identity and DFT calculations indicate that the activation energy of transmetalation increases from Cl to Br to I. These results provide a critical understanding of the role of halogens and the inhibition effect of magnesium salts in KCTP. The significant mechanistic influence of LiCl as an additive in polymerizing an iodinated monomer is also studied. All of our experimental evidence shows that the halides play a noninnocent role in the context of KCTP. Finally, we demonstrate how these new insights can be used to guide future monomer design for obtaining higher quality and more precise π-conjugated polymers.
Results and discussion
The monomers chosen for this study are 2-chloro-5-iodo-3-(3,7-dimethyloctyl)thiophene, 2-bromo-5-iodo-3-(3,7-dimethyloctyl)thiophene, 2,5-dibromo-3-(3,7-dimethyloctyl)thiophene, and 2,5-diiodo-3-(3,7-dimethyloctyl)thiophene. The branched 3,7-dimethyloctyl (DMO) side chain is used to ensure that all resulting polymers are sufficiently soluble in common organic solvents. Electrophilic substitution reactions with the corresponding N-halosuccinimides were used to synthesize all dihalogenated thiophene derivatives (Scheme S1, ESI†). All monomers were characterized using 1H NMR, 13C NMR, and mass spectroscopy. Geminal coupling (2JH–H = −14.08 Hz) is observed for the protons at the 1-position of the DMO side chain due to the chiral centre at the 3-position.11 For example, in 2,5-dibromo-3-(3,7-dimethyloctyl)thiophene, the two methylene protons on the side chain adjacent to the heterocycle are split into two large doublets of doublets flanked by a smaller doublet of doublets on either side (Fig. S1, ESI†).KCTP involves two principal synthetic manipulations: (1) generation of the active monomer by a Grignard metathesis step;12 and (2) initiation and polymerization by application of the catalyst. In our current studies, we strived to eliminate the influence of the Grignard metathesis reactions on the following polymerizations. Generally, monomer activation is carried out by treating a dihalogenated monomer with 0.98 equivalent of i-PrMgCl.13 For 2-chloro-5-iodo-3-(3,7-dimethyloctyl)thiophene and 2-bromo-5-iodo-3-(3,7-dimethyloctyl)thiophene Grignard activation will occur exclusively at the 5-position [denoted as XMg-Th-Cl and XMg-Th-Br (I/Br), respectively]. For 2,5-dibromo-3-(3,7-dimethyloctyl)thiophene and 2,5-diiodo-3-(3,7-dimethyloctyl)thiophene metathesis occurs primarily at the 5-position [∼80%, denoted as XMg-Th-I and XMg-Th-Br (Br/Br), respectively] with a small amount 2-position (∼20%), however this minor species is not a polymerizable monomer due to steric constraints.14
