
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanobowls with controlled openings and interior holes driven by the synergy of hydrogen bonding and π–π interaction†
Hui
Sun
 a,
Danqing
Liu
a and
Jianzhong
Du
*ab
a,
Danqing
Liu
a and
Jianzhong
Du
*ab
aDepartment of Polymeric Materials, School of Materials Science and Engineering, Tongji University, 4800 Caoan Road, Shanghai 201804, China. E-mail: jzdu@tongji.edu.cn; Fax: +86-21-6958-0239; Tel: +86-21-6958-0239
bDepartment of Orthopedics, Shanghai Tenth People's Hospital, Tongji University School of Medicine, Shanghai 200072, China
First published on 21st November 2018
Abstract
Asymmetric nanoparticles such as nanobowls have promising potential in many fields due to their interior asymmetric cavities and specific concave structure. However, the fabrication of nanobowls and control over their openings and interior holes are still challenging. Herein we demonstrate a versatile strategy for preparing nanobowls with precisely controlled openings and interior holes based on the synergy of hydrogen bonding and π–π interaction of homopolymers. We designed and synthesized a series of amphiphilic homopolymers with an amino alcohol moiety and azobenzene pendant (poly(2-hydroxy-3-((4-(phenyldiazenyl)phenyl)amino)propyl methacrylate) (PHAzoMA)). The homopolymers can self-assemble into nanobowls due to the heterogeneous shrinkage of the preformed spheres. Upon increasing the molecular weight of the homopolymers from 10.1 to 76.9 kg mol−1, the sizes of the openings of nanobowls can be precisely controlled from 242 to 423 nm with a linear relationship as a result of the enhancement of the hydrogen bonding and π–π interaction between homopolymer chains. Overall, we have prepared finely controlled nanobowls by the synergy of non-covalent interactions such as hydrogen bonding and π–π interaction of polymers, which opens a new avenue for the preparation of asymmetric nanoparticles.
Introduction
Asymmetric structures widely exist in nature and play important roles in life activities. Asymmetric nanoparticles have shown superior performances in bioimaging, catalysis, detection, etc.1–4 Nanobowls are dense spheres containing an asymmetrically placed single hole and opening on the surface.5–7 They have shown great potential in many fields such as energy storage and nanomotors due to their unique asymmetric cavities, concave structure and high packing density.8–12 However, current preparation of nanobowls highly relies on template methods, which are tedious, time-consuming and not environmentally friendly.8–10 Therefore, alternative methods such as self-assembly of polymers are considered as a facile strategy to prepare nanobowls.5,6,13–17 Recently, Feringa and coworkers prepared bowl-shaped nanoparticles by inducing molecular motor aggregation through mixing selective solvents. They discovered that the formation of nanobowls from small molecules is governed by solvents rather than the molecular structure.6 Nevertheless, the fundamental principles behind the formation of nanobowls from amphiphilic polymers are still unknown. More importantly, the preparation of nanobowls with controlled openings and interior holes remains challenging and the corresponding preparation method has not yet been established.Block copolymers can self-assemble into a wide range of nanostructures including spheres,18 cylinders,19 vesicles,20 disks,21 helices,22etc. Phase separation plays an important role in the formation of these nanostructures.23–25 However, it is difficult for block copolymers to form bowl-shaped nanoparticles due to the clear boundary between hydrophilic and hydrophobic segments.26,27 The driving force for self-assembly is either the immiscibility of one of the blocks in a single-solvent or their mutual incompatibility,18,28,29 which leads to the phase separation between hydrophilic and hydrophobic segments. The clear boundary between hydrophilic and hydrophobic phases impedes the formation of nanobowls since the interior components of nanobowls are composed of both hydrophilic and hydrophobic moieties.5,13 Thus, new building blocks and self-assembly mechanisms for nanobowl fabrication need to be exploited.
Different from amphiphilic block copolymers, there is no clear boundary between hydrophilic and hydrophobic segments in the amphiphilic homopolymers.30–33 So the main driving force for homopolymer self-assembly rests upon intramolecular phase separation and non-covalent interactions including hydrogen bonding, π–π interaction, inclusion complexation, etc.14,34,35 By controlling the non-covalent interactions, unique nanostructures, such as cubes, flower-like nanostructures and nanotubes, can be obtained through homopolymer self-assembly.30,36–38
It has been found that non-covalent interactions such as hydrogen bonding, π–π interaction, and ionic interaction etc. are essential for the formation of bowl-shaped nanoparticles, which enhanced the intermolecular interactions inside the preformed spheres during self-assembly.5,13 When the non-covalent interaction is in the appropriate range to hinder the homogeneous shrinkage of the preformed spheres and to retain the relatively high chain mobility inside the spheres, the subsequent phase separation in the interior of the spheres could lead to the heterogeneous collapse of the spheres to form nanobowls.
To verify the above hypothesis, we extended our experience on self-assembly of amphiphilic homopolymers to nanobowl fabrication.39–41 A series of amphiphilic homopolymers with different molecular weights and pendants were designed and prepared to self-assemble into nanobowls with controlled openings and interior holes. The PHAzoMA homopolymer with an amino alcohol group and azobenzene pendant in the repeating unit was synthesized by reversible addition-fragmentation chain transfer (RAFT) polymerization. As shown in Scheme 1, the PHAzoMA homopolymers can self-assemble into nanobowls composed of hydrophilic and hydrophobic moieties. The synergy between hydrogen bonding and π–π interaction guarantees the intermolecular interaction of the preformed spheres located in an appropriate range that can prevent the homogeneous shrinkage of the spheres and permit chain mobility inside the spheres, leading to the controllable formation of nanobowls. In addition, this synergistic effect between both interactions of homopolymer chains increases with the molecular weight of the homopolymers, resulting in the enlarging of the openings of the nanobowls.
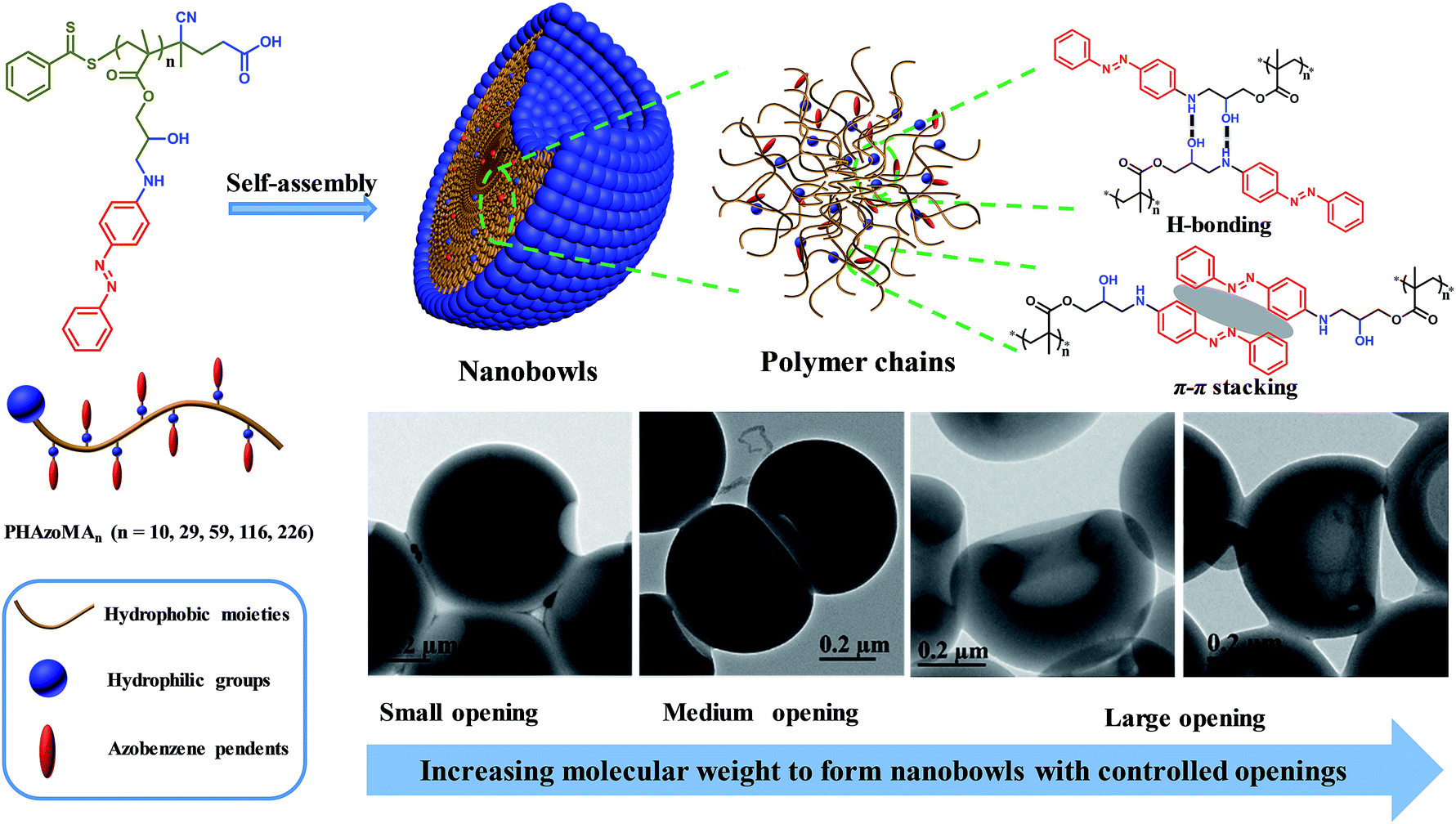 | ||
| Scheme 1 Nanobowls self-assembled from PHAzoMA homopolymers based on the synergy of hydrogen bonding and π–π interaction. | ||
Results and discussion
Synthesis and self-assembly of PHAzoMA homopolymers
The details of the synthesis and characterization of the PHAzoMA homopolymers are provided in the ESI.† Scheme S1 (ESI†) shows the synthetic route of homopolymers, while the 1H NMR spectra and gel permeation chromatography (GPC) traces of the HAzoMA monomer and homopolymers are provided in Fig. S1–S7, ESI.† The properties of PHAzoMA homopolymers are summarized in Table 1. The RAFT process is well controlled with high conversion of the HAzoMA monomer (>94%) and narrow molecular weight distribution (Ð < 1.2). The degrees of polymerization (DPs) of the homopolymers are 10, 29, 59, 116 and 226, respectively, corresponding to the molecular weights ranging from 3.7 to 76.9 kg mol−1 (Table 1).| Homopolymer | Conv.a (%) | DPb | M n,NMR (kg mol−1) | M n,GPC (kg mol−1) | Ð | Morphology |
|---|---|---|---|---|---|---|
| a The conversion of monomer HAzoMA was determined by 1H NMR. b The DP of PHAzoMA was calculated using the conversion: DP = DPtheoretical × conversion (%). c The Mn,NMR was calculated using DP: Mn,NMR = M0 × DP + 279 g mol−1. d The molecular weight dispersity (Ð) of PHAzoMA homopolymers was determined as Mw,GPC/Mn,GPC; the Mw,GPC and Mn,GPC were obtained by GPC using DMF as the eluent. P1, P2 and P3 are homopolymers with similar structures to PHAzoMA but different non-covalent interactions during self-assembly. The molecular structures of P1, P2 and P3 are illustrated in Fig. 6. | ||||||
| PHAzoMA10 | 95 | 10 | 3.7 | 3.3 | 1.18 | HCMs |
| PHAzoMA29 | 97 | 29 | 10.1 | 9.5 | 1.09 | Nanobowls |
| PHAzoMA59 | 98 | 59 | 20.3 | 18.3 | 1.12 | Nanobowls |
| PHAzoMA116 | 97 | 116 | 39.6 | 35.1 | 1.11 | Nanobowls |
| PHAzoMA226 | 94 | 226 | 76.9 | 59.4 | 1.17 | Nanobowls |
| P1 | 100 | 59 | 18.1 | 16.8 | 1.16 | HCMs |
| P2 | 100 | 61 | 19.8 | 18.2 | 1.07 | HCMs |
| P3 | 100 | 73 | 18.3 | 19.5 | 1.11 | Nanobowls |
The PHAzoMA homopolymers could self-assemble into homopolymer complex micelles (HCMs) and nanobowls when adding water to their tetrahydrofuran (THF) solutions, depending on the DPs of homopolymers (Fig. 1). When the DP of PHAzoMA is 10, the homopolymer self-assembles into HCMs with a hydrodynamic diameter (Dh) of 481 nm and polydispersity (PD) of 0.114 (curve (a) in Fig. 1(A)). The TEM study in Fig. 1(B) reveals the solid sphere structure of the HCMs with a diameter of 653 ± 76 nm. As the DP of the PHAzoMA homopolymer increases, an interior hole appears and the membrane collapses to form nanobowls (Fig. 1C–F), which are different from polymersome stomatocytes or collapsed polymer vesicles that have a uniform membrane thickness.42,43 The SEM images in Fig. 2 confirm the collapse of the wall in the thin region.
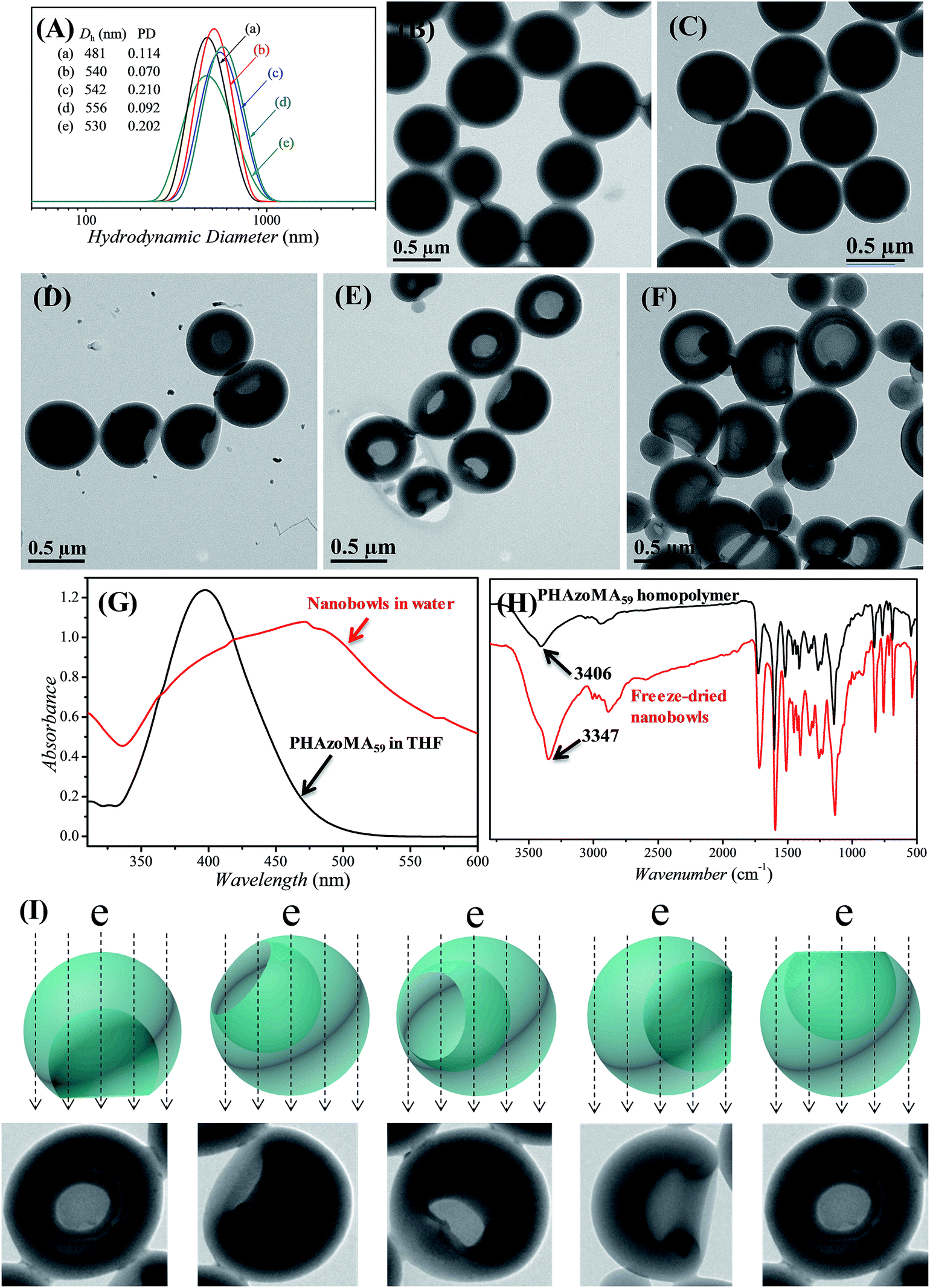 | ||
| Fig. 1 Evolution from PHAzoMA-based homopolymer complex micelles (HCMs) to nanobowls. (A) DLS results of the corresponding nanostructures when the DPs of PHAzoMA are (a) 10, (b) 29, (c) 59, (d) 116 and (e) 226, respectively; (B) TEM images of HCMs when the DP is 10; (C), (D), (E) and (F) TEM images of nanobowls with different sizes of openings and interior holes when the DPs are 29, 59, 116 and 226, respectively; (G) UV-vis spectra of PHAzoMA59 in THF and nanobowls in water, with the red shift of the absorption peak of nanobowls indicating the π–π interaction between azobenzene moieties after self-assembly; (H) FT-IR spectra of PHAzoMA59 and freeze-dried nanobowl powder, with the expanding and the shift of the absorption peak of –OH to a lower wavenumber indicating the formation of hydrogen bonds in nanobowls; (I) images representing the diverse morphologies of nanobowls in TEM images due to different presentation angles. | ||
 | ||
| Fig. 2 SEM images of nanobowls formed from (A) PHAzoMA29 and (B) PHAzoMA116. | ||
In order to determine if there is any THF residue after dialysis, the nanobowls were washed with D2O for three times after dialysis and then dissolved in DMSO-d6 for NMR analysis. The NMR spectra (Fig. S8, ESI†) confirmed the complete removal of THF by dialysis. Thus, control over the size of the openings of the nanobowls is irrelevant to the solvent retention, which is different from the work of Franken et al.,6 and further confirms that the formation of nanobowls is dependent on the molecular structure and non-covalent interactions of homopolymers.
UV-vis and FTIR analyses were conducted to confirm the existence of hydrogen bonding and π–π interaction after self-assembly. As shown in Fig. 1G and H, there is a strong hydrogen bonding effect and π–π stacking interaction when the PHAzoMA59 homopolymer self-assembles into nanobowls. The shift of the absorption peak of –OH to a lower wavenumber (from 3406 to 3347 cm−1) along with the expanding and strengthening of –OH absorbance in the FT-IR spectrum after self-assembly indicates the formation of hydrogen bonds,32 while the red shift of the absorption peak of the azobenzene group (from 397 to 471 nm) in the UV-vis spectrum confirms the existence of π–π stacking interaction.44 The detailed influence of the non-covalent interactions on the formation of nanobowls will be discussed later in this paper.
Control of the sizes of the openings and the interior holes of nanobowls
The size of the opening of the nanobowls can be controlled by varying the DP of the PHAzoMA homopolymers. As illustrated in Fig. 1C–F, when the DP of the PHAzoMA increases to 29, 59, 116 and 226, the sizes of holes of the nanobowls continuously increase. However, the Dhs of the nanobowls almost remain unchanged under the same self-assembly conditions (initial concentration, water addition rate and stirring rate), which are 540, 542, 556 and 530 nm (Fig. 1A), respectively, in accordance with the diameters measured from the TEM images (558 ± 54, 536 ± 48, 530 ± 55 and 541 ± 73 nm, respectively). It is noteworthy that the nanobowls show different morphologies in the TEM images when they are presented at different angles due to their asymmetric structures. The typical morphologies of nanobowls in the TEM images are illustrated in Fig. 1I at different angles.To reveal the relationship between the size of the openings, the interior holes and the DP of the homopolymers, representative TEM images were selected to analyze the electron intensity profiles of the holes. As shown in Fig. 3, when the DP of the PHAzoMA is 29, the width of the openings and depth of the hole are measured to be 242 and 95 nm (Fig. 3A), respectively. As the DP increases to 59, 116 and 226, the width of the holes increases to 265, 354 and 423 nm, respectively, while the depth of the holes goes up to 193, 254 and 363 nm, respectively. The relationship between the average width of the openings and depth of the holes of nanobowls and the DP of the PHAzoMA homopolymer is presented in Fig. 4, showing a linear relationship. As the molecular weight of the homopolymer increases, the interactions of hydrogen bonding and π–π stacking between homopolymer chains increase accordingly, which hinders the homogeneous shrinkage at an earlier stage, resulting in the formation of larger holes. The possible mechanism will be explained hereinafter.
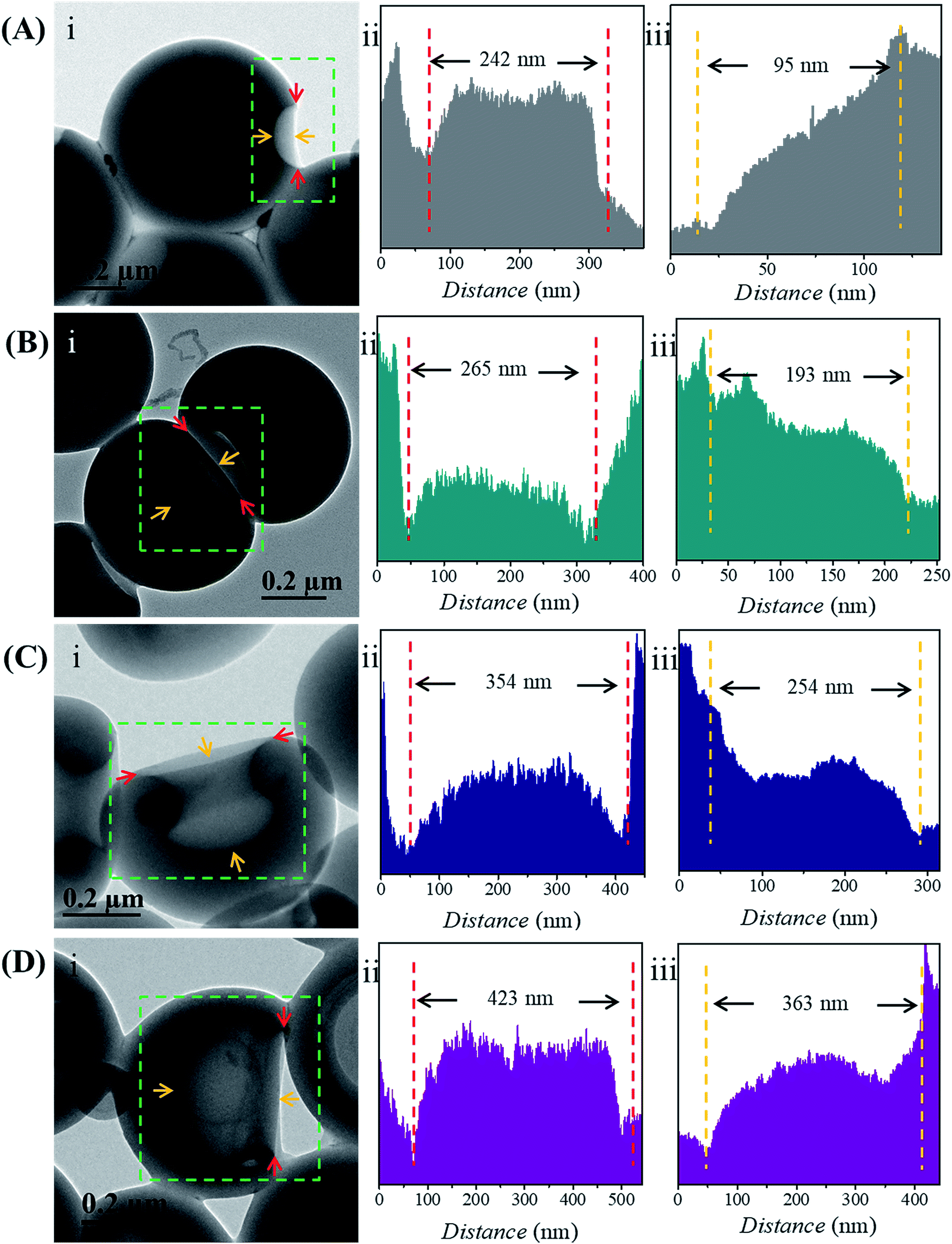 | ||
| Fig. 3 Analysis of the width of the openings and depth of the holes of the nanobowls. In each panel (A–D), the TEM images (i) show the change of the width of the openings and depth of the holes according to the DPs of the PHAzoMA homopolymers: (A) 29, (B) 59, (C) 116 and (D) 226. In each panel (A–D), the intensity profiles of the (ii) width of the openings and (iii) depth of the holes, as measured by TEM across the holes, corresponding to the red and yellow arrows in (i). | ||
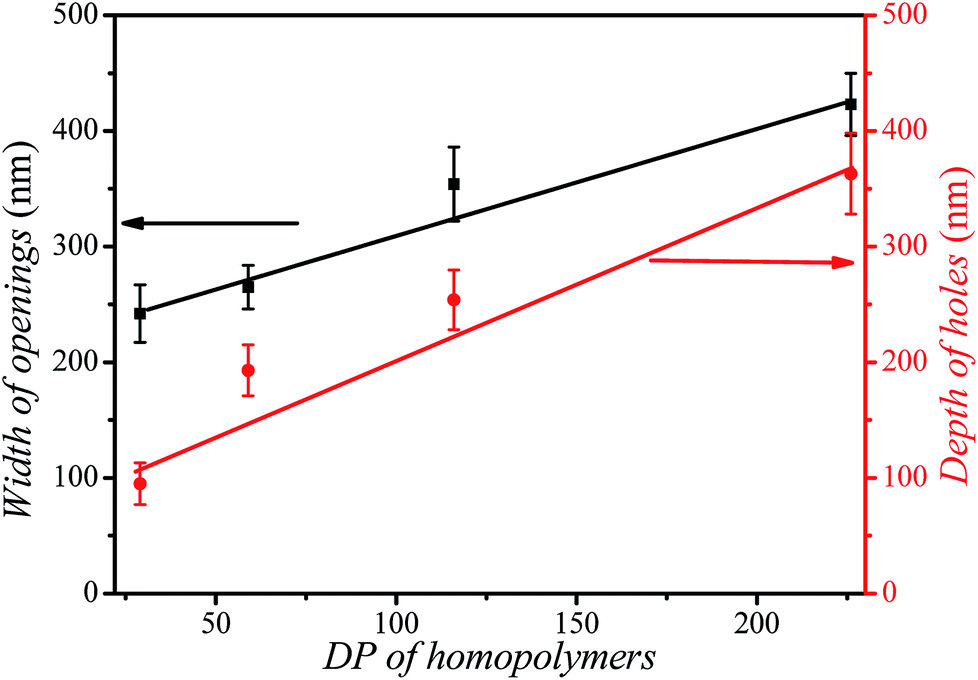 | ||
| Fig. 4 The relationship between the width of the openings (black line), the depth of the holes (red line) of nanobowls and the DP of the PHAzoMA homopolymers. | ||
Formation mechanism of nanobowls
To reveal if the nanobowls are thermodynamically stable, we tested their stability by external disturbances such as ultrasonic treatment, as shown in Fig. S9, ESI.† After sonication for 5 min, the morphology of the nanobowls barely changes, suggesting that nanobowls are thermodynamically stable. Besides, the relatively high Tg (64.3 °C for PHAzoMA29, Fig. S10, ESI†) and strong negative charges (Fig. S11, ESI†) can facilitate the long-term stability of nanobowls in water.To investigate the formation process of nanobowls, solutions of PHAzoMA226 were taken out at different water contents during self-assembly and dispersed in a large amount of water (20 μL of solution in 1 mL of water) to ‘freeze’ the morphology of the formed nano-objects.45 Then the nanostructures were characterized by TEM. As shown in Fig. 5, when the water content is 37.5%, preformed spheres appear. Spheres with small bubbles filled with THF/water are formed at a water content of 50.0% (Fig. 5). The small bubbles inside the spheres coalesce to form large bubbles when the water content increases to 58.3% (Fig. 5), and then the large bubbles further coalesce to form one larger bubble and the collapse of the membranes near the thinner wall leads to the formation of nanobowls (Fig. 5)
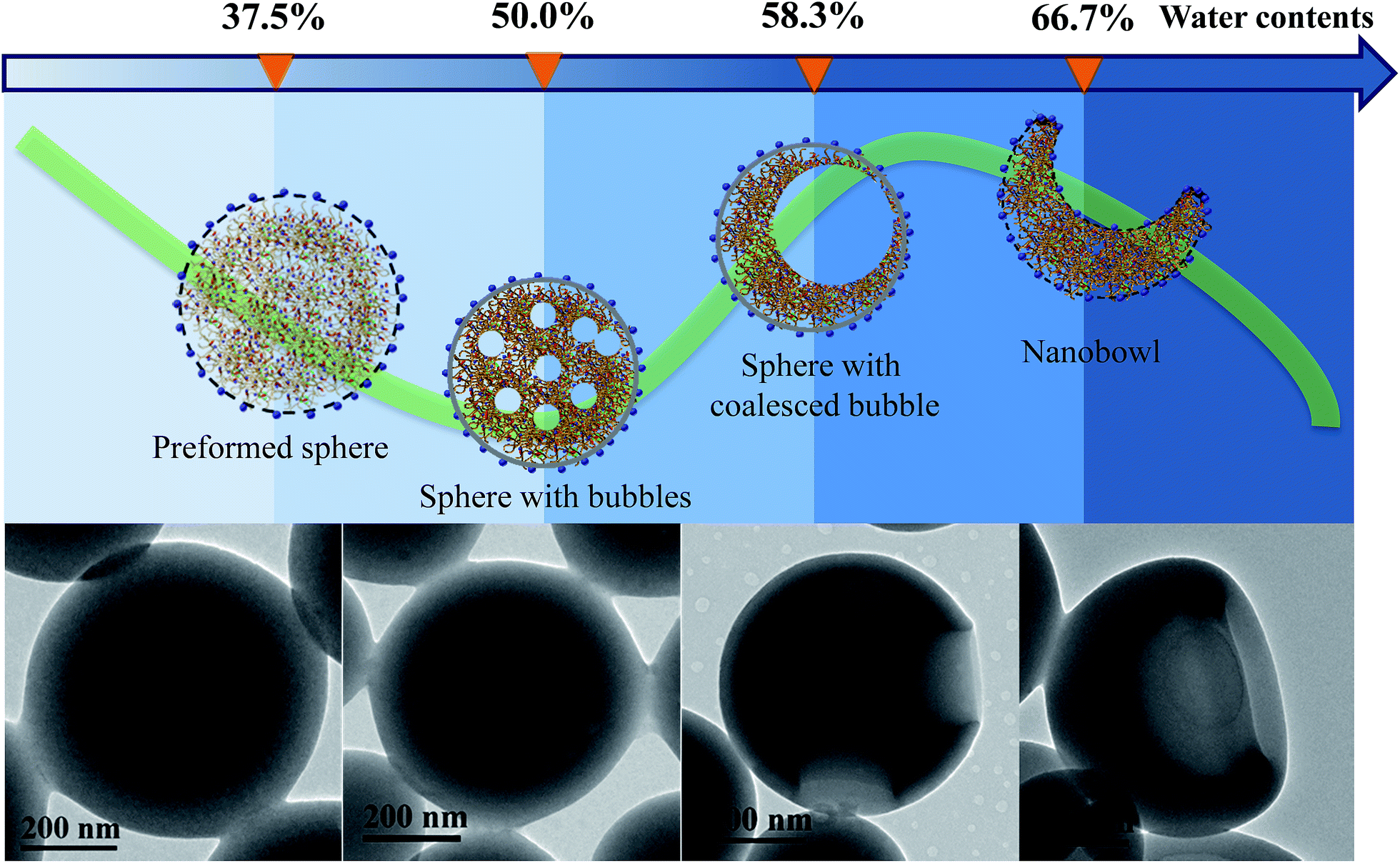 | ||
| Fig. 5 Evolution of nanobowls from PHAzoMA226. With the addition of water, PHAzoMA226 forms preformed spheres, spheres with bubbles, spheres with coalesced bubbles and nanobowls at different water contents. The TEM images show the corresponding nanostructures at the various water contents from left to right. | ||
Based on the above experimental results, the mechanism of the formation of nanobowls is proposed as follows. Upon adding water (non-solvent to PHAzoMA) to the THF solution of the PHAzoMA homopolymers, the fully extended homopolymer chains shrink and aggregate to form preformed spheres composed of soft main chains and rigid azobenzene side chains, with THF and water at the water content of 37.5% (Fig. 5 and S12, ESI†). Further addition of water leads to the size decrease of preformed spheres due to the extraction of THF from them (Fig. S13, ESI†). The phase separation and aggregation of the polymer chains leads to the homogenous shrinkage of the preformed spheres to form HCMs from PHAzoMA10, as a result of the weak interactions between polymer chains due to the low molecular weight and high chain mobility at this stage, as illustrated in Fig. S12 (ESI†) and 1B. However, the interaction between homopolymer chains inside the preformed spheres increases with the molecular weight of the PHAzoMA homopolymers, which hinders further shrinkage of the preformed spheres with further addition of water, as shown in Fig. S13, ESI.† The chain mobility considerably decreases due to the extraction of THF from the surface region of the preformed spheres, resulting in the significant shrinkage of the polymer chains on the surface and the formation of a rigid “skin” at the interface between the sphere and solvent (Fig. S12, ESI†). The rigid skin prevents the homogeneous shrinkage of the sphere but allows the diffusion of water and THF.13 Further extraction of THF from the interior of the sphere would lead to local collapse of the skin to form nanobowls.
As shown in Fig. 5 and S12 (ESI†), when the water content reaches 50%, the phase separation between polymer chains and the solvent (THF/water) results in the formation of a polymer-rich area and bubbles filled with a solvent-rich phase (THF/water). The bubbles coalesce to form large bubbles at a water content of 58.3% (Fig. 5), and then one larger bubble. Since it is difficult for the single bubble to exactly locate in the centre, the asymmetry of forces acting on it will move the bubble in the direction of the thinner wall and thus create asymmetric structures. Once the bubble is close to the edge, the thinner wall of the nanostructure will collapse, resulting in the formation of nanobowls (Fig. 5 and S12, ESI†). For this process to occur, the intermolecular interaction inside the preformed spheres should be within an appropriate range. The inner structure and the origin of a compound sphere and a nanobowl are the same, which results from the homogeneous or heterogeneous shrinkage of the preformed spheres.
As the molecular weight of the PHAzoMA homopolymer increases, the hardened skin forms at an earlier stage of water addition due to the increase in the polymer chain interactions, which leads to the formation of larger bubbles inside the preformed spheres, resulting in the formation of nanobowls with larger holes. This might be the reason why the width of the openings and depth of the holes have a linear relationship with the DP of the homopolymers, as shown in Fig. 4.
To confirm that the synergy between hydrogen bonding and π–π interaction plays a critical role in the formation of bowl-shaped nanoparticles,13 we synthesized homopolymers with different structures that can provide non-covalent interactions, as shown in Fig. 6A. The homopolymers are named P1, P2 and P3. The synthesis and characterization of the corresponding homopolymers followed the same protocols as PHAzoMA. As shown in Table 1, the molecular weights of P1, P2 and P3 are 18.1, 19.8 and 18.3 kg mol−1, respectively, which is similar to that of PHAzoMA59.
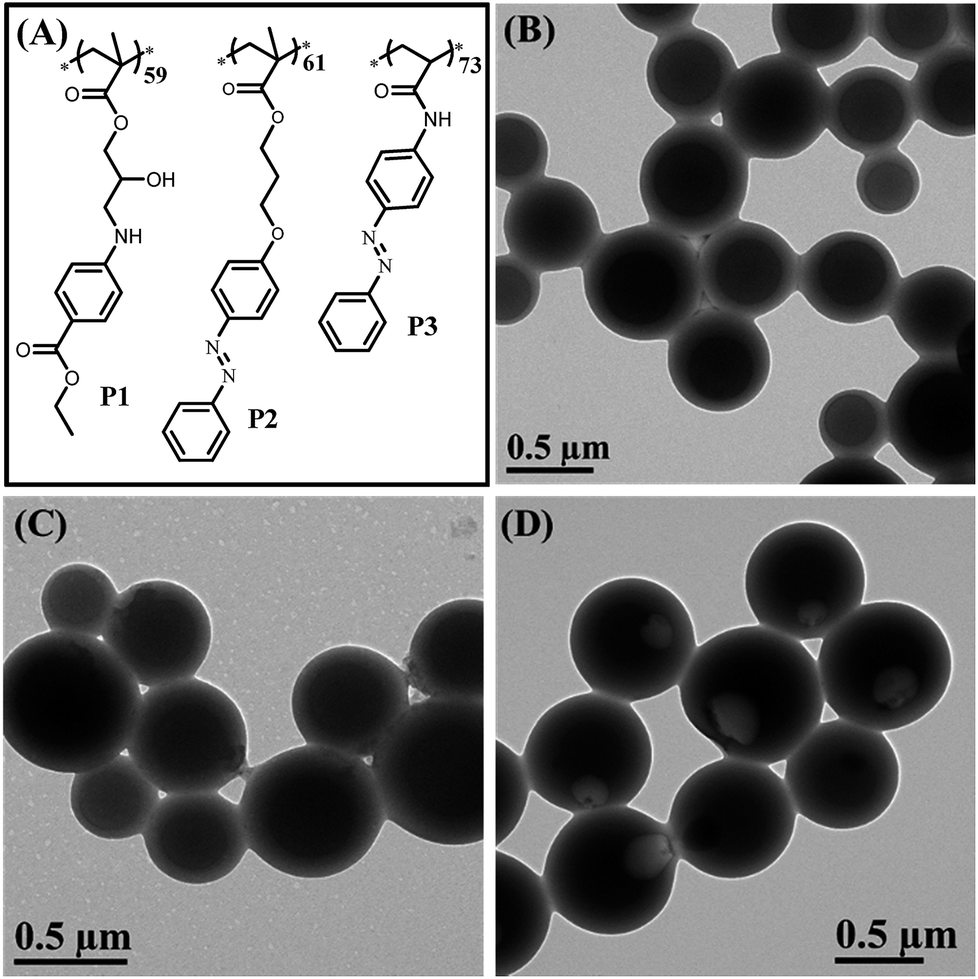 | ||
| Fig. 6 (A) The molecular structures of P1, P2, and P3 that have similar pendant structures and molecular weight to PHAzoMA59, for further confirming the formation mechanism and demonstrating the extensibility of this design strategy to prepare nanobowls; (B), (C) and (D) TEM images of HCMs from P1 and P2, and nanobowls from P3, respectively. | ||
Under the same self-assembly conditions as PHAzoMA, P1 and P2 self-assemble into HCMs, while P3 self-assembles into nanobowls (Fig. 6). The non-covalent interactions and different morphologies self-assembled from P1, P2 and P3 are summarized in Table 2. No obvious π–π interaction within P1 assemblies or hydrogen bonds within P2 assemblies were observed based on the UV-vis and FTIR spectra of P1 and P2 in Fig. S14, ESI.† HCMs are formed because the non-covalent interaction of homopolymers is not strong enough to hinder the homogenous shrinkage of the preformed spheres as there is only one non-covalent interaction for P1 (hydrogen bonding) and P2 (π–π interaction). However, in the presence of both hydrogen bonding and π–π interaction during the self-assembly process (Fig. S14, ESI†), nanobowls are formed from P3. These results prove that the synergistic effect between hydrogen bonding and π–π interaction is essential for the preparation of nanobowls.
| Homopolymer | π–π interaction | Hydrogen bonding | Morphology |
|---|---|---|---|
| P1 | ▲ | HCMs | |
| P2 | ▲ | HCMs | |
| P3 | ▲ | ▲ | Nanobowls |
To further verify the above theory, the photo-isomerization-dependent self-assembly experiments of PHAzoMA29 and P3 were performed. Both polymers were UV-irradiated prior to self-assembly to induce the transformation from trans-azobenzene to cis-azobenzene (Fig. S15, ESI†) which can't form π–π stacking.46 As expected, only HCMs were formed (Fig. S15, ESI†), instead of nanobowls. This result strongly confirmed that the formation of nanobowls is dominated by the synergy between the hydrogen bonding and π–π interaction of homopolymers.
Conclusions
In conclusion, a molecular design principle toward amphiphilic homopolymers was proposed for the preparation of bowl-shaped nanoparticles. Based on this principle, a series of homopolymers with an amino alcohol group and an azobenzene group were synthesized to self-assemble into nanobowls with controlled openings and interior holes as a result of the synergistic effect between the hydrogen bonding and π–π interaction of homopolymers. As the molecular weight of the PHAzoMA increases from 10.1 to 76.9 kg mol−1, the width of the openings and the depth of the holes increase from 242 to 423 nm and 95 to 363 nm, respectively, with a linear correlation. This non-covalent interaction controlled strategy might be extended to the preparation of a range of asymmetrical nanoparticles with controlled surface roughness and internal porosity. The mechanism and approach to precisely control the opening and internal holes of nanobowls may pave a new way for the fabrication and application of asymmetric nano-architectures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. D. is supported by the NSFC (21674081), the Shanghai 1000 Talents Plan (SH01068), and the Fundamental Research Funds for the Central Universities (22120180109 and 1500219107).Notes and references
- L. L. Chng, N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2013, 46, 1825–1837 CrossRef CAS PubMed
.
- L. Guo, Y. Xu, A. R. Ferhan, G. Chen and D.-H. Kim, J. Am. Chem. Soc., 2013, 135, 12338–12345 CrossRef CAS PubMed
- K. Thorkelsson, P. Bai and T. Xu, Nano Today, 2015, 10, 48–66 CrossRef CAS
- Z. Zheng, T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2015, 137, 948–957 CrossRef CAS PubMed
- I. C. Riegel, A. Eisenberg, C. L. Petzhold and D. Samios, Langmuir, 2002, 18, 3358–3363 CrossRef CAS
- L. E. Franken, Y. Wei, J. Chen, E. J. Boekema, D. Zhao, M. C. A. Stuart and B. L. Feringa, J. Am. Chem. Soc., 2018, 140, 7860–7868 CrossRef CAS PubMed
- S. Hyuk Im, U. Jeong and Y. Xia, Nat. Mater., 2005, 4, 671–675 CrossRef CAS PubMed
- F. Pei, T. An, J. Zang, X. Zhao, X. Fang, M. Zheng, Q. Dong and N. Zheng, Adv. Energy Mater., 2016, 6, 1502539 CrossRef
- J. Liang, X. Y. Yu, H. Zhou, H. B. Wu, S. Ding and X. W. Lou, Angew. Chem., Int. Ed., 2014, 53, 12803–12807 CrossRef CAS PubMed
- J. Liang, H. Hu, H. Park, C. Xiao, S. Ding, U. Paik and X. W. Lou, Energy Environ. Sci., 2015, 8, 1707–1711 RSC
- Y. Tu, F. Peng, X. Sui, Y. Men, P. B. White, J. C. M. van Hest and D. A. Wilson, Nat. Chem., 2017, 9, 480–486 CrossRef CAS PubMed
- D. A. Wilson, R. J. Nolte and J. C. van Hest, Nat. Chem., 2012, 4, 268–274 CrossRef CAS PubMed
- X. Y. Liu, J. S. Kim, J. Wu and A. Eisenberg, Macromolecules, 2005, 38, 6749–6751 CrossRef CAS
- X. Liu, J. Liu and M. Jiang, Macromol. Rapid Commun., 2009, 30, 892–896 CrossRef CAS PubMed
- C. Zhang, S. Yang, Y. Zhu, R. Zhang and X. Liu, Carbohydr. Polym., 2015, 133, 637–643 CrossRef CAS PubMed
- D. A. Roberts, M. J. Crossley and S. Perrier, Polym. Chem., 2014, 5, 4016–4021 RSC
- A. Ignacio, B. Miriam, B. M. Isabel, G.-V. Eduardo and S. V. Luis, Chem.–Eur. J., 2010, 16, 1246–1255 CrossRef PubMed
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef CAS PubMed
- P. A. Rupar, L. Chabanne, M. A. Winnik and I. Manners, Science, 2012, 337, 559–562 CrossRef CAS PubMed
- M. Antonietti and S. Forster, Adv. Mater., 2003, 15, 1323–1333 CrossRef CAS
- R. Dabirian, V. Palermo, A. Liscio, E. Schwartz, M. B. J. Otten, C. E. Finlayson, E. Treossi, R. H. Friend, G. Calestani, K. Müllen, R. J. M. Nolte, A. E. Rowan and P. Samorì, J. Am. Chem. Soc., 2009, 131, 7055–7063 CrossRef CAS PubMed
- J. Dupont, G. Liu, K. i. Niihara, R. Kimoto and H. Jinnai, Angew. Chem., Int. Ed., 2009, 48, 6144–6147 CrossRef CAS PubMed
- V. Percec, D. A. Wilson, P. Leowanawat, C. J. Wilson, A. D. Hughes, M. S. Kaucher, D. A. Hammer, D. H. Levine, A. J. Kim, F. S. Bates, K. P. Davis, T. P. Lodge, M. L. Klein, R. H. DeVane, E. Aqad, B. M. Rosen, A. O. Argintaru, M. J. Sienkowska, K. Rissanen, S. Nummelin and J. Ropponen, Science, 2010, 328, 1009–1014 CrossRef CAS PubMed
- J. K. Kim, S. Y. Yang, Y. Lee and Y. Kim, Prog. Polym. Sci., 2010, 35, 1325–1349 CrossRef CAS
- H. Yu, T. Kobayashi and H. Yang, Adv. Mater., 2011, 23, 3337–3344 CrossRef CAS PubMed
- Y. M. Wang, W. L. Mattice and D. H. Napper, Macromolecules, 1992, 25, 4073–4077 CrossRef CAS
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC
- L. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1996, 118, 3168–3181 CrossRef CAS
- K. Hales, Z. Chen, K. L. Wooley and D. J. Pochan, Nano Lett., 2008, 8, 2023–2026 CrossRef CAS PubMed
- E. N. Savariar, S. V. Aathimanikandan and S. Thayumanavan, J. Am. Chem. Soc., 2006, 128, 16224–16230 CrossRef CAS PubMed
- N. Li, G. Ye, Y. He and X. Wang, Chem. Commun., 2011, 47, 4757–4759 RSC
- Y. Q. Zhu, L. Liu and J. Z. Du, Macromolecules, 2013, 46, 194–203 CrossRef CAS
- Y. Q. Zhu, L. Fan, B. Yang and J. Z. Du, ACS Nano, 2014, 8, 5022–5031 CrossRef CAS PubMed
- M. Guo and M. Jiang, Soft Matter, 2009, 5, 495–500 RSC
- J. Zhang, K. Liu, K. Mullen and M. Yin, Chem. Commun., 2015, 51, 11541–11555 RSC
- S. R. Mane and R. Shunmugam, ACS Macro Lett., 2014, 3, 44–50 CrossRef CAS
- J. Zhang, S. You, S. Yan, K. Mullen, W. Yang and M. Yin, Chem. Commun., 2014, 50, 7511–7513 RSC
- Y. Meizhen, F. Chuanliang, S. Jie, Y. Yaming, X. Zejun, Y. Wantai, K. Wolfgang and M. Klaus, Small, 2011, 7, 1629–1634 CrossRef PubMed
- H. Sun, Y. Q. Zhu, B. Yang, Y. F. Wang, Y. P. Wu and J. Z. Du, J. Mater. Chem. A, 2016, 4, 12088–12097 RSC
- H. Sun and J. Z. Du, Nanoscale, 2018, 10, 17354–17361 RSC
- H. Sun, J. H. Jiang, Y. F. Xiao and J. Z. Du, ACS Appl. Mater. Interfaces, 2018, 10, 713–722 CrossRef CAS PubMed
- T. Azzam and A. Eisenberg, Langmuir, 2010, 26, 10513–10523 CrossRef CAS PubMed
- R. S. Rikken, H. Engelkamp, R. J. Nolte, J. C. Maan, J. C. van Hest, D. A. Wilson and P. C. Christianen, Nat. Commun., 2016, 7, 12606 CrossRef CAS PubMed
- C. Kim, M.-J. Yoon, S. H. Hong, M. Park, K. Park and S. Y. Kim, Electron. Mater. Lett., 2016, 12, 388–398 CrossRef CAS
- L. F. Zhang and A. Eisenberg, Macromolecules, 1999, 32, 2239–2249 CrossRef CAS
- A. Natansohn and P. Rochon, Chem. Rev., 2002, 102, 4139–4175 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: The Experimental section and characterization methods, the synthetic route, 1H NMR spectra, GPC traces, zeta potentials, DSC, TEM, and UV-vis and FTIR spectra. See DOI: 10.1039/c8sc03995j |
| This journal is © The Royal Society of Chemistry 2019 |