
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle Ru atoms with precise coordination on a monolayer layered double hydroxide for efficient electrooxidation catalysis†
Zelin
Wang‡
a,
Si-Min
Xu‡
a,
Yanqi
Xu
a,
Ling
Tan
a,
Xian
Wang
a,
Yufei
Zhao
 *a,
Haohong
Duan
*b and
Yu-Fei
Song
*ac
*a,
Haohong
Duan
*b and
Yu-Fei
Song
*ac
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: songyufei@hotmail.com; songyf@mail.buct.edu.cn; zhaoyufei@mail.buct.edu.cn
bDepartment of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: haohong.duan@chem.ox.ac.uk
cBeijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China
First published on 20th November 2018
Abstract
The catalytic properties of single-atom catalysts (SACs) can be influenced largely by the chemical environment exerted by supports. Therefore, a precise location of the single atom is essential for understanding of the reaction mechanism and design of novel SACs. However, the preparation of SACs with a precise location remains a great challenge. Herein, we report a facile one-step method to synthesize single Ru atoms supported on a monolayer NiFe-layered double hydroxide (mono-NiFe). Detailed studies demonstrate that the single Ru atoms are not dispersed randomly in the LDH structure, but are uniquely located on the top of the Fe-metal atom of mono-NiFe via three oxygen atoms. Furthermore, these SACs prove to be highly active for the hydrazine electrooxidation reaction. Density functional theory calculations demonstrate that the single Ru atoms can stabilize the hydrazine electrooxidation intermediate with one unpaired electron (*N2H3 and *N2H), thus lowering the reaction barrier for the rate-determining step. Moreover, the loading amount of single Ru atoms with a precise location can even go up to 7.0 wt% without any aggregation.
Introduction
Single-atom catalysts (SACs) have attracted great attention1–5 because of their unique catalytic properties in heterogeneous reactions such as CO oxidation,6–8 hydrogen production,9 water–gas-shift reactions,10,11 electrocatalysis,12–14etc. Analogous to coordinated ligands in homogenous single-site catalysts, stabilized supports with different anchoring sites along with their numbers and geometries play a significant role in the catalytic properties via strong metal-support interaction (SMSI) in heterogeneous SACs.15 As a result, the preparation of SACs with a precise location to understand the reaction mechanism and further guide the rational design of novel catalysts to enhance the activity is highly desirable.Oxides (hydroxides) show unique structural features such as redox,16 acidic17 and alkaline18 properties which benefit many catalytic reactions, representing important supports for SACs. To date, SACs have been successfully achieved on oxides (hydroxides) such as FeOx,19 ZnO,20 TiO2,3 ZrO2,21 and SiO2.22 Layered double hydroxides (LDHs) are a large family of two-dimensional (2D) brucite Mg(OH)2-like layered anionic clay, with the general formula of [M2+1−xM3+x(OH)2]x+(An−)x/n·mH2O,23–25 where the divalent metal (M2+, such as Mg2+, Zn2+, Ni2+, etc.) in the LDH layer can be well replaced by trivalent ions (M3+, such as Fe3+, V3+, Co3+, Al3+, etc.).26–28 Compared with other oxides (hydroxides), LDHs show the advantage of highly tunable chemical composition,29–31 and thereby can be used as catalysts or precursors in electrocatalysis, etc.32–39 Due to the electrostatic repulsion, M3+ ions are highly dispersed by M2+ ions, leading to the formation of an ordered distribution of both the M2+ and M3+ ions in the LDH framework (Fig. S1†).40 Inspired by the structural feature of LDHs, we envision that single metal atoms may be inserted/anchored orderly on LDHs that inherit the atomic dispersion feature of the LDH structure. Nevertheless, it remains challenging to synthesize single metal atoms on LDHs due to the large tendency of single atom aggregation to form large particles. Very recently, Zhang et al. reported that single Au atoms supported on NiFe–LDH were obtained by an electrodeposition method and the single Au atoms are proposed to be adsorbed upon oxygen atoms, giving excellent oxygen evolution reaction (OER) activity.41 However, the precise location of single metal atoms, for example on the layer or in the layer, bonding to Ni or Fe atoms via oxygen atoms still remains elusive. Meanwhile, the limitation of a high loading amount of single metal atoms remains a great challenge for their further applications, and only a few papers have achieved it so far.3,42
Herein, we report a facile one-step coprecipitation method to synthesize single Ru atoms supported on monolayer NiFe–LDH (denoted as Ru1/mono-NiFe). Detailed characterization and density functional theory (DFT) calculations verify that single Ru atoms are uniquely anchored on the top of the Fe atoms via coordination with three oxygen atoms (Scheme 1). Compared with monolayer NiFe–LDH, the Ru1/mono-NiFe catalyst exhibits high catalytic activity for the hydrazine electrooxidation reaction. DFT calculations suggest that the excellent activity is attributed to the fact that the single Ru atoms can stabilize the hydrazine electrooxidation intermediates with one unpaired electron (*N2H3 and *N2H) and decrease the band gap energy (Eg). We further demonstrate that the loading amount of the single Ru atoms on the LDH with a precise location can go up to 7.0 wt%, and LDHs with different compositions (MgAl–LDH and NiAl–LDH) can be applied as supports for SACs due to the advantage of the unique LDH structure.
