
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMild complexation protocol for chiral CpxRh and Ir complexes suitable for in situ catalysis†
B.
Audic
 a,
M. D.
Wodrich
b and
N.
Cramer
*a
a,
M. D.
Wodrich
b and
N.
Cramer
*a
aLaboratory of Asymmetric Catalysis and Synthesis, EPFL SB ISIC LCSA, BCH 4305, CH-1015 Lausanne, Switzerland. E-mail: nicolai.cramer@epfl.ch
bLaboratory for Computational Molecular Design, EPFL SB ISIC LCMD, BCH 5312, CH-1015 Lausanne, Switzerland. E-mail: matthew.wodrich@epfl.ch
First published on 31st October 2018
Abstract
A practical complexation method for chiral cyclopentadienyl (Cpx) iridium and rhodium complexes is described. The procedure uses the free CpxH with stable and commercially available rhodium(I) and iridium(I) salts without base or additive. The conditions are mild and do not require the exclusion of air and moisture. A salient feature is the suitability for in situ complexations enhancing the user-friendliness of Cpx ligands in asymmetric catalysis. DFT-calculations confirm an intramolecular proton abstraction pathway by either the bound acetate or methoxide. Furthermore, the superior facial selectivity of the proton abstraction step enabled the development of TMS-containing trisubstituted Cpx ligands which display improved enantioselectivities for the benchmarking dihydroisoquinolone synthesis.
Introduction
Cyclopentadienyl (Cp) coordinated transition-metal complexes are a very important class of catalysts enabling a broad range of versatile and atom-economic transformations.1 During the last decade, the dominance of Cp and Cp* (pentamethylcyclopentadienyl) has been challenged by new designer Cp ligands. Modulation of the electronic and steric properties of the Cp ligands has become a key focus for reaction optimization. These ligands display superior performance such as enhanced reactivity,2 improved regioselectivity3 and diastereoselectivity.4 Moreover, the rise of chiral Cp ligands (Cpx) enabled the development of enantioselective processes.5While the synthesis of simple classical Cp* and Cp complexes with late transition-metals is well established,6 accessing the corresponding metal Cp complexes of more elaborate – especially chiral – Cp derivatives remains problematic and suffers from a number of undesirable attributes (Fig. 1). For instance, the increased value of tailored cyclopentadienes requiring multi-step syntheses imposes the use of the CpH derivative as the limiting reactant, making protocols that use multiple CpH equivalents prohibitively inefficient. A frequently largely reduced crystallinity and/or enhanced solubility limit purification options of reaction mixtures from suboptimal reaction outcomes. Until now, the most reliably and broadly applicable method involves the use of stoichiometric amounts of highly toxic thallium bases,5f often paired with toxic benzene as solvent.7 Alkali bases were inferior and caused often complex reaction outcomes. A complementary method used chloride based metal salts without a base.8 The released hydrochloric acid during the complexation is incompatible with some sensitive functional groups of designer Cp scaffolds. We have developed a complementary complexation strategy capitalizing on β-carbon elimination of cyclopentadienyl carbinols as Cp-ligand precursors.9 While this method consists of an improvement, it requires the synthesis of the ligand precursor by an additional synthetic step. Moreover, it is limited to highly substituted and hindered Cp variants. In consequence, the development of a robust and general complexation platform for a broad range of tailored chiral Cpx ligands remains an important task in improving utility and user friendliness of the resulting Cp metal catalysts.
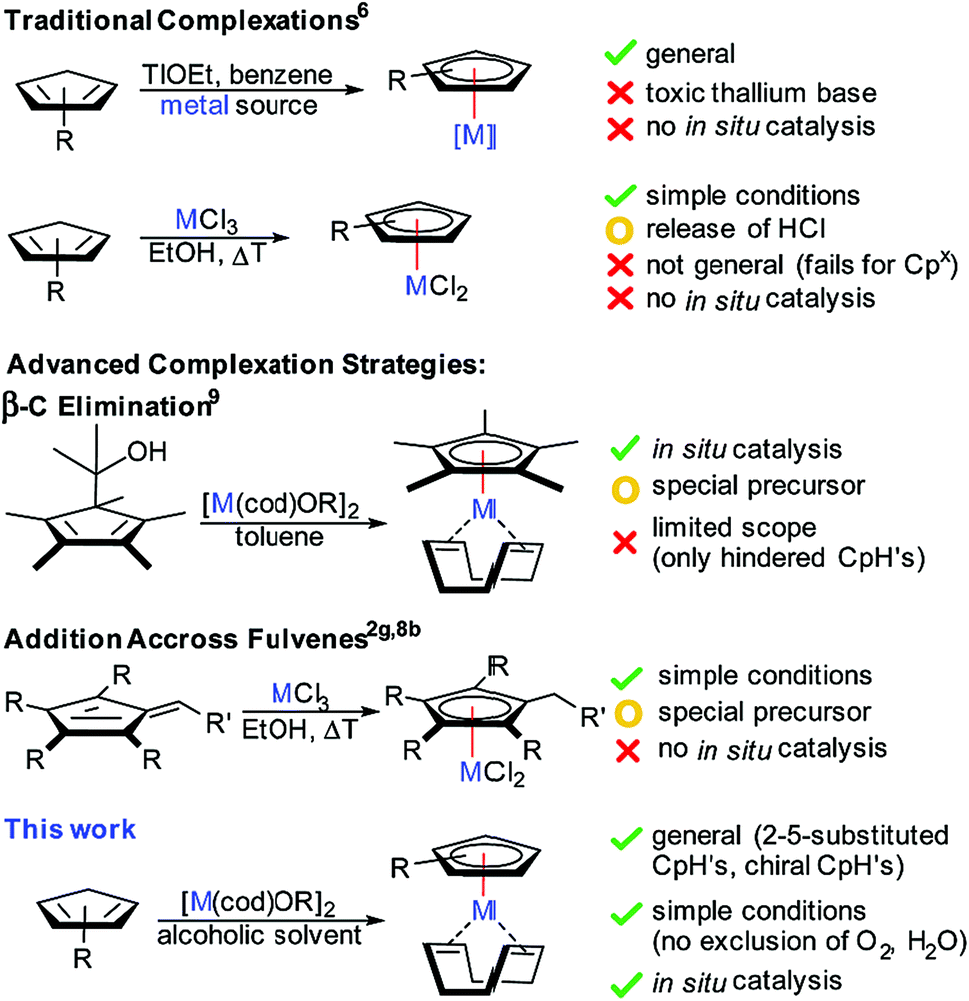 | ||
| Fig. 1 Characteristics and drawbacks of complexation strategies for cyclopentadienyl ligands. | ||
Herein, we report a mild and convenient method to access key Rh(I) and Ir(I) Cp complexes upon simply mixing a stable and easily accessible metal precursor with CpxH in protic solvents. The protocol qualifies as well for user friendly in situ catalyst assembly.