When designing studies to test the influence of halogens on KCTP the catalyst must be carefully considered. The Ni(dppe)Cl2 pre-catalyst is known to be sparingly soluble in THF, resulting in sluggish initiation that could potentially complicate kinetic studies. To address this issue, pre-initiation with five equivalents of monomer7a and utilization of a THF-soluble o-tolyl functionalized Ni(o-tolyl)(dppe)Cl external initiator15 were both tested as alternative rapid initiation routes. Pre-initiation was found to yield pronounced tailing and several sets of end groups in the resulting polymers. Conversely, the use of an external initiator led to polymers with a unimodal molecular weight distribution and well-defined end groups (Fig. S2, ESI†). Since the Ni(o-tolyl)(dppe)Cl external initiator leads to better quality polymers it was used in all of the subsequent polymerization studies.
The polymerization of XMg-Th-Br (I/Br) proceeds rapidly at room temperature after addition of the external initiator. All evidence supports a controlled chain-growth mechanism. Notably the monomer consumption reaches 80% within one minute (Fig. 1). Crude polymer samples were analyzed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF/MS).16 All aliquots collected during the polymerization have a unimodal size distribution and shift entirely to higher molecular weight regions (Fig. S3, ESI†). The degree of polymerization (DP) increases linearly as a function of monomer conversion without deviation (Fig. S4, ESI†). Analogous to the brominated monomer, the polymerization of XMg-Th-Cl is also complete within minutes. However, low molecular weight and broad dispersity (Fig. S5, ESI†) both indicate a non-living polymerization when changing the halogen from Br to Cl.7d,8
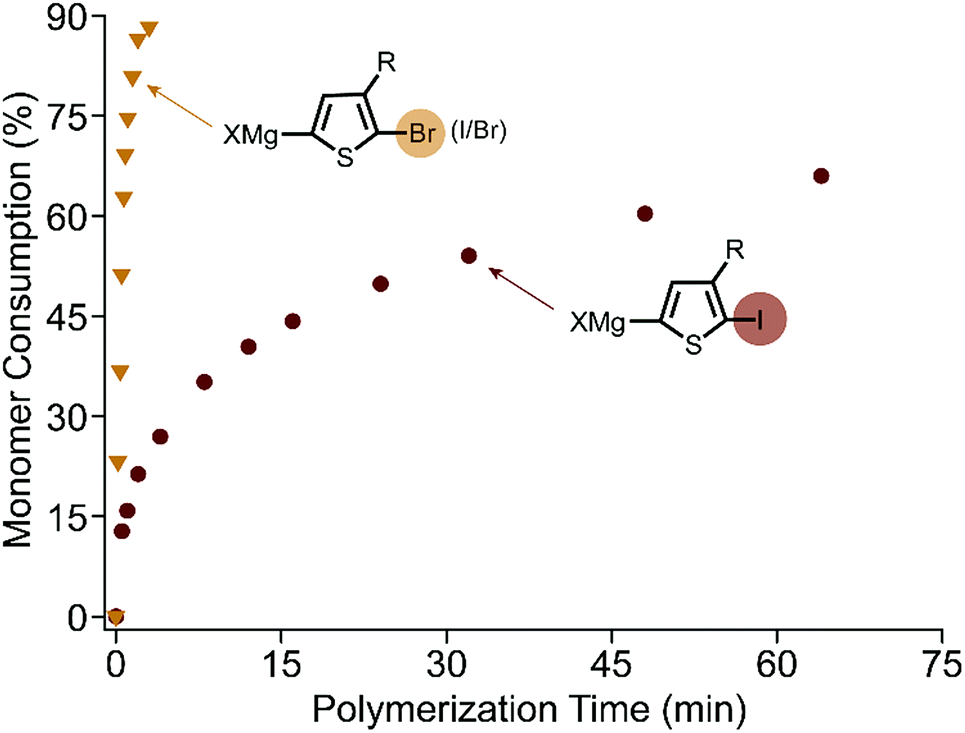 | ||
| Fig. 1 Kinetic plots of monomer consumption during polymerization of XMg-Th-Br (I/Br) and XMg-Th-I. At 2.5% catalyst loading, 0.1 M monomer concentration. | ||
In contrast, XMg-Th-I has vastly slower polymerization kinetics. The DP increases linearly with monomer consumption (Fig. S6, ESI†), consistent with chain-growth character. Surprisingly, the polymerization proceeds quickly at the start but then slows down considerably as it progresses (Fig. 1). Overall, the polymerization of XMg-Th-I is much slower than both XMg-Th-Br (I/Br) and XMg-Th-Cl, with only 60% of the monomer being consumed after 100 minutes. Considering that kinetic differences could arise from XMg-Th-Br (I/Br) and XMg-Th-Cl both exclusively metathesizing on one side of the asymmetric monomer, while a mixture of activated