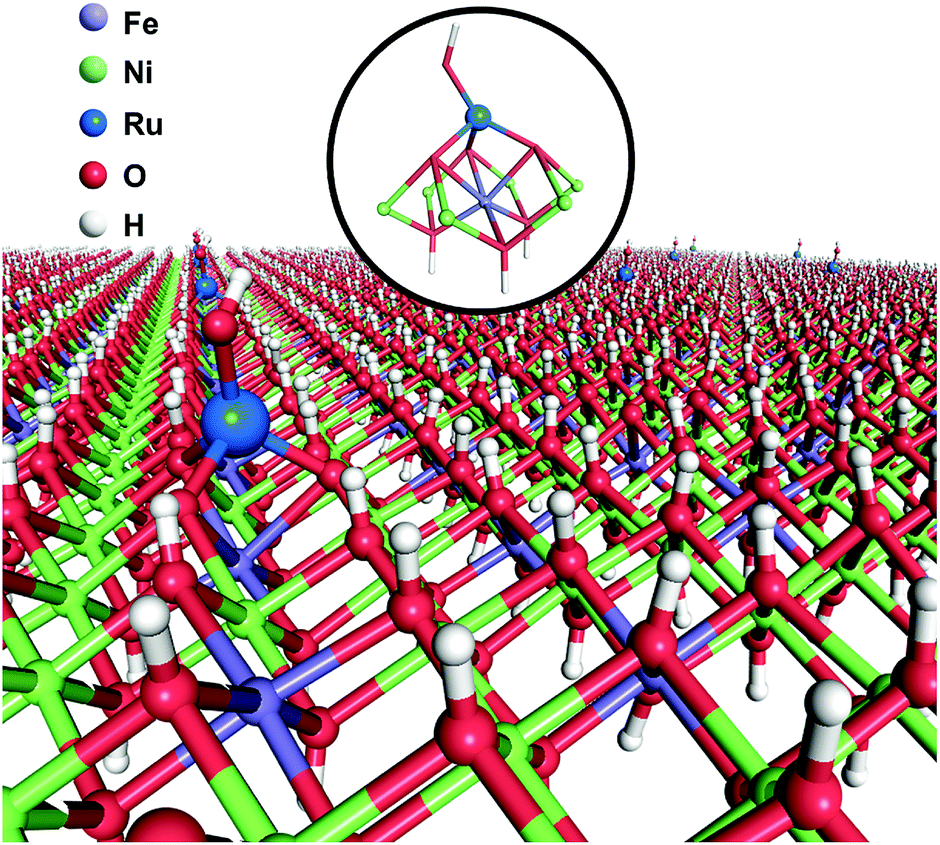 | ||
| Scheme 1 Schematic illustration of Ru1/mono-NiFe as obtained using a one-step coprecipitation method. | ||
Results and discussion
Single Ru atoms dispersed on monolayer NiFe–LDH (Ru1/mono-NiFe) were synthesized via a one-step coprecipitation method in formamide solution.23,36 By adding different amounts of RuCl3, the loading amount of Ru1/mono-NiFe can be determined to be 0.3 and 1.6 wt% through inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis (denoted as Ru1/mono-NiFe-x (x = 0.3, 1.6)). Transmission electron microscopy (TEM) images (Fig. 1A and B) reveal that the morphologies of monolayer NiFe–LDH (mono-NiFe) and Ru1/mono-NiFe-0.3 are similar, giving a lateral size of ∼30 nm and thickness of ∼1 nm. The lattice fringe spacing of 0.15 nm corresponding to the (110) plane of mono-NiFe can be observed in Fig. 1C, which reveals the successful preparation of the LDH structure.43,44 To characterize the dispersion properties of Ru atoms on the monolayer LDH, high-angle annular dark-field scanning TEM microscopy (HAADF-STEM) images are obtained which show isolated spots with higher intensity (Fig. 1D), indicating that the Ru atoms are dispersed as single atoms without aggregation. Meanwhile, the energy-dispersive X-ray spectroscopy (EDS) mapping images suggest the uniform distributions of Ni, Fe and Ru over the entire mono-NiFe nanosheets (Fig. S2†). Moreover, the thickness of the Ru1/mono-NiFe-0.3 nanosheets was determined to be ∼1 nm (Fig. 1E and F) by atomic force microscopy (AFM), corresponding to the thickness of a monolayer plus absorbed species (NO3−, H2O and formamide layers of 0.4–0.5 nm).26,43 The Tyndall effect indicates the presence of highly dispersed colloidal mono-NiFe and Ru1/mono-NiFe-0.3 nanosheets (Fig. 1G). Mono-NiFe has a high BET surface area of 310.6 m2 g−1, which is in accordance with the reported surface area of monolayer LDHs.23,45 The high surface area of Ru1/mono-NiFe-0.3 of 358 m2 g−1 (Fig. S3†) is expected to provide more active sites, which can be beneficial for the further electrocatalysis. | ||
| Fig. 1 TEM images of (A) mono-NiFe and (B) Ru1/mono-NiFe-0.3; (C) HRTEM image of Ru1/mono-NiFe-0.3; (D) HAADF-STEM image of Ru1/mono-NiFe-0.3 (the circles are drawn around some of the isolated Ru atoms); (E) AFM image of Ru1/mono-NiFe-0.3 nanosheets; (F) the corresponding height curves of AFM in (E); (G) photographs of the dispersion of mono-NiFe and Ru1/mono-NiFe-0.3 in water. | ||
In order to verify the local structure of the single Ru atoms dispersed on Ru1/mono-NiFe, X-ray absorption near edge structure (XANES) and Extended X-ray absorption fine structure (EXAFS) were performed. In XANES curves (Fig. 2A and S4†), the Ru K-edge absorption positions of Ru1/mono-NiFe-0.3 and Ru1/mono-NiFe-1.6 are nearly the same as that of RuCl3, indicating the +3 oxidation state of Ru in Ru1/mono-NiFe-x (x = 0.3, 1.6). The EXAFS spectra of Ru1/mono-NiFe-x (x = 0.3, 1.6) exhibit extended periods and a reduction in the oscillation amplitudes, which are quite different results from those of Ru–Foil and RuO2 (Fig. 2B). This can be attributed to the different coordination environments of the Ru atoms. With a further increase of the loading amount of Ru in mono-NiFe from 0.3 wt% to 1.6 wt%, k2χ EXAFS displays a similar spectrum, indicating a similar coordination environment of Ru atoms in mono-NiFe. Wavelet transform (WT) analysis is a powerful tool to separate backscattering atoms to provide both resolution in the radial distance k-space