
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective synthesis of gem-disubstituted N-Boc diazaheterocycles via decarboxylative asymmetric allylic alkylation†
Alexander W.
Sun‡
 ,
Stephan N.
Hess‡
and
Brian M.
Stoltz
*
,
Stephan N.
Hess‡
and
Brian M.
Stoltz
*
Warren and Katherine Schlinger Laboratory for Chemistry and Chemical Engineering, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA. E-mail: stoltz@caltech.edu
First published on 31st October 2018
Abstract
An enantioselective synthesis of diverse N4-Boc-protected α,α-disubstituted piperazin-2-ones using the palladium-catalyzed decarboxylative allylic alkylation reaction has been achieved. Using a chiral Pd-catalyst derived from an electron deficient PHOX ligand, chiral piperazinones are synthesized in high yields and enantioselectivity. The chiral piperazinone products can be deprotected and reduced to valuable gem-disubstituted piperazines. This reaction is further extended to enable the enantioselective synthesis of α,α-disubstituted tetrahydropyrimidin-2-ones, which are hydrolyzed into corresponding chiral β2,2-amino acids.
Introduction
Nitrogen containing heterocycles are ubiquitous components of biologically active small molecules.1 For instance, piperazine, a representative diazaheterocycle, is the third most prevalent heterocycle in small molecule pharmaceuticals1 and can be found in bioactive molecules such as the antiviral natural product herquline A,2 the blockbuster psychiatric drug aripiprazole,3 and the HIV integrase inhibitor dolutegravir4 (as the oxidized counterpart, piperazin-2-one) (Fig. 1).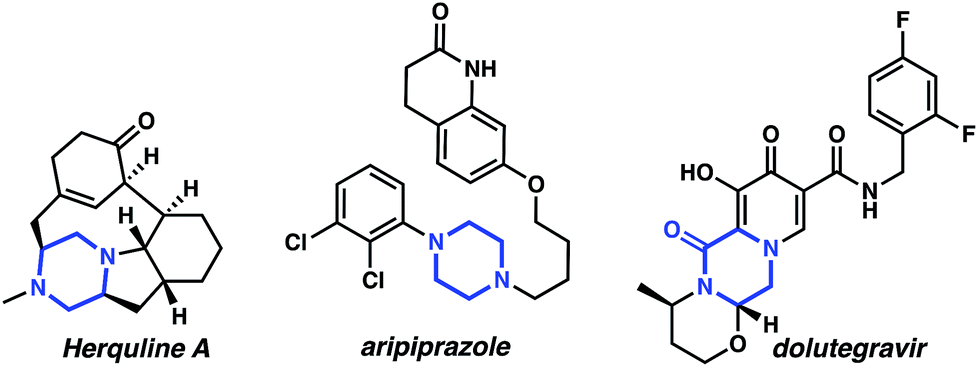 | ||
| Fig. 1 Representative piperazine and piperazine-2-one pharmaceuticals and natural products. | ||
In the field of medicinal chemistry and fragment-based drug discovery, an emerging paradigm to enhance drug-like properties of small molecules involves reducing molecular flatness by incorporating chiral centers.5 This method of increasing molecular complexity can enhance binding affinity and specificity for a target, as receptor–ligand binding is often defined by three dimensional contacts.5b Furthermore, depending on the chemical composition of the chiral center, other properties such as metabolism, aqueous solubility, and cell-penetration may be optimized.5a The prevalence of piperazine in bioactive molecules makes it an excellent scaffold upon which to introduce chirality and increase molecular complexity. Chiral gem-disubstitution on any of four carbon atoms in the piperazine ring could dramatically alter physicochemical properties (Fig. 2). Despite this potential, there are no gem-disubstituted piperazines among the current 98 FDA approved piperazine-containing non-natural product derived drugs.6 In contrast, a significant number contain mono-substituted piperazines, which can be prepared through a variety of strategies,7 including cyclization of chiral precursors,7b–d enantioselective hydrogenation of pyrazines,7e,f palladium-catalyzed cyclizations,7g–i or asymmetric lithiation.7j,k
 | ||
| Fig. 2 Advantages of chiral gem-disubstituted piperazines. | ||
The dearth of piperazine gem-disubstitution in the pharmacopoeia highlights the significant challenge of installing fully substituted chiral centers into diazaheterocycles.8 The state of the art is perhaps best exemplified by the Bode laboratory's extensive development of tin (SnAP) and silicon (SLAP) coupling reagents to generate spiro-substituted piperazines (Scheme 1a).9 However, this method is not yet enantioselective and involves potentially toxic tin reagents. Other non-enantioselective methods to synthesize gem-disubstituted piperazines include [4 + 2] cycloadditions of 1,2 diamines with propargyl alcohols developed by Rawal (Scheme 1b),7g and the ring expansion of oxetane spirocycles by Carreira (Scheme 1c).10
