
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA bismuth oxide/graphene oxide nanocomposite membrane showing super proton conductivity and low methanol permeability†
Bailing
Liu
a,
Dongming
Cheng
a,
Haotian
Zhu
a,
Jing
Du
a,
Ke
Li
a,
Hong-Ying
Zang
 *ac,
Huaqiao
Tan
a,
Yonghui
Wang
a,
Wei
Xing
*b and
Yangguang
Li
*a
*ac,
Huaqiao
Tan
a,
Yonghui
Wang
a,
Wei
Xing
*b and
Yangguang
Li
*a
aKey Lab of Polyoxometalate Science of Ministry of Education, Key Laboratory of Nanobiosensing and Nanobioanalysis at Universities of Jilin Province, Faculty of Chemistry, Northeast Normal University, Changchun 130024, P. R. China. E-mail: zanghy100@nenu.edu.cn; liyg658@nenu.edu.cn; Fax: +86-431-85684009; Tel: +86-431-85099108
bState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin 130022, PR China. E-mail: xingwei@ciac.ac.cn
cSchool of Chemistry and Environmental Engineering, Changchun University of Science and Technology, Changchun 130024, P. R. China
First published on 17th October 2018
Abstract
Proton exchange membrane fuel cells are still limited as state-of-art proton exchange membranes perform poorly at high and low temperature and are easily damaged by harsh electrochemical conditions such as reactive peroxide species. One effective solution to this issue is to develop new types of proton conductive materials that are capable of working in a broad temperature range. A simple vacuum-assisted filtration method is employed to obtain a well-ordered new proton-conducting membrane by immobilizing nanosized bismuth oxide clusters [H6Bi12O16] (NO3)10·6(H2O) {H6Bi12O16} onto graphene oxide (GO) supports (named as {H6Bi12O16}/GO). {H6Bi12O16}/GO is stable in acidic media and has high proton conductivity over the temperature range from −40 to 80 °C. The proton conductivity of the {H6Bi12O16}/GO membrane is 0.564 S cm−1 at 80 °C in aqueous solution (in plane), and 0.1 S cm−1 at 80 °C and 97% RH (out of plane), respectively. Without loss of high proton conductivity, the membrane also exhibited 100-fold lower methanol permeability than a Nafion 117 membrane. Moreover, {H6Bi12O16}/GO displayed good catalytic decomposition of hydrogen peroxide and superior humidity response and recovery properties. These advantages mean that {H6Bi12O16}/GO holds great promise as a solid-state electrolyte that can potentially be applied in energy conversion devices in the future.
Introduction
Proton exchange membrane fuel cells have been regarded as promising energy conversion devices due to their high efficiency, environmentally friendly characteristics and extensive applications, including in portable electronic devices, electric vehicles, and smart grids.1 The proton exchange membrane is one of the key components that affect the overall performance of fuel cells.Currently, the most widely used perfluorinated sulfonated polymer, Nafion, is deemed to be the benchmark for proton-conducting materials. Its unique oriented ionic nanochannels can facilitate fast proton transportation, with a proton conductivity of up to 0.1 S cm−1 at 80 °C under 100% RH.2 However, it suffers from several drawbacks, such as high cost, significant decline of proton conductivity above 80 °C or below its freezing point, poor endurance of membranes caused by freeze/thaw cycles and high methanol permeability. More importantly, the oxygen reduction reaction in proton exchange membrane fuel cells produces strong oxidizing groups such as hydrogen peroxide/radicals (˙OH or ˙OOH) that can attack the internal skeleton structure of Nafion and initiate decomposition, which may dramatically increase the risk of fuel cell breakdown and reduce their service life.3
Recently, much effort has been devoted to creating new materials conquering these disadvantages, such as metal–organic frameworks, polyoxometalate-based hybrids, etc.4 As we know, there exists a distinct trade-off effect between proton transportation and methanol permeation. An effective method to reduce methanol permeability without lowering proton conductivity is to introduce hydrophilic groups (e.g., hydroxyl groups or sulfonic, phosphonic or carboxylic acids) which can help construct proton conducting pathways via intermolecular and intramolecular hydrogen bonds.5
Polyoxometalate (POM) anions are a type of structurally well-defined metal-oxide nanoclusters with excellent hydrophilicity and good catalytic decomposition of hydrogen peroxide.6 In the primary structure of POMs, the oxygen atoms on the surface of a POM are coordinated by protons, while the secondary structure involves hydrogen bonds between water molecules and the oxygen atoms of POMs, which helps to construct a 3D proton transfer pathway. POMs have widely been acknowledged as an important and efficient proton conductor, but their high water solubility limits their application in fuel cells.7 Traditionally, employing large counterions to replace protons can effectively reduce POMs' solubility, but this strategy sacrifices the proton conductivity. To guarantee the proton source and lower the solubility of POMs, we need to find a new strategy and utilize unconventional POM clusters to obtain the desired materials. We found that bismuth oxide cationic nanoclusters have abundant oxygen atoms on the surface, protonated water molecules surrounding them and low water solubility. The cationic nature of bismuth oxide enables it to interact with anions, especially ones with large specific surface area.8
GO is a 2D layered material with large specific surface area and abundant hydrophilic groups (such as hydroxyl groups, carboxyl groups and epoxide groups) on the surface, which could be beneficial for both the improvement of proton conductivity and hindering the infiltration of methanol.9 GO can assemble into lamellar microstructures showing unique ordered 2D ion transport channels and water retention ability.10 Multilayer stacking provides more optional pathways for proton transportation, which might also promote rapid proton transfer.
Merging the merits of the above two materials, an innovative proton-conducting composite membrane has been prepared by immobilizing nanosized bismuth oxide [H6Bi12O16] (NO3)10·6(H2O) {H6Bi12O16} onto a graphene oxide (GO) support (named as {H6Bi12O16}/GO), which enables the material to possess superior proton conductivity, catalytic decomposition of hydrogen peroxide, low methanol permeability, and suitability in a wide temperature range, even at sub-zero conditions. GO walls decorated with more hydrophilic bismuth oxide cation clusters with abundant protons would retain higher water content and present continuous hydrogen bonding pathways for interlayer galleries, which speeds up proton migration and hydrogen bond breaking/reformation. The material has numerous nanogaps and abundant hydrogen bonding networks, which is beneficial for alleviating the methanol crossover and facilitates fast proton transfer. Meanwhile, {H6Bi12O16}/GO has unique electrochemical redox activity for H2O2, improving the service life of the membrane. In this way, the material showed a high proton conductivity of 0.1 S cm−1 at 97% RH and 80 °C. In an aqueous solution, a {H6Bi12O16}/GO composite membrane showed five times higher proton conductivity (0.564 S cm−1) than Nafion (∼0.1 S cm−1) at 80 °C (Scheme 1).
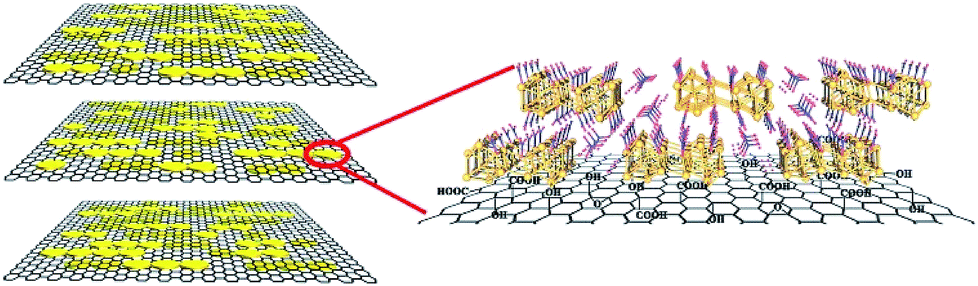 | ||
| Scheme 1 (Left) A structural model of downsized {H6Bi12O16} nanoparticles dispersed on the graphene oxide surface. The yellow cylinders represent the positively charged bismuth oxygen clusters balanced on the GO forming a {H6Bi12O16}/GO nanocomposite. (Right) Magnified chemical-interaction structure diagram for the {H6Bi12O16}/GO nanocomposite. | ||
Results and discussion
Structural characterization of {H6Bi12O16} and the {H6Bi12O16}/GO composite
The {H6Bi12O16}/GO nanocomposite membrane was synthesized via a co-precipitation mechanism (Fig. 1a). The [H6Bi12O16]10+ polycation nanoclusters interacted with GO via strong electrostatic force and hydrogen bonds. {H6Bi12O16} clusters are composed of two [H3Bi6O8]5+ groups joined across the symmetry centres through two bridging O atoms, together with nitrate anions and water molecules (Fig. S1†).11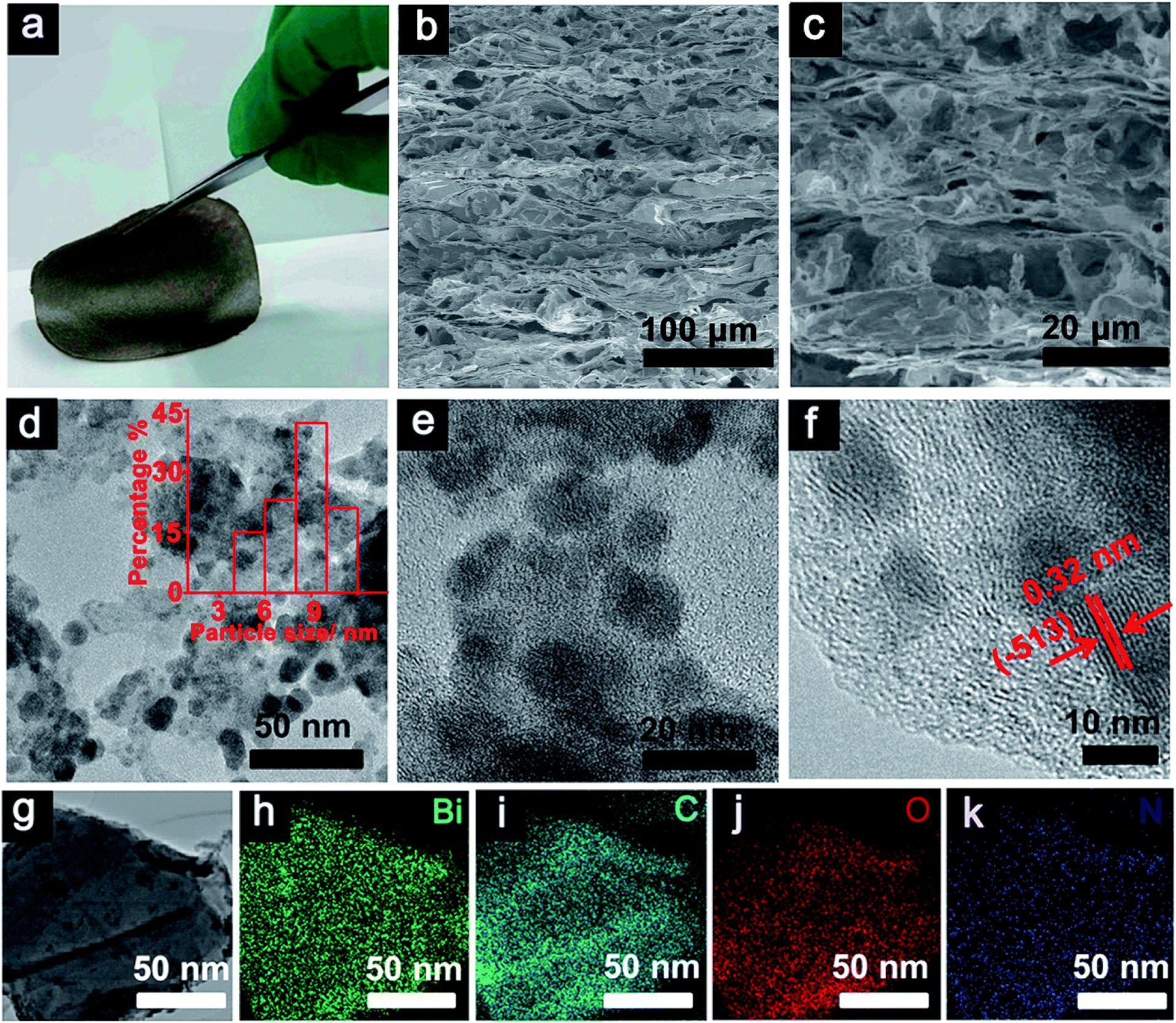 | ||
| Fig. 1 (a) The {H6Bi12O16}/GO membrane was obtained via vacuum-assisted filtration. (b and c) Scanning electron microscope (SEM) cross-sectional images of {H6Bi12O16}/GO. (d–f) TEM images of {H6Bi12O16}/GO. (g–k) Corresponding elemental mapping images of Bi, C, O and N of {H6Bi12O16}/GO. | ||
The scanning electron microscopy (SEM) images showed the cross-sectional morphology of the {H6Bi12O16}/GO membrane. It is clearly observed from Fig. 1b and c that {H6Bi12O16} cluster nanoparticles were randomly distributed on the surface and edge of the multilayer stacked graphene oxide sheets with an average size of ca. 8.66 nm. This result was further verified by the TEM images in Fig. 1d–f. For comparison, pure {H6Bi12O16} bulk material is comprised of regular nanoplates with an average size of 0.28 μm (Fig. S2†). The introduction of GO is helpful to improve the specific surface area of the composite materials and reduce the size of particles.12 At the same time, it is also confirmed that we have successfully downsized the bulk {H6Bi12O16} crystalline material to {H6Bi12O16} nanocrystals tethered onto GO supports. The high-resolution TEM images of {H6Bi12O16} clusters presented lattice fringes with a d-spacing of 0.32 nm, associated with their (−5 1 3) crystallographic plane (Fig. S3†). The high-resolution TEM image of the {H6Bi12O16}/GO exhibited the presence of GO layers and clear lattice fringes of bismuth oxide {H6Bi12O16} nanocrystals with interplanar distances of 0.32 nm (Fig. 1d–f), which is consistent with the {H6Bi12O16} bulk material. This can further explain that the {H6Bi12O16} nanocrystals spread on the graphene oxide sheets forming a bismuth oxide/GO composite. Fig. 1g–k show the corresponding elemental mapping images of {H6Bi12O16}/GO, which confirmed the distribution of Bi, C, O and N across the entire range. The EDX results further confirm the composition of {H6Bi12O16}/GO as depicted in Fig. S4.†
The powder X-ray diffraction (XRD) patterns of GO, {H6Bi12O16} and {H6Bi12O16}/GO are shown in Fig. 2a. The XRD pattern of the {H6Bi12O16}/GO composite contains both peaks of GO and {H6Bi12O16}, indicating the successful combination of the two components; characteristic 2θ peaks located at 6.519°, 11.634°, 24.894°, 27.770° and 42.548° were indexed to the (1 0 0), (1 1 0), (−3 2 2), (−5 1 3) and (−8 0 6) facets of {H6Bi12O16} (JCPDF 70-1226). The peak of GO at 11.8° indicates that the interlayer spacing is 0.82 nm. The chemical and thermal stability of the composite was characterized by XRD as well. The XRD patterns remain unchanged after soaking in boiling water and 8 M nitric acid. The temperature-varied XRD patterns demonstrate that the composite is stable up to 120 °C (Fig. S5†).13
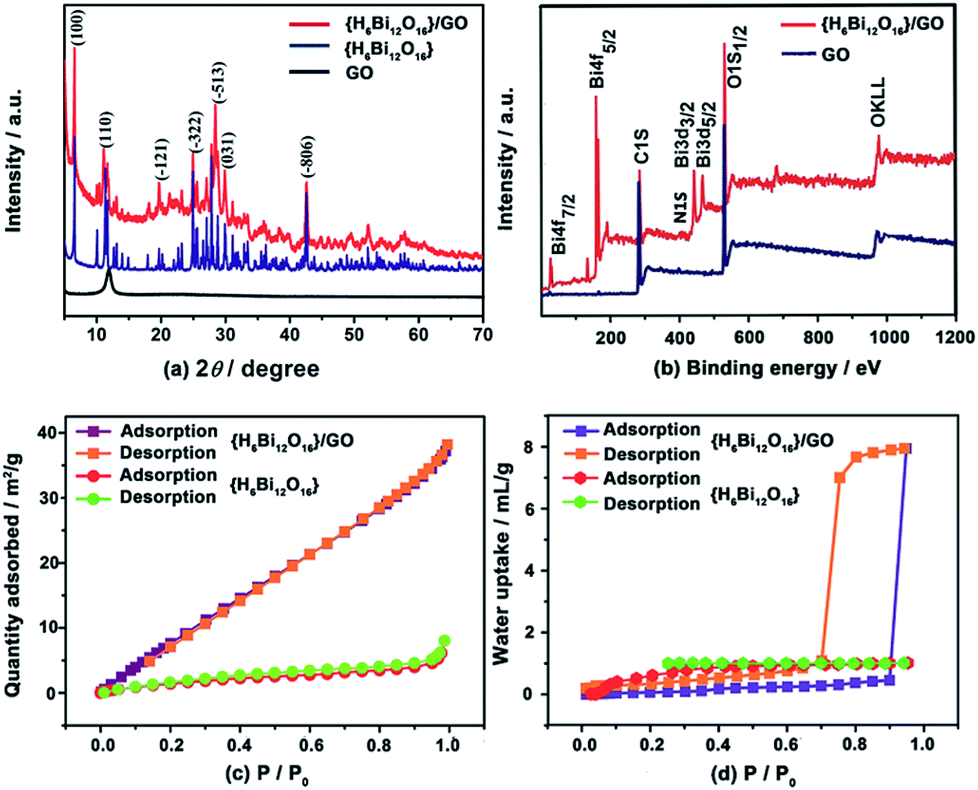 | ||
| Fig. 2 (a) Powder XRD spectra of {H6Bi12O16}/GO (red curve), {H6Bi12O16} (blue curve) and GO (purple curve). (b) XPS spectra of survey {H6Bi12O10}/GO and GO. (c) N2 adsorption/desorption isotherms of {H6Bi12O16}/GO and {H6Bi12O16}. (d) Water adsorption/desorption isotherms of {H6Bi12O16} and {H6Bi12O16}/GO. | ||
The FT-IR spectra (Fig. S6†) and Raman spectra (Fig. S7†) also confirmed the combination of {H6Bi12O16}/GO. X-ray photoelectron spectroscopy (XPS) measurements were performed to investigate the chemical composition and valence states of the {H6Bi12O16}/GO composite. The full scan XPS of {H6Bi12O16}/GO indicated the presence of Bi, C, O and N elements (Fig. 2b).
The binding energy of Bi 4f and C 1s indicates that Bi and C are present in the composite (Fig. S8†). Furthermore, the BET surface areas of the {H6Bi12O16} and {H6Bi12O16}/GO were measured to be 7 m2 g−1 and 49.6 m2 g−1, respectively, suggesting that GO can slightly increase the specific surface area of the composite (Fig. 2c). The N2 adsorption isotherm of {H6Bi12O16}/GO was not a hysteresis loop, suggesting its nonporous structure.14
Thermogravimetric analysis (TGA) of {H6Bi12O16} and {H6Bi12O16}/GO showed excellent thermal stability for these materials (Fig. S9 and S10†). {H6Bi12O16}/GO showed a much higher weight loss than {H6Bi12O16}, proving that more water molecules or hydroxyl groups exist in {H6Bi12O16}/GO. Water vapour adsorption of {H6Bi12O16} and {H6Bi12O16}/GO dried samples was carried out at 25 °C to test their water adsorption and water retention abilities. As shown in Fig. 2d, the water adsorption increased slowly with the increase of water vapour relative pressure for {H6Bi12O16} and {H6Bi12O16}/GO. Surprisingly, when the relative pressure continued to rise to above 0.9, the amount of adsorption increased rapidly for {H6Bi12O16}/GO, and the maximum water uptake is 177.5 cm3 g−1, corresponding to approximately 14.4 wt% water molecules adsorbed, which might be attributed to surface adsorption.15
The water adsorption isotherm of {H6Bi12O16}/GO with large hysteresis loops appeared to be a type-IV isotherm, showing that the material had excellent hydrophilic properties and high water holding capacity.16 The huge water adsorption at high relative pressure might be attributed to the capillary action. {H6Bi12O16} nanoparticles assembled next to each other on the hydrophilic surface of GO, leading to many pinholes that caused capillary action. Due to the synergistic effect of GO and {H6Bi12O16}, the composite showed better water adsorption and retention compared with bulk {H6Bi12O16} under high relative pressure. We speculate that {H6Bi12O16}/GO contains a large number of free-dangling water retention hydrophilic groups (–OH, –COOH), which would make the matrix store more water by capillary action to eliminate grain boundaries between nanoparticles under high relative humidity. This is beneficial for promoting proton transfer via the continuous long-range order of the hydrogen bonding network.
Proton conductivity of the {H6Bi12O16}/GO membrane
The proton conductivity of the {H6Bi12O16}/GO was evaluated by AC impedance spectroscopy using the vacuum-assisted filtration method to assemble well-ordered membranes attached to the surface between four platinum electrodes.17 The {H6Bi12O16}/GO membranes exhibit higher proton conductivities than the pristine {H6Bi12O16} (Fig. 3a and S11, ESI†). The proton conductivity of {H6Bi12O16}/GO membranes is 0.14 S cm−1 in an aqueous solution at 30 °C. As the temperature was elevated to 80 °C, the proton conductivity of the {H6Bi12O16}/GO membrane exhibited large enhancement, its proton conductivity reaching 0.564 S cm−1 (Fig. 3a & S12†). We proposed that in aqueous solution, more loosely bonded water molecules on the surface of the composite membrane behave like free liquid water, eliminating innumerable grain boundaries of {H6Bi12O16}/GO in the nanocomposite, achieving consecutive in-plane proton transfer paths for rapid proton migration. The XRD patterns of the {H6Bi12O16}/GO membrane did not show any difference before and after the proton conductivity test (Fig. S5†). As shown in Table S2,† the {H6Bi12O16}/GO composite film exhibits good ion exchange capacity (IEC) and water uptake capacity (WU), as reported in the ESI.†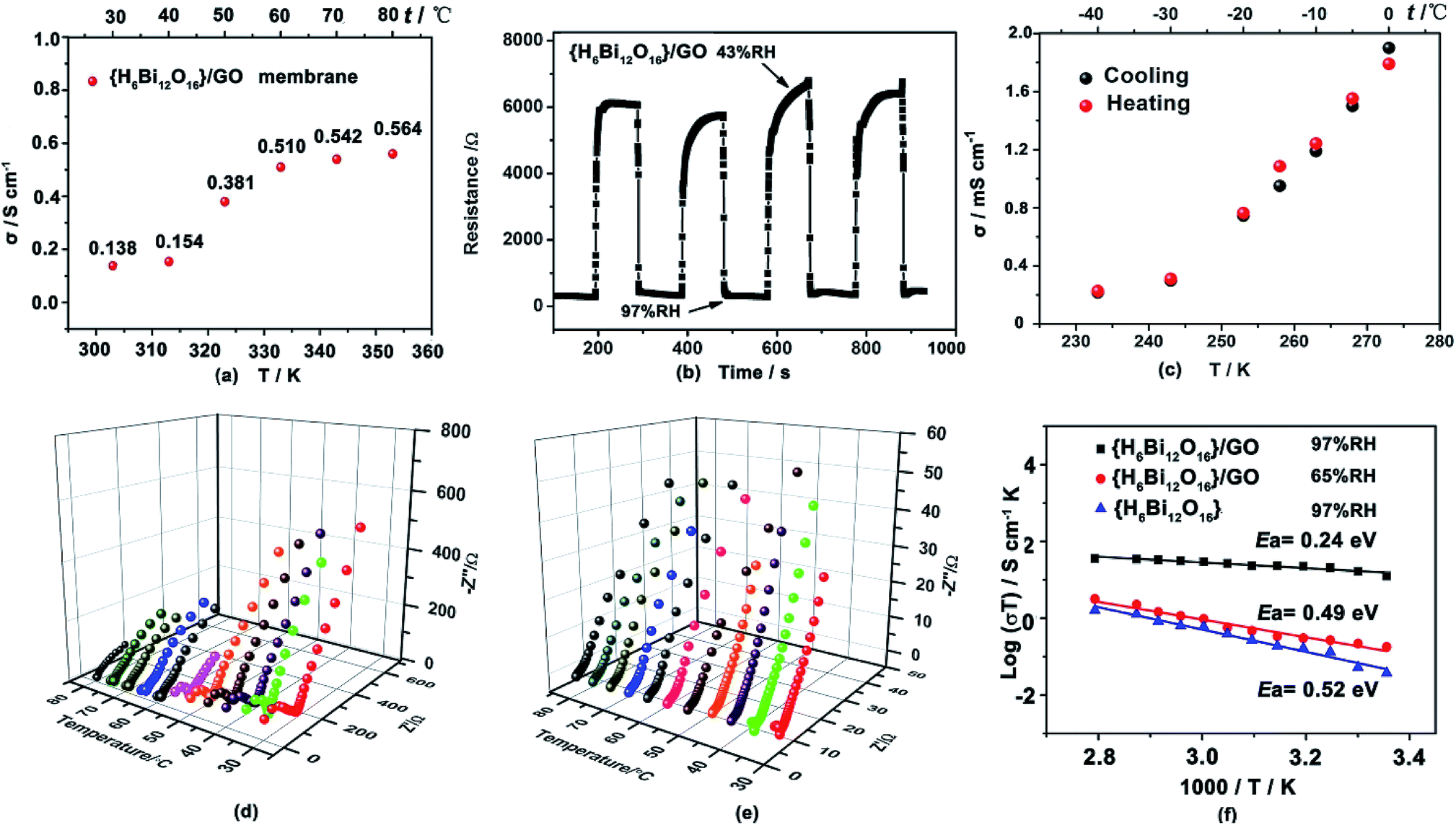 | ||
| Fig. 3 (a) Proton conductivity (σ) of the {H6Bi12O16}/GO membrane at various temperatures in aqueous solution. (b) Typical response curves of the{H6Bi12O16}/GO film upon exposure to 43% RH and 97% RH at 1000 Hz. (c) Negligible changes of proton conductivity of humidified {H6Bi12O16}/GO during heating–cooling cycles at sub-zero conditions. (d) Nyquist plots for {H6Bi12O16} at 97% RH and various temperatures. (e) Nyquist plots for {H6Bi12O16}/GO at 97% RH and various temperatures. (f) Arrhenius-type plot of the conductivity of {H6Bi12O16}/GO and {H6Bi12O16} at various temperatures and humidity conditions. | ||
To further explore the humidity sensitivity of {H6Bi12O16}/GO under harsh working conditions, the resistance responses to humidity based on the {H6Bi12O16}/GO membrane were measured at 1000 Hz.18Fig. 3b shows four cycles of response and recovery processes between 43% and 97% RH for the {H6Bi12O16}/GO membrane. Apparently, the composite membrane presented an impedance change between ∼6000 Ω and ∼200 Ω for humidity increments from 43% to 97% RH, showing a high magnitude response to different relative humidities. Moreover, the {H6Bi12O16}/GO membrane presented much faster water adsorption and desorption process response times t97% of 2 s and 4.3 s, respectively. In comparison, the Nafion 117 film revealed that for hysteresis as large as 97% RH (Fig. S13†), the response times for both adsorption and desorption procedures were much longer. This can be attributed to the surface oxygen atoms of the {H6Bi12O16}/GO membrane, providing a larger number of active sites for humidity sensitivity. In particular, the adsorption process could not reach an equilibrium state, even after 10 hours. To further elucidate the correlation between proton conductivity in the vertical direction of {H6Bi12O16}/GO and different relative humidities, we measured the proton transfer performance of {H6Bi12O16}/GO under varying relative humidity values, ranging from 35% to 97% RH at 30 °C. The proton conductivity was evaluated by ac impedance spectroscopy using a compacted pellet of the powder sample attached to the surface of two gold electrodes. As shown in Fig. S14,† proton conductivity increases with the rise of relative humidity. The proton conductivity of pure {H6Bi12O16} material ranges from 1.0 × 10−8 S cm−1 to 7.4 × 10−4 S cm−1 from 35% RH to 97% RH (Fig. S12†). As for the {H6Bi12O16}/GO composite, its proton conductivity can only reach 9.74 × 10−7 S cm−1 under a relative humidity of 35%, due to the lack of adequate water molecules to build abundant hydrogen bonding networks under low relative humidity (Fig. S14†). But when RH increased to 97%, the value increased rapidly to 5.5 × 10−2 S cm−1, which is almost 105 times higher than that under low relative humidity at 30 °C. The value is much higher than that of most MOF/POM-based materials at ambient temperature. We proposed that the obvious improvement in proton conductivity of {H6Bi12O16}/GO resulted from the following factors: firstly, {H6Bi12O16}/GO materials can provide a higher concentration of protons as well as proton donor and receptor sites for proton transportation. Secondly, the bismuth oxide surface contained multiple water layers, including molecularly adsorbed water hydroxyls, a hydrogen bonded layer and a first physisorbed layer. This effectively reduced the energy barrier and facilitated the fast transportation of protons in the composite. Thirdly, graphene oxide nanosheets had high surface area to promote nanocrystallization of {H6Bi12O16} particles and cause nanogaps. Meanwhile GO provides hydrophilic sites and constructs continuous hydrogen bonding networks for proton transfer between {H6Bi12O16} particles. All these reduced grain boundary resistance between pure bismuth oxides effectively constructing long-range hydrogen-bonding networks. Under high relative humidity, the gaps of {H6Bi12O16} nanoparticles were filled with water molecules due to capillary action, which facilitates quick proton transfer via continuous hydrogen bonding networks between {H6Bi12O16} particles.
Proton conducting materials that suitably work below the freezing point have gradually received attention due to real application demand.19 The {H6Bi12O16}/GO composite showed a remarkable proton conductivity of 2.17 × 10−4 S cm−1 at −40 °C (Fig. S15 and S16†). We also confirmed the cycle stability of the {H6Bi12O16}/GO material via performing heating and cooling cycle measurements (Fig. 3c), and little variation in its proton conductivity was found from −40 °C to 0 °C. We proposed that the crystalline water is retained between particle gaps under the freezing point and forms a more extensive hydrogen-bond network for improving the proton conductivity to a large extent. {H6Bi12O16}/GO represents a new type of solid state proton-conductive material with relatively high proton conductivity at sub-zero temperatures.
Proton-conducting materials as electrolytes for fuel cells were required to be electric insulators to inhibit interelectric breakdown.20 We investigated the electron conductivity of {H6Bi12O16}/GO by direct-current measurements, and the electron conductivity was negligible.21 Thus, {H6Bi12O16}/GO itself was an electric insulator. Simultaneously, we conducted ionic conductivity measurements of {H6Bi12O16}/GO in deuterated water under different RH% conditions to prove that the ionic conductivity is due to proton transfer (Fig. S17†).22 To prove that {H6Bi12O16}/GO is not a physical mixture of GO and {H6Bi12O16}, we also compared the proton conductivity of a physical mixture of {H6Bi12O16} and GO (Fig. S18†). The observed proton conductivity of this physically mixed material was much lower than that of our synthesized {H6Bi12O16}/GO composite.23
Activation energy (Ea) was another important factor for measuring proton migration barriers.24 As shown in Fig. 3d–f, we deduced that the activation energy values (Ea) for pristine {H6Bi12O16} and {H6Bi12O16}/GO are ca. 0.52 eV and 0.24 eV from Arrhenius plots in the temperature range of 30–80 °C under 97% RH, respectively. For pristine {H6Bi12O16}, it can be concluded that proton conduction was probably dominated by H3O+ dynamic motion or rotation on the surface, leading to a high activation energy following the Vehicle mechanism.25 For {H6Bi12O16}/GO, protons propagate through the continuous hydrogen-bonding networks constructed by hydroxyl and carboxyl groups along with adsorbed water molecules, obeying the Grotthuss mechanism. The Grotthuss mechanism may involve two modes of protonated water clusters, Eigen cation H3O+–(H2O)3 and Zundel ion H2O⋯H+⋯OH2 (Fig. S19†).26 Under 65% RH, proton conductivities at various temperatures are shown in Fig. S20;† the activation energy of {H6Bi12O16}/GO is 0.49 eV, which suggests that a mixed Grotthuss and Vehicle mechanism may exist under lower relative humidity. To investigate the durability of the composite material, we measured the proton conductivity of the {H6Bi12O16}/GO sample at 80 °C under 97% RH for 12 h (Fig. S21†). The proton conductivity of {H6Bi12O16}/GO remained at approximately 0.1 S cm−1, suggesting its long-term durability under high relative humidity,27 which is comparable to that of the commercial Nafion membrane (0.1 S cm−1, 80 °C, 100% RH). This value is also higher than those of previously reported studies regarding GO and/or MOF-modified membranes (Table S1†). Meanwhile, the structural integrity was evaluated from the PXRD patterns before and after the proton conductivity measurement (Fig. S5†).
Cyclic voltammetry was performed to prove that the composite can electrocatalyse the decomposition of hydrogen peroxide.28 As shown in Fig. 4, two well-defined redox couples can be observed in PBS solution at pH 7.4. {H6Bi12O16}/GO shows promising electro-catalytic activity towards reduction of H2O2, as revealed by the changes in peak current upon the addition of H2O2. As the concentration of hydrogen peroxide increases continuously, the cathodic current increases, while the related anodic current decreases. This indicates that in the fuel cell application, the hydrogen peroxide formed during the oxygen reduction reaction will be decomposed by {H6Bi12O16}/GO and lower the effect on the efficiency of the fuel cell.29 It is noteworthy that most proton exchange membrane materials cannot electrocatalyse the decomposition of hydrogen peroxide. Above all, the {H6Bi12O16}/GO composite showed super proton conductivity even at sub-zero temperatures and catalysed the decomposition of hydrogen peroxide.
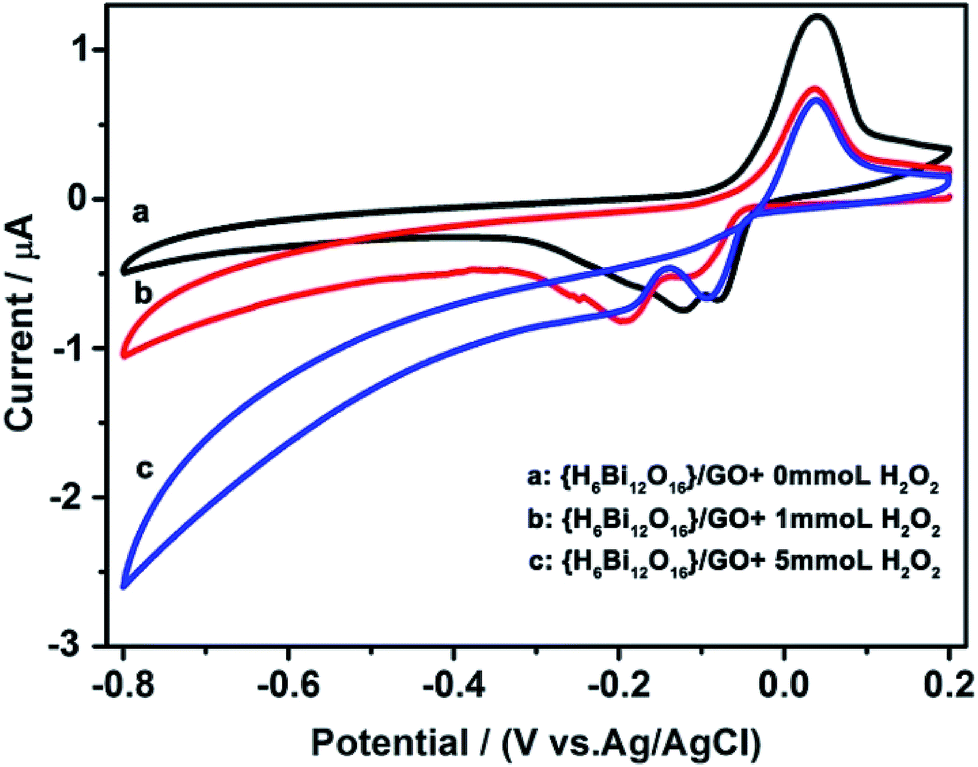 | ||
| Fig. 4 Electrocatalytic reduction of H2O2 with {H6Bi12O16}/GO in a 0.1 M KH2PO4–Na2HPO4 aqueous solution (pH = 7.4). Scan rate: 50 mV s−1. | ||
As an important indicator to evaluate the resistance for fuel permeation of membranes applied in direct methanol fuel cells, methanol permeability (P) tests were carried out with comparison between {H6Bi12O16}/GO and Nafion.30 As shown in Fig. 5, the methanol permeability of {H6Bi12O16}/GO decreased with increasing thickness of membranes as a general rule. Moreover, membranes of {H6Bi12O16}/GO with thickness from 30 μm to 200 μm have a lower methanol permeability compared to those of Nafion with similar thicknesses (approximately 2 orders of magnitude). Combined with the above results, the introduction of graphene oxide leads to the {H6Bi12O16}/GO composite membrane exhibiting obviously lower methanol permeability than the Nafion membrane, and the methanol permeability of the membranes remained almost unchanged with increasing the reaction time, remaining at ∼10−10 cm2 s−1 (Fig. S22 & Table S3†).
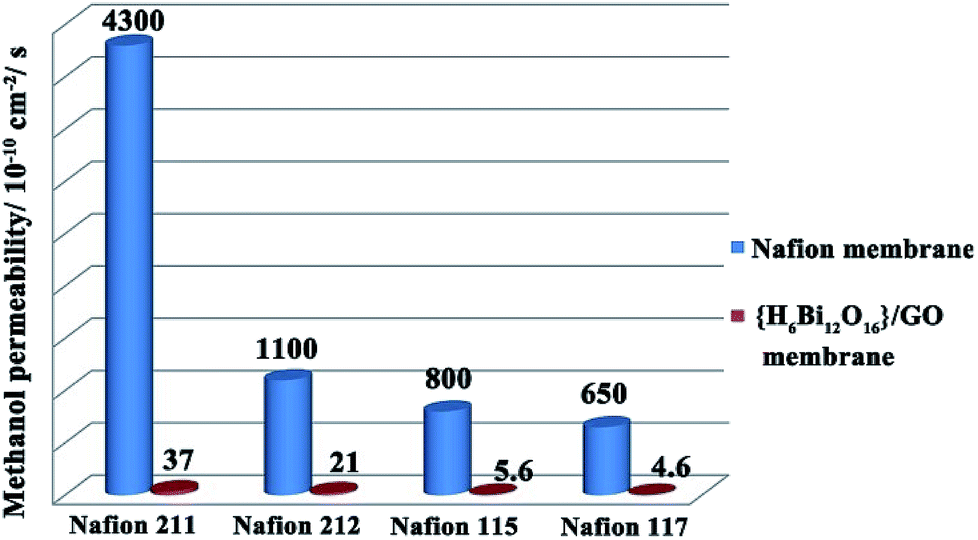 | ||
| Fig. 5 Comparison of methanol permeability of the {H6Bi12O16}/GO composite membrane and different thickness Nafion membranes. | ||
We discussed that ultralow methanol diffusion can be achieved through the interlayer spacing of multilayer stacked GO nanosheets to prevent methanol from entering, while facilitating fast transport of protons by the construction of highly continuous ionic channels.31 On the other hand, we assumed that the composite material induced hydrogen-bond barrier from hydrophilic groups results in more tortuous channels, preventing methanol crossover.
The high proton conductivity combined with the excellent resistance against methanol permeation make the {H6Bi12O16}/GO suitable for use in DMFCs. The {H6Bi12O16}/GO composite material cannot endure super strong mechanical extrusion during membrane electrode assembly and so we mixed {H6Bi12O16}/GO with polyvinyl alcohol (PVA). The cell performance of the {H6Bi12O16}/GO/PVA membranes was evaluated at 80 °C under fuel cell operating conditions of 1 M methanol solution at the anode side and oxygen gas at the cathode side. The DMFCs using the {H6Bi12O16}/GO/PVA membranes have better performance because of the enhanced proton conductivity and reduced methanol permeability of the {H6Bi12O16}/GO/PVA membranes (Fig. 6). The open-circuit voltage (OCV) and the maximum power density of the DMFCs using the {H6Bi12O16}/GO/PVA membranes reach 0.89 V and 3.2 mW cm−2, respectively. It is worth mentioning that the open-circuit voltage (OCV) is higher than that of most reported ones due to the excellent methanol resistance of {H6Bi12O16}/GO.32 In comparison, pure PVA films did not exhibit DMFC performance.
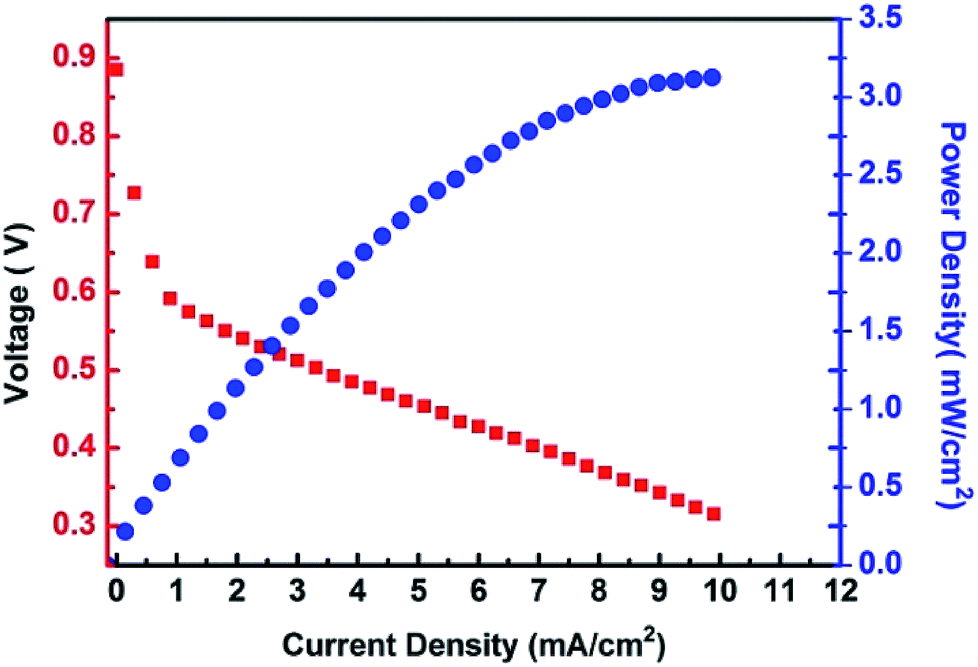 | ||
| Fig. 6 Polarization and power density curves of DMFCs using {H6Bi12O16}/GO/PVA membranes. | ||
Conclusions
In summary, inspired by polyoxometalates, we utilized hydrophilic bismuth oxide {H6Bi12O16} cationic nanocrystals in situ interacting with hydrophilic GO to form a {H6Bi12O16}/GO nanocomposite as an efficient proton-conducting material. The {H6Bi12O16}/GO nanocomposite can effectively overcome the trade-off effect between proton conductivity and methanol permeability, that is, lower methanol permeability without the loss of superior proton conductivity, and meanwhile show catalytic decomposition of hydrogen peroxide, as well as quick humidity response and recovery properties. The {H6Bi12O16}/GO composite membrane showed better proton conductivity than Nafion in aqueous solution with a proton conductivity (in plane) of 0.564 S cm−1 at 80 °C and also showed good performance at sub-zero temperatures with a value of 2.17 × 10−4 S cm−1 at −40 °C. {H6Bi12O16}/GO followed the Grotthuss mechanism under 97% RH. The composite membrane showed 100-fold lower methanol permeability than a Nafion 117 membrane. Its ability to catalyse hydrogen peroxide may provide researchers a new way of extending the service life of the membrane in fuel cells. This work provides a promising blueprint for the design of new proton-conducting materials, conquering the drawbacks of traditional membranes.Experimental section
Synthesis of the {H6Bi12O16}/GO membrane
Bi(NO3)3·5H2O (12.1 g, 0.025 mol) was homogenously dispersed in 10 mL nitric acid (6 mol L−1) with vigorous stirring at room temperature. Then, 15 mL graphene oxide (10 mg mL−1) solution was added dropwise into the above solution, while adjusting the pH of the solution to 0 using 6 M NaOH. The solution was stirred for 2 hours to obtain a uniformly dispersed {H6Bi12O16}/GO mixed solution. The above mixed solution was filtered by vacuum-assisted filtration and the {H6Bi12O16}/GO membrane was obtained by a direct self-assembly method, as illustrated in Fig. 7.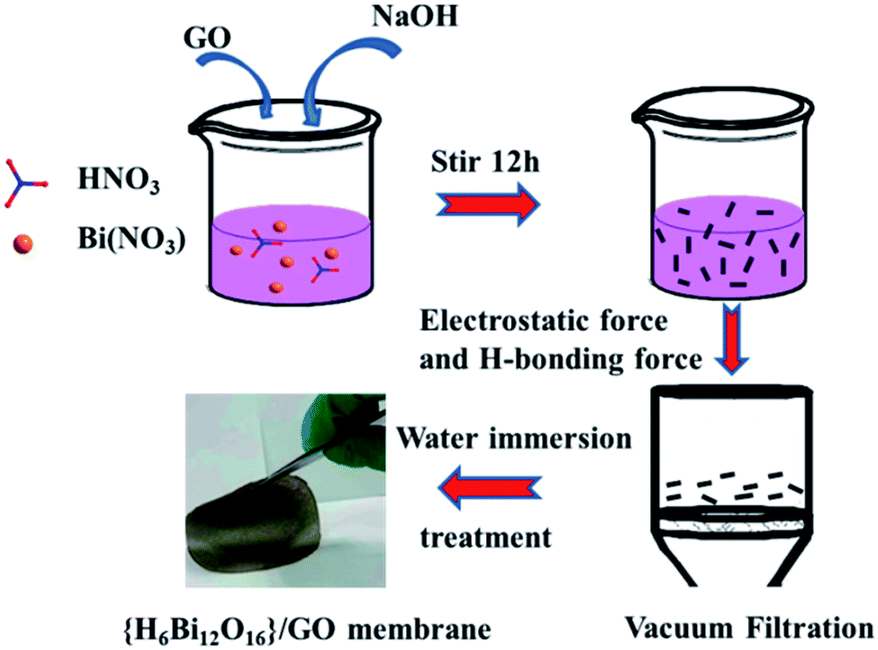 | ||
| Fig. 7 Scheme of the synthetic procedure for the {H6Bi12O16}/GO composite membrane. | ||
Conflicts of interest
There are no conflicts of interest.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21471028, 26171036, and 21871042), National Key Basic Research Program of China (No. 2013CB834802), and Natural Science Foundation of Jilin Province (No. 20150101064JC). We would like to acknowledge the support from the Jilin Provincial Department of Education. Many thanks for useful discussion and advice from Professor Carsten Streb.Notes and references
- (a) K. Firouz Tadavani, A. Abdolmaleki, M. R. Molavian, S. Borandeh, E. Sorvand and M. Zhiani, Energy Fuels, 2017, 31, 11460–11470 CrossRef CAS; (b) E. E. Josberger, P. Hassanzadeh, Y. X. Deng, J. Sohn, M. J. Rego, C. T. Amemiya and M. Rolandi, Sci. Adv., 2016, 2, e1600112 Search PubMed; (c) J. H. Wee, Renewable Sustainable Energy Rev., 2007, 11, 1720–1738 CrossRef CAS.
- (a) K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535–4586 CrossRef CAS PubMed; (b) A. Kraytsberg and Y. Ein-Eli, Energy Fuels, 2014, 28, 7303–7330 CrossRef CAS.
- (a) R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, X. J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K. Kimijima and N. Iwashita, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS PubMed; (b) Y. S. Wei, X. P. Hu, Z. Han, X. Y. Dong, S. Q. Zang and T. C. Mak, J. Am. Chem. Soc., 2017, 139, 3505–3512 CrossRef CAS PubMed.
- (a) S. Hu, M. Lozada-Hidalgo, F. C. Wang, A. Mishchenko, F. Schedin, R. R. Nair, E. W. Hill, D. W. Boukhvalov, M. I. Katsnelson, R. A. W. Dryfe, I. V. Grigorieva, H. A. Wu and A. K. Geim, Nature, 2014, 516, 227–229 CrossRef CAS PubMed; (b) N. T. T. Nguyen, H. Furukawa, F. Gandara, C. A. Trickett, H. M. Jeong, K. E. Cordova and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 15394–15397 CrossRef CAS PubMed; (c) D. D. Ordinario, L. Phan, W. G. T. Walkup, J. M. Jocson, E. Karshalev, N. Husken and A. A. Gorodetsky, Nat. Chem., 2014, 6, 596–602 CrossRef CAS PubMed; (d) K. Niinomi, S. Miyazawa, M. Hibino, N. Mizuno and S. Uchida, Inorg. Chem., 2017, 56, 15187–15193 CrossRef CAS PubMed; (e) H. Y. Zang, J. J. Chen, D. L. Long, L. Cronin and H. Miras, Chem. Sci., 2016, 7, 3798–3804 RSC; (f) Y. X. Ye, X. Z. Wu, Z. Z. Yao, L. Wu, Z. T. Cai, L. H. Wang, X. L. Ma, Q. H. Chen, Z. J. Zhang and S. C. Xiang, J. Mater. Chem. A, 2016, 4, 4062–4070 RSC.
- (a) Y. Li, L. Liang, C. Liu, Y. Li, W. Xing and J. Q. Sun, Adv. Mater., 2018, 30, 1707146–1707153 CrossRef PubMed; (b) G. W. He, X. Y. He, X. L. Wang, C. Y. Chang, J. Zhao, Z. Y. Li, H. Wu and Z. G. Jiang, Chem. Commun., 2016, 52, 2173–2176 RSC.
- (a) S. Kim, B. Joarder, J. A. Hurd, J. Zhang, K. W. Dawson, B. S. Gelfand, N. E. Wong and G. K. H. Shimizu, J. Am. Chem. Soc., 2018, 140, 1077–1082 CrossRef CAS PubMed; (b) L. Y. Zhang, T. T. Cui, X. Cao, C. J. Zhao, Q. Chen, L. X. Wu and H. L. Li, Angew. Chem., Int. Ed., 2017, 56, 9013–9017 CrossRef CAS PubMed; (c) J. Miao, Y. L. Chen, Y. W. Li, J. J. Cheng, Q. Y. Wu, K. W. Ng, X. Cheng, R. Chen, C. Cheng and Z. K. Tang, ACS Appl. Nano Mater., 2018, 1, 564–571 CrossRef CAS.
- (a) P. Yang, M. Alsufyani, A. H. Emwas, C. Q. Chen and N. M. Khashab, Angew. Chem., Int. Ed., 2018, 57, 1–6 CrossRef; (b) Y. Kim, K. Ketpang, S. Jaritphun, J. S. Parkb and S. Shanmugam, J. Mater. Chem. A, 2015, 3, 8148–8155 RSC; (c) S. Uchida, R. Hosono, R. Eguchi, R. Kawahara, R. Osuga, J. N. Kondo, M. Hibinob and N. Mizuno, Phys. Chem. Chem. Phys., 2017, 19, 29077–29083 RSC; (d) J. C. Liu, Q. Han, L. J. Chen, J. W. Zhao, C. Streb and Y. F. Song, Angew. Chem., Int. Ed., 2018, 57, 8416–8420 CrossRef CAS PubMed.
- (a) S. Y. Chou, W. H. Chung, L. W. Chen, Y. M. Dai, W. Y. Lin, J. H. Lin and C. C. Chen, RSC Adv., 2016, 6, 82743–82758 RSC; (b) S. Sanna, V. Esposito, M. Christensen and N. Pryds, APL Mater., 2016, 4, 121101 CrossRef; (c) P. I. Molina, K. Kozma, M. Santala, C. Falaise and M. Nyman, Angew. Chem., Int. Ed., 2017, 56, 16277–16281 CrossRef CAS PubMed; (d) O. Sadeghi, M. Amiri, E. Reinheimer and M. Nyman, Angew. Chem., Int. Ed., 2018, 57, 6247–6250 CrossRef CAS PubMed; (e) X. H. Gao, H. B. Wu, L. X. Zheng, Y. J. Zhong, Y. Hu and X. W. Lou, Angew. Chem., Int. Ed., 2014, 53, 5917–5921 CrossRef CAS PubMed.
- (a) M. R. Karim, K. Hatakeyama, M. Koinuma and S. Hayami, J. Mater. Chem. A, 2017, 5, 7243–7256 RSC; (b) L. Cao, X. He, Z. Jiang, X. Li, Y. Li, Y. Ren, L. Yang and H. Wu, Chem. Soc. Rev., 2017, 46, 6725–6745 RSC; (c) H. Beydaghi, M. Javanbakht and E. Kowsari, Polymer, 2016, 87, 26–37 CrossRef CAS; (d) P. Sun, H. Liu, K. Wang, M. Zhong, D. Wu and H. Zhu, Chem. Commun., 2015, 51, 3251–3254 RSC.
- (a) G. He, C. Chang, M. Xu, S. Hu, L. Li, J. Zhao, Z. Li, Z. Li, Y. Yin, M. Gang, H. Wu, X. Yang, M. D. Guiver and Z. Y. Jiang, Adv. Funct. Mater., 2015, 25, 7502–7511 CrossRef; (b) K. Hatakeyama, M. R. Karim, C. Ogata, H. Tateishi, A. Funatsu, T. Taniguchi, M. Koinuma, S. Hayami and Y. Matsumoto, Angew. Chem., Int. Ed., 2014, 53, 6997–7000 CrossRef CAS PubMed; (c) K. Raidongia and J. X. Huang, J. Am. Chem. Soc., 2012, 134, 16528–16531 CrossRef CAS PubMed; (d) S. Kim, S. Zhou, Y. Hu, M. Acik, Y. J. Chabal, C. Berger, W. D. Heer, A. Bongiorno and E. Riedo, Nat. Mater., 2012, 11, 544–549 CrossRef CAS PubMed; (e) P. Z. Sun, R. Z. Ma and T. Sasaki, Chem. Sci., 2018, 9, 33–43 RSC.
- F. Lazarini, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 3169–3173 CrossRef.
- (a) L. W. Su, J. P. Hei, X. B. Wu, L. B. Wang and Z. Zhou, Adv. Funct. Mater., 2017, 27, 1605544–1605552 CrossRef; (b) B. Y. Guan, X. Y. Yu, H. B. Wu and X. W. Lou, Adv. Mater., 2017, 29, 1703614–1703634 CrossRef PubMed.
- G. Xu, K. Otsubo, T. Yamada, S. Sakaida and H. Kitagawa, J. Am. Chem. Soc., 2013, 135, 7438–7441 CrossRef CAS PubMed.
- (a) M. Kruk and M. Jaroniec, Chem. Mater., 2001, 13, 3169–3183 CrossRef CAS; (b) Y. G. Jin, S. Z. Qiao, L. Zhang, Z. P. Xu, S. Smart, J. C. Dinizda Costa and G. Q. Lu, J. Power Sources, 2008, 185, 664–669 CrossRef CAS.
- F. Yang, G. Xu, Y. B. Dou, B. Wang, H. Zhang, H. Wu, W. Zhou, J. R. Li and B. L. Chen, Nat. Energy, 2017, 2, 877–883 CrossRef CAS.
- Y. W. Liu, S. M. Liu, X. Y. Lai, J. Miao, D. F. He, N. Li, F. Luo, Z. Shi and S. X. Liu, Adv. Funct. Mater., 2015, 25, 4480–4485 CrossRef CAS.
- J. Wang, H. Zhang, X. Yang, S. Jiang, W. Lv, Z. Y. Jiang and S. Z. Qiao, Adv. Funct. Mater., 2011, 21, 971–978 CrossRef CAS.
- (a) M. Liu, L. Chen, S. Lewis, S. Y. Chong, M. A. Little, T. Hasell, I. M. Aldous, C. M. Brown, M. W. Smith, C. A. Morrison, L. J. Hardwick and A. I. Cooper, Nat. Commun., 2016, 7, 12750–12759 CrossRef CAS PubMed; (b) L. Vilciauskas, M. E. Tuckerman, G. Bester, S. J. Paddison and K. D. Kreuer, Nat. Chem., 2012, 4, 461–466 CrossRef CAS PubMed; (c) L. Yang, S. Wang, K. Blinn, M. Liu, Z. Liu, Z. Cheng and M. Liu, Science, 2009, 326, 126–129 CrossRef CAS PubMed; (d) X. Y. Dong, J. J. Li, Z. Han, P. G. Duan, L. K. Li and S. Q. Zang, J. Mater. Chem. A, 2017, 5, 3464–3474 RSC.
- (a) Y. Li, M. F. Jiao, H. J. Zhao and M. J. Yang, Sens. Actuators, B, 2018, 273, 133–142 CrossRef CAS; (b) D. Z. Zhang, X. Q. Zong, Z. L. Wu and Y. Zhang, Sens. Actuators, B, 2018, 266, 52–62 CrossRef CAS.
- H. Zhong, Z. Fu, J. M. Taylor, G. Xu and R. Wang, Adv. Funct. Mater., 2017, 27, 1701465–1701472 CrossRef.
- S. Horike, D. Umeyama, M. Inukai and S. Kitagawa, J. Am. Chem. Soc., 2012, 134, 7612–7615 CrossRef CAS PubMed.
- (a) H. Xu, S. S. Tao and D. L. Jiang, Nat. Mater., 2016, 15, 722–726 CrossRef CAS PubMed; (b) B. Chai and X. Wang, RSC Adv., 2015, 5, 7589–7596 RSC.
- D. W. Kang, K. S. Lim, K. J. Lee, J. H. Lee, W. R. Lee, J. H. Song, K. H. Yeom, J. Y. Kim and C. S. Hong, Angew. Chem., Int. Ed., 2016, 55, 16123–16126 CrossRef CAS PubMed.
- Y. Guo, Z. Q. Jiang, W. Ying, L. P. Chen, Y. Z. Liu, X. B. Wang, Z. J. Jiang, B. L. Chen and X. S. Peng, Adv. Mater., 2018, 30, 1705155–1705163 CrossRef PubMed.
- (a) A. Braun and Q. Chen, Nat. Commun., 2017, 8, 15830–15838 CrossRef CAS PubMed; (b) P. Sun, K. Wang and H. W. Zhu, Adv. Mater., 2016, 28, 2287–2310 CrossRef CAS PubMed; (c) Y. G. Jin, S. Z. Qiao, J. C. Diniz da Costa, B. J. Wood, B. P. Ladewig and G. Q. Lu, Adv. Funct. Mater., 2007, 17, 3304–3311 CrossRef CAS.
- (a) K. D. Kreuer, S. J. Paddison, E. Spohr and M. Schuster, Chem. Rev., 2004, 104, 4637–4678 CrossRef CAS PubMed; (b) M. J. Wei, J. Q. Fu, Y. D. Wang, J. Y. Gu, B. L. Liu, H. Y. Zang, E. L. Zhou, K. Z. Shao and Z. M. Su, J. Mater. Chem. A, 2017, 5, 1085–1093 RSC.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535–4585 CrossRef CAS PubMed.
- J. P. Bai, F. Su, H. T. Zhu, H. Sun, L. C. Zhang, M. Y. Liu, W. S. You and Z. M. Zhu, Dalton Trans., 2015, 44, 6423–6430 RSC.
- W. P. Mounfield III, A. Garg, Y. S. Horn and Y. R. Leshkov, Chem, 2018, 4, 16–26 Search PubMed.
- H. W. Huang, Y. He, X. W. Li, M. Li, C. Zeng, F. Dong, X. Du, T. R. Zhang and Y. H. Zhang, J. Mater. Chem. A, 2015, 48, 24547–24556 RSC.
- R. K. Joshi, P. Carbone, F. C. Wang, V. G. Kravets, Y. Su, I. V. Grigorieva, H. A. Wu, A. K. Geim and R. R. Nair, Science, 2014, 343, 752–754 CrossRef CAS PubMed.
- (a) S. Mohanapriya and V. Raj, Ionics, 2018, 24, 2729–2743 CrossRef CAS; (b) A. Khabibullin, S. D. Minteer and I. Zharov, J. Mater. Chem. A, 2014, 2, 12761–12769 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03726d |
| This journal is © The Royal Society of Chemistry 2019 |