
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceB- and N-embedded color-tunable phosphorescent iridium complexes and B–N Lewis adducts with intriguing structural and optical changes†
Qiuxia
Li
a,
Chao
Shi
 *ab,
Manli
Huang
c,
Xing
Wei
d,
Hong
Yan
*d,
Chuluo
Yang
*c and
Aihua
Yuan
*ab
*ab,
Manli
Huang
c,
Xing
Wei
d,
Hong
Yan
*d,
Chuluo
Yang
*c and
Aihua
Yuan
*ab
aSchool of Material Science and Engineering, Jiangsu University of Science and Technology, Zhenjiang 212003, P. R. China. E-mail: shichao@just.edu.cn; aihua.yuan@just.edu.cn
bSchool of Environmental and Chemical Engineering, Jiangsu University of Science and Technology, Zhenjiang 212003, P. R. China
cDepartment of Chemistry, Hubei Key Lab on Organic and Polymeric Optoelectronic Materials, Wuhan University, Wuhan 430072, P. R. China. E-mail: clyang@whu.edu.cn
dState Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing 210093, P. R. China. E-mail: hyan1965@nju.edu.cn
First published on 18th January 2019
Abstract
A novel family of B- and N-embedded phosphorescent iridium complexes has been prepared. Single crystal structures indicate that the B-embedded polycyclic unit exhibits better planarity than the N-embedded polycyclic unit, which leads to different π–π-stacking and electrical characteristics. More importantly, by controlling the number of boron or nitrogen atoms embedded, solution-processed OLED devices incorporating these emitters as emitting layers can achieve a phosphorescence color variation from green to deep red (638 nm) and show low-efficiency roll-off and turn-on voltage. In particular, the B-embedded complex Ir-BB shows good color purity with a narrow full width at half maximum (1211 cm−1) and CIE coordinates (0.67, 0.31) in the deep red light region. Notably, B-embedded iridium complexes can also react with two different Lewis bases (pyridine and DMAP) to form intriguing B–N Lewis adducts through different coordination modes. During this process, significantly different structural and optical changes are triggered by the structure and electronic properties of Lewis bases, as confirmed by X-ray crystallographic, 1H NMR and spectral analysis.
Introduction
Boron (B) or nitrogen (N) atom-embedded planar π-conjugated organic semiconductors1–16 have attracted much attention due to intriguing structural and electronic features, which are very promising in the fields of organic solid luminescent materials, magnetic materials, two-photon materials, carrier-transporting materials and chemosensors. Recently, a limited number of reports appeared on B- and N-embedded dioxygen-bridged polycyclic unit-based small organic molecules,15,16 which were used as host materials in phosphorescent organic light-emitting diodes (OLEDs) and hole-transporting materials in organic field-effect transistors (OFETs). However, little attention has been paid to tuning photophysical properties of phosphorescent metal complexes by the use of B- or N-embedded polycyclic units. More importantly, how the rigidity, planarity and electronic effects of these units affect the structure, carrier transport and luminescence of metal complexes is well worth studying.On the other hand, phosphorescent iridium(III) complexes17–29 have attracted a great deal of interest as emitting materials owing to their rich and diverse chemical structures, high luminescence efficiency, appropriate phosphorescence lifetime and high structural stability. In particular, modification of the cyclometalating ligand (C^N ligand) can control the emission color throughout the entire visible region that can be beneficial for realizing full-color and white-light displays. However, deep red phosphorescence with good color purity has been difficult to achieve due to a very wide full width at half maximum (FWHM) for most iridium(III) complexes.28,29 To design a novel family of color-tunable efficient phosphorescent materials, we have been interested in B- or N-embedded dioxygen-bridged polycyclic units with good rigidity, and electron-withdrawing or electron-donating features that may help reduce the barrier height for electron or hole injection.
In addition, tricoordinate boron-containing units possess certain Lewis acidity and can react with Lewis bases to form the corresponding tetracoordinate adducts,5,7,30–34 which could induce new electronic and optical properties. However, most studies on planar tricoordinate boron-containing compounds are mainly focused on pure organic π-conjugated molecules,1–12 which often show fluorescence rather than phosphorescence. Furthermore, how the structure and electrical properties of Lewis bases affect the excited state energy of the adduct still needs further research. So it is anticipated that introduction of B- and N-embedded dioxygen-bridged polycyclic units into phosphorescent iridium(III) complexes may improve the structure and the optical properties of these complexes and allow better understanding the roles of the units.