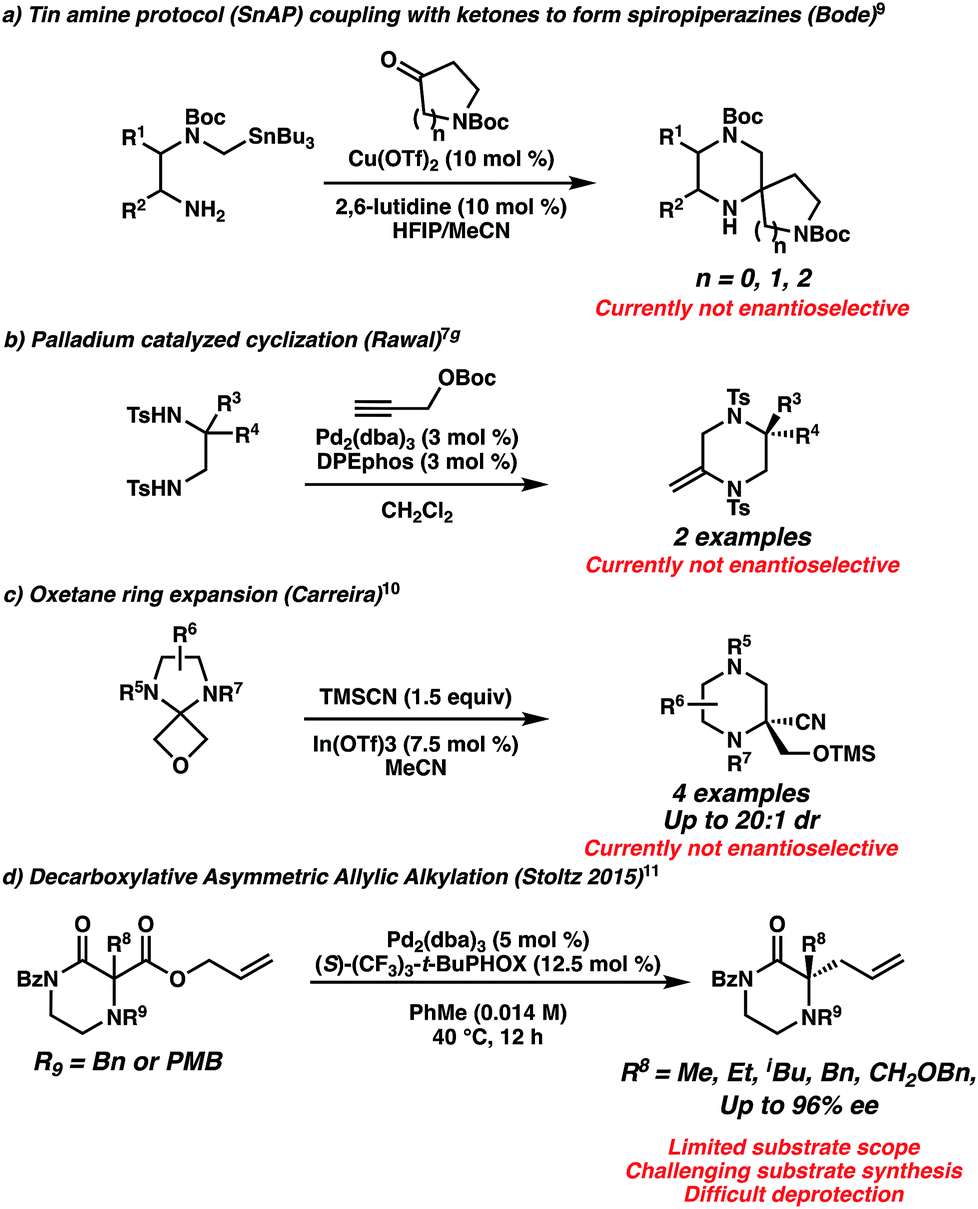 | ||
| Scheme 1 Existing approaches for gem-disubstituted piperazines and piperazinones. | ||
To our knowledge, the only method enabling the highly enantioselective synthesis of gem-disubstituted piperazines was published by our group in 2015.11 We leveraged the palladium-catalyzed decarboxylative asymmetric allylic alkylation reaction to synthesize chiral α,α-disubstituted N4-benzylated piperazin-2-ones (Scheme 1d). These piperazin-2-ones could then be reduced into desirable chiral gem-disubstituted piperazines. However, we encountered two issues during the course of our attempts to utilize this method to synthesize disubstituted piperazines for medicinal chemistry purposes: first, the optimal nucleophilic benzyl-protected N4 significantly complicated substrate synthesis via nucleophilic side reactions, leading to low yields of products. As a result, we failed to synthesize many desired substrates via standard enolate functionalization of the dicarbonyl precursor. Instead, we resorted to a low-yielding 5-step convergent sequence beginning from an amino acid (Scheme 2a). As a result, our substrate scope displayed limited functional group diversity. Second, in the course of working with a N4-PMB-protected piperazin-2-one, we failed to remove the recalcitrant PMB group using a variety of common cleavage conditions that were orthogonal to the sensitive allyl functionality.
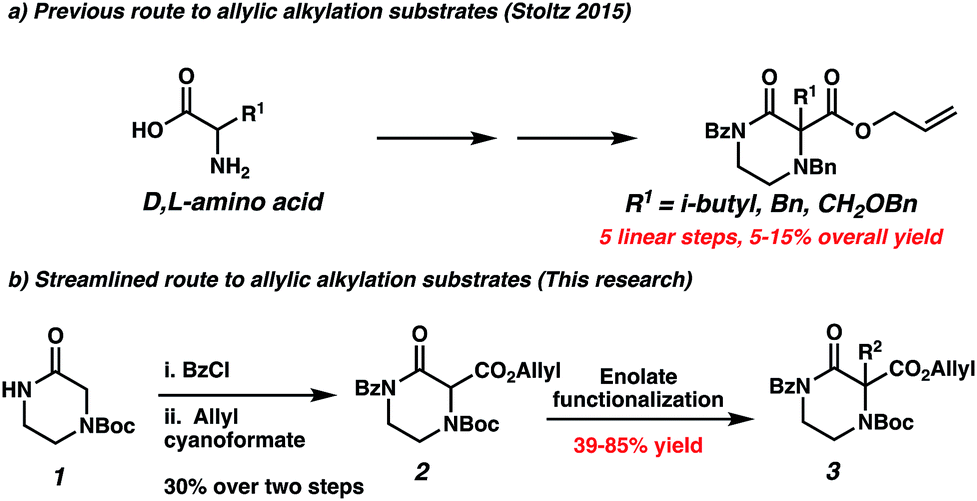 | ||
| Scheme 2 Comparison of routes to piperazin-2-one decarboxylative alkylation substrates. | ||
Results and discussion
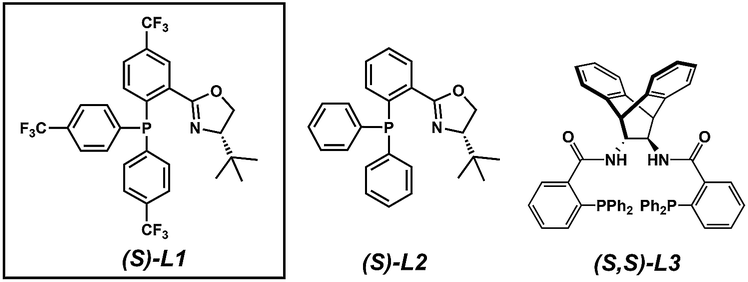
The substrate-dependent drawbacks of our approach leave room for an improved method to synthesize chiral disubstituted piperazines. In an effort to expand upon our group's longstanding interest in transition-metal catalyzed allylic alkylation, we sought to identify an alternative protecting group that could overcome the aforementioned limitations by enabling both facile substrate synthesis and protecting group cleavage. After extensive explorations directed toward this goal, we were pleased to discover that replacing the N4-benzyl protecting group with a simple Boc (tert-butoxycarbonyl) protecting group addressed the issue of difficult substrate synthesis. Presumably, the electron-withdrawing Boc group attenuates the nucleophilicity of N4, enabling divergent access to a wider array of substrates 3via enolate functionalization of dicarbonyl compound 2, which could be synthesized in only two steps from Boc-piperazinone 1 (Scheme 2b, see the ESI† for full synthetic details).With a streamlined approach toward a wide range of decarboxylative alkylation substrates, we began optimizing the reaction conditions: with model substrate 3m, conditions based on our previous report11 using 10 mol% (S)-(CF3)3-t-BuPHOX ligand and 4 mol% Pd2(pmdba)3 in 0.014 M toluene gave the product 3m in only 76% ee (Table 1, entry 1). Other commonly used allylic alkylation ligands such as (S)-t-BuPHOX (L2) and (S,S)-ANDEN-Ph Trost (L3) were tested, both giving suboptimal enantioselectivities (entries 2 and 3). We then examined the effect of solvent on the enantioselectivity (entries 4–6), ultimately finding that 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hexanes–toluene provided high yield and ee.