of Ru K-edge EXAFS.17,46 As shown in Fig. 2C, the characteristic peak around 9 Å−1 of the Ru–Foil sample can be assigned to the Ru–Ru bond, and the peaks at 4.1 and 9 Å−1 are assigned to the first Ru–O and second Ru–Ru shells of RuO2. In contrast, Ru1/mono-NiFe-0.3 affords only one peak at 4.1 Å−1, indicating the existence of only the Ru–O shell. The Fourier transform EXAFS (FT-EXAFS) oscillation spectra of Ru1/mono-NiFe-1.6 and Ru1/mono-NiFe-0.3 show only the peak of the Ru–O shell at 1.5 Å and no metallic Ru–Ru shell peak at 2.4 Å can be observed (Fig. 2D). All these above results provide definitive proof for the successful synthesis of single Ru atoms supported on monolayer LDH nanosheets (Fig. 2E).
 | ||
| Fig. 2 (A) The Ru K-edge XANES spectra; (B) the Ru K-edge EXAFS k2χ functions for Ru1/mono-NiFe-1.6, Ru1/mono-NiFe-0.3, RuCl3, RuO2 and Ru–Foil; (C) the wavelet transforms for the k2-weighted EXAFS signals of Ru1/mono-NiFe-0.3, RuO2 and Ru–Foil; (D) the magnitude of k2-weighted Fourier transforms of the Ru K-edge EXAFS spectra for Ru1/mono-NiFe-0.3, Ru1/mono-NiFe-1.6, RuO2 and Ru–Foil; (E) schematic illustration of Ru1/mono-NiFe-x. | ||
Since different locations of single Ru atoms can affect profoundly the corresponding catalytic properties, the precise location of the Ru atoms in Ru1/mono-NiFe was investigated systematically. We anticipate the possible locations of Ru atoms as the following three models. For Model 1, the Ru atoms are inserted into the LDH monolayer and occupy the position of Fe atoms (Fig. S5B†); for Model 2, the Ru atoms are localized upon the surface of the LDH layer and the Ru atoms vertically face to the O atoms of the LDH face to the O atoms (Fig. S5C†); for Model 3, the Ru atoms are localized on the LDH surface and vertically face to the metal (Ni or Fe atoms) via coordination with three oxygen atoms (Fig. S5D†). As illustrated in Fig. S6A and E,† FT-EXAFS spectra of NiFe–LDH show two peaks corresponding to the first metal–O shell at ∼1.5 Å and the second metal–metal shell at ∼2.7 Å (metal = Ni or Fe). However, for Ru1/mono-NiFe-x (x = 0.3, 1.6) (Fig. S6B–D†), the absence of the peak around 7.5 Å−1 due to the Ru-metal shell clearly rules out the possibility of Ru being inserted into the LDH layer. The coordination number of Ru–O (∼4.1, Table S1†) implies the existence of a tetrahedral RuO4 site, as evidenced by FT-EXAFS results (Fig. S6E†). This result suggests that the dispersion of Ru ions in the LDH layer with octahedral coordination is impossible. Compared to the second shell in the Ru R-space of Ru1/mono-NiFe-1.6 at 2.2 Å (Fig. S6F†), the tetrahedral RuO4 anchored on the metal site (Model 3) shows a better fitting result than RuO4 anchored on the O site (Model 2). However, it is hard from the XAFS data to identify whether the Ru atoms are located on the top of Fe sites or Ni sites. In order to obtain a better understanding of the coordination environment of Ru atoms, we calculated the relative energy of Ru atoms located on the Ni, O and Fe sites on the LDH, and the corresponding result is 0 eV, −0.15 eV, and −0.48 eV (Fig. S7A–D†) respectively, suggesting that the Ru atoms located on the top of Fe sites are more stable. Therefore, we can make a conclusion that the Ru atom is anchored on metal sites in the LDH, and located on the trivalent Fe site in the LDH layer (Fig. S7E†). The formation of this structure is reasonable since Ru is in the same subgroup as Fe in the periodic table. Moreover, the oxidation state of Fe (+3) is the same as that of Ru (+3).
The XANES spectra and EXAFS oscillations of Ni and Fe for mono-NiFe and Ru1/mono-NiFe-0.3 are similar (Fig. S8†). The coordination numbers of Ni–O in mono-NiFe and Ru1/mono-NiFe-0.3 are the same (5.8). In terms of the Fe–O shell, the coordination numbers in mono-NiFe and Ru1/mono-NiFe-0.3 are 5.7 (Fig. S8C and F, Table S2†), indicating the existence of oxygen vacancies in mono-NiFe and Ru1/mono-NiFe-0.3 (Fig. S9†).44,47 X-ray photoelectron spectroscopy (XPS) results indicate that after loading the Ru atoms on mono-NiFe, the binding energy of Ni 2p and Fe 2p cores shows no obvious difference (Fig. S10†). For the Ru 3p core (Fig. S11†), there is no clear peak for Ru1/mono-NiFe-0.3, mainly due to the low Ru content of only 0.3 wt%. With a further increase of the loading amount of Ru, Ru1/mono-NiFe-1.6 exhibits a clear peak at ∼463.7 eV, which can be assigned to Ru3+.48
Hydrazine electrooxidation has attracted broad attention due to its high theoretical cell voltage, absence of CO2 emission and large hydrogen density delivery.49–51 A single Ru atom catalyst with a precise location offers an ideal model to study the reaction mechanism. The electrochemical performance of mono-NiFe, Ru1/mono-NiFe-0.3 and Ru1/mono-NiFe-1.6 toward hydrazine electrooxidation was systematically investigated. In contrast, bulk-NiFe with a size of ∼1 μm and thickness of ∼50 nm was synthesized as a reference to evaluate the hydrazine electrooxidation performance (Fig. S12†). The LSV curves (Fig. 3A) demonstrate that the electrocatalytic abilities are in the following sequence: Ru1/mono-NiFe-1.6 > Ru1/mono-NiFe-0.3 > mono-NiFe > bulk-NiFe > carbon paper. As can be seen in Fig. 3B, at a current density of 10 mA cm−2, Ru1/mono-NiFe-1.6 requires a potential of 1260 mV, which is 41 mV, 104 mV and 134 mV lower than that of Ru1/mono-NiFe-0.3, mono-NiFe and bulk-NiFe, respectively. The Tafel slope of Ru1/mono-NiFe-0.3 is 107 mV dec−1, which is smaller than that of mono-NiFe (119 mV dec−1). Moreover, the Nyquist plots (Fig. 3D) show that Ru1/mono-NiFe-1.6 and Ru1/mono-NiFe-0.3 have a much smaller semicircle in the Nyquist plot, corresponding to a smaller charge transfer resistance than that of mono-NiFe and bulk-NiFe. This result suggests that the presence of single Ru atoms in Ru1/mono-NiFe benefits the charge transport compared to pure mono-NiFe. Furthermore, the double-layer (dl) capacitance takes the following order: Ru1/mono-NiFe-0.3 (264 μF cm−2) ≈ mono-NiFe (256 μF cm−2) > bulk-NiFe (107 μF cm−2). On the basis of the above results, we can make a conclusion that compared with mono-NiFe, the enhanced hydrazine electrooxidation activity of Ru1/mono-NiFe-0.3 is mainly due to the single Ru atoms rather than the electrochemical surface area (Fig. S13†). In other words, the single Ru atoms are the key factor for the improvement of the catalytic performance. In addition, Ru1/mono-NiFe-0.3 shows ∼98% faradaic efficiency toward hydrazine electrooxidation (Fig. S14†), indicating a satisfactory energy conversion from electrical energy to chemical energy. The stability of Ru1/mono-NiFe-0.3 and Ru1/mono-NiFe-1.6 shows no obvious decline after 600 cycles (Fig. S15 and S16†). Additionally, the XPS signals of Ru, Fe and Ni in Ru1/mono-NiFe-0.3 and Ru1/mono-NiFe-1.6 show almost no change after hydrazine electrooxidation (Fig. S17–S19†), indicating the stability of the catalysts.
 | ||
| Fig. 3 (A) LSV curves of Ru1/mono-NiFe-1.6, Ru1/mono-NiFe-0.3, mono-NiFe, bulk-NiFe and carbon paper in 1 M KOH with 0.2 M hydrazine at a scan rate of 5 mV s−1; (B) the required potentials of different electrocatalysts at a current density of 10 mA cm−2; (C) Tafel plots of mono-NiFe and Ru1/mono-NiFe-0.3; (D) electrochemical impedance spectra of Ru1/mono-NiFe-1.6, Ru1/mono-NiFe-0.3, mono-NiFe and bulk-NiFe, respectively. | ||
To elucidate the influence of the electronic structure of single Ru atoms on the LDH toward hydrazine oxidation, DFT calculations were performed on bulk-NiFe (model of a defect-free multilayer LDH), mono-NiFe (monolayer LDH with oxygen vacancies), and Ru1/mono-NiFe (single RuO4 located on the Fe site of mono-NiFe). The optimized geometries of these three models are displayed in Fig. S20.†
The density of states for bulk-NiFe, mono-NiFe, and Ru1/mono-NiFe is shown in Fig. 4A. The valence band maximum (VBM) for bulk-NiFe is mainly composed of the O-2p orbital, while the conduction band minimum (CBM) is mainly constituted by the Fe-3d orbital, revealing that the Fe is the electrocatalytic active site in bulk-NiFe. Then, with doping the oxygen vacancy in mono-NiFe, two intermediate energy levels near the VBM occur, which is attributed to the unpaired electron resulting from the oxygen vacancy. The existence of intermediate energy levels leads to the reduction of the band gap energy (Eg) from 1.10 eV in bulk-NiFe to 0.60 eV in mono-NiFe. For Ru1/mono-NiFe, Ru-4d becomes a major component for both the VBM and CBM of Ru1/mono-NiFe, further decreasing the Eg to 0.38 eV. The corresponding isosurface for Ru1/mono-NiFe also gives an obviously increased charge density with respect to the frontier orbital of mono-NiFe (Fig. 4B). In general, the decreased Eg for Ru1/mono-NiFe facilitates the electron conduction and reduces the energy loss during the electrocatalytic process.
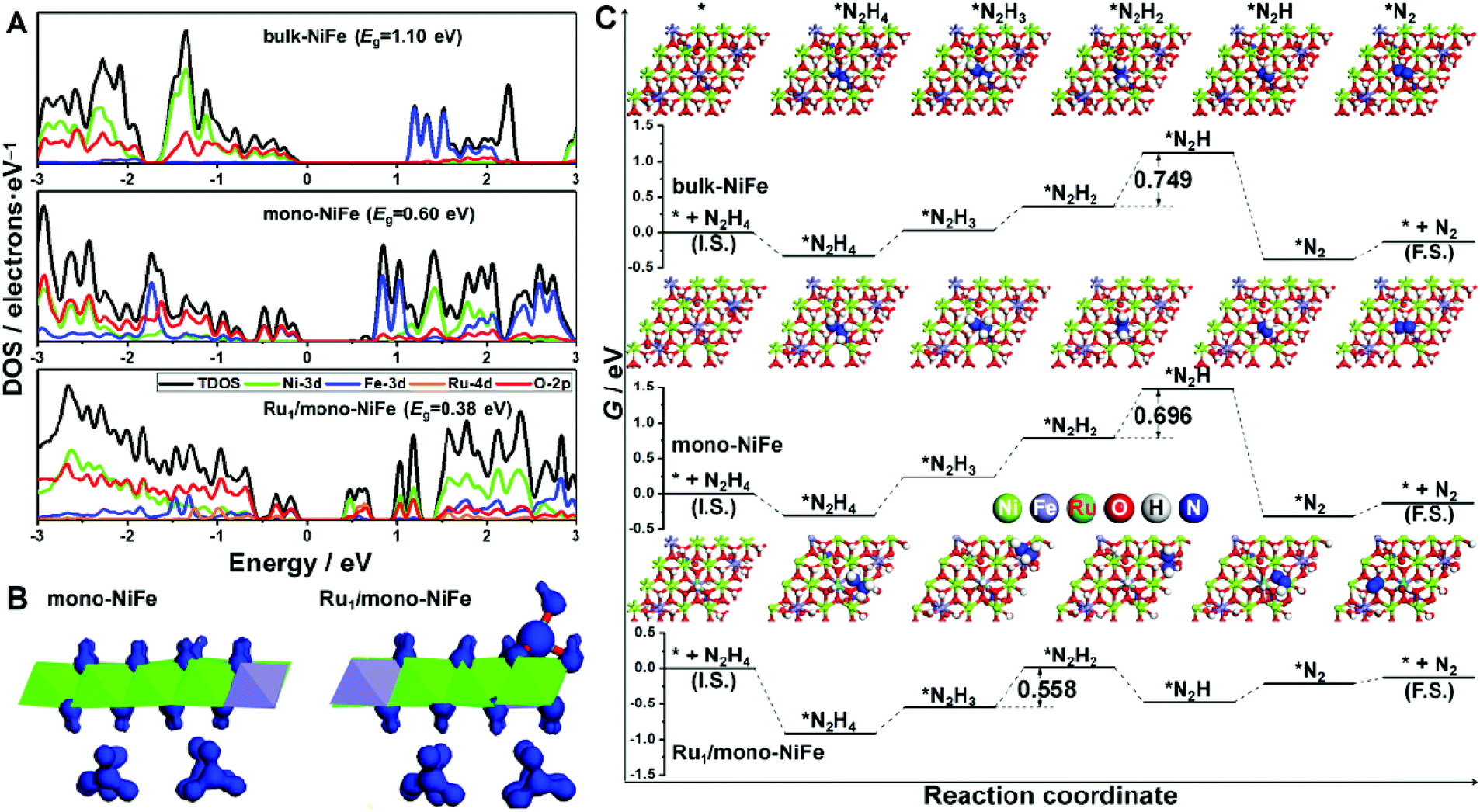 | ||
| Fig. 4 (A) Density of states plots for bulk-NiFe, mono-NiFe and Ru1/mono-NiFe, respectively; (B) isosurfaces for mono-NiFe and Ru1/mono-NiFe; (C) standard free energy diagrams for the hydrazine electrooxidation on bulk-NiFe, mono-NiFe, and Ru1/mono-NiFe, respectively; the number in the figure represents the Gibbs free energy change of the corresponding rate-determining step in hydrazine electrooxidation. | ||
Moreover, the thermodynamic mechanism for hydrazine electrooxidation was investigated. Firstly, mono-NiFe with Ru located on the Ni or O site was constructed and named Ru1(above Ni)/mono-NiFe and Ru1(above O)/mono-NiFe, for comparison. The geometries of the reaction intermediates for hydrazine electrooxidation (*, *N2H4, *N2H3, *N2H2, *N2H, and *N2, * is the reaction site) were optimized and are displayed in Fig. 4C and S21† (top view), and Fig. S22† (side view). Then, the Gibbs free energy change (ΔG) for each elementary step of hydrazine electrooxidation was calculated with eqn (5)–(10) (ESI†) and is listed in Fig. 4C, S21 and Table S3.† It is found that the reaction ΔG values for Ru1(above Ni)/mono-NiFe and Ru1(above O)/mono-NiFe are larger than that of Ru1/mono-NiFe, further revealing that the precise location of Ru on the Fe site promotes the hydrazine electrooxidation (Fig. S21†), which is derived from the electronic effect of Ru that can increase the electropositivity of Fe (Fig. S23†). As shown in Fig. 4C, the rate-determining step for hydrazine electrooxidation on bulk-NiFe and mono-NiFe is the dehydrogenation of *N2H2 to *N2H, while the most difficult step for Ru1/mono-NiFe is the formation of *N2H2. As a result, it is intriguing that the introduction of single Ru atoms changes the rate-determining step in the hydrazine oxidation. The introduction of Ru can stabilize the intermediates with one unpaired electron, i.e., *N2H3 and *N2H, which is derived from the d-electron arrangement of Ru. Above all, the Gibbs free energy barriers for hydrazine electrooxidation were calculated to be 0.749 eV, 0.696 eV, and 0.558 eV on bulk-NiFe, mono-NiFe and Ru1/mono-NiFe, respectively. Therefore, the introduction of single Ru atoms located on the Fe site improves the hydrazine electrocatalytic activity of mono-NiFe, well explaining the experimentally excellent hydrazine electrooxidation activity of Ru1/mono-NiFe.
Furthermore, preparations of single Ru atoms with a higher loading amount of 3.8 wt% and 7.0 wt% and precise location were also achieved by using mono-NiFe as the support. The real loading amount was confirmed by ICP analysis and the precise location of single Ru atoms on the LDH surface is the same as that of Ru1/mono-NiFe-x (x = 0.3, 1.6) as confirmed by XAFS analysis (Fig. 5 and S24†). Besides, single Ru atoms loaded on different LDH supports such as monolayer NiAl–LDH (Ru1/mono-NiAl) and monolayer MgAl–LDH (Ru1/mono-MgAl) were successfully applied for the synthesis of SACs with a precise location on the Al atoms, as verified by the presence of only the Ru–O shell peak at 1.5 Å in XAFS and FT-XAFS spectra (Fig. 5 and S25†). Additionally, a single Ru atom loaded LDH (Ru1/NiFe-x) was also successfully synthesized through a hydrothermal method (Fig. S26†). These findings suggest LDHs are effective supports for the preparation of SACs with a precise location.
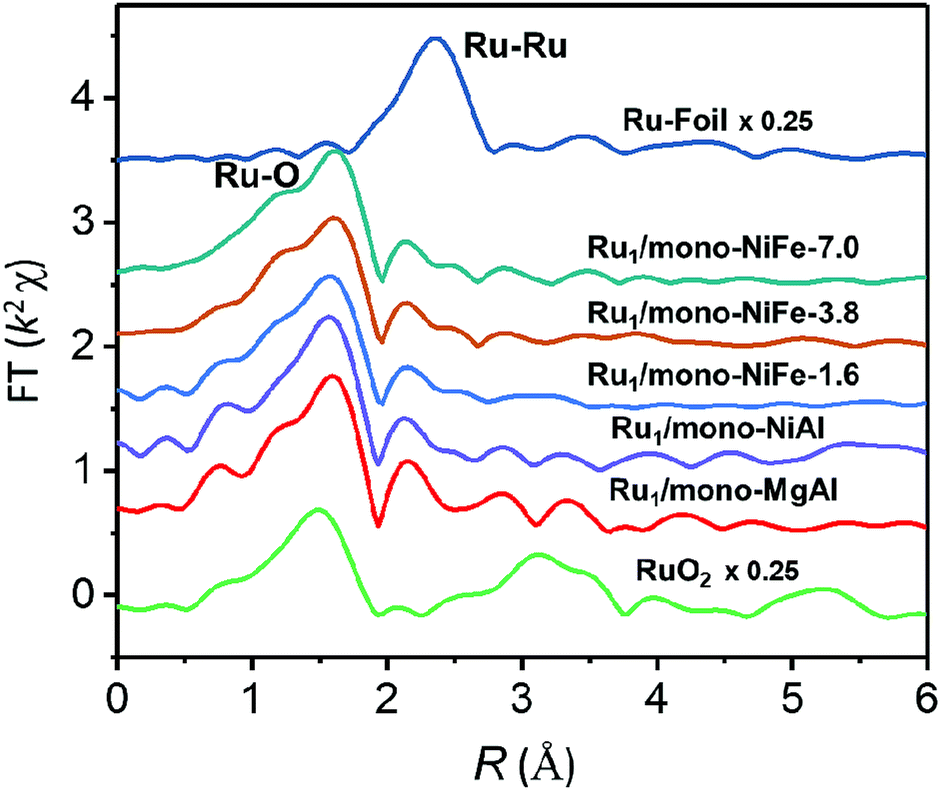 | ||
| Fig. 5 Magnitude of k2-weighted Fourier transforms of the Ru K-edge EXAFS spectra for Ru1/mono-NiFe-7.0, Ru1/mono-NiFe-3.8, Ru1/mono-NiFe-1.6, Ru1/mono-NiAl, Ru1/mono-MgAl, RuO2 and Ru–Foil, respectively. | ||
Conclusions
To summarize, single Ru atoms supported on mono-NiFe have been successfully synthesized using a facile, one-step coprecipitation method. As evidenced by XAFS, STEM, HRTEM, and AFM results, the single Ru atoms are uniquely localized on the top of the Fe atoms via three oxygen atoms on mono-NiFe. The synthesis of single Ru atoms on LDHs with a precise location can be extended to a higher Ru loading amount to 7.0 wt% and to a broad selection of LDHs (like monolayer NiAl–LDH and monolayer MgAl–LDH) as supports. DFT calculations further reveal that the precise location of single Ru atoms lowers the reaction barrier and Eg, explaining the enhanced hydrazine electrooxidation. This work demonstrates the importance of the precise location of single atoms on catalytic properties and thereby has broad implications on designing and characterisation of SACs with a precise location. Further studies on other noble metals (Ir, Rh, Pd, Au, etc.) atomically dispersed on LDH supports are currently underway in our lab.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (U1707603, 21625101, 21521005, U1507102, and 21878008), the National Key Research and Development Program of China (2017YFB0307303), the 973 program (Grant no. 2014CB932104), the Beijing Natural Science Foundation (2182047) and the Fundamental Research Funds for the Central Universities (XK1802-6). We acknowledge the National Supercomputing Center in Shenzhen for providing the materials studio (version 6.1, CASTEP). The XAFS experiments were conducted in 1W1B beam line of Beijing Synchrotron Radiation Facility (BSRF).Notes and references
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740 CrossRef CAS PubMed.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. Zheng, Science, 2016, 352, 797 CrossRef CAS PubMed.