regioisomers are possible for the diiodinated monomer metathesis, we further investigated the polymerization of XMg-Th-Br (Br/Br) to directly compare between halogen-symmetric monomers. The polymerization rate measured for XMg-Th-Br (Br/Br) was comparable to that using XMg-Th-Br (I/Br) (Fig. S7, ESI†). This indicates that the halogen at 2-position of the activated monomer is accountable for the difference in polymerization kinetics, and not the monomer activation process or the presence of an unreactive regioisomer.
The KCTP catalytic cycle involves four steps: transmetalation (TM), reductive elimination (RE), ring walking (RW) and oxidative addition (OA) (Scheme 1).6c,16 After the addition of Ni(o-tolyl)(dppe)Cl external initiator, one active monomer is transmetalated to the Ni centre forming the Ni(II)-thienyl-(o-tolyl) complex 2. Reductive elimination (RE) occurs to form a new carbon–carbon bond and the Ni catalyst remains associated as Ni(0)-π complex 3. To achieve a chain-growth mechanism, the Ni(0) catalyst migrates intramolecularly to the terminal carbon–halogen bond followed by oxidative addition (OA) affording Ni(II)-thienyl halide complex 1. The four consecutive steps of the catalytic cycle continue until the active monomers are consumed and the catalyst then rests as the Ni(II)-thienyl halide complex 1.
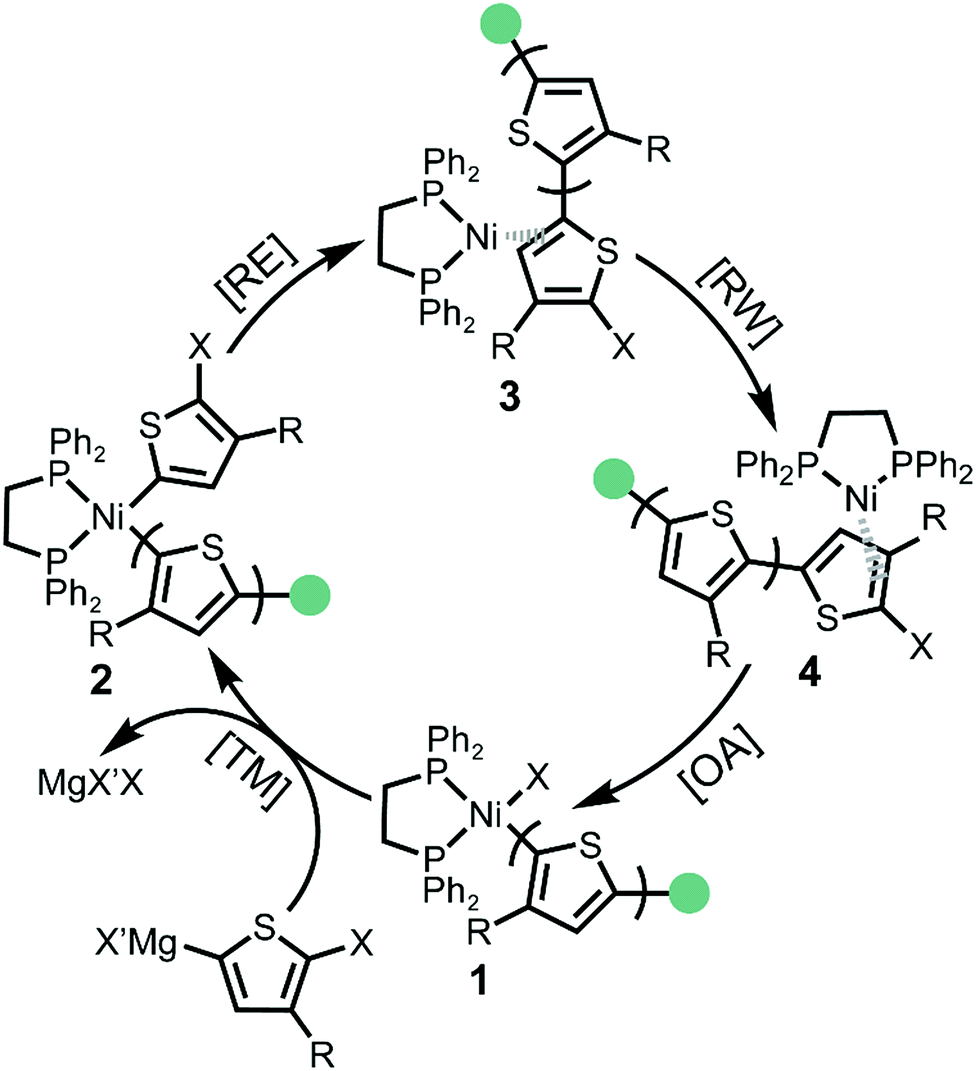 | ||
| Scheme 1 Proposed mechanism for Kumada catalyst transfer polycondensation. [OA]: oxidative addition; [TM]: transmetalation, [RE]: reductive elimination; [RW]: ring walking. The green circle represents an o-tolyl group. | ||
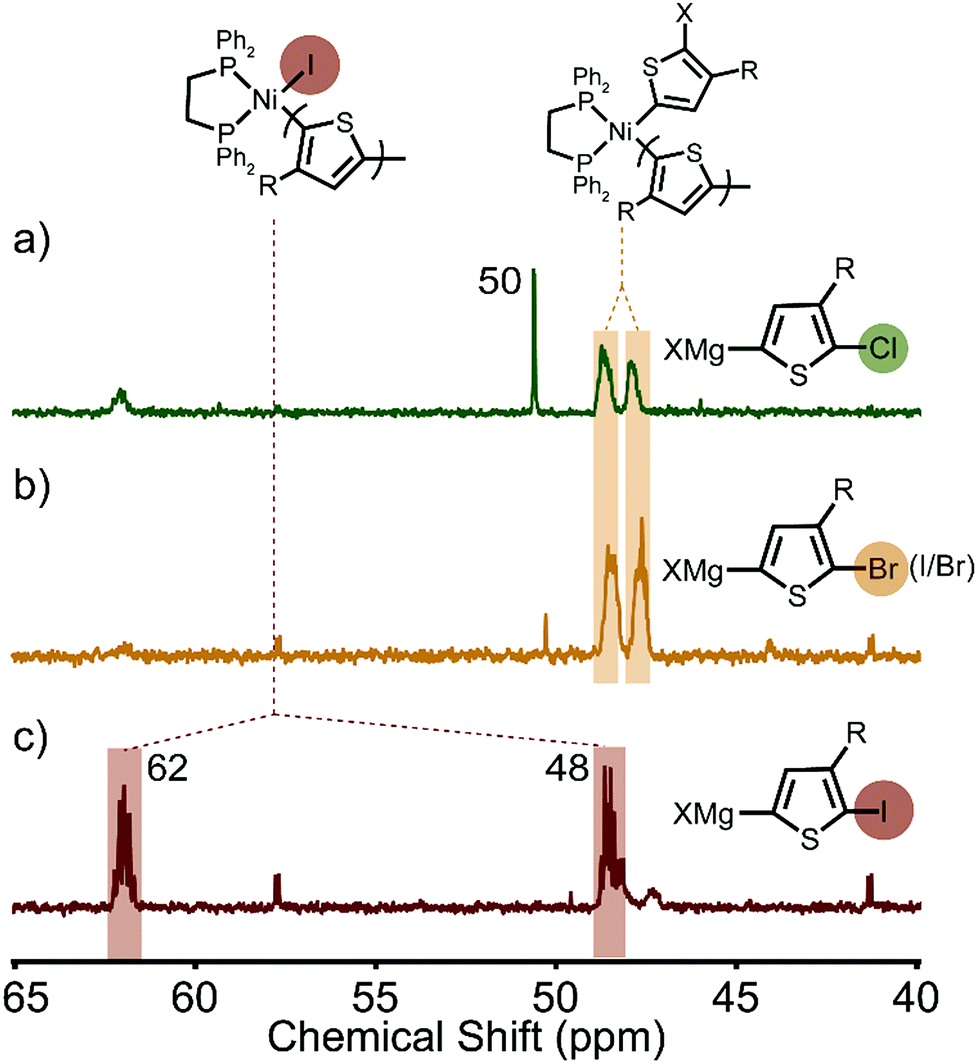
After the polymerizations of XMg-Th-Cl and XMg-Th-I, two peaks are observed by 31P NMR in a region anticipated for the Ni(II)-thienyl halide complex 1 (Fig. 2). Surprisingly, we find that the spectra of the post-polymerization complexes are identical in these two cases (48 and 62 ppm, Fig. 2a and c). After the polymerizations of XMg-Th-Br (I/Br) these two peaks are also observed, along with two additional up-field shifted peaks (43 and 59 ppm, Fig. 2b). We assign the signals at 48 and 62 ppm to Ni(II)-thienyl iodide and the signals at 43 and 59 ppm to Ni(II)-thienyl bromide.7a Further support of this assignment is provided when magnesium iodide was added to the polymerization of XMg-Th-Br (I/Br) after collecting the post-polymerization spectrum. This experiment causes two of the original peaks (43 and 59 ppm) to disappear and two peaks with the same chemical shifts as those found after the polymerizations of XMg-Th-Cl and XMg-Th-I (48 and 62 ppm) to strengthen (Fig. 2d).