Herein, we first designed and synthesized a no-atom embedded phosphorescent iridium(III) complex, Ir-OO, as a model complex (Scheme 1) and then prepared a series of B- and N-embedded phosphorescent iridium(III) complexes, containing single-atom embedded complexes (Ir-ON and Ir-OB) and two-atom embedded complexes (Ir-NN and Ir-BB) (Scheme 1). The results demonstrate that the B-embedded polycyclic unit can exhibit better planarity than the N-embedded polycyclic unit, which leads to different π–π-stacking and electrical characteristics. More importantly, by controlling the number of boron or nitrogen atoms embedded, OLED devices incorporating these emitters as emitting layers can achieve a phosphorescence color variation from green to deep red and show low-efficiency roll-off and turn-on voltage. In particular, the B-embedded complex Ir-BB shows good color purity with a narrow full width at half maximum (1211 cm−1) and CIE coordinates (0.67, 0.31) in the saturated deep red light region. Notably, tricoordinate planar B-embedded complexes can also form their corresponding tetracoordinate B–N Lewis adducts (Ir-OB·L and Ir-BB·L, Scheme 1) with addition of Lewis bases L (pyridine and 4-dimethylaminopyridine (DMAP)). During this process, significantly different structural and optical changes are triggered by the structure and electronic properties of Lewis bases, as confirmed by X-ray crystallographic, 1H NMR and spectral analysis.
 | ||
| Scheme 1 Chemical structures of B- and N-embedded phosphorescent iridium complexes (Ir-ON, Ir-NN, Ir-OB and Ir-BB) and the model complex Ir-OO, and the conversion from tricoordinate B-embedded complexes (Ir-OB and Ir-BB) to their corresponding tetracoordinate Lewis adducts (Ir-OB·L and Ir-BB·L) in the presence of Lewis base L. | ||
Results and discussion
Synthesis and structure
A ligand (L-1) without atom-embedment was gained by reaction of 2-(3,5-difluorophenyl)pyridine and phenol with excess base. The N-embedded ligand (L-2) was finally prepared through an efficient Suzuki–Miyaura cross-coupling reaction by using N-embedded polycyclic unit-based boron ester as a boron reagent, while the B-embedded ligand (L-3) was synthesized by a one-step debromoborylation reaction of 2-(4-bromo-3,5-diphenoxyphenyl)pyridine (Fig. S1†). Then, all complexes can be prepared according to well-established methods35–37 for the synthesis of the traditional complex [Ir(C^N)2(acac)] by adding two same (for Ir-NN and Ir-BB) or different (for Ir-ON and Ir-OB) C^N ligands (Fig. S2†). All the new iridium(III) complexes were completely identified by 1H NMR, 13C NMR, HR-MS, elemental analysis and single-crystal X-ray crystallography. All the crystal structures (Fig. 1, and S4–S8, and Table S1†) show that the iridium center can be coordinated by two same (for Ir-NN, Ir-BB and Ir-OO) or different (for Ir-ON and Ir-OB) C^N ligands and one acetylacetone anion ligand in a distorted octahedral coordination geometry. Notably, the B-embedded dioxygen-bridged polycyclic unit in the crystal structure Ir-BB or Ir-OB shows higher planarity with smaller torsion angles (Ir-OB: 0.127° and −2.586°, Ir-BB: 0.024° and −4.044°) (Fig. 1) than (Ir-ON: 21.030° and 31.125°, Ir-NN: 22.691° and 14.953°) (Fig. 1) the N-embedded dioxygen-bridged polycyclic unit in the crystal structure Ir-ON or Ir-NN, and even higher than (18.9°) a B-embedded dioxygen-bridged polycyclic unit-based helical structure reported