- K. Mori, T. Taga and H. Yamashita, ACS Catal., 2017, 7, 3147 CrossRef CAS.
- J. Liu, ACS Catal., 2016, 7, 34 CrossRef.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634 CrossRef CAS PubMed.
- J. Lin, B. Qiao, J. Liu, Y. Huang, A. Wang, L. Li, W. Zhang, L. F. Allard, X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2012, 51, 2920 CrossRef CAS PubMed.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Science, 2017, 358, 1419 CrossRef CAS PubMed.
- L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y. W. Li, C. Shi, X. D. Wen and D. Ma, Nature, 2017, 544, 80 CrossRef CAS PubMed.
- S. Yao, X. Zhang, W. Zhou, R. Gao, W. Xu, Y. Ye, L. Lin, X. Wen, P. Liu, B. Chen, E. Crumlin, J. H. Guo, Z. Zuo, W. Li, J. Xie, L. Lu, C. J. Kiely, L. Gu, C. Shi, J. A. Rodriguez and D. Ma, Science, 2017, 357, 389 CrossRef CAS PubMed.
- M. Yang, L. F. Allard and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2013, 135, 3768 CrossRef CAS PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944 CrossRef CAS PubMed.
- L. Wang, H. Li, W. Zhang, X. Zhao, J. Qiu, A. Li, X. Zheng, Z. Hu, R. Si and J. Zeng, Angew. Chem., Int. Ed., 2017, 56, 4712 CrossRef CAS PubMed.
- L. Wang, W. Zhang, S. Wang, Z. Gao, Z. Luo, X. Wang, R. Zeng, A. Li, H. Li, M. Wang, X. Zheng, J. Zhu, W. Zhang, C. Ma, R. Si and J. Zeng, Nat. Commun., 2016, 7, 14036 CrossRef CAS PubMed.
- S. Li, Y. Xu, Y. Chen, W. Li, L. Lin, M. Li, Y. Deng, X. Wang, B. Ge, C. Yang, S. Yao, J. Xie, Y. Li, X. Liu and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 10761 CrossRef CAS PubMed.
- M. Xu, S. Yao, D. Rao, Y. Niu, N. Liu, M. Peng, P. Zhai, Y. Man, L. Zheng, B. Wang, B. Zhang, D. Ma and M. Wei, J. Am. Chem. Soc., 2018, 140, 11241 CrossRef CAS PubMed.
- H. Duan, J. Dong, X. Gu, Y. K. Peng, W. Chen, T. Issariyakul, W. K. Myers, M. J. Li, N. Yi, A. F. R. Kilpatrick, Y. Wang, X. Zheng, S. Ji, Q. Wang, J. Feng, D. Chen, Y. Li, J. C. Buffet, H. Liu, S. C. E. Tsang and D. O'Hare, Nat. Commun., 2017, 8, 591 CrossRef PubMed.
- W. Bing, L. Zheng, S. He, D. Rao, M. Xu, L. Zheng, B. Wang, Y. Wang and M. Wei, ACS Catal., 2018, 8, 656 CrossRef CAS.
- H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, S. Miao, J. Liu and T. Zhang, Nat. Commun., 2014, 5, 5634 CrossRef CAS PubMed.
- R. Lang, T. Li, D. Matsumura, S. Miao, Y. Ren, Y. T. Cui, Y. Tan, B. Qiao, L. Li, A. Wang, X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2016, 55, 16054 CrossRef CAS PubMed.
- B. Qiao, J. Liu, Y.-G. Wang, Q. Lin, X. Liu, A. Wang, J. Li, T. Zhang and J. Liu, ACS Catal., 2015, 5, 6249 CrossRef CAS.
- X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng, M. Wei, D. Tan, R. Si, S. Zhang, J. Li, L. Sun, Z. Tang, X. Pan and X. Bao, Science, 2014, 344, 616 CrossRef CAS PubMed.
- J. Yu, Q. Wang, D. O'Hare and L. Sun, Chem. Soc. Rev., 2017, 46, 5950 RSC.
- G. Fan, F. Li, D. G. Evans and X. Duan, Chem. Soc. Rev., 2014, 43, 7040 RSC.
- R. Ma and T. Sasaki, Adv. Mater., 2010, 22, 5082 CrossRef CAS PubMed.
- Y. Zhao, X. Jia, G. Chen, L. Shang, G. I. Waterhouse, L.-Z. Wu, C.-H. Tung, D. O'Hare and T. Zhang, J. Am. Chem. Soc., 2016, 138, 6517 CrossRef CAS PubMed.
- K. Fan, H. Chen, Y. Ji, H. Huang, P. M. Claesson, Q. Daniel, B. Philippe, H. Rensmo, F. Li, Y. Luo and L. Sun, Nat. Commun., 2016, 7, 11981 CrossRef CAS PubMed.
- R. Ma, K. Takada, K. Fukuda, N. Iyi, Y. Bando and T. Sasaki, Angew. Chem., Int. Ed., 2008, 47, 86 CrossRef CAS PubMed.
- K. Motokura, D. Nishimura, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 5662 CrossRef CAS PubMed.
- Y. Zhu, Z. An, H. Song, X. Xiang, W. Yan and J. He, ACS Catal., 2017, 7, 6973 CrossRef CAS.
- X. Ma, Z. An, Y. Zhu, W. Wang and J. He, ChemCatChem, 2016, 8, 1773 CrossRef CAS.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452 CrossRef CAS PubMed.
- G. Chen, T. Wang, J. Zhang, P. Liu, H. Sun, X. Zhuang, M. Chen and X. Feng, Adv. Mater., 2018, 30, 1706279 CrossRef PubMed.
- W.-J. Liu, L. Dang, Z. Xu, H.-Q. Yu, S. Jin and G. W. Huber, ACS Catal., 2018, 8, 5533 CrossRef CAS.
- J. Y. Chen, L. Dang, H. Liang, W. Bi, J. B. Gerken, S. Jin, E. E. Alp and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 15090 CrossRef CAS PubMed.
- J. Yu, B. R. Martin, A. Clearfield, Z. Luo and L. Sun, Nanoscale, 2015, 7, 9448 RSC.
- C. Chen, C. F. H. Byles, J. C. Buffet, N. H. Rees, Y. Wu and D. O'Hare, Chem. Sci., 2016, 7, 1457 RSC.
- X. Deng, J. Huang, H. Wan, F. Chen, Y. Lin, X. Xu, R. Ma and T. Sasaki, J. Energy Chem., 2018 DOI:10.1016/j.jechem.2018.07.007.
- J. Yu, J. Liu, A. Clearfield, J. E. Sims, M. T. Speiegle, S. L. Suib and L. Sun, Inorg. Chem., 2016, 55, 12036 CrossRef CAS PubMed.
- P. J. Sideris, U. G. Nielsen, Z. Gan and C. P. Grey, Science, 2008, 321, 113 CrossRef CAS PubMed.
- J. Zhang, J. Liu, L. Xi, Y. Yu, N. Chen, S. Sun, W. Wang, K. M. Lange and B. Zhang, J. Am. Chem. Soc., 2018, 140, 3876 CrossRef CAS PubMed.
- H. Li, L. Wang, Y. Dai, Z. Pu, Z. Lao, Y. Chen, M. Wang, X. Zheng, J. Zhu, W. Zhang, R. Si, C. Ma and J. Zeng, Nat. Nanotechnol., 2018, 13, 411 CrossRef CAS PubMed.
- Y. Zhao, Q. Wang, T. Bian, H. Yu, H. Fan, C. Zhou, L.-Z. Wu, C.-H. Tung, D. O'Hare and T. Zhang, Nanoscale, 2015, 7, 7168 RSC.
- Y. Zhao, X. Zhang, X. Jia, G. I. N. Waterhouse, R. Shi, X. Zhang, F. Zhan, Y. Tao, L.-Z. Wu, C.-H. Tung, D. O'Hare and T. Zhang, Adv. Energy Mater., 2018, 8, 1703585 CrossRef.
- Q. Wang and D. O'Hare, Chem. Commun., 2013, 49, 6301 RSC.
- M. Munoz, P. Argoul and F. Farges, Am. Mineral., 2003, 88, 694 CrossRef CAS.
- Y. Zhao, G. Chen, T. Bian, C. Zhou, G. I. Waterhouse, L.-Z. Wu, C.-H. Tung, L. J. Smith, D. O'Hare and T. Zhang, Adv. Mater., 2015, 27, 7824 CrossRef CAS PubMed.
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072 CrossRef CAS.
- Z. Li, M. Shao, H. An, Z. Wang, S. Xu, M. Wei, D. G. Evans and X. Duan, Chem. Sci., 2015, 6, 6624 RSC.
- L. Zhou, M. Shao, C. Zhang, J. Zhao, S. He, D. Rao, M. Wei, D. G. Evans and X. Duan, Adv. Mater., 2017, 29, 1604080 CrossRef PubMed.
- Y. Meng, X. Zou, X. Huang, A. Goswami, Z. Liu and T. Asefa, Adv. Mater., 2014, 26, 6510 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section and extended characterization results, including STEM, BET, XAFS, XPS data, electrochemical data, and DFT results. See DOI: 10.1039/c8sc04480e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |