
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient room-temperature production of high-quality graphene by introducing removable oxygen functional groups to the precursor†
Hongwu
Chen
a,
Wencheng
Du
a,
Jing
Liu
a,
Liangti
Qu
ab and
Chun
Li
 *a
*a
aDepartment of Chemistry, MOE Key Laboratory of Bioorganic Phosphorus Chemistry & Chemical Biology, Tsinghua University, Beijing 100084, P. R. China. E-mail: chunli@mail.tsinghua.edu.cn
bKey Laboratory for Advanced Materials Processing Technology, Ministry of Education of China, State Key Laboratory of Tribology, Department of Mechanical Engineering, Tsinghua University, Beijing 100084, P. R. China
First published on 8th November 2018
Abstract
The excellent solution-processability of graphene oxide (GO) has provided a collection of strategies for the construction of functional graphene assemblies. To improve the performance of graphene-based materials, structurally intact GO should be prepared as a precursor for high-quality graphene; however, solution chemical methods have been constantly challenged by a structural integrity versus fabrication yield trade-off. Here, we report a wet chemical method for the high-efficiency production of a high-quality graphene oxide precursor, with all steps conducted at room-temperature. The functionalization of graphite was performed under temperature and water content control in a concentrated sulfuric acid–potassium permanganate system, and the resulting GO showed a monolayer yield of over 120%. We show that the increased production yield comes from the high functionalization degree and, more interestingly, the functional groups on GO were proven to be removable upon reduction with hydroiodic acid, which produced high-quality graphene-based materials.
Introduction
Explorations into the physical properties of monolayer pristine graphene1–3 have revealed the enormous potential of the material: it has an ultra-high tensile strength (∼125 GPa) and Young's modulus (∼1 TPa),2 excellent thermal conductivity (up to 5300 W m−1 K−1)3 and special performance relating to many other properties.4–6 However, the utility of pristine graphene is limited by its poor solution-processability,7 which hinders the preparation of functional graphene-based assemblies. Therefore, an approach that introduces high-concentration dispersible graphene oxide (GO) as a precursor for graphene was established during the past decade,8–10 providing an alternative choice for applied graphene science. Following this strategy, resulting functional graphene-based materials in the form of fibers,11 bulk film12 and hydrogels13,14 have displayed profuse applications in structural materials,15,16 supercapacitors and batteries,17 nano-filtration membranes,18etc. To improve the conductivities and mechanical properties of graphene-based assemblies, which is of high importance for most applications, the synthesis of GO with less structural defects has proven to be crucial for solution-based methods.19,20Generally, there are two routes for the solution chemical synthesis of GO: electrochemical oxidation and chemical oxidation. Though believed to be eco-friendly, the challenge posed by the electrochemical approach is the low yield of single layer GO with a considerable functionalization degree.21–23 Another way is the solution chemical synthesis of GO based on the Charpy–Hummers oxidation method.24,25 Having been developed and studied for over a century, the acid–oxidant solution is still proven to be the most efficient system for the intercalation and oxidation of graphite. The very first protocol proposed by Hummers and Offeman25 involved a first oxidation step at 35 °C and a water-enhanced process, with the temperature ramping up to the boiling point of water. The final product, with a bright yellow appearance, was found to be highly dispersible in water, with a defect-rich nanostructure.26 However, over-oxidation in the second step of functionalization27,28 results in the formation of unrepairable structural defects that severely deteriorate the material performance of the reduced GO (rGO). Modifications to the traditional recipe have been reported continuously.29–32 For instance, Xu et al. reduced the amount of KMnO4 used for oxidation and obtained structurally intact GO, but only in 20% monolayer yield;30 Eigler et al. obtained rGO with remarkable Hall mobility by preforming the overall reaction process at a temperature < 10 °C, however, the yield was also not satisfactory (43%).31 Until now, the trade-off between structural integrity and yield still remains a challenge in the chemical oxidation method.8
In this work, we present a reaction recipe that could remove this trade-off, namely, achieving both high-yield for GO (>120%) and intact structures of the resulting monolayer rGO with a Raman ID/IG ratio approaching 2.4. We found that with a combination of a room-temperature oxidation procedure and a functionalization effect of ∼4 wt% water existing in commercial concentrated H2SO4, the GO basal plane was covered with removable functional groups (mainly hydroxyl and epoxy groups), which could enhance the exfoliation yield and dispersibility, and give graphene material with fewer defects after HI reduction, generating graphene film with conductivity up to 780 S cm−1. GO samples synthesized with this recipe also showed similar processability (for the fabrication of films, fibers and ultra-light aerogels) as samples produced via the classic Charpy–Hummers method. Furthermore, the presented approach is quite energy-saving, and could meet the requirements for the scalable production of graphene in industry.
Results and discussion
In the classic Charpy–Hummers method, graphite was subjected to two-step oxidation—after the addition of KMnO4 and during the addition of water.27 In this study, we propose an optimized recipe by skipping the second step, characterized by oxidation and defect formation; instead, water in commercial concentrated H2SO4 (∼4 wt%) was shown to be sufficient for the bulk functionalization of the graphene basal plane, with all the steps performed at 20 °C. GO prepared via the optimized procedure is denoted as GO-20 and, for comparison, GO samples were also synthesized via the classic Charpy–Hummers method (CGO) without the addition of NaNO3 (ref. 25 and 33) and the low temperature method (GO-5) first specified by Eigler et al.31 (Experimental section). In the method reported by Eigler et al., both steps were transferred from high temperature (>35 °C) to nearly 5 °C.31The graphite powder samples (325 mesh and 200 mesh) used for the preparation of GO were characterized via scanning electron microscopy (SEM) and X-ray diffraction (XRD) methods (Fig. S1†). The results showed the different flake sizes for 325 mesh (∼40 μm) and 200 mesh (∼65 μm) graphite and the similar crystallinities for both samples. In this study, detailed characterization was carried out for GO fabricated from 325 mesh graphite for a clarification of the mechanism, and we also show that the method developed herein could be employed for the synthesis of large graphene sheets from 200 mesh graphite (ESI†).
Dimension characterization of GO sheets
SEM images of GO sheets assembled on a SiO2/Si substrate and the statistical results for flake area (A) versus perimeter (P) obtained from over 500 individual GO flakes are displayed in Fig. 1a, b, S2 and S3.† The average lateral size (L), calculated from the square root of the mean flake area (L = A1/2), shows similar distribution behavior for all GO samples, from 4.0 μm for CGO to 2.8 μm for GO-20 to 3.1 μm for GO-5. The slightly decreased size of the GO sheets was attributed to the mild sonication step (10 min) that is frequently used for the exfoliation of graphite oxide,34 however, the size distribution is comparable to results reported previously for 325 mesh GO samples.19,20 Apart from the lateral size, the shape distribution of the GO flakes could also be related to the sample treatment process.35 The fitting line using the formula P = αA1/2 in Fig. 1b (with α as the fitting parameter) clearly indicated that the CGO sheets possessed higher circularity (0.77 ± 0.17) than the GO-20 (0.63 ± 0.19) and GO-5 samples (0.66 ± 0.19). This is probably because boundary cutting during the high temperature oxidation process reshapes the GO sheets from irregular graphite starting materials, as observed in the case of long-term sonication treatment.35 | ||
| Fig. 1 Characterization of GO flakes fabricated via various methods: (a) an SEM image of GO-20 flakes on a SiO2/Si substrate; (b) a plot of perimeter against flake area of the GO sheets, the fitting lines represent the shape distribution of the GO sheets; (c) an AFM image of a GO-20 sample deposited on a mica surface; (d) a comparison of the single layer yields of GO samples prepared via different methods; and (e) photographs of GO aqueous dispersions (1.0 mg mL−1). | ||
Atomic force spectroscopy (AFM) studies were carried out to characterize the thickness of the GO sheets (Fig. 1c). The results show that the thickness of the GO flakes is mainly located in the range of 0.9–1.1 nm, which is consistent with the typical thickness of GO monolayers.36,37 We further calculate the monolayer yields of the GO samples (Fig. 1d). The results show that the yield of GO-20 is comparable to CGO (120% and 130%, respectively), and displayed huge improvement (50%) over that of GO-5. In the following discussion, we will show that the high-yield production of GO-20 is a result of its specific chemical structure.
Chemical structures of GO samples
All GO samples were purified via centrifugation to remove graphite particles and un-exfoliated graphite oxide aggregates (Experimental section). XRD patterns of the GO films (fabricated via a vacuum filtration method) confirm the removal of the graphite phase from the GO samples from the absent graphite crystalline peak at 26–27° (Fig. S4†).20 A demonstration of the excellent solution dispersibility of the GO-20 sample is shown in Fig. 1e. In addition, with the solid content of the aqueous dispersion concentrated at 2 wt%, we proved that the high viscosity of the GO-20 dispersion could render it with the ability to be 3D printed (Fig. S5†).Element and functional group analyses were performed via X-ray photoelectron spectroscopy (XPS) and Fourier-transform infrared spectroscopy (FT-IR) (Fig. 2a–d). From element analysis of XPS spectra, manganese species were present below the detection limit (<0.1 at%, Fig. 2a), suggesting the purity of the GO samples. Deconvolution of the C 1s peaks give the proportion of oxygen-containing groups (Fig. 2b and Table S1†);19 for the CGO sample, the fraction of carboxyl groups (289.0 eV, 3.1%) is higher than in the GO-20 and GO-5 samples (1.3% and 1.4%, respectively). The C/O atomic ratio reflects a higher oxygen-functionalization degree for CGO and GO-20 (2.0 and 2.1), whereas GO-5 showed a lower oxygen content (C/O 2.4). FT-IR spectroscopy provided more reliable qualitative results relating to the percentages of functional groups because of its non-destructive nature.38,39 In Fig. 2d, the absorbance peaks for sp2 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (1580 cm−1), CO (∼1720 cm−1) and C–O (∼1040 cm−1) bonds38 are extracted from the overall spectra (Fig. S6†) and integrated for comparison. The results show that the CGO and GO-20 samples have similar proportions of pristine graphitic areas, whereas GO-5 preserves relatively more conjugated CC bonds, indicating the higher functionalization degrees of the GO-20 and CGO specimens. From other aspects, GO-20 and GO-5 show lower ratios of CO/C–O functionalities when compared with CGO, suggesting that hydroxyl and epoxy groups (C–O) are predominant in the GO-20 specimen, different to CGO.
C (1580 cm−1), CO (∼1720 cm−1) and C–O (∼1040 cm−1) bonds38 are extracted from the overall spectra (Fig. S6†) and integrated for comparison. The results show that the CGO and GO-20 samples have similar proportions of pristine graphitic areas, whereas GO-5 preserves relatively more conjugated CC bonds, indicating the higher functionalization degrees of the GO-20 and CGO specimens. From other aspects, GO-20 and GO-5 show lower ratios of CO/C–O functionalities when compared with CGO, suggesting that hydroxyl and epoxy groups (C–O) are predominant in the GO-20 specimen, different to CGO.
 | ||
| Fig. 2 Characterization of the chemical structures of the GO samples: (a) XPS spectra of the GO samples; (b) C 1s spectra of various GO samples; (c and d) FT-IR spectra of the GO samples and the ratios of functional groups derived from FT-IR data; (e) UV-vis absorbance spectra of dilute GO aqueous dispersions; (f) TGA analysis (TG and DTG curves) of GO aerogels; and (g) the proposed structural features of all the GO samples. The color key given in the inset of (a) applies to all parts of figure. | ||
Characterization of the conjugated structures and thermal stabilities (Fig. 2e and f) provide further evidence regarding the functionalization features of the GO specimens. Ultraviolet-visible spectroscopy (UV-vis) details the average sizes of the conjugated areas of the GO samples from the position of the maximum absorbance peak at ∼230 nm (corresponding to π–π* excitation).40 From the UV-vis spectra of CGO and GO-20, the π–π* absorbance peak located at 231 nm is in line with the characteristics of a densely functionalized GO basal plane.20 In contrast, the redshift of this peak to 234 nm in the GO-5 sample suggests that a larger aromatic domain was preserved during oxidation (Fig. 2e). Furthermore, thermal gravimetric analysis (TGA) reveals that a CGO aerogel has a higher maximum decomposition temperature (187 °C) compared to GO-20 (153 °C) and GO-5 (135 °C) (Fig. 2f), indicating the stronger interlayer interactions in CGO, as a result of higher fraction of COOH groups.32,41
According to the widely-accepted Lerf–Klinowski structural model of GO,42,43 major functional groups intruding on the graphene skeleton include hydroxyl, epoxy, carbonyl and carboxyl groups. Among these, carboxyl and carbonyl groups, which are formed via the intense oxidation cleavage of C–C bonds, are usually accompanied by the formation of structural defects.10 Generally, C–O related functional groups can be removed upon chemical reduction (henceforth called removable functional groups), finally leading to a recovered graphitic structure. Chemical characterization reveals that GO-20 has a higher C/O ratio and lower fraction of un-oxidized CC skeleton when compared to the GO-5 sample, and a lower CO defect region compared to the CGO sample (Fig. 2g), which indicates a higher proportion of removable functional group coverage in GO-20 that both preserves the intact structure and enhances the exfoliation rate, resulting in a high production yield.
Structural characteristics of rGO samples
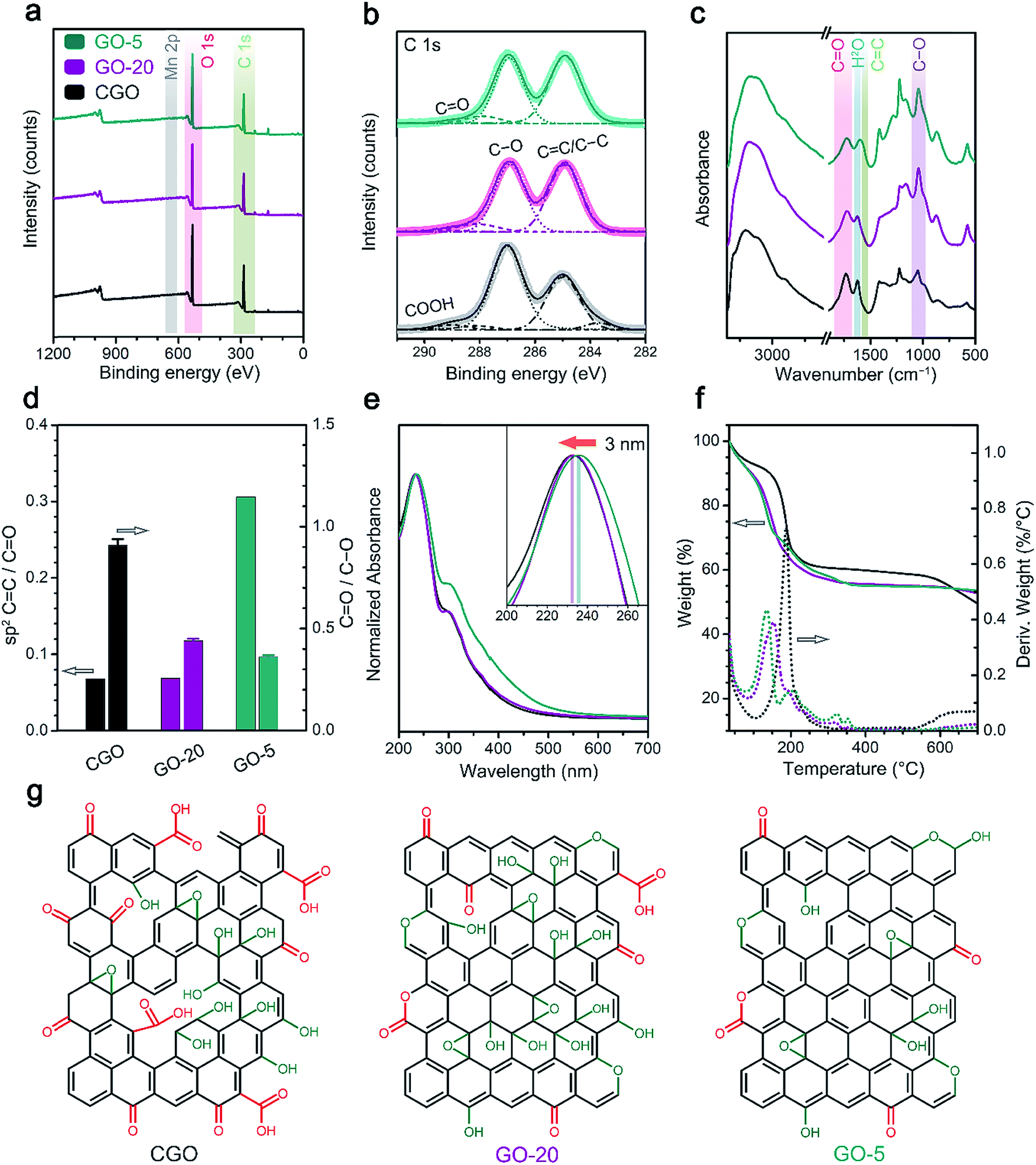
The chemical reduction of GO samples with HI acid is accompanied by the removal of a large proportion of functional groups and the restoration of a conjugated structure.44,45 The characterization of rGO from the ratio of remaining functional groups to structural defects gave more information on the quality of the graphene materials and the chemical features of the GO precursors (Fig. 3 and S7†).20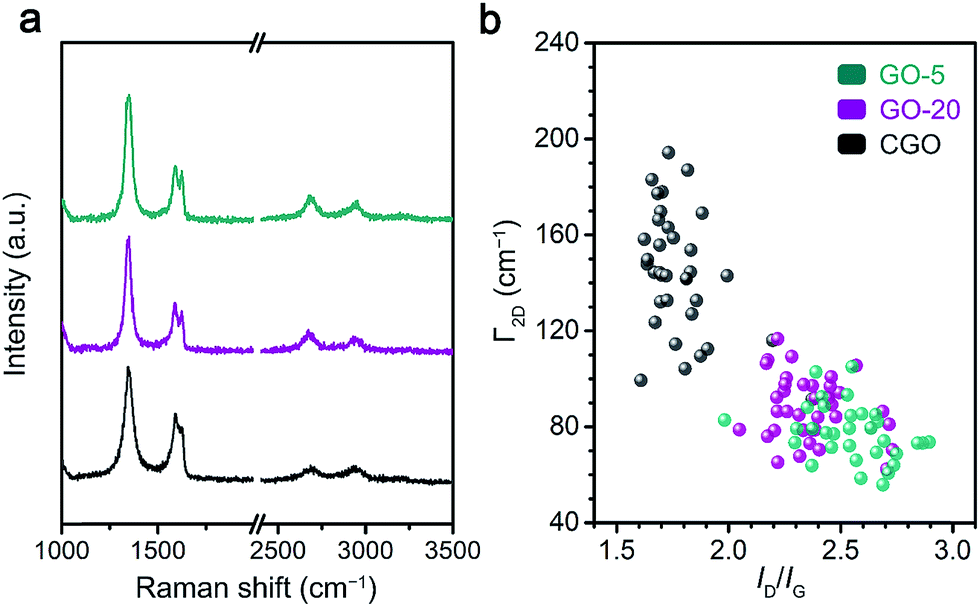 | ||
| Fig. 3 Raman characterization of rGO single layers: (a) Raman spectra of single layer rGO sheets; and (b) the distribution of ID/IG and the FWHM of the 2D band for different rGO samples derived from at least 40 individual rGO sheets for each specimen. | ||
Raman spectroscopy was employed to probe the structural defects on single layer rGO sheets reduced by HI/CH3COOH vapor (Experimental section). Major Raman bands for a typical rGO monolayer include the G band (∼1580 cm−1, originating from the E2g mode for pristine graphene), D band (∼1340 cm−1, corresponding to a TO phonon mode near the K point for defective graphene) and 2D band (∼2680 cm−1, an overtone of the D band).46 Based on statistical results obtained from at least 40 rGO sheets (Fig. 3b and Table S2†), the more defective nature of the reduced CGO sample could be deduced from the lower ID/IG value (1.78 ± 0.16), broader 2D band (FWHM = 144 ± 26 cm−1) and blueshifting of the G band (centered at 1591.2 ± 2.6 cm−1) compared with reduced GO-20 and GO-5 (Table S2†). Furthermore, the average distance (LD) between structural defects on rGO sheets follows a non-linear relationship with the ID/IG value.47,48 Given that a graphene monolayer chemically reduced from a GO precursor displays a positive correlation between LD and ID/IG that follows formula (1) below,20
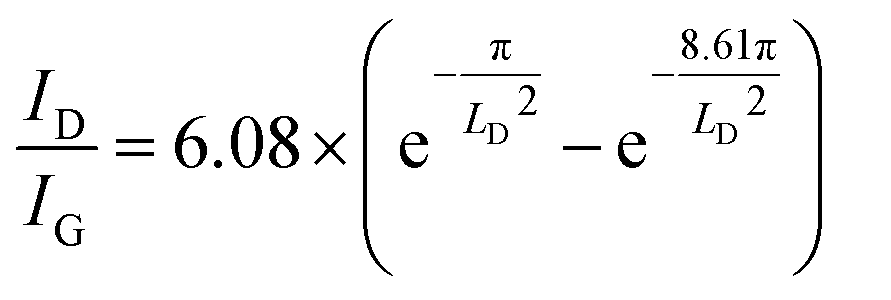 | (1) |
In the Lerf–Klinowski model, the flake or defect boundaries are concentrated with carboxyl or carbonyl groups. Therefore, the highly defective nature of reduced CGO samples is further proved via FT-IR spectra (Fig. S7a†): the relative amount of CO functional groups in reduced CGO films is conspicuously higher than in reduced GO-20 and GO-5 films. XPS analysis of rGO specimens (Fig. S7b–d†) also revealed that reduced CGO films still preserve a higher oxygen functional group content (carboxyl groups: 7.1%) than reduced GO-20 and GO-5 films (∼4.5%). Given the higher functionalization degree of GO-20 compared to GO-5, the similar C/O ratios of the rGO samples show that the majority of chemical groups on the GO-20 basal plane are removable during HI reduction.
Dominant factors in the high-yield fabrication of structurally intact graphene
From detailed chemical and structural characterization of GO and rGO samples, the high-yield production of graphene with good structural integrity is attributed to the bulk functionalization of the GO basal plane with removable hydroxyl and epoxy groups. As a benefit of this strategy, the trade-off between preparation yield and structural integrity is removed. However, the dominant factors that result in the non-destructive oxidation of the graphene skeleton in this reaction system still need to be clarified.Herein, the synergistic effects of oxidation temperature and water content in the reaction media are proven to be the critical factor in the selective functionalization of GO with respect to the kinds of chemical groups and structural defects. First, the temperature variation during chemical oxidation was recorded (Fig. 4a). The slow addition of KMnO4 at a speed of 3 min g−1 was initiated as the system temperature cooled down to 20 °C during the synthesis of CGO as well as GO-20, and 5 °C in the case of GO-5. Note that the only difference between the fabrication of the GO-20 and GO-5 samples was the system temperature during the oxidation step, and the temperature rise during the addition of the oxidant was shown to be negligible (ΔT < 3 °C) (Fig. 4a), leading to the conclusion that the higher functionalization degree of GO-20 resulted from the elevated reaction temperature.
 | ||
| Fig. 4 Mechanism underlying the high-yield preparation of high-quality graphene samples: (a) the temperatures of the reaction systems recorded during the chemical oxidation of graphite, showing the differences in temperature variation for the different reaction recipes involved in this study; (b) a comparison of the yield of GO and the appearance of graphite oxide after oxidation for samples with no water and a certain amount of water (∼4.0 wt%) existing in H2SO4; (c) the difference between the chemical structures of GO samples revealed by FT-IR; (d) the reaction mechanism that leads to the different yields and oxidation degrees of the final GO products. | ||
The addition of water (previously identified as the second oxidation step in CGO synthesis)27,28 served as the important factor in the formation of structural defects. In the preparation of GO-20 and GO-5, this step was skipped by terminating the reaction in excess water at room temperature (Experimental section). However, the sharp temperature rise up to 89 °C during the hydrolysis process in CGO preparation coupled with the vigorous oxidation effects of MnO4− in H2SO4 media were the critical factors that brought about the higher oxidization degree and structural defects.
Furthermore, the water in commercial concentrated H2SO4 (4.2 ± 0.2 wt%, determined via an acid-base titration method) also proved to be crucial in the bulk functionalization of GO-20. Herein, excess P4O10 was employed as a strong desiccator for the removal of water in sulfuric acid, resulting in the formation of oleum:
| P4O10 + 6H2SO4 → 4H3PO4 + 6SO3 |
The yield of GO prepared in H2SO4 dehydrated with P4O10 (denoted as GO–P4O10) was reduced to ∼60% compared to GO-20 (Fig. 4b), despite the reaction conditions for both remaining otherwise identical. The proportion of CO functional groups in GO–P4O10 was shown to be significantly lower than in GO-20 from FT-IR characterization (Fig. 4c), and the higher ID/IG value for reduced monolayer GO–P4O10 also revealed the intact structure of the graphene skeleton (Fig. S8†). A comparison of the different reaction recipes involved in this study is summarized in Fig. 4d, in which the interplay between reaction temperature and H2O in the reaction system results in the unique chemical structure of GO-20, which could be used in the fabrication of high-quality graphene-based materials.
Applications of high-quality GO and rGO materials
We have proposed a reaction recipe that is suitable for the high-yield and scalable production of high-quality graphene with arbitrary size. However, from industrial considerations related to the production of graphene, energy efficiency is one of the major concerns.7 The energy–yield index (EI) formula, which features considerations of both energy input and production yield, is specified below (see ESI† for the calculation of individual items):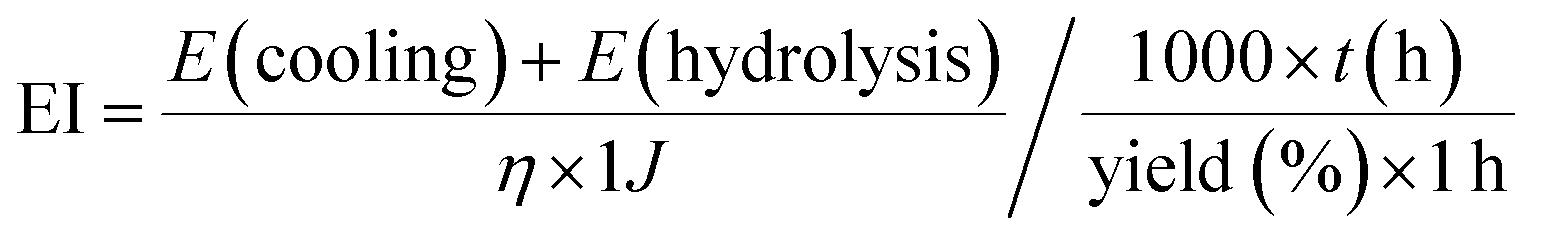 | (2) |
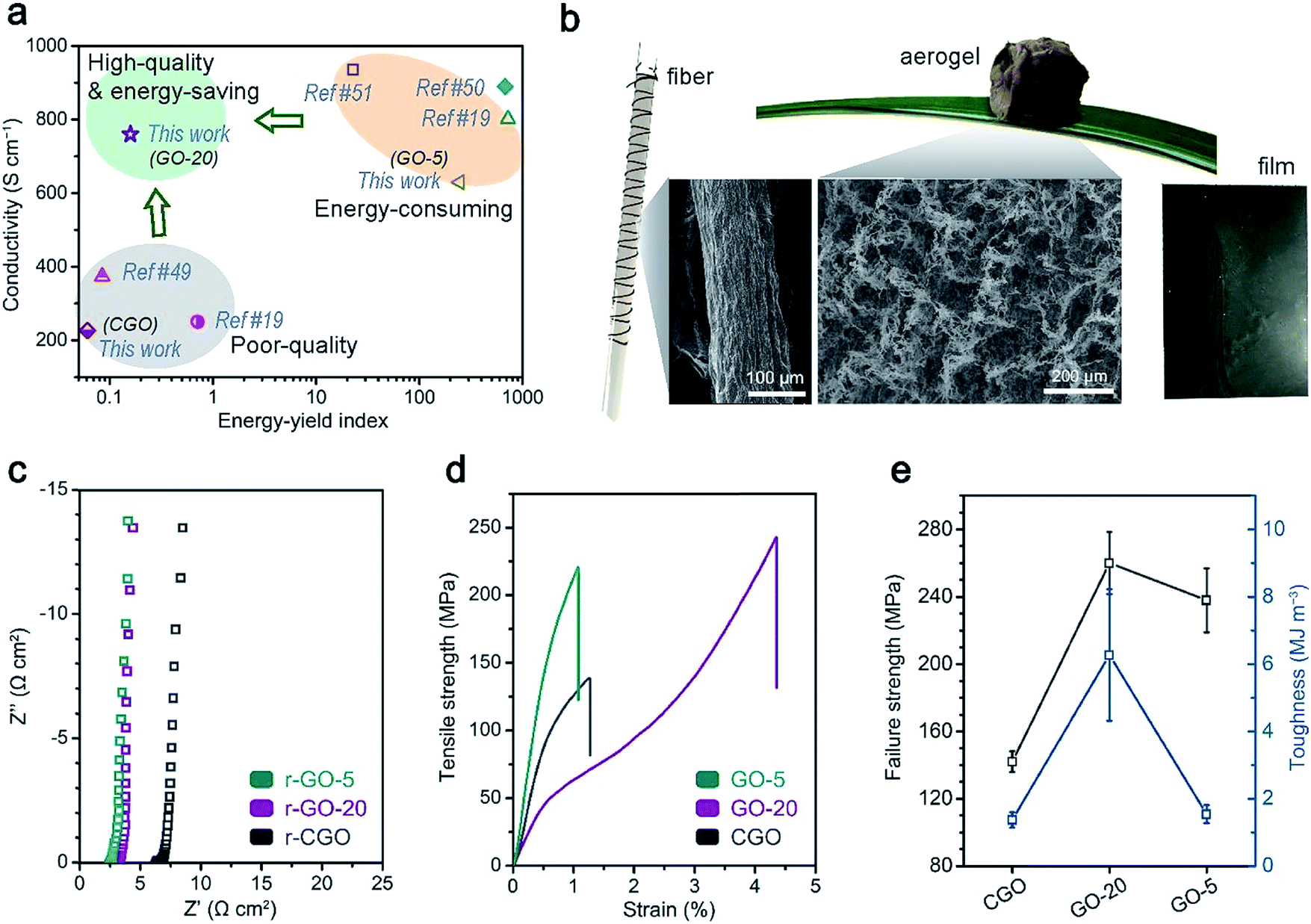 | ||
| Fig. 5 A comparison of the applications of the GO samples, which require good structural integrity. (a) An overall comparison of GO synthesis approaches in terms of electrical conductivity and the fabrication energy–yield index (the definition is specified in formula (2)); (b) a demonstration of the solution-processability of the GO-20 sample: ultralight aerogels, compact films and fibers could be facilely prepared using a GO-20 aqueous dispersion; (c) the reduced GO films were used as flexible current collectors: EIS characterization is shown; and (d and e) the mechanical properties of the GO films: (d) strength and strain curves and (e) the statistical failure strength and toughness performances of the GO films are shown. | ||
For the application of graphene-based assemblies, structurally intact GO and rGO materials have shown large potential in various application backgrounds that require excellent performance in terms of electrical and mechanical properties. The direct manifestation of high-quality graphene-based samples could be demonstrated in the application of rGO film as a folding-resistant current collector and GO film as a structural material (Fig. 5c–e).37 The electrochemical performances of electrochemical double layer capacitors (EDLCs) that featured activated carbon (AC) as the electroactive material and rGO film as the current collector were characterized (Fig. 5c and S9†). The low internal resistance (2.7 Ω cm2) between the reduced GO-20 film and AC electrode was assisted by the high conductivity (∼780 S cm−1), which leads to the high rate performance of the EDLC (ESI†). On the other hand, GO-20 film material also displays superior mechanical properties compared to CGO and even GO-5 film (Fig. 5d and e): the ultimate strength (254 ± 17 MPa), which is comparable to GO-5 film (238 ± 19 MPa), could be attributed to stronger interactions from hydroxyl/hydroxyl groups or the conjugated area, which appear weaker in CGO film.20 Intriguingly, the superior toughness of GO-20 film (6.3 ± 2.0 MJ m−3) over GO-5 and CGO film (∼1.4 MJ m−3) may come from stress dissipating effects mediated by hydrogen bonds between GO sheets, which is more prevalent in GO-20 samples.
Conclusions
The wet chemical synthesis of structurally intact graphene in high yields has been continuously studied over the past decade. We show that the most efficient oxidation system, developed by Hummers et al., could be modified to achieve our goal—i.e. to remove the trade-off between structural integrity and production yield. Synergistic effects from oxidation temperature and water content in the reaction media are shown to result in the bulk functionalization of the graphene basal plane with removable oxygen-containing groups, which enhanced the yield and simultaneously preserved the intact structure. Additionally, the GO sample fabricated with the optimized recipe displayed good dispersibility and solution-processability for the preparation of functional graphene-based assemblies with excellent material performance. The methodology revealed in this study, introducing removable functional groups to the precursor, might make this a propitious starting point for the mass production of high-quality graphene materials.Experimental section
Raw materials
Graphite powder (325 mesh and 200 mesh) was purchased from Qingdao Huatai Lubricant Sealing S&T Co. Ltd. (Qingdao, China); sulfuric acid (95–98%) and potassium permanganate were obtained from Beijing Chemical Reagent Co. (Beijing, China). Hydroiodic acid (57%) was from Adamas Reagent.Preparation of GO precursor
GO samples were prepared mainly via three different procedures: a modified Charpy–Hummers' method (CGO),25,33 a room-temperature modified Hummers' method (GO-20) and a low-temperature modified Hummers' method (GO-5). For the synthesis of CGO and GO-20, graphite powder (1.0 g) was dispersed in concentrated sulfuric acid (30 mL) in a 250 mL flask, then the suspension was stirred vigorously at 20 °C for 30 min. KMnO4 (3.0 g) was then added to the reaction system over 10 min. For CGO preparation, the system was heated to 35 °C and kept for 1 h, followed by the addition of 30 mL of H2O over 10 min; whereas for the production of GO-20, the oxidation process was performed at 20 °C for 3 h. The preparation of GO-5 employed the same reaction system but with all procedures conducted at 5°C—including the addition of KMnO4 (3.0 g) and the oxidation process (without the addition of water, for 12 h). All reactions were terminated by pouring the slurries into 500 mL of deionized water (20 °C). The purification of the graphite oxide samples and the exfoliation procedures are discussed in detail in the ESI.†Reduction of the GO precursors
To obtain monolayer rGO sheets assembled on flat SiO2/Si surfaces (for Raman characterization), a GO dispersion (10 μg mL−1) was deposited on a SiO2/Si substrate, which was followed by chemical reduction with HI/CH3COOH vapor at 60 °C for 6 h. The reduction of the GO films was performed in 15 wt% HI acid aqueous solution at 20 °C for 12 h and was followed by repeated ethanol washing (24 h) to remove I2 adsorbed on the rGO films.Fabrication of graphene-based assemblies
Compact GO films were prepared via filtration-induced assembly (20 mL of 1.0 mg mL−1 GO dispersion) on poly(tetrafluoride ethylene) (PTFE) membranes (with an average pore size of 0.22 μm). Once prepared, the films were stored in an allochroic silica gel-filled desiccator. GO fibers were fabricated via a wet spinning process, with the concentration of GO stock being 10.0 mg mL−1, and the coagulation bath consisted of 5 wt% CaCl2 dissolved in a mixture of ethanol and deionized water (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). An ultra-light GO aerogel was prepared via the freeze-drying of a 1.0 mg mL−1 GO aqueous dispersion.
:3). An ultra-light GO aerogel was prepared via the freeze-drying of a 1.0 mg mL−1 GO aqueous dispersion.
Characterization of GO and rGO samples
Field-emission scanning electron microscopy (FEI Sirion 200, USA) was used to collect the high-resolution SEM images of GO on SiO2/Si substrates for size and shape characterization. Atomic force microscopy (Shimadzu, SPM-9600) was employed for the characterization of the thicknesses of GO sheets (deposited on freshly-cleaved mica surfaces). X-ray photoelectron spectra were acquired using an X-ray photoelectron spectrometer (ESCALAB 250, Thermo Fisher Scientific, USA) with an aluminum Kα X-ray source (incident energy 1486.6 eV) under 30.0 eV transmission energy. Attenuated total reflectance Fourier transform infrared spectra were collected using a UATR 2 infrared spectrometer (Perkin Elmer, USA). The ultraviolet-visible spectroscopy characterization of GO aqueous suspensions was performed using a Lambda 35 spectrophotometer (Perkin Elmer, USA) over the range of 200–700 nm with reference to deionized water. Thermal gravimetric analysis was conducted on a Q50 analyzer (TA instruments, USA) from room temperature to 800 °C under an N2 atmosphere, and the heating rate was set to 5 K min−1. Raman characterization was carried out on a LabRAM HR Evolution spectrometer (HORIBA Jobin Yvon, France) using a 532 nm laser with a 2.5% filter and 30 s acquisition time. Details about mechanical testing, conductivity measurements and other characterization are available in the ESI.†Author contributions
H. C., W. D. and J. L. performed the experiments. H. C. collected and analyzed the data. H. C. and C. L. wrote the manuscript. L. Q. revised the manuscript. C. L. conceived the whole project. All authors have discussed the results and made comments on the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work is financially supported by the Natural Science Foundation of China (51673108, 51433005, 21674056, 51673026) and the National Key R&D Program of China (2016YFA0200202, 2017YFB1104300).Notes and references
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Electric field effect in atomically thin carbon films, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Measurement of the elastic properties and intrinsic strength of monolayer graphene, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- A. A. Balandin, Thermal properties of graphene and nanostructured carbon materials, Nat. Mater., 2011, 10, 569–581 CrossRef CAS PubMed.
- F. Bonaccorso, Z. Sun, T. Hasan and A. C. Ferrari, Graphene photonics and optoelectronics, Nat. Photonics, 2010, 4, 611–622 CrossRef CAS.
- D. Chen, H. Feng and J. Li, Graphene oxide: Preparation, functionalization, and electrochemical applications, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- C. Chung, Y.-K. Kim, D. Shin, S.-R. Ryoo, B. H. Hong and D.-H. Min, Biomedical applications of graphene and graphene oxide, Acc. Chem. Res., 2013, 46, 2211–2224 CrossRef CAS PubMed.
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, A roadmap for graphene, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- S. Park and R. S. Ruoff, Chemical methods for the production of graphenes, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS PubMed.
- D. Li, M. B. Mueller, S. Gilje, R. B. Kaner and G. G. Wallace, Processable aqueous dispersions of graphene nanosheets, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- S. Eigler, Controlled chemistry approach to the oxo-functionalization of graphene, Chem.–Eur. J., 2016, 22, 7012–7027 CrossRef CAS PubMed.
- Z. Xu, H. Sun, X. Zhao and C. Gao, Ultrastrong fibers assembled from giant graphene oxide sheets, Adv. Mater., 2013, 25, 188–193 CrossRef CAS PubMed.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Preparation and characterization of graphene oxide paper, Nature, 2007, 448, 457–460 CrossRef CAS PubMed.
- Y. Xu, K. Sheng, C. Li and G. Shi, Self-assembled graphene hydrogel via a one-step hydrothermal process, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed.
- Z. Tang, S. Shen, J. Zhuang and X. Wang, Noble-metal-promoted three-dimensional macroassembly of single-layered graphene oxide, Angew. Chem., Int. Ed., 2010, 49, 4603–4607 CrossRef CAS PubMed.
- S. Wan and Q. Cheng, Fatigue-resistant bioinspired graphene-based nanocomposites, Adv. Funct. Mater., 2017, 27, 1703459 CrossRef.
- S. Wan, J. Peng, L. Jiang and Q. Cheng, Bioinspired graphene-based nanocomposites and their application in flexible energy devices, Adv. Mater., 2016, 28, 7862–7898 CrossRef CAS PubMed.
- Y. Sun, Q. Wu and G. Shi, Graphene based new energy materials, Energy Environ. Sci., 2011, 4, 1113–1132 RSC.
- Y. Han, Z. Xu and C. Gao, Ultrathin graphene nanofiltration membrane for water purification, Adv. Funct. Mater., 2013, 23, 3693–3700 CrossRef CAS.
- M. Zhang, Y. Wang, L. Huang, Z. Xu, C. Li and G. Shi, Multifunctional pristine chemically modified graphene films as strong as stainless steel, Adv. Mater., 2015, 27, 6708–6713 CrossRef CAS PubMed.
- H. Chen, M. Wu and C. Li, Structural integrity versus lateral size: Enhancing graphene-based film materials by reducing planar defects rather than flake boundary, Carbon, 2018, 139, 216–225 CrossRef CAS.
- H. Chen, C. Li and L. Qu, Solution electrochemical approach to functionalized graphene: history, progress and challenges, Carbon, 2018, 140, 41–56 CrossRef CAS.
- P. Yu, S. E. Lowe, G. P. Simon and Y. L. Zhong, Electrochemical exfoliation of graphite and production of functional graphene, Curr. Opin. Colloid Interface Sci., 2015, 20, 329–338 CrossRef CAS.
- S. Yang, M. R. Lohe, K. Müllen and X. Feng, New-generation graphene from electrochemical approaches: production and applications, Adv. Mater., 2016, 28, 6213–6221 CrossRef CAS PubMed.
- G. Charpy, Sur la formation de l’oxyde graphitique et la définition du graphite, C. R. Hebd. Séances Acad. Sci., 1909, 148, 920–923 CAS.
- W. S. Hummers and R. E. Offeman, Preparation of graphitic oxide, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- K. Erickson, R. Erni, Z. Lee, N. Alem, W. Gannett and A. Zettl, Determination of the local chemical structure of graphene oxide and reduced graphene oxide, Adv. Mater., 2010, 22, 4467–4472 CrossRef CAS PubMed.
- J. H. Kang, T. Kim, J. Choi, J. Park, Y. S. Kim, M. S. Chang, H. Jung, K. T. Park, S. J. Yang and C. R. Park, Hidden second oxidation step of hummers method, Chem. Mater., 2016, 28, 756–764 CrossRef CAS.
- M. S. Chang, Y. S. Kim, J. H. Kang, J. Park, S. J. Sung, S. H. So, K. T. Park, S. J. Yang, T. Kim and C. R. Park, Guidelines for tailored chemical functionalization of graphene, Chem. Mater., 2017, 29, 307–318 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, Improved synthesis of graphene oxide, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- Y. Xu, K. Sheng, C. Li and G. Shi, Highly conductive chemically converted graphene prepared from mildly oxidized graphene oxide, J. Mater. Chem., 2011, 21, 7376–7380 RSC.
- S. Eigler, M. Enzelberger-Heim, S. Grimm, P. Hofmann, W. Kroener, A. Geworski, C. Dotzer, M. Roeckert, J. Xiao, C. Papp, O. Lytken, H.-P. Steinrueck, P. Mueller and A. Hirsch, Wet chemical synthesis of graphene, Adv. Mater., 2013, 25, 3583–3587 CrossRef CAS PubMed.
- J. Chen, Y. Zhang, M. Zhang, B. Yao, Y. Li, L. Huang, C. Li and G. Shi, Water-enhanced oxidation of graphite to graphene oxide with controlled species of oxygenated groups, Chem. Sci., 2016, 7, 1874–1881 RSC.
- J. Chen, B. Yao, C. Li and G. Shi, An improved Hummers method for eco-friendly synthesis of graphene oxide, Carbon, 2013, 64, 225–229 CrossRef CAS.
- C. E. Halbig, T. J. Nacken, J. Walter, C. Damm, S. Eigler and W. Peukert, Quantitative investigation of the fragmentation process and defect density evolution of oxo-functionalized graphene due to ultrasonication and milling, Carbon, 2016, 96, 897–903 CrossRef CAS.
- A. Liscio, K. Kouroupis-Agalou, X. D. Betriu, A. Kovtun, E. Treossi, N. M. Pugno, G. De Luca, L. Giorgini and V. Palermo, Evolution of the size and shape of 2D nanosheets during ultrasonic fragmentation, 2D Mater, 2017, 4, 025017 CrossRef.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Graphene and graphene oxide: Synthesis, properties, and applications, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- Y. Wen, M. Wu, M. Zhang, C. Li and G. Shi, Topological design of ultrastrong and highly conductive graphene films, Adv. Mater., 2017, 29, 1702831 CrossRef PubMed.
- M. Acik, G. Lee, C. Mattevi, A. Pirkle, R. M. Wallace, M. Chhowalla, K. Cho and Y. Chabal, The role of oxygen during thermal reduction of graphene oxide studied by infrared absorption spectroscopy, J. Phys. Chem. C, 2011, 115, 19761–19781 CrossRef CAS.
- M. Acik, G. Lee, C. Mattevi, M. Chhowalla, K. Cho and Y. J. Chabal, Unusual infrared-absorption mechanism in thermally reduced graphene oxide, Nat. Mater., 2010, 9, 840–845 CrossRef CAS PubMed.
- P. V. Kumar, N. M. Bardhan, S. Tongay, J. Wu, A. M. Belcher and J. C. Grossman, Scalable enhancement of graphene oxide properties by thermally driven phase transformation, Nat. Chem., 2014, 6, 151–158 CrossRef CAS PubMed.
- Y. Qiu, F. Collin, R. H. Hurt and I. Kuelaots, Thermochemistry and kinetics of graphite oxide exothermic decomposition for safety in large-scale storage and processing, Carbon, 2016, 96, 20–28 CrossRef CAS PubMed.
- H. Y. He, J. Klinowski, M. Forster and A. Lerf, A new structural model for graphite oxide, Chem. Phys. Lett., 1998, 287, 53–56 CrossRef CAS.
- A. Lerf, H. Y. He, M. Forster and J. Klinowski, Structure of graphite oxide revisited, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- S. Pei, J. Zhao, J. Du, W. Ren and H.-M. Cheng, Direct reduction of graphene oxide films into highly conductive and flexible graphene films by hydrohalic acids, Carbon, 2010, 48, 4466–4474 CrossRef CAS.
- I. K. Moon, J. Lee, R. S. Ruoff and H. Lee, Reduced graphene oxide by chemical graphitization, Nat. Commun., 2010, 1, 73 Search PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Raman spectrum of graphene and graphene layers, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- M. M. Lucchese, F. Stavale, E. H. M. Ferreira, C. Vilani, M. V. O. Moutinho, R. B. Capaz, C. A. Achete and A. Jorio, Quantifying ion-induced defects and Raman relaxation length in graphene, Carbon, 2010, 48, 1592–1597 CrossRef CAS.
- L. G. Cancado, A. Jorio, E. H. Martins Ferreira, F. Stavale, C. A. Achete, R. B. Capaz, M. V. O. Moutinho, A. Lombardo, T. S. Kulmala and A. C. Ferrari, Quantifying defects in graphene via Raman spectroscopy at different excitation energies, Nano Lett., 2011, 11, 3190–3196 CrossRef CAS PubMed.
- L. Peng, Z. Xu, Z. Liu, Y. Wei, H. Sun, Z. Li, X. Zhao and C. Gao, An iron-based green approach to 1 h production of single-layer graphene oxide, Nat. Commun., 2015, 6, 5716 CrossRef CAS PubMed.
- M. Wu, Y. Li, B. Yao, J. Chen, C. Li and G. Shi, A high-performance current collector-free flexible in-plane micro-supercapacitor based on a highly conductive reduced graphene oxide film, J. Mater. Chem. A, 2016, 4, 16213–16218 RSC.
- L. Dong, Z. Chen, S. Lin, K. Wang, C. Ma and H. Lu, Reactivity-controlled preparation of ultralarge graphene oxide by chemical expansion of graphite, Chem. Mater., 2017, 29, 564–572 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03695k |
| This journal is © The Royal Society of Chemistry 2019 |