by the Hatakeyama group.15 Another interesting structural feature is the packing style of Ir-NN and Ir-BB (Fig. 1). Ir-NN shows a larger overlapping area of π–π-stacking relative to Ir-BB, and the intermolecular distance (3.95 Å) of Ir-NN is longer than that of Ir-BB (3.52 Å). In contrast, Ir-OB, Ir-ON and Ir-OO show little π–π-stacking (Fig. S4–S6†) owing to the distorted structure of the 3,5-diphenoxybenzene unit, which can cause greater steric hindrance than B- or N-embedded planar dioxygen-bridged polycyclic units. Unexpectedly, the orientations of the two phenoxy groups are completely opposite between Ir-ON and Ir-OB (Fig. 1). | ||
| Fig. 1 The X-ray crystal structures of Ir-ON, Ir-NN, Ir-OB and Ir-BB and the packing diagram of Ir-NN and Ir-BB. | ||
Photophysical and electrochemical properties
As shown in Fig. 2a and S3,† according to the UV/visible absorption spectra of all iridium complexes (Ir-OO, Ir-ON, Ir-OB, Ir-NN and Ir-BB) and the corresponding ligands (L-1, L-2 and L-3) in degassed CH2Cl2, it is found that the intense absorption bands below 340 nm can be assigned to the spin-allowed π → π* (LC) transitions of the C^N ligands. The weak low-energy absorption band in the region of 350–560 nm is assigned to various charge-transfer (CT) transitions (MLCT, LLCT or ILCT). Notably, the lowest energy bands of B-embedded iridium(III) complexes (Ir-OB and Ir-BB) are clearly red-shifted relative to those of N-embedded iridium(III) complexes (Ir-ON and Ir-NN) – that is, introduction of boron atoms can greatly lower the excited-state energy level. A similar change trend is also found in their PL spectra in CH2Cl2. The phosphorescence color can be tuned from green (Ir-OO, 544 nm) to deep red (Ir-BB, 638 nm) by embedding two boron atoms, and the red shift range is nearly 100 nm. In addition, the B-embedded iridium(III) complexes (Ir-OB: ΦPL = 31%, τ = 1.6 μs; Ir-BB: ΦPL = 21%, τ = 1.4 μs) show higher phosphorescence efficiencies and longer emission lifetimes (Table 1) than N-embedded iridium(III) complexes (Ir-ON: ΦPL = 18%, τ = 1.2 μs; Ir-NN: ΦPL = 11%, τ = 1.2 μs) in CH2Cl2. In contrast, in CBP: m-MTDATA mixed films, B-embedded iridium(III) complexes (Ir-OB: ΦPL = 44%, τ = 8.4 μs; Ir-BB: ΦPL = 34%, τ = 7.3 μs) exhibit lower phosphorescence efficiencies and shorter emission lifetimes (Table 1) than N-embedded iridium(III) complexes (Ir-ON: ΦPL = 53%, τ = 13 μs; Ir-NN: ΦPL = 50%, τ = 15 μs). The electrochemical properties of all complexes were studied by cyclic voltammetry in CH2Cl2 (Fig. S10† and Table 1). The oxidation potentials of the iridium center for all B- and N-embedded iridium(III) complexes (Ir-ON: 0.64 V, Ir-NN: 0.93 V, Ir-OB: 0.99 V and Ir-BB: 0.95 V) show an obvious redshift than that of Ir-OO (0.35 V). In addition, the oxidation potentials of B-embedded polycyclic units (Ir-OB: 0.31 V and Ir-BB: 0.28 V) exhibit a small redshift relative to that of the N-embedded polycyclic unit (Ir-ON: 0.21 V and Ir-NN: 0.22 V). Therefore, introduction of boron and nitrogen atoms can greatly increase the HOMO energy level, especially for complexes Ir-ON (HOMO value: −5.01 eV) and Ir-NN (HOMO value: −5.02 eV), which show HOMO values about 0.13 eV more than that of Ir-OO (HOMO value: −5.15 V). | ||
| Fig. 2 (a) Absorption and (b) PL spectra of Ir-OO, Ir-ON, Ir-NN, Ir-OB and Ir-BB in degassed CH2Cl2 (inset: luminescence photographs of iridium complexes). (c) EL spectra and (d) CIE coordinates and (e) the efficiency versus current density relationship for devices of all complexes. | ||
| Complexes |
λ
abs nm (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) ε) |
PLa/PLc (nm) | E ox1/2 (V) | E g (eV) | HOMO/LUMOb (eV) | Φ PL /ΦPLc (%) | τ /τc (μs) |
|---|---|---|---|---|---|---|---|
| a Recorded in degassed CH2Cl2 at a concentration of 10−5 M at 298 K with an excitation wavelength of 370 nm. b The HOMO (eV) = −e(Eox1/2 + 4.8) and Eg = 1240/λ, where λ is the absorption wavelength threshold. LUMO (eV) = Eg + HOMO. c Measured for 10 wt% doped CBP: m-MTDATA mixed films. | |||||||
| Ir-OO | 231 (5.20), 259 (5.11), 366 (4.24), 429 (3.87), 484 (3.36), 497 (2.94) | 544/538 | 0.35 | 2.40 | −5.15/−2.75 | 15/68 | 1.1/5.6 |
| Ir-ON | 230 (5.24), 259 (5.29), 364 (4.66), 419 (4.51), 481 (3.84), 512 (2.45) | 577/568 | 0.21/0.64 | 2.36 | −5.01/−2.65 | 18/53 | 1.2/13 |
| Ir-NN | 230 (5.47), 259 (5.58), 363 (4.98), 419 (4.91), 486 (3.93), 516 (2.52) | 583/579 | 0.22/0.34/0.93 | 2.33 | −5.02/−2.69 | 11/50 | 1.2/15 |
| Ir-OB | 231 (5.20), 260 (5.11), 327 (4.87), 416 (4.08), 523 (3.84), 559 (2.49) | 630/622 | 0.31/0.99 | 2.21 | −5.11/−2.90 | 31/44 | 1.6/8.4 |
| Ir-BB | 233 (5.28), 262 (5.21), 332 (5.11), 422 (4.36), 499 (4.11), 560 (3.38) | 638/639 | 0.28/0.95 | 2.15 | −5.08/−2.93 | 21/34 | 1.4/7.3 |
Solution-processed OLED device characterization
All iridium(III) complexes were tested in unoptimized solution-processed OLED devices with the same simple architecture of ITO/PEDOT: PSS (50 nm)/CBP (45%): m-MTDATA (45%): Ir complex (10%) (60 nm)/DPEPO (10 nm)/TmPyPB (50 nm)/Liq (1 nm)/Al (100 nm) (Fig. 2, S11, and S12, and Table S3†). Electroluminescence spectra (538–638 nm) (Fig. 2c) of all complexes resemble their PL spectra measured in CH2Cl2 (Fig. 2b). Notably, Ir-BB shows deep red (638 nm) emission, which is more red shifted than the non-embedded boron complex Ir(ppy-B(Mes)2)2acac (609 nm) reported by the Wong group.28 Furthermore, it also shows good color purity with CIE coordinates (0.67, 0.31) in the saturated deep red light region and a narrow FWHM (1211 cm−1) (Fig. 2c and d) and is superior to other iridium complexes reported.28,29 In addition, all complex-based devices show low-efficiency roll-off (Fig. 2e) and turn-on voltage (2.9–3.4 V) (Table S3†). This can be attributed to B- or N-embedded polycyclic units which can reduce the barrier for electron or hole injection. Nevertheless, they all display relatively lower maximum external quantum efficiencies (EQEs) (5.1–7.5%) than other excellent iridium38–41 and platinum42 complexes, and thermally activated delayed fluorescent (TADF) materials43–50 reported in the orange–red region (EQE > 10%). The relatively poor device performance is most likely due to the undesirable solubility of these iridium complexes in the solution process.B–N Lewis adducts
To investigate the Lewis acidity of B-embedded iridium(III) complexes (Ir-OB and Ir-BB), we choose two different Lewis bases (pyridine and DMAP) for comparison. Fortunately, single crystals of the B–N Lewis adducts Ir-BB·pyridine and Ir-BB·DMAP were obtained (Fig. 3 and S9, and Table S2†). It is found that the original planar B-embedded polycyclic units have formed shallow bowl-shaped structures (Fig. 3) due to B–N coordination. Interestingly, as Ir-BB has two boron-containing planes, two Lewis base molecules have different coordination modes: firstly, only one base molecule coordinates at the angle between the two planes (Fig. 3a), and the other base molecule binds on the outside of one plane (Fig. 3a, mode 1), such as in Ir-BB·pyridine (Fig. 3b). Secondly, these two base molecules both bind at the angle between the two planes (Fig. 3d, mode 2), such as in Ir-BB·DMAP (Fig. 3e). Unexpectedly, the obvious π–π interaction between two pyridine moieties (the distance is 3.42 Å) can be observed in the crystal structure of Ir-BB·DMAP (Fig. 3e), but not in the crystal structure of Ir-BB·pyridine (Fig. 3b) due to different coordination modes. It is notable that the B–N distance of Ir-BB·DMAP(1.638 Å and 1.672 Å) is shorter than that of Ir-BB·pyridine (1.658 Å and 1.688 Å) (Fig. 3b and e). This comparison demonstrates a rather stronger B–N coordination in Ir-BB·DMAP due to the higher Lewis basicity of DMAP relative to pyridine. Such interesting structural changes can also be monitored by 1H NMR spectroscopy in d6-DMSO solution. With addition of excess pyridine (41 eq.) and DMAP (10 eq.), all the original nuclear magnetic peaks of Ir-BB disappear and some new peaks emerge, which are assigned to Ir-BB·pyridine (Fig. 3c) and Ir-BB·DMAP (Fig. 3f), respectively.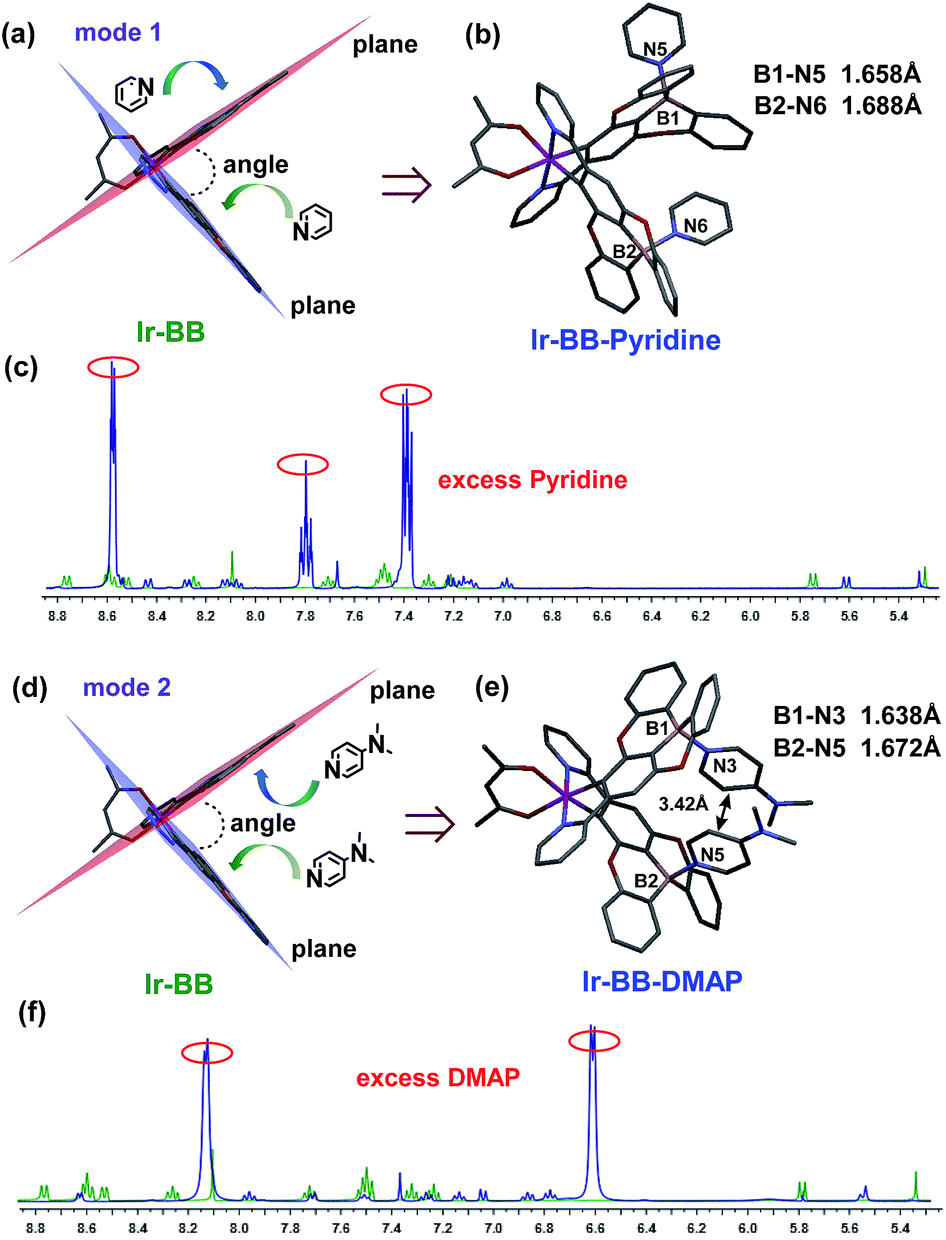 | ||
| Fig. 3 The X-ray crystal structures of (b) Ir-BB·pyridine and (e) Ir-BB·DMAP were formed through the coordination of Ir-BB and two pyridine molecules by mode 1 (a) and two DMAP molecules by mode 2 (d), respectively. Titrations of Ir-BB in d6-DMSO with excess pyridine (c) and DMAP (f) monitored by 1H NMR spectroscopy. | ||
Additionally, the optical changes caused by the newly formed B–N Lewis adducts can be monitored by UV and PL titration experiments (Fig. 4). Upon addition of pyridine or DMAP, all the absorption bands (LC and CT) of Ir-BB are gradually decreased (Fig. 4a and b), which should be attributed to the fact that the newly formed tetracoordinate borides break the π-conjugation of the ligand, thereby weakening the UV absorption. Interestingly, with addition of pyridine, the absorption bands (CT) show a small red shift (Fig. 4a) in contrast to a significant blue shift (Fig. 4b) in the presence of DMAP. A similar change trend is also found in the PL spectrum. The emission peak of Ir-BB is gradually decreased and redshifted from 647 nm to 667 nm (Fig. 4c) with the addition of pyridine, and its phosphorescence lifetime is also changed from 873 ns to 677 ns. In contrast, a significant blue-shift emission peak of Ir-BB·DMAP appears at 575 nm (Fig. 4d) with a shorter phosphorescence lifetime of 183 ns with the addition of DMAP. This comparison indicates that the structure and electronic properties of Lewis bases can affect the excited state energy of the adducts.
 | ||
| Fig. 4 Titrations of Ir-BB in DMSO ([Ir-BB]0 = 3 × 10−5 M) with pyridine (a and c) and DMAP (b and d) monitored by UV-vis absorption (a and b) and phosphorescence (λex = 370 nm) spectroscopy (c and d). | ||
In addition, Ir-OB displays a similar structural and optical change in the presence of Lewis bases (Fig. S13 and S14†). The binding constants of Ir-OB toward pyridine and DMAP (DMSO) are estimated to be 19.6 M−1 and 8.0 × 103 M−1 (Fig. S15 and S16†), respectively. These values are much lower than those of trinaphthylborane (5.1 × 103 M−1 for pyridine and 6.6 × 106 M−1 for DMAP) reported by the Yamaguchi group.7 The lower Lewis acidity of Ir-OB should be attributed to the larger steric hindrance.
Theoretical calculations
To understand the roles of B- and N-embedded polycyclic units and how the addition of these two Lewis bases affects the excited state behavior of B–N Lewis adducts, DFT calculations at the B3LYP/6-31G(d) level were performed with all the complexes discussed above (Fig. 5, and S17–S19, and Tables S4 and S5†). The HOMOs of the triplet excited state (T1) of Ir-OO, Ir-OB and Ir-BB are mainly distributed on the iridium(III) center, phenyl ring and two oxygen atoms of the C^N ligand, whereas the HOMOs of Ir-ON and Ir-NN are mainly located on the whole N-embedded C^N ligand but little on the metal iridium (Fig. S17†). The LUMOs of Ir-OO, Ir-OB and Ir-BB are distributed on the whole C^N ligand, and the LUMOs of Ir-ON and Ir-NN reside on 2-phenyl pyridine and the embedded nitrogen atom of the C^N ligand. As a result, the transition character between B and N-embedded complexes is quite different: the transitions of N-embedded iridium(III) complexes exhibit only 3ILCT (for Ir-NN) or 3ILCT and 3LLCT (for Ir-ON) but little 3MLCT, because the charges on the metal iridium cannot be easily transferred to the N-embedded C^N ligand with rich charges. In contrast, the transition of B-embedded iridium(III) complexes do not only show 3ILCT (for Ir-BB) or 3ILCT and 3LLCT (for Ir-OB) but also show 3MLCT (Fig. S17†), which can be attributed to the electron-withdrawing ability of the B-embedded C^N ligand that can easily induce the charge transfer from the metal to the ligand. | ||
| Fig. 5 The optimized geometries and main orbital transitions of (a) Ir-BB·pyridine and (b) Ir-BB·DMAP in the lowest triplet excited state (T1). | ||
For B–N Lewis adducts, taking Ir-BB·pyridine and Ir-BB·DMAP as an example, the T1 of two adducts are both dominated by the HOMO → LUMO transition (Fig. 5 and Table S5†). The HOMOs of these two adducts are similar and mainly reside on the iridium(III) center, phenyl ring and two oxygen atoms of the C^N ligand. Interestingly, the LUMO of Ir-BB·DMAP resides on the partial C^N ligand moieties (Fig. 5b), whereas the LUMO of Ir-BB·pyridine is completely located on the Lewis base pyridine moieties (Fig. 5a). As a result, Ir-BB·DMAP has MLCT transition character in T1, while the T1 of Ir-BB·pyridine exhibits intramolecular charge transfer character from the Lewis acid Ir-BB to Lewis base pyridine. This result can explain the above significantly different optical changes triggered by the structure and electronic properties of Lewis bases.
Conclusions
In conclusion, we have prepared a novel family of B- and N-embedded phosphorescent iridium(III) complexes. By controlling the number of boron or nitrogen atoms embedded, OLED devices incorporating these emitters as emitting layers can lead to a phosphorescence color change from green to deep red. In particular, the B-embedded complex Ir-BB shows good color purity with a narrow full width at half maximum (1211 cm−1) and CIE coordinates (0.67, 0.31) in the saturated deep red light region. We also investigate B-embedded complex-based B–N Lewis adducts containing different Lewis bases and understand the correlations between the intriguing structure and optical properties. This work provides a new strategy for the investigation and application of B- and N-embedded phosphorescent materials and also extends the scope of B- and N-embedded semiconductors from pure organic molecules to metal complexes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21601069, 51672114, 21531004 and 91433201), Natural Science Foundation of Jiangsu Province of China (BK20160552 and BK20161357), Natural Science Foundation of the Higher Education Institutions of Jiangsu Province (16KJB150011), Postdoctoral Research Foundation of China (2017M611746) and Jiangsu Postdoctoral Research Foundation (1701115B).Notes and references
- S. Nakatsuka, H. Gotoh, K. Kinoshita, N. Yasuda and T. Hatakeyama, Angew. Chem., Int. Ed., 2017, 56, 5087 CrossRef CAS PubMed.
- Z. X. Giustra and S.-Y. Liu, J. Am. Chem. Soc., 2018, 140, 1184 CrossRef CAS PubMed.
- X.-Y. Wang, F.-D. Zhuang, R.-B. Wang, X.-C. Wang, X.-Y. Cao, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2014, 136, 3764 CrossRef CAS PubMed.
- X.-Y. Wang, H.-R. Lin, T. Lei, D.-C. Yang, F.-D. Zhuang, J.-Y. Wang, S.-C. Yuan and J. Pei, Angew. Chem., Int. Ed., 2013, 52, 3117 CrossRef CAS PubMed.
- S. Saito, K. Matsuo and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 9130 CrossRef CAS PubMed.
- K. Matsuo, S. Saito and S. Yamaguchi, Angew. Chem., Int. Ed., 2016, 55, 1 CrossRef PubMed.
- K. Matsuo, S. Saito and S. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 12580 CrossRef CAS PubMed.
- T. Hatakeyama, S. Hashimoto, S. Seki and M. Nakamura, J. Am. Chem. Soc., 2011, 133, 18614 CrossRef CAS PubMed.
- T. Hatakeyama, S. Hashimoto, T. Oba and M. Nakamura, J. Am. Chem. Soc., 2012, 134, 19600 CrossRef CAS PubMed.
- T. Hashimoto, T. Ikuta, K. Shiren, S. Nakatsuka, J. Ni, M. Nakamura and T. Hatakeyama, Chem. Mater., 2014, 26, 6265 CrossRef.
- C. Dou, S. Saito, K. Matsuo, I. Hisaki and S. Yamaguchi, Angew. Chem., Int. Ed., 2012, 51, 12206 CrossRef CAS PubMed.
- Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529 CrossRef CAS PubMed.
- K. Suzuki, S. Kubo, K. Shizu, T. Fukushima, A. Atsushi-Wakamiya, Y. Murata, C. Adachi and H. Kaji, Angew. Chem., Int. Ed., 2015, 54, 15231 CrossRef CAS PubMed.
- W. Zhang, G. Li, L. Xu, Y. Zhuo, W. Wan, N. Yan and G. He, Chem. Sci., 2018, 9, 4444 RSC.
- K. Hirai, K. Nakajima, S. Nakatsuka, K. Shiren, J. Ni, S. Nomura, T. Ikuta and T. Hatakeyama, Angew. Chem., Int. Ed., 2015, 54, 13581 CrossRef PubMed.
- A. Wakamiya, H. Nishimura, T. Fukushima, F. Suzuki, A. Saeki, S. Seki, I. Osaka, T. Sasamori, M. Murata, Y. Murata and H. Kaji, Angew. Chem., Int. Ed., 2014, 53, 5800 CrossRef CAS PubMed.
- M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750 CrossRef CAS PubMed.
- A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, T. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971 CrossRef CAS PubMed.
- C. Shi, H. Sun, X. Tang, W. Lv, H. Yan, Q. Zhao, J. Wang and W. Huang, Angew. Chem., Int. Ed., 2013, 52, 13434 CrossRef CAS PubMed.
- K.-H. Kim, S. Lee, C.-K. Moon, S.-Y. Kim, Y.-S. Park, J.-H. Lee, J. W. Lee, J. Huh, Y. You and J.-J. Kim, Nat. Commun., 2014, 5, 4769 CrossRef CAS PubMed.
- K. Y. Zhang, Q. Yu, H. Wei, S. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770 CrossRef CAS PubMed.
- S. Kesarkar, W. Mróz, M. Penconi, M. Pasini, S. Destri, M. Cazzaniga, D. Ceresoli, P. R. Mussini, C. Baldoli, U. Giovanella and A. Bossi, Angew. Chem., Int. Ed., 2016, 55, 2714 CrossRef CAS PubMed.
- M. J. Jurow, C. Mayr, T. D. Schmidt, T. Lampe, P. I. Djurovich, W. Brütting and M. E. Thompson, Nat. Mater., 2016, 15, 85 CrossRef CAS PubMed.
- J. Lee, H.-F. Chen, T. Batagoda, C. Coburn, P. I. Djurovich, M. E. Thompson and S. R. Forrest, Nat. Mater., 2016, 15, 92 CrossRef CAS PubMed.
- P.-N. Lai, C. H. Brysacz, M. K. Alam, N. A. Ayoub, T. G. Gray, J. Bao and T. S. Teets, J. Am. Chem. Soc., 2018, 140, 10198 CrossRef CAS PubMed.
- X. Li, J. Zhang, Z. Zhao, L. Wang, H. Yang, Q. Chang, N. Jiang, Z. Liu, Z. Bian, W. Liu, Z. Lu and C. Huang, Adv. Mater., 2018, 30, 1705005 CrossRef PubMed.
- C. Shi, H. Sun, Q. Jiang, Q. Zhao, J. Wang, W. Huang and H. Yan, Chem. Commun., 2013, 49, 4746 RSC.
- B. G. Zhou, C.-L. Ho, W.-Y. Wong, Q. Wang, D. Ma, L. Wang, Z. Lin, T. B. Marder and A. Beeby, Adv. Funct. Mater., 2008, 18, 499 CrossRef.
- P. Tao, W.-L. Li, J. Zhang, S. Guo, Q. Zhao, H. Wang, B. Wei, S.-J. Liu, X.-H. Zhou, Q. Yu, B.-S. Xu and W. Huang, Adv. Funct. Mater., 2016, 26, 881 CrossRef CAS.
- K. Ansorg, H. Braunschweig, C.-W. Chiu, B. Engels, D. Gamon, M. Hgüel, T. Kupfer and K. Radacki, Angew. Chem., Int. Ed., 2011, 50, 2833 CrossRef CAS PubMed.
- D. Frath, J. Massue, G. Ulrich and R. Ziessel, Angew. Chem., Int. Ed., 2014, 53, 2290 CrossRef CAS PubMed.
- Y.-J. Shiu, Y.-C. Cheng, W.-L. Tsai, C.-C. Wu, C.-T. Chao, W. Lu, Y. Chi, Y.-T. Chen, S.-H. Liu and P.-T. Chou, Angew. Chem., Int. Ed., 2016, 55, 3017 CrossRef CAS PubMed.
- S. Osumi, S. Saito, C. Dou, K. i. Matsuo, K. Kume, H. Yoshi-kawa, K. Awagaa and S. Yamaguchi, Chem. Sci., 2016, 7, 219 RSC.
- K. Liu, R. A. Lalancette and F. Jäkle, J. Am. Chem. Soc., 2017, 139, 18170 CrossRef CAS PubMed.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304 CrossRef CAS PubMed.
- X. Xu, X. Yang, J. Dang, G. Zhou, Y. Wu, H. Li and W.-Y. Wong, Chem. Commun., 2014, 50, 2473 RSC.
- X. Xu, H. Guo, J. Zhao, B. Liu, X. Yang, G. Zhou and Z. Wu, Chem. Mater., 2016, 28, 8556 CrossRef CAS.
- R. Wang, D. Liu, H. Ren, T. Zhang, H. Yin, G. Liu and J. Li, Adv. Mater., 2011, 23, 2823 CrossRef CAS PubMed.
- Y. Zheng, A. S. Batsanov, M. A. Fox, H. A. Al-Attar, K. Abdullah, V. Jankus, M. R. Bryce and A. P. Monkman, Angew. Chem., Int. Ed., 2014, 53, 11616 CrossRef CAS PubMed.
- Z. Chen, L. Wang, C.-L. Ho, S. Chen, S. Suramitr, A. Plucksacholatarn, N. Zhu, S. Hannongbua and W.-Y. Wong, Adv. Opt. Mater., 2018, 6, 1800824 CrossRef.
- B. Jiang, C. Zhao, X. Ning, C. Zhong, D. Ma and C. Yang, Adv. Opt. Mater., 2018, 6, 1800108 CrossRef.
- G. Cheng, Q. Wan, W.-H. Ang, C.-L. Kwong, W.-P. To, P.-K. Chow, C.-C. Kwok and C.-M. Che, Adv. Opt. Mater., 2018 DOI:10.1002/adom.201801452.
- W. Zeng, H.-Y. Lai, W.-K. Lee, M. Jiao, Y.-J. Shiu, C. Zhong, S. Gong, T. Zhou, G. Xie, M. Sarma, K.-T. Wong, C.-C. Wu and C. Yang, Adv. Mater., 2018, 30, 1704961 CrossRef PubMed.
- K. Wu, T. Zhang, Z. Wang, L. Wang, L. Zhan, S. Gong, C. Zhong, Z.-H. Lu, S. Zhang and C. Yang, J. Am. Chem. Soc., 2018, 140, 8877 CrossRef CAS PubMed.
- C. Chen, R. Huang, A. S. Batsanov, P. Pander, Y.-T. Hsu, Z. Chi, F. B. Dias and M. R. Bryce, Angew. Chem., Int. Ed., 2018, 57, 16407 CrossRef CAS PubMed.
- C. Li, R. Duan, B. Liang, G. Han, S. Wang, K. Ye, Y. Liu, Y. Yi and Y. Wang, Angew. Chem., Int. Ed., 2017, 56, 11525 CrossRef CAS PubMed.
- H. Wang, L. Meng, X. Shen, X. Wei, X. Zheng, X. Lv, Y. Yi, Y. Wang and P. Wang, Adv. Mater., 2015, 27, 4041 CrossRef CAS PubMed.
- S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang and C. Adachi, Angew. Chem., Int. Ed., 2014, 53, 6402 CrossRef CAS PubMed.
- Q. Zhang, H. Kuwabara, W. J. P. Jr, S. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070 CrossRef CAS PubMed.
- D.-H. Kim, A. D'Aléo, X.-K. Chen, A. D. S. Sandanayaka, D. Yao, L. Zhao, T. Komino, E. Zaborova, G. Canard, Y. Tsuchiya, E. Choi, J. W. Wu, F. Fages, J.-L. Brédas, J.-C. Ribierre and C. Adachi, Nat. Photonics, 2018, 12, 98 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1832468–1832470 and 1860052–1860055. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04252g |
| This journal is © The Royal Society of Chemistry 2019 |