Results and discussion
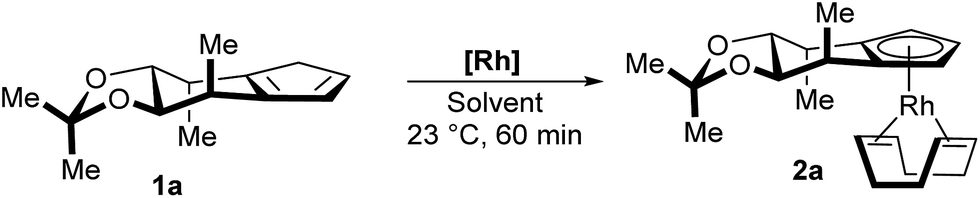
We started our investigations towards a mild and general complexation with cyclopentadiene 1a. A set of common rhodium precursors was screened for reactivity under a variety of conditions and additives (Table 1). The common [Rh(cod)Cl]2 did not react at all (entries 1 and 2). As well, no reaction was observed for [Rh(cod)OMe]2 in toluene (entry 3). In contrast, running the reaction in methanol produced the desired Cp complex 2a in modest yield (entry 4). Switching the rhodium source to [Rh(cod)OAc]2 resulted in a fast and clean conversion yielding 2a in 88% yield (entry 5). Ethanol and TFE proved to be as efficient as methanol (entries 6 and 9), whereas bulkier alcohols display largely decreased reaction rate and yields (entries 7 and 8). HFIP mainly led to degradation (entry 10). The use of THF as solvent completely inhibited the reaction (entry 11). Notably, the complexation is user friendly and has low technical requirements. It does not require the use of a glovebox and no precaution to exclude oxygen and humidity from the reaction vessel has to be taken. The final complexes bearing the stabilizing cyclooctadiene (cod) ligand are stable towards convenient standard flash chromatography on silica gel.|
|
||||
|---|---|---|---|---|
| Entry | [Rh] | Solvent | Conversionb (%) | Yieldb (%) |
| a Conditions: 40 μmol 1a, 24 μmol [Rh], 0.2 M in indicated solvent at 23 °C for 60 min. b Determined by 1H-NMR with an internal standard. c Partial cleavage of the acetal (30%) was observed. | ||||
| 1 | [Rh(cod)Cl]2 | toluene | <5 | 0 |
| 2 | [Rh(cod)Cl]2 | MeOH | <5 | 0 |
| 3 | [Rh(cod)OMe]2 | toluene | <5 | 0 |
| 4 | [Rh(cod)OMe]2 | MeOH | 85 | 28c |
| 5 | [Rh(cod)OAc]2 | MeOH | 100 | 88 |
| 6 | [Rh(cod)OAc]2 | EtOH | 100 | 89 |
| 7 | [Rh(cod)OAc]2 | iPrOH | 50 | 41 |
| 8 | [Rh(cod)OAc]2 | tBuOH | 10 | 5 |
| 9 | [Rh(cod)OAc]2 | TFE | 100 | 88 |
| 10 | [Rh(cod)OAc]2 | HFIP | 100 | 30 |
| 11 | [Rh(cod)OAc]2 | THF | <5 | 0 |
Initially, a variety of achiral cyclopentadiene substrates were tested (Scheme 1). For instance, tBu2CpH, Me4CpH and Cp*H participated in a smooth and rapid complexation, giving the desired CpRhI(cod) complexes 2b–2d in excellent yields. Solely, bulky and electron-deficient Ph4CpH required longer reaction time and gave reduced yields of 2e. With the observed success for the achiral Cp ligands, we have successfully tested the complete portfolio of the most useful chiral Cpx ligands for this transformation. In detail, mannitol-derived Cp ligand 2a4f was isolated in 85% yield. The mild conditions at ambient temperature were compatible with the acetal group. Similarly, readily accessible cyclopentane-fused cyclopentadiene 1f7a and the most versatile methoxy-bearing CpxH 1g10 engaged in complexations giving 2f and 2g in very high yields. Moderate solubility of the CpxH 2g required some heating. Bulkier isopropoxy-derivative (2h) reacts at ambient temperature, whereas substrates with larger groups (2i–2j) require gentle heating for optimal reactivity without compromising the high yield. Bulky tri-substituted cyclopentadiene 1k gave corresponding complex 2k in significantly improved yields compared to the thallium-process.11 Notably, the complexation protocol works as well very smoothly for penta-substituted Cpx* substrate (2l). Such true chiral Cp* analogues have been so far refractory substrates for complexation, hampering application in catalysis. Previously, any classical complexation methods failed for this substrate, making β-carbon elimination strategy from tert-carbinols the only viable method.9 Importantly, the illustrated complexation strategy is equally viable to access the corresponding CpxIr complexes. The complexation employing commercially available [Ir(cod)OMe]2 complex provided the desired complexes (3a, 3f-3g) in good yields. Compared to illustrate examples with rhodium, the complexation is slower with iridium and requires gentle heating. For more reluctant cases, the reaction can be performed with more reactive [Ir(cod)OAc]2, which is prepared in a single step from [Ir(cod)Cl]2.12
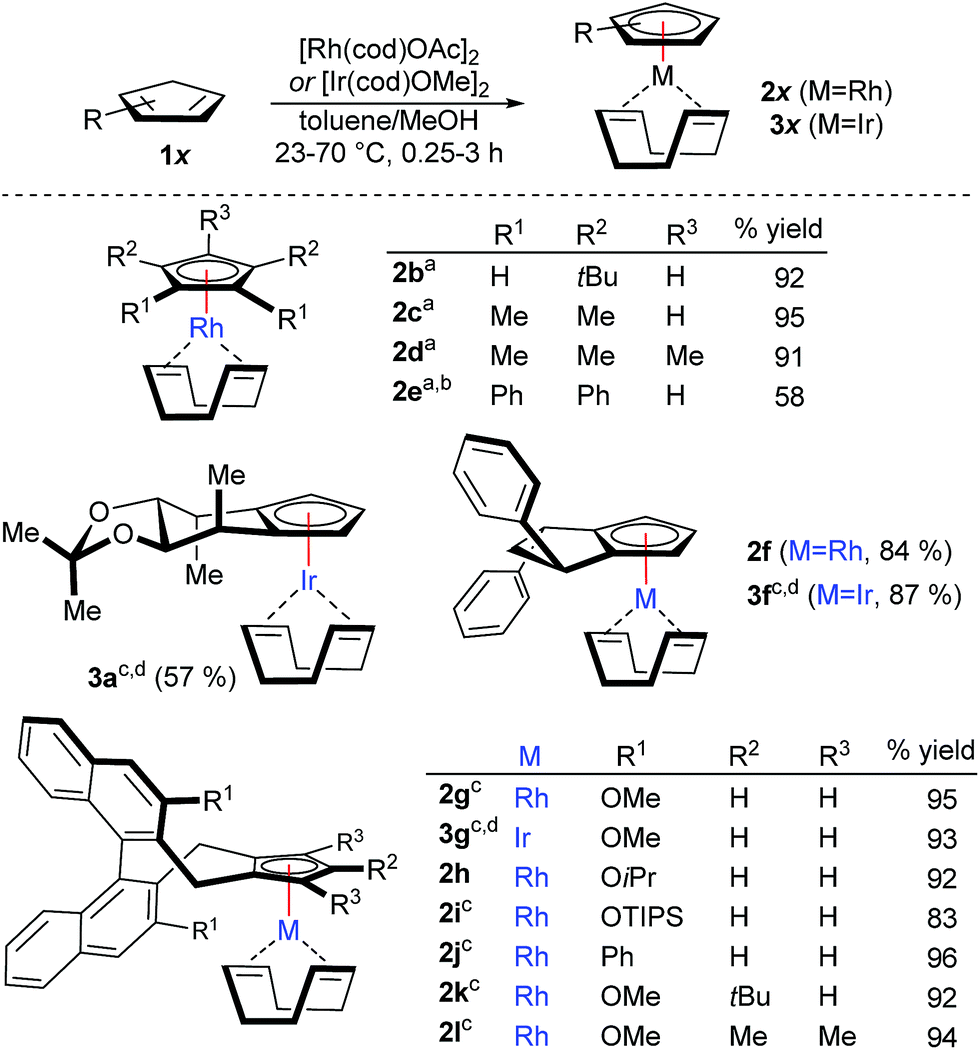 | ||
Scheme 1 Conditions: 0.1 mmol 1x, 0.06 mmol [Rh(cod)OAc]2 or [Ir(cod)OMe]2 in toluene/MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (0.2 M) at 23 °C. (a) 0.4 mmol 1x, 0.1 mmol [Rh(cod)OAc]2; (b) 24 h; (c) 70 °C; (d) [Ir(cod)OMe]2 instead of [Rh(cod)OAc]2. :1) (0.2 M) at 23 °C. (a) 0.4 mmol 1x, 0.1 mmol [Rh(cod)OAc]2; (b) 24 h; (c) 70 °C; (d) [Ir(cod)OMe]2 instead of [Rh(cod)OAc]2. | ||
To foster further understanding of the mechanism of complexation, DFT computations (at the PBE0-dDsC/TZ2P//M06/def2-SVP theoretical level, see ESI for computational detail†) of different possible pathways were performed.13 Two potential pathways exist, which involve reactions of pentamethylcyclopentadiene with either a Rh(OAc) (pathway A, Scheme 2) or alternatively a Rh(OMe) (pathway B, Scheme 2) fragment. Each pathway begins with the thermodynamically favorable cleavage of the solvated Rh(cod)OAc dimer (as determined from the crystal structure)14 to from a Rh(cod)(OAc)(MeOH) monomer (Int1). From this point the two pathways diverge. Methanol can either be directly dissociated in process endergonic process costing ∼12 kcal mol−1 (Int1 → TS1, pathway A). Int2 then forms reaction complex Int3 in the presence of the Cp*-precursor olefin. Proton abstraction from the diene by the bound acetate (TS2) is energetically costly and represents the highest point on the pathway A reaction profile at +24.1 kcal mol−1. The final products, Cp*Rh(cod) and acetic acid, are then liberated in a exergonic process. According to the computations, the early parts of pathway A appear to be quite facile, with proton abstraction representing the likely rate-determining step.13a
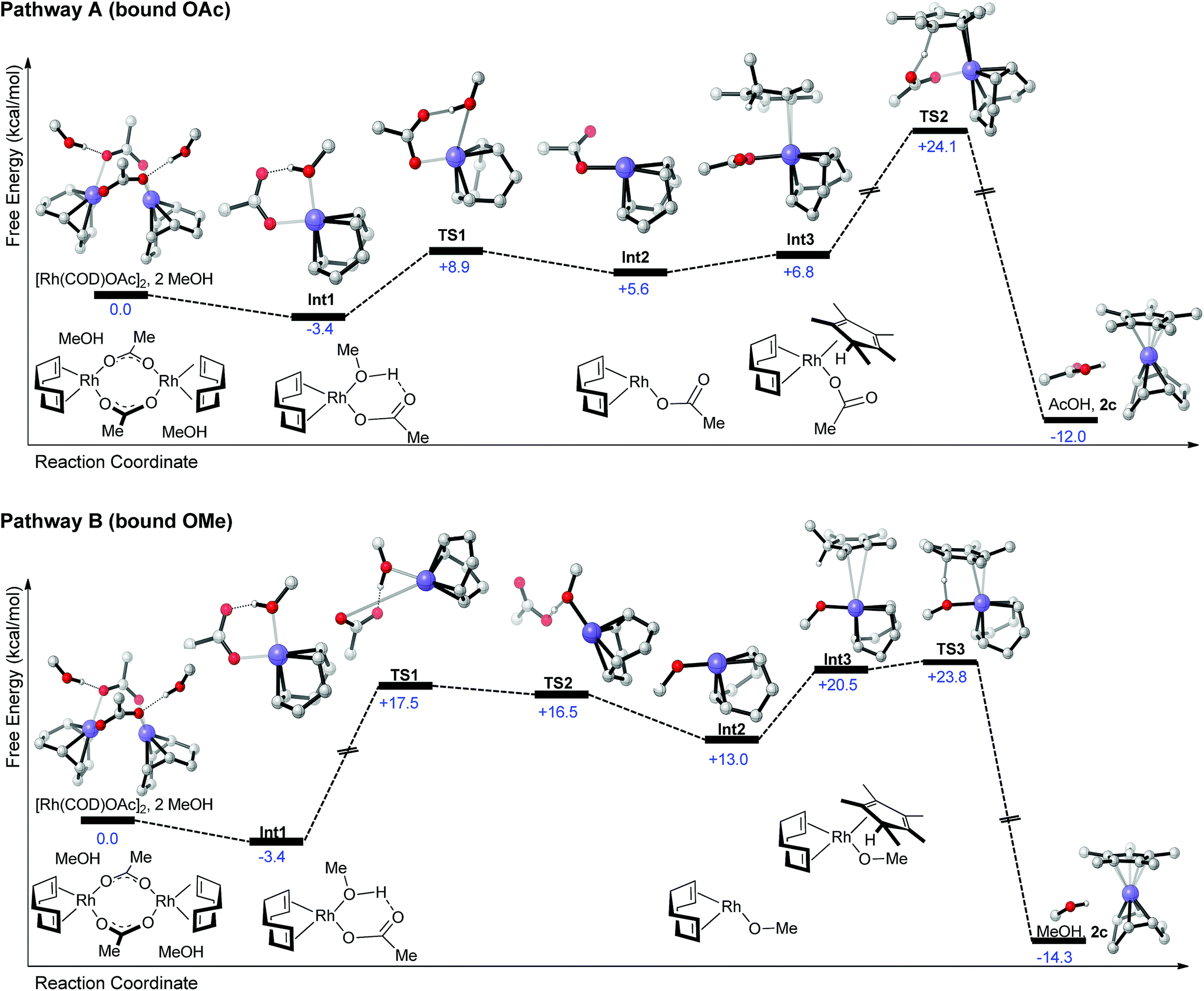 | ||
| Scheme 2 Potential reaction profiles proceeding form the [Rh(COD)OAc]2 to RhCp*(COD). Each of the reaction pathways is characterized by roughly similar energetic profiles. Computations at the PBE0-dDsC/TZ2P//M06/def2-SVP level (see ESI for details†). | ||
On the other hand, pathway B is characterized by a series of relatively high-energy transition states located at the beginning of the reaction profile (TS1/TS2). These costly steps are associated with the initial dissociation of the acetate ion (TS1) and proton transfer from methanol (TS2) to form a Rh(cod)OMe species (Int2) and a liberated acetic acid molecule. Creation of the Int3 reaction complex by association of the Cp*-precursor olefin followed by a proton abstraction process (TS2) is then accomplished with a low energy barrier and results in direct formation of the Cp*Rh(cod) product complex and a solvent molecule. Overall, the highest point of both reaction profiles is very close in energy (+24.1 vs. +23.8 kcal mol−1) and maybe both accessible under the mild reaction conditions utilized. Since the transformation works cleaner and in higher yields with [Rh(cod)OAc]2 compared to [Rh(cod)OMe]2 (which would only allow for pathway B), involvement of pathway A is assumed. Alternatively, the better solubility and potentially easier dimer of [Rh(cod)OAc]2 may account for the superiority of this precursor complex.
Besides the illustrated practical utility of the complexation strategy, our method offers two significant additionally advances. First, it enables new opportunities in complex ligand design previously unfeasible substitutions. Secondly, the facile complexation allows for the first time true in situ complexation and catalysis. In our attempt to pursue the concept of late-stage diversification and adjustability of the steric demand of a common ligand platform, we found that deprotonation of CpxH with BuLi and subsequent trapping with TMSCl uniformly provided a single regio- and diastereomer (Scheme 3). X-Ray crystal structure analysis of intermediate 1m confirmed that the TMS group occupies the position off the C2-symmetry axis of 1a at the more accessible face (See ESI†).15
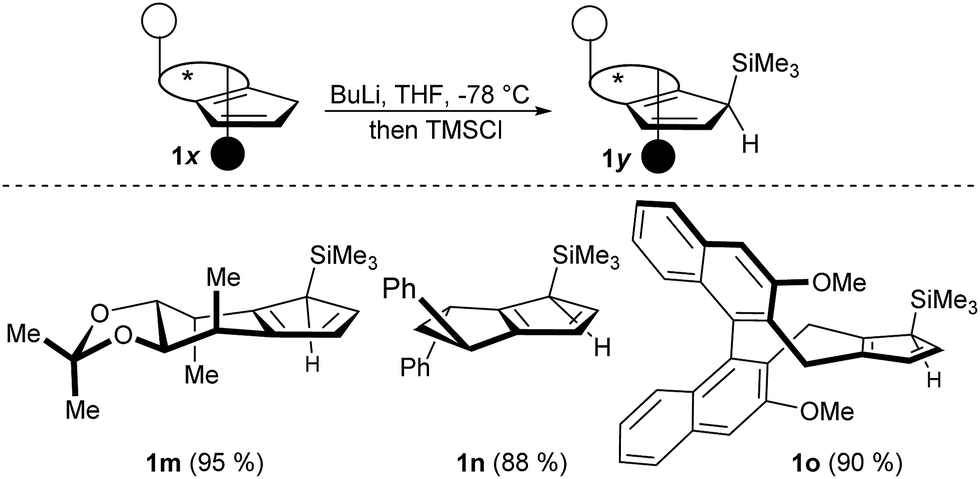 | ||
| Scheme 3 Regio- and diastereoselective silylation yields trisubstituted Cpx ligands 1m–1o. | ||
However, complexation of non-C2-symmetric ligands poses inherent selectivity issue due to a notoriously difficult facial discrimination in the complexation step of the flat Cp anion.7d In consequence, the typical thallium ethoxide complexation method is not suitable due to its lack of facial discrimination of the deprotonation. This results in inseparable mixture of diastereoisomers. Along the same lines, chloride containing rhodiumIII salts mainly resulted in desilylated complexation product.16 We reasoned that with previously developed complexation method, the rhodium precursor would selectively approach from the face opposed to the TMS group. Indeed, the combination of [Rh(cod)OAc]2 as metal precursor in tBuOH as solvent cleanly yields the desired trimethylsilyl-bearing Cpx complexes 2m–2oas single diastereoisomers (Scheme 4). Similarly, the same method is applicable to access the corresponding iridium complexes 3m–3o. No proto-desilylated complexes (3a, 3f–3g) were observed. Interestingly, despite the increased steric bulk of the TMS-cyclopentadienes, the complexations proceed at ambient temperature, pointing to a low energy barrier. Both, the regioselectivity of the silylation reaction of the parent chiral cyclopentadiene and the subsequent diastereoselective complexation were unequivocally confirmed by single crystal X-ray diffraction analysis of iridium complexes 3m and 3o (Fig. 2).17
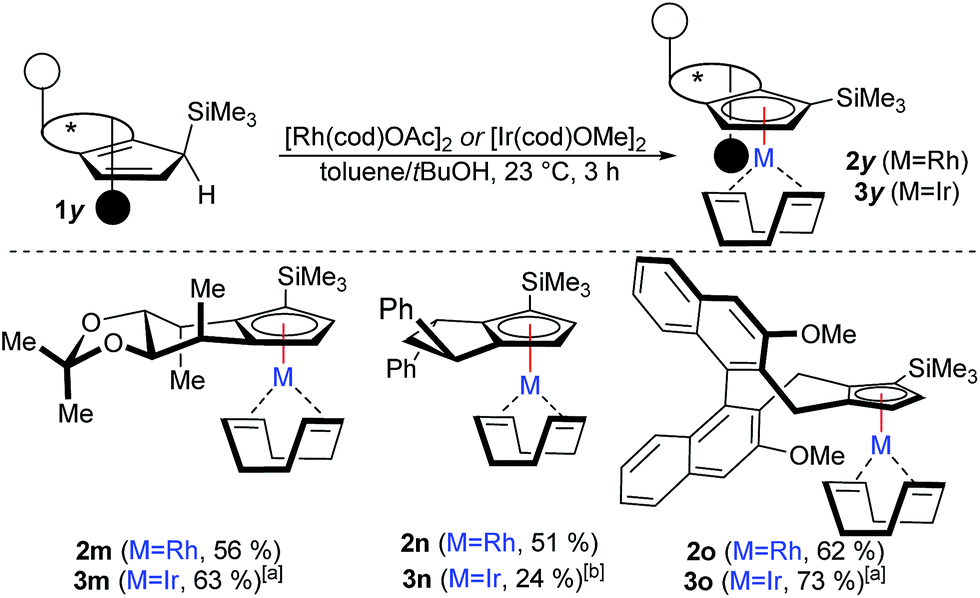 | ||
| Scheme 4 Diastereoselective complexation with TMS-Cpx ligands: 0.1 mmol 1y, 0.06 mmol [Rh(cod)OAc]2, toluene/tBuOH at 23 °C for 3 h. (a) With [Ir(cod)OMe]2 (b) with [Ir(cod)OAc]2. | ||
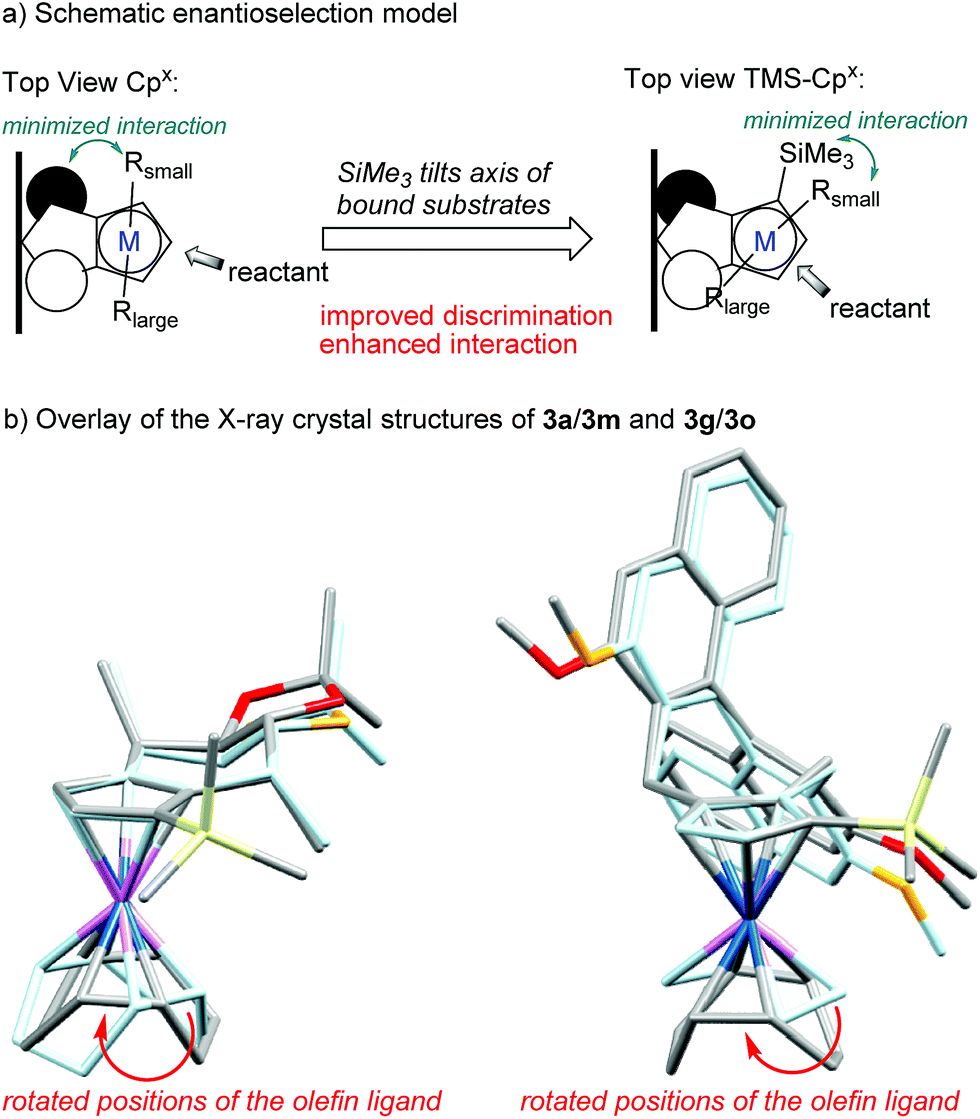 | ||
| Fig. 2 Overlay of the X-ray crystal structures reveals the influence of the TMS group on the axis of the olefin ligands (faint blue: parent Cpx-complexes without TMS group). | ||
The proximity of the bulky trimethylsilyl group to the steering pseudoaxial methyl group of 3m leads to extended sidewalls. Comparison of the X-ray crystal structures of 3a 5i and 3m visualizes well the enhanced shielding caused by the installed ligand feature (Fig. 2). While the TMS group slightly influences the chiral backbone, it has a profound effect on the orientation of alkene ligands on the metal (axis with respect to the chiral selector backbone). Consequently, on a substrate bound to the metal, the small fragment (Rsmall) would minimize the interactions with the TMS function, while the bulkier part (Rlarge) would stay in the open area of the Cpx. The bulk of the silyl group forces an orientation closer to the chiral selector which could result in an enhanced interaction and improved discrimination. Hence, the introduction of the TMS as extended side wall could help pre-organization around the metal center thus improving enantio- and regioselectivity compared to the parent ligand.
The performance of complex 2m was benchmarked in the enantioselective dihydroisoquinolinone synthesis.18 Compared to the previously best performing catalyst (having the diphenyl acetal which is cumbersome to access), an equivalent selectivity was obtained for styrene as the coupling partners (Scheme 5). Dihydroisoquinolinone 6b, a precursor for chiral 1,3,2,-diazaphospholenes,19 was obtained in 80% yield with high 93:7 er. Importantly, a significant selectivity improvement was observed in reactions with challenging cyclic olefins. Complex 2m outperformed all previously reported complexes.4f,7d Reaction with 1,3-cyclohexadiene afforded 6c in 95:5 er (91.5:8.5 previously). Similarly, reactions with cyclopentene and dihydrofurane gave 6d (95:5 er) and 6e (97:3 er) in significantly improved enantioselectivity. Notably, norbornene reacted with an excellent selectivity of 99:1 er, outperforming Perekalin's planar chiral catalyst.20 Importantly, the selectivity improved as well for challenging vinyltrimethylsilane (90:10 er), while maintaining a similar level of regioselectivity. Even with the new catalytic system, terminal alkyl alkenes remain notoriously challenging substrates. Notably a regioselectivity of 2.4:1 was observed, while previously a 1:1 mixture was generated.21 It seems likely that with further fine-tuning of this bulky trisubstituted Cpx ligand class, regio- and enantioselectivities could be further boosted.
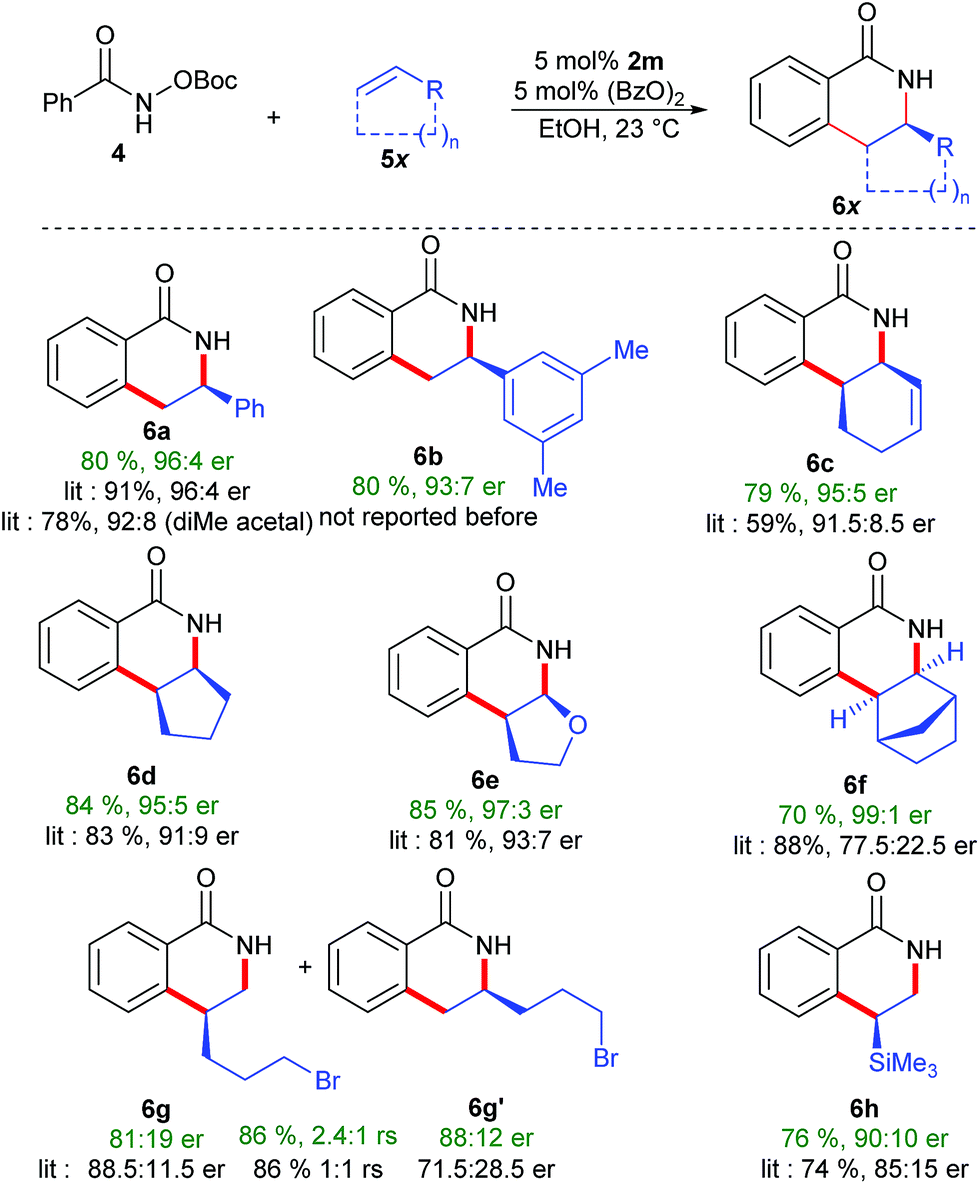 | ||
| Scheme 5 Conditions: 0.1 mmol 4, 0.2 mmol 5, 5 μmol 2m, 5 μmol (BzO)2 in EtOH (1 M) at 23 °C. | ||
The development of protocols for an in situ complexation, meaning the addition of the CpxH ligand (or a suitable surrogate) and the metal salt altogether with the substrates and additives for the desired catalysis is a high priority, due to its simplicity and accessibility to non-specialists. The outlined complexation has several advantageous characteristics. It proceeds sufficiently fast in almost quantitative manner under mild conditions. The only generated by-products are either catalytic amounts of alcohols (common solvents) or carboxylic acids. In particular for rhodium(III)-catalyzed functionalizations, carboxylic acids or carboxylates are frequently added additives that are essential for reactivity.22 We examined four different bench-mark reactions for their in situ complexation suitability (Scheme 6). For instance, the enantioselective formation of dihydroisoquinolone 6a by the in situ approach proceeded in comparable yield and enantioselectivity compared to the usage of the preformed Cpx-rhodium catalyst.4f In a similar manner, the in situ complexation with the BINOL-OiPr Cpx1h, follow by the oxidation to Rh(III), allows the synthesis of insoindolone 8.10a Besides substrates with internal oxidants, we investigated the compatibility of common oxidants such as Ag2CO3 and Cu(OAc)2. In this respect, the desymmetrization of phosphinamide 9 proceeded smoothly.23 Replacing the common [Rh(cod)OAc]2 by [Rh(cod)OBz]2 did not influence the assembly of the catalyst but is essential for obtaining high enantioselectivity. Moreover, the cyclooctadiene of [Rh(cod)OAc]2 the can be replaced by two ethylene groups ([Rh(C2H4)2OAc]2). This resulted in a more reactive catalyst for You's reaction of pyrazolone 11 with diphenyl acetylene.24 Again, the in situ method provided comparable results.
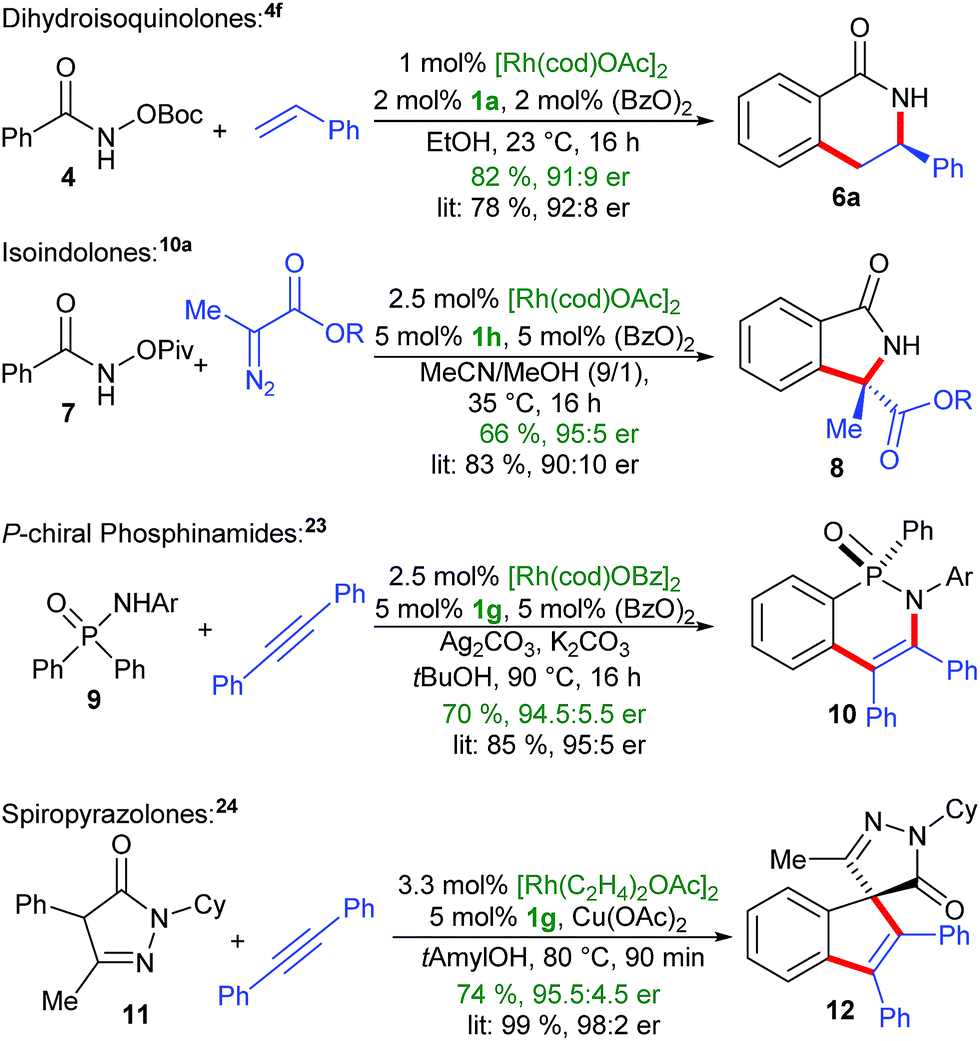 | ||
| Scheme 6 Application of in situ CpxRh-catalyst assembly under different benchmark reactions. Ar = 3,5-CF3-C6H3, R = CH(iPr)2. | ||
Conclusions
In summary, we disclosed a convenient and general complexation method to access chiral Cpx iridium and rhodium complexes. The method has large advance over previous methods as it uses the free CpxH in combination with stable and commercial rhodium(I) and iridium(I) salts without the addition of base or any additive. Moreover, the conditions are mild and the reactions can be conductive without precautions to exclude air and moisture. DFT-calculations revealed two potential pathways with a low energy barrier in which either a bound methoxide or acetate is involved in an intramolecular proton abstraction of the coordinated cyclopentadiene. Moreover, the selectivity and mildness of the complexation method enabled the development of new TMS-containing Cpx ligands which displayed superior selectivity for benchmarking dihydroisoquinolone synthesis. Another salient feature is its suitability for in situ complexation which significantly enhances the user-friendliness of Cpx ligands. This holds the potential to open the versatile catalytic application with designer ligands to a broader community.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the EPFL and the Swiss National Science Foundation (Consolidator Grant No. 157741). M. D. W. thanks Prof. C. Corminboeuf for financial support and access to computational resources. We thank Dr R. Scopelliti and Dr F. Fadaei Tirani for X-Ray crystallographic analysis of compounds 1m, 3a, 3m and 3o.Notes and references
- J. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, CA, 2010 Search PubMed.
- (a) T. Piou, F. Romanov-Michailidis, M. Romanova-Michaelides, K. E. Jackson, N. Semakul, T. D. Taggart, B. S. Newell, C. D. Rithner, R. S. Paton and T. Rovis, J. Am. Chem. Soc., 2017, 139, 1296–1310 CrossRef CAS PubMed; (b) A. Davis, C. Wang and T. Rovis, Synlett, 2015, 26, 1520–1524 CrossRef PubMed; (c) T. Piou and T. Rovis, J. Am. Chem. Soc., 2014, 136, 11292–11295 CrossRef CAS PubMed; (d) T. Piou and T. Rovis, Nature, 2015, 527, 86–90 CrossRef CAS PubMed; (e) F. Romanov-Michailidis, K. F. Sedillo, J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2015, 137, 8892–8895 CrossRef CAS PubMed; (f) T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 11846–11848 RSC; (g) Y. Shibata and K. Tanaka, Angew. Chem., Int. Ed., 2011, 50, 10917–10921 CrossRef CAS PubMed.
- (a) T. K. Hyster, D. M. Dalton and T. Rovis, Chem. Sci., 2014, 6, 254–258 RSC; (b) T. K. Hyster and T. Rovis, Chem. Sci., 2011, 2, 1606–1610 RSC; (c) J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2014, 136, 2735–2738 CrossRef CAS PubMed; (d) M. D. Wodrich, B. Ye, J. F. Gonthier, C. Corminboeuf and N. Cramer, Chem.–Eur. J., 2014, 20, 15409–15418 CrossRef CAS PubMed.
- T. Piou, F. Romanov-Michailidis, M. A. Ashley, M. Romanova-Michaelides and T. Rovis, J. Am. Chem. Soc., 2018, 140, 9587–9593 CrossRef CAS PubMed.
- (a) C. G. Newton, D. Kossler and N. Cramer, J. Am. Chem. Soc., 2016, 138, 3935–3941 CrossRef CAS PubMed; (b) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308–1318 CrossRef CAS PubMed; (c) G. Erker and A. A. H. van der Zeijden, Angew. Chem., Int. Ed., 1990, 29, 512–514 CrossRef; (d) A. Gutnov, B. Heller, C. Fischer, H.-J. Drexler, A. Spannenberg, B. Sundermann and C. Sundermann, Angew. Chem., Int. Ed., 2004, 43, 3795–3797 CrossRef CAS PubMed; (e) T. K. Hyster, L. Knörr, T. R. Ward and T. Rovis, Science, 2012, 338, 500–503 CrossRef CAS PubMed; (f) B. Ye and N. Cramer, Science, 2012, 338, 504–506 CrossRef CAS PubMed; (g) D. Kossler and N. Cramer, J. Am. Chem. Soc., 2015, 137, 12478 CrossRef CAS PubMed; (h) G. Song, W. W. N. O and Z. Hou, J. Am. Chem. Soc., 2014, 136, 12209–12212 CrossRef CAS PubMed; (i) M. Dieckmann, Y.-S. Jang and N. Cramer, Angew. Chem., Int. Ed., 2015, 54, 12149–12152 CrossRef CAS PubMed.
- (a) R. Uson, L. A. Oro, J. A. Cabeza, H. E. Bryndza and M. P. Stepro, in Inorg. Synth., John Wiley & Sons, Inc., 2007, pp. 126–130 Search PubMed; (b) M. Arthurs, H. K. Al-Daffaee, J. Haslop, G. Kubal, M. D. Pearson, P. Thatcher and E. Curzon, J. Chem. Soc., Dalton Trans., 1987, 2615–2619 RSC; (c) B. M. Trost and C. M. Older, Organometallics, 2002, 21, 2544–2546 CrossRef CAS; (d) M. A. Mantell, J. W. Kampf and M. Sanford, Organometallics, 2018, 37, 3240–3242 CrossRef CAS.
- (a) S.-G. Wang, S. H. Park and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 5459–5462 CrossRef CAS PubMed; (b) B. Ye and N. Cramer, J. Am. Chem. Soc., 2013, 135, 636–639 CrossRef CAS PubMed; (c) J. Zheng, W.-J. Cui, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2016, 138, 5242–5245 CrossRef CAS PubMed; (d) Z.-J. Jia, C. Merten, R. Gontla, C. G. Daniliuc, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2017, 56, 2429–2434 CrossRef CAS PubMed.
- (a) W.-H. Wang, Y. Suna, Y. Himeda, J. T. Muckerman and E. Fujita, Dalton Trans., 2013, 42, 9628–9636 RSC; (b) S. Yoshizaki, Y. Shibata and K. Tanaka, Angew. Chem., Int. Ed., 2017, 56, 3590–3593 CrossRef CAS PubMed.
- G. Smits, B. Audic, M. D. Wodrich, C. Corminboeuf and N. Cramer, Chem. Sci., 2017, 8, 7174–7179 RSC.
- (a) B. Ye, P. A. Donets and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 507–511 CrossRef CAS PubMed; (b) B. Ye and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 7896–7899 CrossRef CAS PubMed; (c) Y.-S. Jang, M. Dieckmann and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 15088–15092 CrossRef CAS PubMed; (d) T. J. Potter, D. N. Kamber, B. Q. Mercado and J. A. Ellman, ACS Catal., 2017, 7, 150–153 CrossRef CAS PubMed; (e) J. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2014, 53, 13244–13247 CrossRef CAS PubMed; (f) J. Zheng, S.-B. Wang, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2015, 137, 4880–4883 CrossRef CAS PubMed.
- Y. Sun and N. Cramer, Chem. Sci., 2018, 9, 2981–2985 RSC.
- R. N. Haszeldine, R. J. Lunt and R. V. Parish, J. Chem. Soc. A, 1971, 3696–3698 RSC.
- (a) J. E. Bercaw, N. Hazari and J. A. Labinger, Organometallics, 2009, 28, 5489–5492 CrossRef CAS; (b) T. S. Ahmed, I. A. Tonks, J. A. Labinger and J. E. Bercaw, Organometallics, 2013, 32, 3322–3326 CrossRef CAS.
- C. M. Filloux and T. Rovis, J. Am. Chem. Soc., 2015, 137, 508–517 CrossRef CAS PubMed.
- H.-L. Teng, Y. Luo, B. Wang, L. Zhang, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2016, 55, 15406–15410 CrossRef CAS PubMed.
- (a) J. A. Klang and D. B. Collum, Organometallics, 1988, 7, 1532–1537 CrossRef CAS; (b) C. H. Winter, S. Pirzad, D. D. Graf, D. H. Cao and M. J. Heeg, Inorg. Chem., 1993, 32, 3654–3659 CrossRef CAS.
- CCDC 1869889 (3a), CCDC 1869894 (1m), CCDC 1869888 (3m) and CCDC 1869893 (3o) contains the supplementary crystallographic data for this paper.†.
- N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449–6457 CrossRef CAS PubMed.
- S. Miaskiewicz, J. H. Reed, P. A. Donets, C. C. Oliveira and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 4039–4042 CrossRef CAS PubMed.
- E. A. Trifonova, N. M. Ankudinov, A. A. Mikhaylov, D. A. Chusov, Y. V. Nelyubina and D. S. Perekalin, Angew. Chem., Int. Ed., 2018, 57, 7714–7718 CrossRef CAS PubMed.
- B. Ye, PhD thesis, Ecole Polytechnique Fédérale de Lausanne, 2015.
- Recent examples: (a) S. Wu, X. Wu, C. Fu and S. Ma, Org. Lett., 2018, 20, 2831–2834 CrossRef CAS PubMed; (b) F. Xu, W.-F. Kang, Y. Wang, C.-S. Liu, J.-Y. Tian, R.-R. Zhao and M. Du, Org. Lett., 2018, 20, 3245–3249 CrossRef CAS PubMed; (c) Y. Li, D. Shi, Y. Tang, X. He and S. Xu, J. Org. Chem., 2018, 83, 9464–9470 CrossRef CAS PubMed.
- Y. Sun and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 364–367 CrossRef CAS PubMed.
- J. Zheng, S.-B. Wang, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2017, 56, 4540–4544 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization of all new compounds. CCDC 1869889, 1869894, 1869888 and 1869893. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04385j |
| This journal is © The Royal Society of Chemistry 2019 |