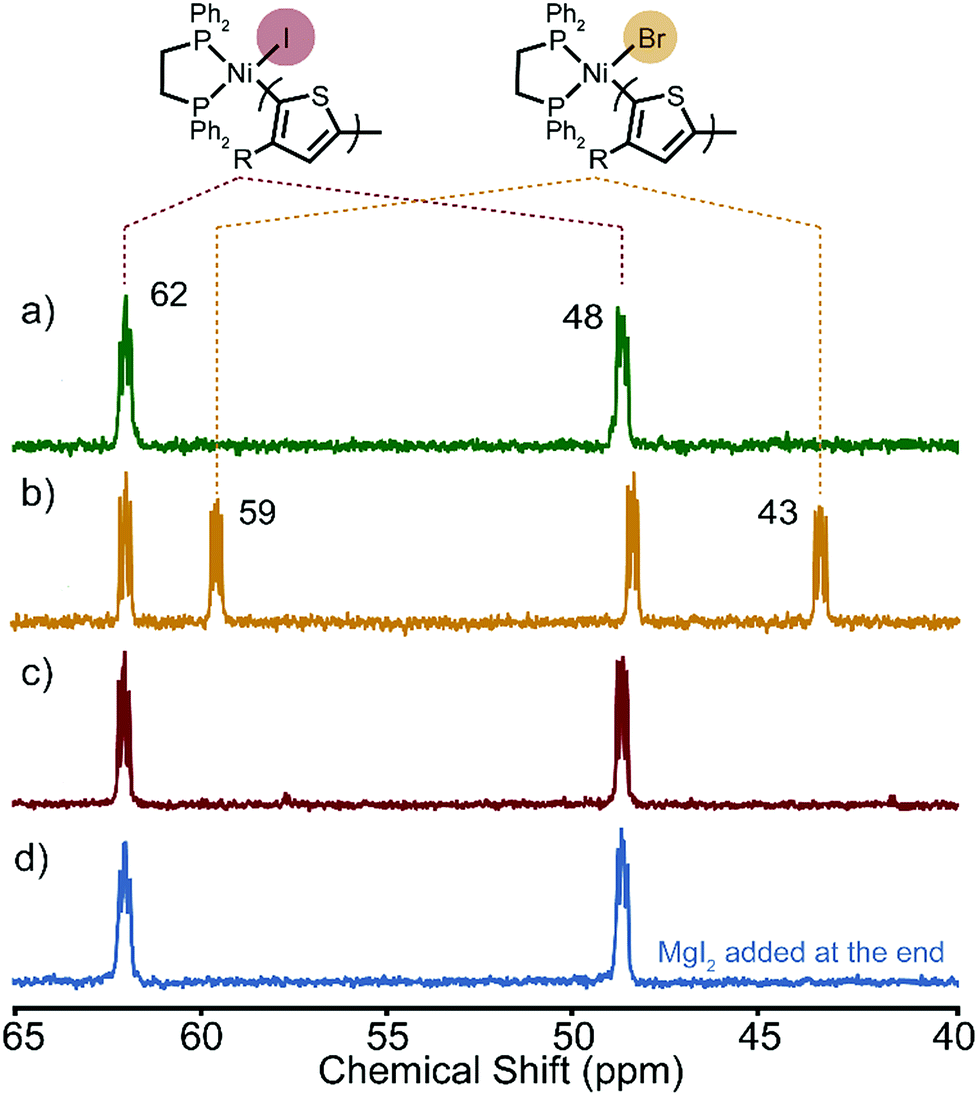 | ||
| Fig. 2 31P NMR spectra of the resting state(s) after the polymerization of (a) XMg-Th-Cl, (b) XMg-Th-Br (I/Br), (c) XMg-Th-I, and (d) XMg-Th-Br (I/Br) with MgI2 added at the end. | ||
The unexpected presence of Ni(II)-thienyl iodide after polymerizing XMg-Th-Cl or XMg-Th-Br (I/Br) is a result of halogen exchange (HE) between the original Ni(II)-thienyl chloride or Ni(II)-thienyl bromide and an iodide source, formed through either the decomposition of isopropyl iodide14 or the nucleophilic substitution reaction between i-PrMgCl and isopropyl iodide in the course of the metathesis. This observation indicates the preferential formation of Ni(II)-thienyl iodide when iodide is present (Fig. 2). This observation seems to contradict the halide affinity for Ni(II) diphosphine complexes (Cl > Br > I).17 However, it should be noted that these halides are counterions of Mg2+. For instance, when only free halides are considered, density functional theory (DFT) calculations show that the equilibrium favors the formation of Ni(II)-thienyl chloride by 93 kJ mol−1. When magnesium salts are considered instead, the equilibrium favors the formation of Ni(II)-thienyl iodide by 17 kJ mol−1 (Scheme 2). This tendency is likely due to the much stronger interactions between Mg2+ (hard acid) and chloride (hard base).18
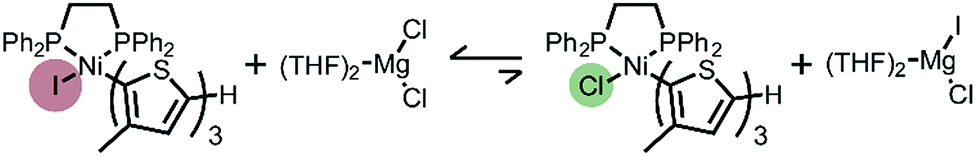 | ||
| Scheme 2 Halogen exchange equilibrium between complex 1 and magnesium salts. | ||
Distinct spectra are also observed during in situ31P NMR spectroscopic studies carried out on the polymerizations. For reference, the 31P NMR spectrum of the external initiator has two doublets centred at 32 and 53 ppm (Jpp = 40 Hz, Fig. S8, ESI†). Once mixed with a monomer solution the 31P NMR spectrum rapidly changes depending on the type of activated monomer. We first evaluated XMg-Th-Br (I/Br), where two proximate, broad multiplets (47–48 ppm) were observed to be indicative of two phosphorus nuclei in similar chemical environments (Fig. 3b). We assign the catalyst resting state to Ni(II)-dithienyl complex 2 (Scheme 1), which is consistent with a previous report by McNeil and coworkers that RE is the rate-determining step for this polymerization.7a
 | ||
| Fig. 3 31P NMR spectra of the resting state(s) during the polymerization of (a) XMg-Th-Cl, (b) XMg-Th-Br (I/Br), and (c) XMg-Th-I. | ||
In the case of the XMg-Th-Cl polymerization, the Ni(II)-dithienyl complex 2 is observed, however an additional distinct and sharp singlet appears around 50 ppm (Fig. 3a), which is indicative of phosphorus nuclei in an identical chemical environment. The observed singlet does not match any intermediates in the catalytic cycle (complex 1–4 in Scheme 1) suggesting that some of the active catalyst is diffusing away from the propagating polymer chain, likely due to the difficult OA or other side reactions.7a,17 This is consistent with a mixture of polymers with different end-group compositions being observed in this polymerization (Fig. S9, ESI†).15b,19 We therefore conclude that the catalyst rests as dissociated Ni(phosphine) complex and Ni(II)-dithienyl complex 2 in the polymerization of XMg-Th-Cl.
The greatest mixture of intermediates is observed when polymerizing XMg-Th-I. The major signals (48 and 62 ppm) are assigned to Ni(II)-thienyl iodide complex 1 (Scheme 1, Fig. 3c), and match the signals observed after polymerization. In addition, one set of minor signals is attributed to the formation of Ni(II)-(o-tolyl) iodide (41 and 57 ppm), which is confirmed experimentally by a separately prepared Ni complex (Fig. S8, ESI†) and can only be formed by HE between the added external initiator and an iodide source. We can therefore conclude that the catalyst rests mostly as Ni(II)-thienyl iodide complex 1 in the polymerization of XMg-Th-I based on its stronger NMR signals.
Thus far, we have established the halogen-dependence of catalyst resting states by 31P NMR, and that TM becomes more difficult when iodide is used. This raises the question of how halogen atoms influence TM.20 Activation energies (AE) for TM of a fully dissociated monomer calculated by DFT increase from 44 kJ mol−1 (Cl) to 59 kJ mol−1 (Br) to 79 kJ mol−1 (I) indicating the greater difficulty of TM going from Cl to Br to I (Fig. 4). There are two possible explanations for this increase in AE. First, this trend can be interpreted as a result of increased steric congestion around the Ni centre. Steric mapping shows that the percent buried volume (%VBur) around the Ni centre in complex 1 increases moving from Cl to Br to I (Fig. S10, ESI†).21 Alternatively, weaker Mg–I interactions serve to further increase the AE for I (soft base) compared to Br and Cl.
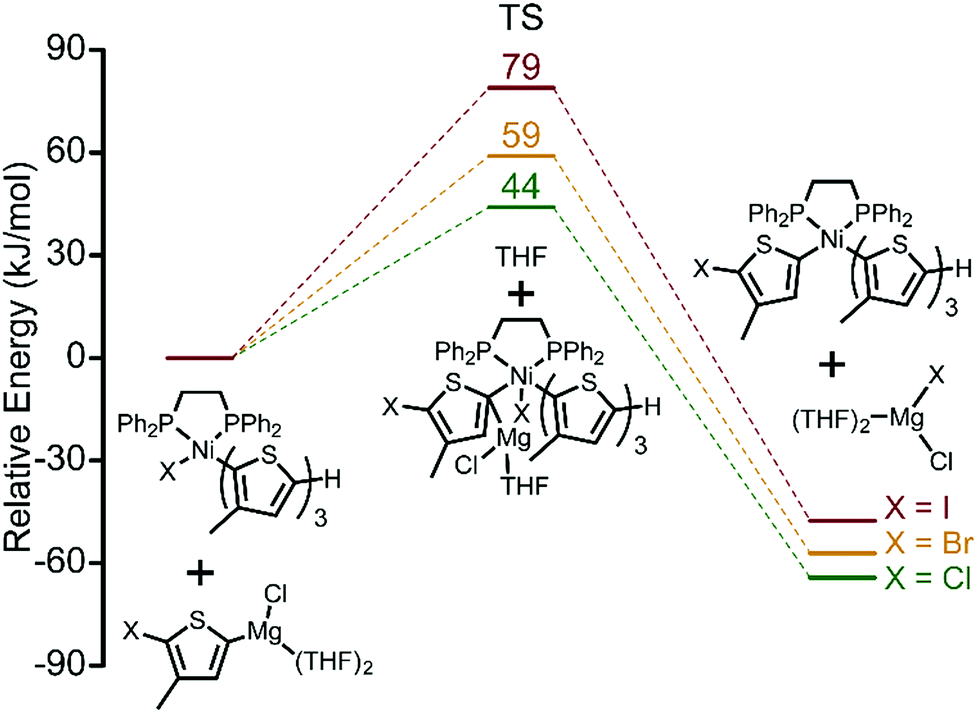 | ||
| Fig. 4 DFT calculations of transmetalation transition states for the three monomers, XMg-Th-I, XMg-Th-Br, and XMg-Th-Cl. | ||
Significant product inhibition is also observed in the iodothiophene polymerization (Fig. 1). To investigate these effects, a series of polymerizations were carried out with different additives. The polymerization decelerates when adding increasing amounts of magnesium chloride (Fig. S11, ESI†) and accelerates when adding an increasing amount of o-tolyl magnesium chloride (Fig. S12, ESI†). Due to the inhibitive effect of adding of MgCl2 to the polymerization, we can rule out the influence of a Schlenk equilibrium which would be expected to increase the percentage of Grignard form.22 We therefore hypothesize that the produced magnesium salts, which are a product of TM, form a more stable complex with the Grignard reagents which decreases the reactivity of the activated monomers.23 As the polymerization proceeds, the concentration of magnesium salt builds up and decelerates the polymerization to a large extent. These different features over the course of the polymerization are also observed when polymerizing 2,5-diiodo-3-(3,7-dimethyloctyl)selenophene (Fig. S13, ESI†) and 2,5-diiodo-3-(3-ethylheptyl)tellurophene24 showing the general nature of these findings. In contrast, when o-tolyl magnesium chloride serves as a sacrificial reagent to coordinate with produced magnesium salts in solution, the polymerization rate increases. This is the first study to emphasize the role of product inhibition in KCTP, which is consistent with the distinct polymerization kinetics observed.
This suggests that the polymerization of XMg-Th-I is very sensitive to product complexation in solution. Previous reports have demonstrated that the actual structure of Grignard reagents is far more complicated than the generic formula R-MgX indicates and LiCl is believed to de-aggregate these complex aggregates.25In situ31P NMR shows that when a turbo Grignard (i-PrMgCl·LiCl) is used for iodinated monomer metathesis, the catalyst resting state changes from Ni(II)-thienyl iodide complex 1 to Ni(II)-dithienyl complex 2 (Fig. S14, ESI†). McNeil and coworkers have previously reported that LiCl has no influence on the catalyst resting state in polymerizations of XMg-Th-Br (Br/Br).7a In contrast, here we demonstrate the unique mechanistic influence of LiCl as an additive in KCTP to accelerate the TM of an iodinated monomer, where the catalyst otherwise rests as pre-TM complex 1. Combined with in situ31P NMR studies and DFT calculations, this further proves that the distinct kinetic features originate from the iodine atom and Grignard aggregation, together with magnesium by-product inhibition.
We previously reported that 3-(2-ethylhexyl)-5-chloromagnesio-2-iodotellurophene polymerizes very slowly (requiring over 12 hours to complete), and is accompanied by early chain termination and re-initiation under typical KCTP conditions.24 It is well known that branched side chains increase monomer bulkiness and slow down the overall polymerization.26 The present study shows that iodine also contributes to slowing down the polymerization to a large extent. This led us to wonder whether the use of bromine instead of iodine would improve the polymerization of 2-ethylhexyltellurophene. We therefore synthesized the asymmetric monomer 2-bromo-3-(2-ethylhexyl)-5-iodotellurophene, (Scheme S2, ESI†). A test polymerization targeting DP = 30 was attempted and found to give a unimodal molecular distribution, low polydispersity (Đ = 1.2) and high yield (85%) (Fig. 5a). The MALDI-TOF/MS spectrum shows only one set of end groups corresponding to o-tolyl and H (Fig. 5b). All evidence strongly supports a controlled chain growth mechanism. This successful polymerization emphasizes the importance of the halogen functionality, particularly for monomers with bulky side chains.
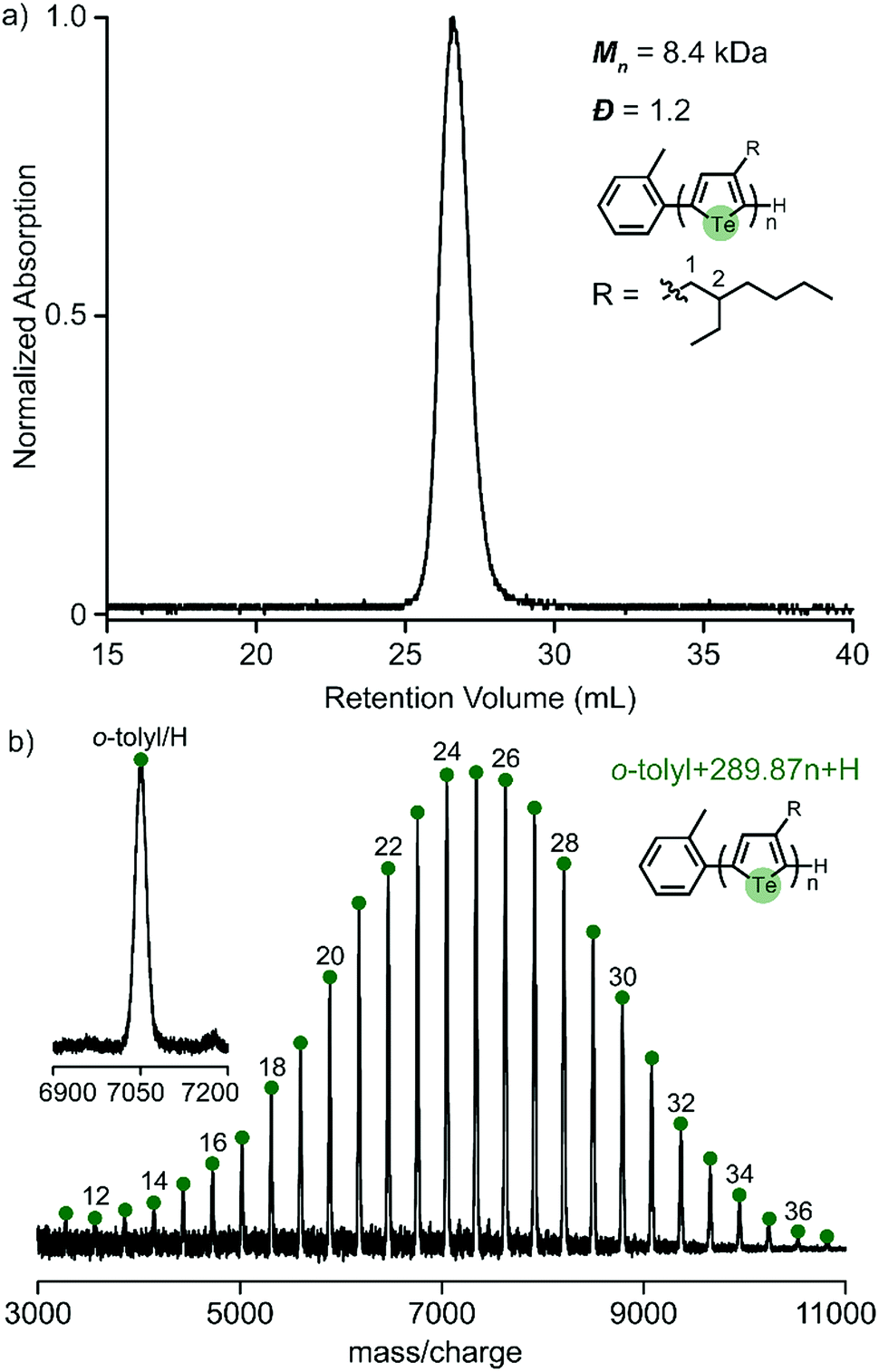 | ||
| Fig. 5 (a) GPC elution profile and (b) MALDI-TOF/MS spectra of poly(3-ethylhexyltellurophene) synthesized from a newly designed monomer. The inset shows the observed and predicted mass/charge values for a labelled peak (found: 7049.41; prediction: 7049.02). | ||
Conclusions
In summary, we have discovered the important role that halogens and magnesium salts play in Kumada catalyst transfer polycondensation. Non-living behaviour is observed in the polymerization of a chlorinated monomer, while both brominated and iodinated monomers are polymerized in a controlled manner but with distinct kinetics. In situ31P NMR reveals that the catalyst resting state changes from a Ni(II)-dithienyl complex to a Ni(II)-thienyl iodide complex when Br is replaced with I. The addition of LiCl leads to a change in catalyst resting state when polymerizing the iodinated monomer, highlighting the significant mechanistic influence of LiCl in KCTP. Based on these studies, a new tellurophene monomer with a bulky side chain is designed and demonstrated to undergo controlled polymerization for the first time. These insights are likely not limited to controlled polymerizations utilizing cross-coupling reactions, but are potentially more widely applicable to Kumada cross coupling reactions in general.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. S. S. is grateful from support from the NSERC of Canada, The Canadian foundation for Innovation, the Ontario research fund and the Ontario Centres of Excellence. S. Y. is grateful for the support of Colin Hahnemann Bayley Fellowship. This research was enabled in part by support provided by SHARCNET and Compute Canada.Notes and references
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- (a) M. A. Baker, C.-H. Tsai and K. J. T. Noonan, Chem.–Eur. J., 2018, 24, 13078–13088 CrossRef CAS PubMed; (b) R. Grisorio and G. P. Suranna, Polym. Chem., 2015, 6, 7781–7795 RSC; (c) K. Okamoto and C. K. Luscombe, Polym. Chem., 2011, 2, 2424–2434 RSC; (d) M. Al-Hashimi, Y. Han, J. Smith, H. S. Bazzi, S. Y. A. Alqaradawi, S. E. Watkins, T. D. Anthopoulos and M. Heeney, Chem. Sci., 2016, 7, 1093–1099 RSC.
- (a) A. Yokoyama, R. Miyakoshi and T. Yokozawa, Macromolecules, 2004, 37, 1169–1171 CrossRef CAS; (b) E. E. Sheina, J. Liu, M. C. Iovu, D. W. Laird and R. D. McCullough, Macromolecules, 2004, 37, 3526–3528 CrossRef CAS.
- (a) K.-B. Seo, I.-H. Lee, J. Lee, I. Choi and T.-L. Choi, J. Am. Chem. Soc., 2018, 140, 4335–4343 CrossRef CAS PubMed; (b) K. Fuji, S. Tamba, K. Shono, A. Sugie and A. Mori, J. Am. Chem. Soc., 2013, 135, 12208–12211 CrossRef CAS PubMed; (c) Y. Qiu, J. Mohin, C.-H. Tsai, S. Tristram-Nagle, R. R. Gil, T. Kowalewski and K. J. T. Noonan, Macromol. Rapid Commun., 2015, 36, 840–844 CrossRef CAS.
- (a) T. Yokozawa and Y. Ohta, Chem. Rev., 2016, 116, 1950–1968 CrossRef CAS PubMed; (b) A. K. Leone and A. J. McNeil, Acc. Chem. Res., 2016, 49, 2822–2831 CrossRef CAS PubMed.
- (a) C. R. Bridges, T. M. McCormick, G. L. Gibson, J. Hollinger and D. S. Seferos, J. Am. Chem. Soc., 2013, 135, 13212–13219 CrossRef CAS PubMed; (b) V. Senkovskyy, R. Tkachov, H. Komber, M. Sommer, M. Heuken, B. Voit, W. T. S. Huck, V. Kataev, A. Petr and A. Kiriy, J. Am. Chem. Soc., 2011, 133, 19966–19970 CrossRef CAS PubMed; (c) Z. J. Bryan and A. J. McNeil, Macromolecules, 2013, 46, 8395–8405 CrossRef CAS; (d) M. C. Stefan, A. E. Javier, I. Osaka and R. D. McCullough, Macromolecules, 2008, 42, 30–32 CrossRef; (e) A. Sui, X. Shi, Y. Wang, Y. Geng and F. Wang, Polym. Chem., 2015, 6, 4819–4827 RSC; (f) M. Heeney, W. Zhang, D. J. Crouch, M. L. Chabinyc, S. Gordeyev, R. Hamilton, S. J. Higgins, I. McCulloch, P. J. Skabara, D. Sparrowe and S. Tierney, Chem. Commun., 2007, 5061–5063 RSC; (g) S. R. Lee, Z. J. Bryan, A. M. Wagner and A. J. McNeil, Chem. Sci., 2012, 3, 1562 RSC.
- (a) E. L. Lanni and A. J. McNeil, J. Am. Chem. Soc., 2009, 131, 16573–16579 CrossRef CAS PubMed; (b) E. L. Lanni and A. J. McNeil, Macromolecules, 2010, 43, 8039–8044 CrossRef CAS; (c) R. Tkachov, V. Senkovskyy, H. Komber, J.-U. Sommer and A. Kiriy, J. Am. Chem. Soc., 2010, 132, 7803–7810 CrossRef CAS; (d) T. Hardeman, J. De Becker and G. Koeckelberghs, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3701–3706 CrossRef CAS.
- S. Tamba, K. Shono, A. Sugie and A. Mori, J. Am. Chem. Soc., 2011, 133, 9700–9703 CrossRef CAS PubMed.
- M. Kobayashi, J. Chen, T. C. Chung, F. Moraes, A. J. Heeger and F. Wudl, Synth. Met., 1984, 9, 77–86 CrossRef CAS.
- R. L. Elsenbaumer, K. Y. Jen and R. Oboodi, Synth. Met., 1986, 15, 169–174 CrossRef CAS.
- A. Grisin, S. Oliver, M. D. Ganton, J. Bacsa and P. A. Evans, Chem. Commun., 2015, 51, 15681–15684 RSC.
- R. H. Lohwasser and M. Thelakkat, Macromolecules, 2011, 44, 3388–3397 CrossRef CAS.
- R. Miyakoshi, A. Yokoyama and T. Yokozawa, J. Am. Chem. Soc., 2005, 127, 17542–17547 CrossRef CAS PubMed.
- R. Tkachov, V. Senkovskyy, H. Komber and A. Kiriy, Macromolecules, 2011, 44, 2006–2015 CrossRef CAS.
- (a) E. A. Standley, S. J. Smith, P. Muller and T. F. Jamison, Organometallics, 2014, 33, 2012–2018 CrossRef CAS; (b) H. A. Bronstein and C. K. Luscombe, J. Am. Chem. Soc., 2009, 131, 12894–12895 CrossRef CAS PubMed.
- A. K. Leone, P. K. Goldberg and A. J. McNeil, J. Am. Chem. Soc., 2018, 140, 7846–7850 CrossRef CAS PubMed.
- P. S. Jarrett and P. J. Sadler, Inorg. Chem., 1991, 30, 2098–2104 CrossRef CAS.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 581–587 CrossRef CAS.
- A. A. Pollit, N. K. Obhi, A. J. Lough and D. S. Seferos, Polym. Chem., 2017, 8, 4108–4113 RSC.
- A. K. Vitek, A. K. Leone, A. J. McNeil and P. M. Zimmerman, ACS Catal., 2018, 8, 3655–3666 CrossRef CAS.
- L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef CAS.
- D. Seyferth, Organometallics, 2009, 28, 1598–1605 CrossRef CAS.
- H. R. Hays, J. Org. Chem., 1968, 33, 4201–4205 CrossRef CAS.
- S. Ye, M. Steube, E. I. Carrera and D. S. Seferos, Macromolecules, 2016, 49, 1704–1711 CrossRef CAS.
- R. L.-Y. Bao, R. Zhao and L. Shi, Chem. Commun., 2015, 51, 6884–6900 RSC.
- J. Hollinger and D. S. Seferos, Macromolecules, 2014, 47, 5002–5009 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04808h |
| This journal is © The Royal Society of Chemistry 2019 |