:1 hexanes–toluene provided high yield and ee.
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Solvent | Yieldb (%) | eec (%) |
| a Screens performed on a 0.04 mmol scale. b All reported yields are for isolated products. c The ee values were determined by chiral SFC analysis. Bz = benzoyl, Boc = tert-butoxycarbonyl, pmdba = bis(4-methoxybenzylidene)acetone. | ||||
| 1 | L2 | Tol | 75 | 76 |
| 2 | L2 | Tol | 70 | 1 |
| 3 | L3 | Tol | 83 | 52 |
| 4 | L1 | THF | — | 13 |
| 5 | L1 | MTBE | — | 80 |
| 6 | L1 | 2:1 Hex/Tol |
93 | 93 |
|
||||
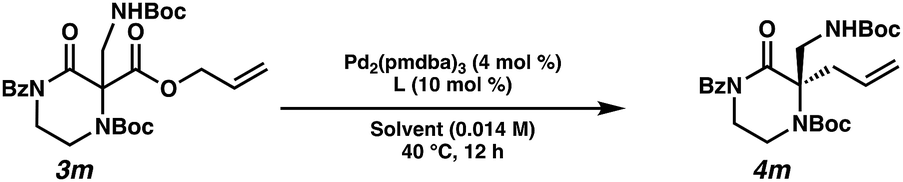
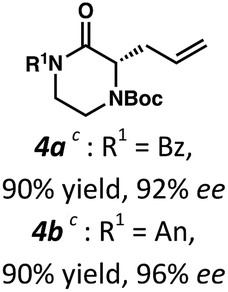
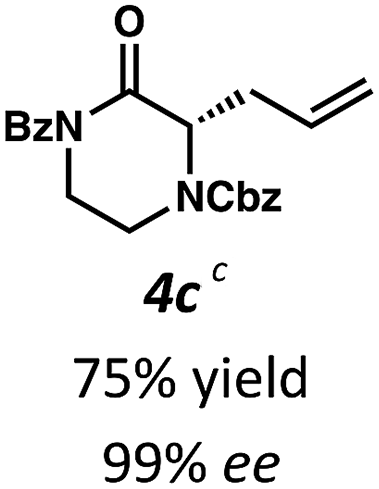
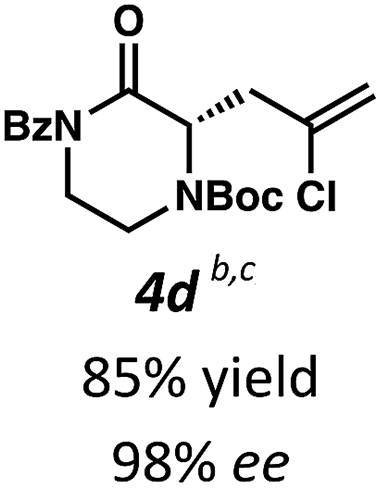
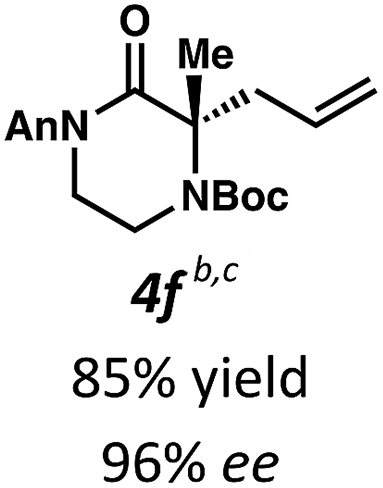
Employing these satisfactory reaction conditions, we then explored the substrate scope of the decarboxylative alkylation of various N4-Boc protected piperazinones (Table 2). We first tested the α-monosubstituted piperazinone 3a, finding that typical conditions in a 0.033 M solution of toluene afforded the product 4a in high yield and enantioselectivity. Another monosubstituted piperazinone 4b with a N1-anisoyl protecting group, was also obtained in high yield and ee. Furthermore, replacement of the N4-Boc protecting group with Cbz (3c), and 2-chloro substitution of the allyl group (3d) similarly provided good results.
|
|
|||
|---|---|---|---|
|
a Conditions: piperazin-2-one 3 (1.0 equiv.), Pd2(dba)3 (4 mol%), (S)-(CF3)3-tBuPHOX (10 mol%), in 2:1 hexanes/toluene (0.014 M) at 40 °C for 12–24 h.
b Pd2(pmdba)3 (4 mol%) instead of Pd2(dba)3.
c Toluene (0.033 M) instead of 2:1 hexanes/toluene. All reported yields are for isolated products. The ee values were determined by chiral SFC analysis. An = 4-methoxybenzoyl, dba = dibenzylideneacetone.
|
|||
|
|
|
|
|
|
|

|
|
|
|
|
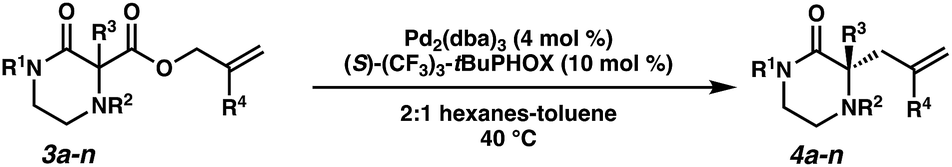
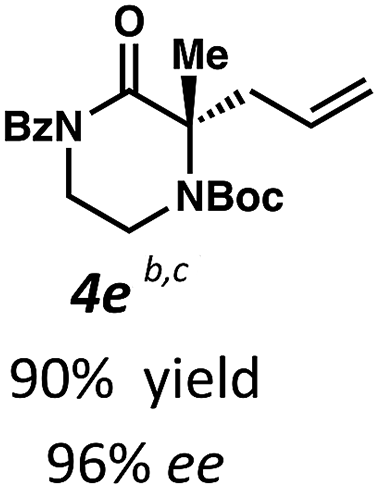
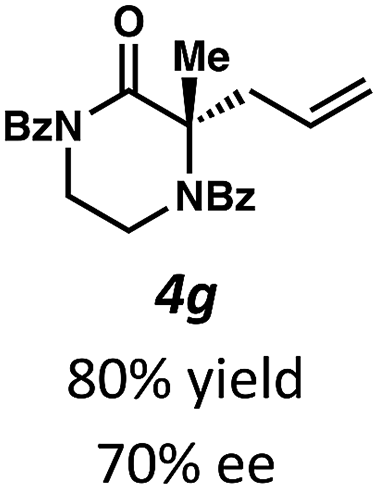
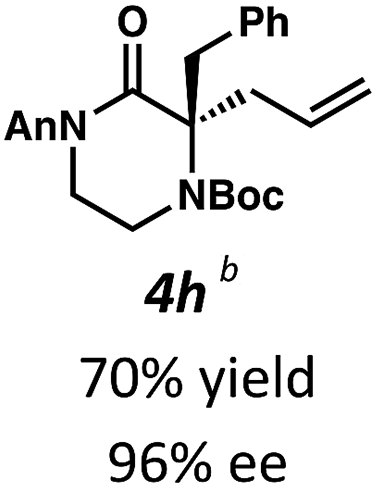
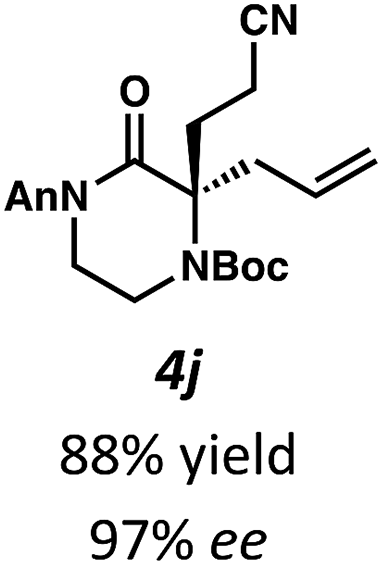
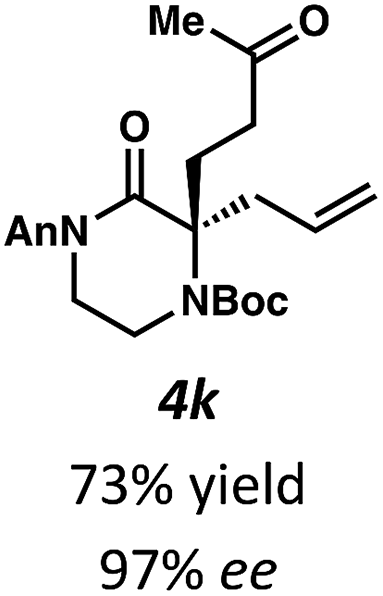
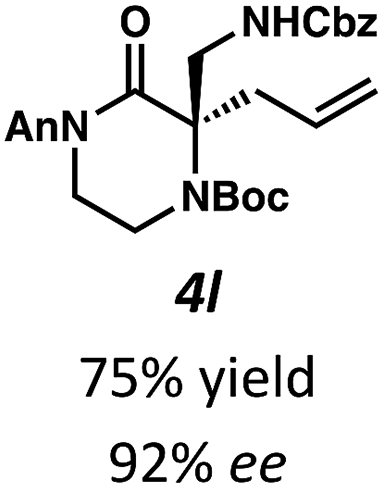
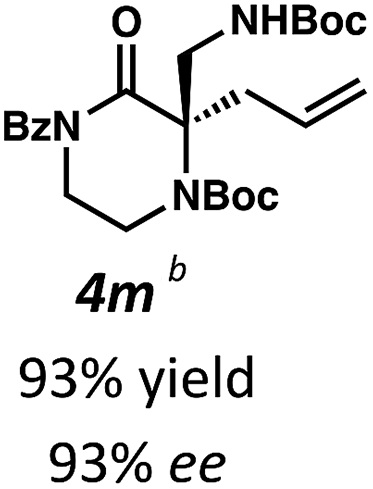
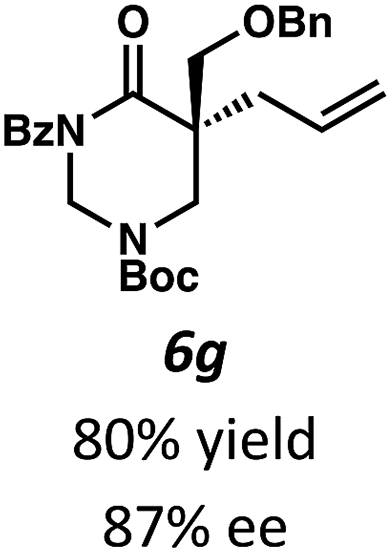
Next, we examined α,α-disubstituted substrates, finding that for the simple cases of methyl substitution (3e, 3f), performing the reaction at 0.033 M concentration in toluene gave high yields and ee, with no improvement with the use of 2:1 hexanes–toluene at 0.014 M concentration. The exception was N4-benzoyl protected substrate 3g, which in our previous work gave 52% ee in toluene;11 under our optimized conditions in 2:1 hexanes–toluene, the substrate gave an improved albeit modest ee of 70%. Larger substituents such as α-benzylated compound 3h or benzyloxymethyl ether 3i required the use of 2:1 hexanes–toluene to achieve high enantioselectivity. This unique mixed-solvent requirement was observed in all substituents other than methyl. A wide range of functional groups was tolerated: notably, the nitrile, ketone, and methylcarbamate substrates, which could not be accessed in our previously described efforts, gave high ee and yields (3j–m).
With the N-Boc piperazin-2-one substrates providing excellent results, we wondered whether an isomeric substrate class, the N-Boc tetrahydropyrimidin-2-ones (5), would perform similarly as well (Scheme 3C). The resulting α,α-disubstituted tetrahydropyrimidin-2-one products (6) are especially interesting as they may be hydrolyzed to afford valuable β2,2-amino acids (7), which are used in peptidomimetic therapeutics and in the design of unnatural peptides and proteins with unique secondary and tertiary structures.12 In fact, there is strong precedent for an allylic alkylation approach toward β2,2-amino acids. In 2018 the Shibasaki group described the decarboxylative allylic alkylation of an N,O heterocycle to set the quaternary stereocenter, followed by N–O bond heterolysis to generate β2,2-amino acids (Scheme 3A).13a Later in 2018, Cossy and co-workers described a similar approach involving intermolecular allylic alkylation (Scheme 3B).13b Notably, the substrate scope for these two examples is limited to mostly α-benzylic and α-aryl compounds. If decarboxylative alkylation could be achieved with the versatile tetrahydropyrimidin-2-one scaffold, a broader range of chiral β2,2-amino acids might be accessible (Scheme 3C). Additionally, tetrahydropyrimidinones may be reductively transformed into hexahydropyrimidines, which are found in bioactive molecules such as the antibiotic hexetidine and the macrocyclic natural product verbamethine.14
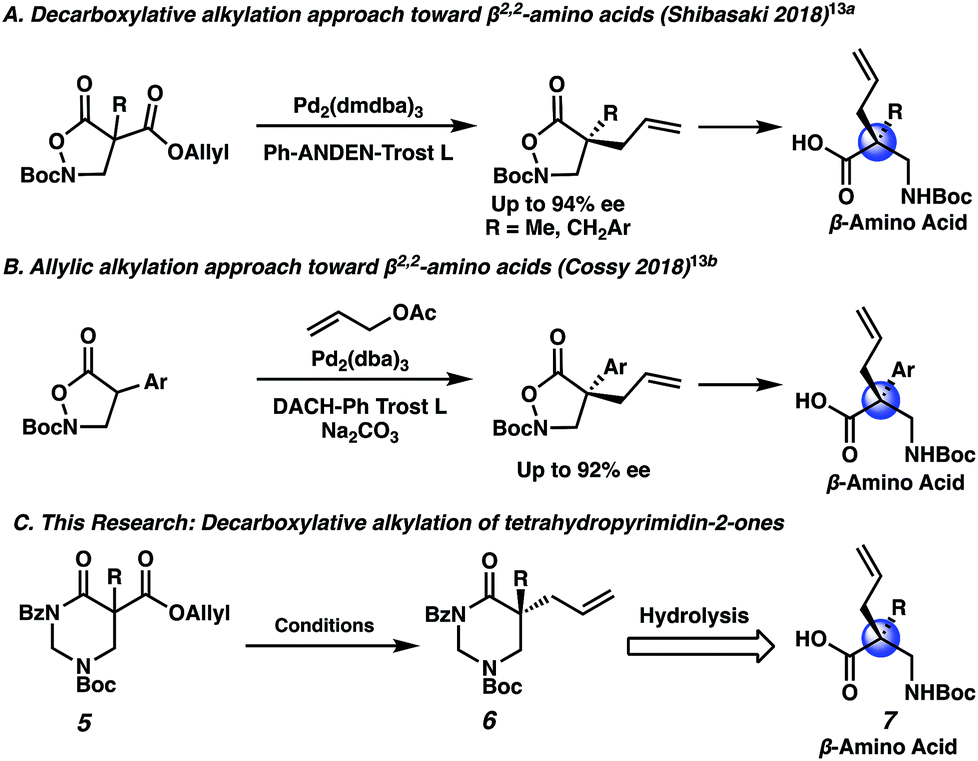 | ||
| Scheme 3 Synthetic approaches toward β2,2-amino acids using the allylic alkylation reaction. | ||
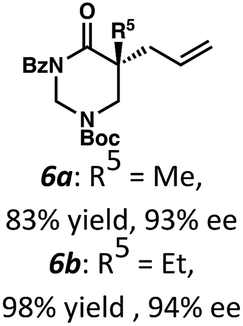
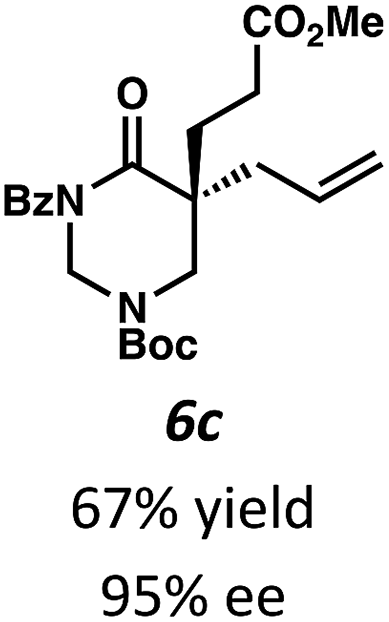
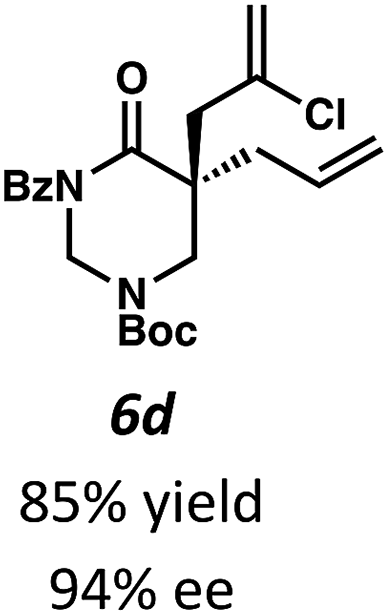
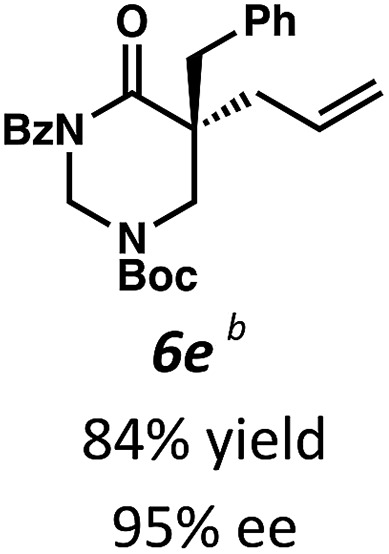
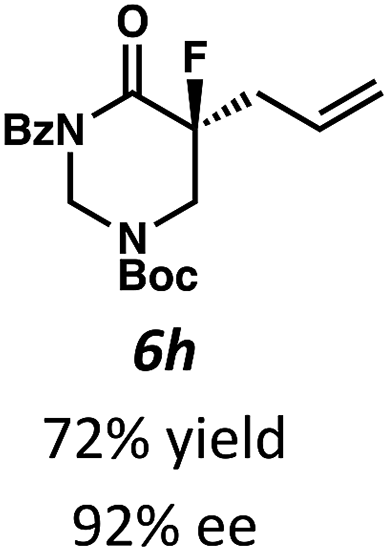
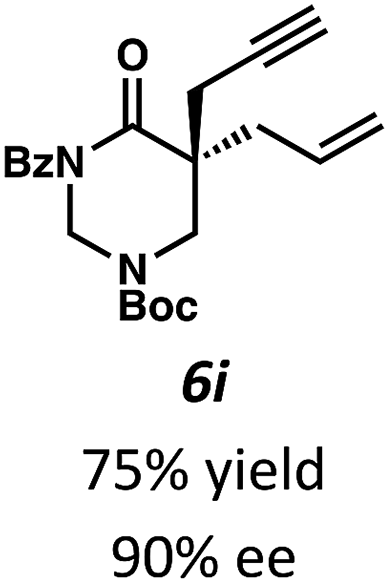
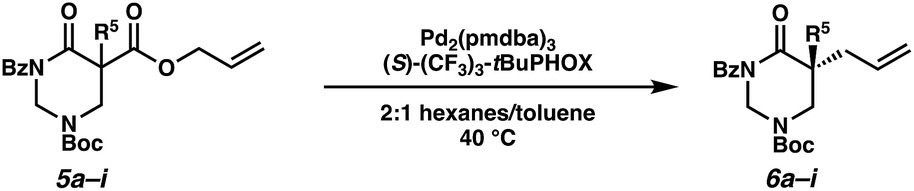
Beginning our investigations with methyl substrate 5a (Table 3), we were pleased to find that our optimized conditions in 2:1 hexanes–toluene furnished the desired decarboxylative alkylation product 6a in high yield and ee. The reaction proved amenable to a variety of α-substituents, including ethyl (6b), methyl ester (6c), 2-chloroallyl (6d), and benzyl (6e). Furthermore, the reaction is scalable, with the benzyl substrate 5e giving good results in a 1 gram reaction. In contrast, the nitrile and benzyloxymethyl ether products 6f and 6g were isolated with reduced enantioselectivities. Pleasingly, the fluorine and propargyl substrates 5h and 5i performed well, and may serve as a precursor to a novel fluorinated β2,2-amino acid, or for biorthogonal click reactions, respectively.
|
|
|||
|---|---|---|---|
|
a Conditions: tetrahydropyrimidin-2-one 5 (1.0 equiv.), Pd2(pmdba)3 (4 mol%), (S)-(CF3)3-tBuPHOX (10 mol%), in 2:1 hexanes/toluene (0.014 M) at 40 °C for 12–24 h. All reported yields are for isolated products.
b Reaction performed with 1 gram of substrate 5e. The ee values were determined by chiral SFC analysis.
|
|||
|
|
|
|

|
|
|
|
Having demonstrated excellent results for the decarboxylative alkylation of piperazin-2-ones and tetrahydropyrimidin-2-ones, we began exploring their synthetic utility. In contrast to the relative difficulty of removing the N4-benzyl type protecting groups in our previous work, both Boc groups of 4m were easily removed by treatment with excess TFA to give aminopiperazinone 8 (Scheme 4a). With LiOH, the benzoyl group could be orthogonally removed to provide the free amide 9 (Scheme 4b). The amide 9 could then be reduced with LiAlH4 to the corresponding chiral aminopiperazine 10. To further illustrate the synthetic versatility realized by Boc-deprotection of N4, we hydrogenated the allyl olefin of methyl piperazinone 4e before cleaving the Boc group to obtain 11. We then applied Gaunt's15 method of C–H carbonylation on the aliphatic amine of 11 to forge the fused β-lactam 12, which bears resemblance to the core of various β-lactam antibiotics and β-lactamase inhibitors (Scheme 4c). Lastly, we transformed four tetrahydropyrimidinone substrates, 6c–e and 6h, into their corresponding acyclic forms (Scheme 4d, e). Using a two-step protocol involving TFA-mediated Boc cleavage followed by saponification with LiOH, we successfully obtained the crude β2,2-amino acid (Scheme 4d); to facilitate silica gel chromatographic isolation, we chose to mask the free amine and carboxylic acid with Boc and benzyl groups, respectively, resulting in protected β2,2-amino acid 13. We note that in this four step sequence, we only performed one chromatographic separation to isolate the protected β-amino acid. In contrast, novel unprotected β2,2-amino acids 14–16 bearing pendant carboxylic acid, chloroallyl, and fluorine atom functional groups could be obtained directly by purification with reverse phase preparatory HPLC following the two-step deprotection sequence (Scheme 4e).
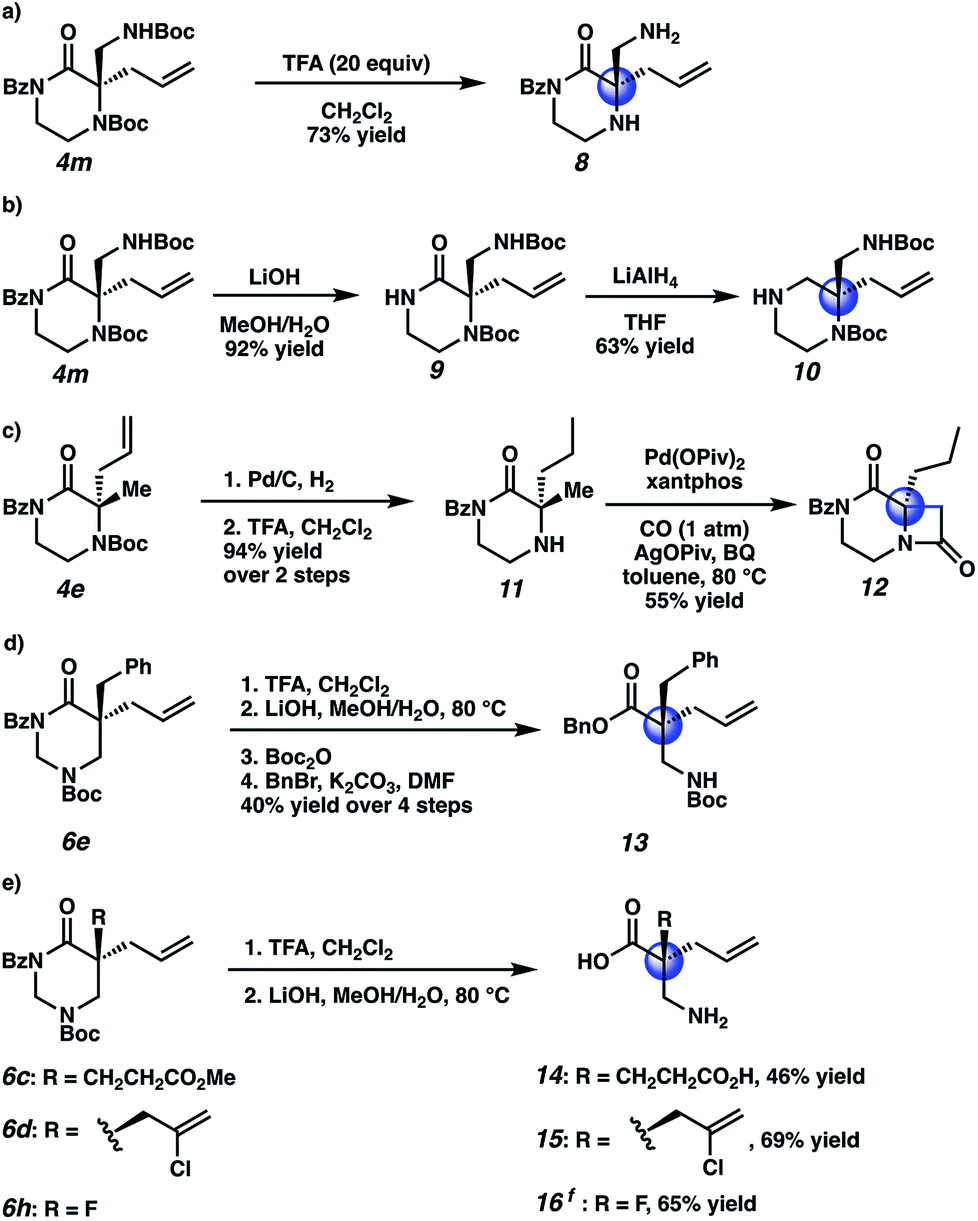 | ||
| Scheme 4 Product derivatization: (a) Boc cleavage to the free amine. (b) Benzoyl cleavage and reduction to the piperazine. (c) Boc cleavage, allyl hydrogenation, and C–H carbonylation of the free amine. (d, e) hydrolysis and subsequent protection of the β2,2-amino acid. fKOH, 1:1 MeOH/H2O, rt; then, HCl, MeOH, rt. BQ = benzoquinone. Piv = pivalate. | ||
Conclusions
In summary, we have developed a decarboxylative asymmetric allylic alkylation reaction to synthesize unprecedented chiral α,α-disubstituted piperazin-2-ones and tetrahydropyrimidin-2-ones. This work represents a significant improvement upon existing technology, featuring ease of substrate synthesis, facile N4-Boc deprotection, and broader substrate scope. The resulting piperazinones can be transformed into valuable chiral gem-disubstituted piperazines that are anticipated to provide stereodefined access to new chemical space in drug discovery. Similarly, the tetrahydropyrimidinone scaffold could be hydrolyzed to provide access to corresponding protected and free β2,2-amino acids. We anticipate this decarboxylative allylic alkylation protocol will find utility in the field of medicinal chemistry as well as in natural products synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The NIH-NIGMS (R01GM080269) and Caltech are thanked for support of our research program. Additionally, A. W. S. thanks the NIH-NIGMS for a predoctoral fellowship (Ruth L. Kirschstein Institutional National Research Service Award F30GM120836) and a UCLA-Caltech Medical Scientist Training Program Fellowship (T32GM008042). S. N. H. thanks the Bayer Foundation for an Otto Bayer Scholarship. Dr David VanderVelde is thanked for assistance with structural assignments via NMR analysis. Dr Scott Virgil, Dr Marchello Cavitt, Dr Brendan O'Boyle, Dr Justin Hilf, Dr Corey Reeves, and Kevin Yang are thanked for helpful discussions.Notes and references
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- (a) S. Omura, A. Hirano, Y. Iwai and R. Masuma, J. Antibiot., 1979, 32, 786–790 CrossRef CAS PubMed; (b) T. Chiba, Y. Asami, T. Suga, Y. Watanabe, T. Nagai, F. Momose, K. Nonaka, M. Iwatsuki, H. Yamada, S. Ōmura and K. Shiomi, Biosci., Biotechnol., Biochem., 2017, 81, 59–62 CrossRef CAS PubMed; (c) X. Yu, F. Liu, Y. Zou, M.-C. Tang, L. Hang, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2016, 138, 13529–13532 CrossRef CAS PubMed.
- M. A. Grady, T. L. Gasperoni and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2003, 2, 427–428 CrossRef CAS PubMed.
- (a) H. Wang, M. D. Kowalski, A. S. Lakdawala, F. G. Vogt and L. Wu, Org. Lett., 2015, 17, 564–567 CrossRef CAS PubMed; (b) R. E. Ziegler, B. K. Desai, J.-A. Jee, B. F. Gupton, T. D. Roper and T. F. Jamison, Angew. Chem., Int. Ed., 2018, 57, 7181–7185 CrossRef CAS PubMed.
- (a) K. Hirata, M. Kotoku, N. Seki, T. Maeba, K. Maeda, S. Hirashima, T. Sakai, S. Obika, A. Hori and Y. Hase, et al. , ACS Med. Chem. Lett., 2016, 7, 23–27 CrossRef CAS PubMed; (b) A. W. Hung, A. Ramek, Y. Wang, T. Kaya, J. A. Wilson, P. A. Clemons and D. W. Young, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6799–6804 CrossRef CAS PubMed; (c) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed; (d) F. Lovering, Med. Chem. Commun., 2013, 4, 515–519 RSC; (e) S. Monteleone, J. E. Fuchs and K. R. Liedl, Front. Pharmacol., 2017, 8, 552 CrossRef PubMed.
- Using the drugbank.ca database, we curated a structure search of piperazine-containing small molecule drugs. Out of 98 drugs, none were gem-disubstituted on the piperazine ring.
- For a review, see: (a) K. E. Gettys, Z. Ye and M. Dai, Synthesis, 2017, 49, 2589–2604 CrossRef CAS; (b) R. Holl, D. Schepmann and B. Wünsch, Med. Chem. Commun., 2012, 3, 673–679 RSC; (c) G. Sudhakar, S. Bayya, K. J. Reddy, B. Sridhar, K. Sharma and S. R. Bathula, Eur. J. Org. Chem., 2014, 1253–1265 CrossRef CAS; (d) S. Chamakuri, P. Jain, S. K. Reddy Guduru, J. W. Arney, K. R. MacKenzie, C. Santini and D. W. Young, J. Org. Chem., 2018, 83, 6541–6555 CrossRef CAS PubMed; (e) R. Kuwano and Y. Ito, J. Org. Chem., 1999, 64, 1232–1237 CrossRef CAS; (f) W.-X. Huang, L.-J. Liu, B. Wu, G.-S. Feng, B. Wang and Y.-G. Zhou, Org. Lett., 2016, 18, 3082–3085 CrossRef CAS PubMed; (g) T. D. Montgomery and V. H. Rawal, Org. Lett., 2016, 18, 740–743 CrossRef CAS PubMed; (h) J. S. Nakhla and J. P. Wolfe, Org. Lett., 2007, 9, 3279–3282 CrossRef CAS PubMed; (i) B. M. Cochran and F. E. Michael, Org. Lett., 2008, 10, 329–332 CrossRef CAS PubMed; (j) B. P. McDermott, A. D. Campbell and A. Ertan, Synlett, 2008, 875–879 CrossRef CAS; (k) J. D. Firth, P. O'Brien and L. Ferris, J. Am. Chem. Soc., 2016, 138, 651–659 CrossRef CAS PubMed.
- (a) J. Meyers, M. Carter, N. Y. Mok and N. Brown, Future Med. Chem., 2016, 8, 1753–1767 CrossRef PubMed; (b) C. M. Marson, in Advances in Heterocyclic Chemistry, ed. E. F. V. Scriven and C. A. Ramsden, Academic Press, 2017, pp. 13–33 Search PubMed; (c) C.-V. T. Vo and J. W. Bode, J. Org. Chem., 2014, 79, 2809–2815 CrossRef CAS PubMed.
- (a) W.-Y. Siau and J. W. Bode, J. Am. Chem. Soc., 2014, 136, 17726–17729 CrossRef CAS PubMed; (b) K. Geoghegan and J. W. Bode, Org. Lett., 2015, 17, 1934–1937 CrossRef CAS PubMed; (c) S.-Y. Hsieh and J. W. Bode, Org. Lett., 2016, 18, 2098–2101 CrossRef CAS PubMed.
- S. A. Ruider, S. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2013, 52, 11908–11911 CrossRef CAS PubMed.
- K. M. Korch, C. Eidamshaus, D. C. Behenna, S. Nam, D. Horne and B. M. Stoltz, Angew. Chem., Int. Ed., 2015, 54, 179–183 CrossRef CAS PubMed.
- (a) B. Weiner, W. Szymański, D. B. Janssen, A. J. Minnaard and B. L. Feringa, Chem. Soc. Rev., 2010, 39, 1656–1691 RSC; (b) D. L. Steer, R. A. Lew, P. Perlmutter, A. I. Smith and M.-I. Aguilar, Curr. Med. Chem., 2002, 9, 811–822 CrossRef CAS PubMed; (c) D. Seebach, A. K. Beck and D. J. Bierbaum, Chem. Biodiversity, 2004, 1, 1111–1239 CrossRef CAS PubMed.
- (a) J.-S. Yu, H. Noda and M. Shibasaki, Angew. Chem., Int. Ed., 2018, 57, 818–822 CrossRef CAS PubMed; (b) M. Nascimento de Oliveira, S. Arseniyadis and J. Cossy, Chem.–Eur. J., 2018, 24, 4810–4814 CrossRef CAS PubMed.
- (a) A. Guggisberg, K. Drandarov and M. Hesse, Helv. Chim. Acta, 2000, 83, 3035–3042 CrossRef CAS; (b) K. Drandarov, A. Guggisberg and M. Hesse, Helv. Chim. Acta, 2002, 85, 979–989 CrossRef CAS.
- J. R. Cabrera-Pardo, A. Trowbridge, M. Nappi, K. Ozaki and M. J. Gaunt, Angew. Chem., Int. Ed., 2017, 56, 11958–11962 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, 1H NMR, 13C NMR, and IR spectra. See DOI: 10.1039/c8sc03967d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |