
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSyntheses of defined sulfated oligohyaluronans reveal structural effects, diversity and thermodynamics of GAG–protein binding†
Sebastian
Köhling‡
a,
Joanna
Blaszkiewicz‡
a,
Gloria
Ruiz-Gómez
b,
María Isabel
Fernández-Bachiller
a,
Katharina
Lemmnitzer
c,
Nydia
Panitz
d,
Annette G.
Beck-Sickinger
d,
Jürgen
Schiller
 c,
M. Teresa
Pisabarro
*b and
Jörg
Rademann
*a
c,
M. Teresa
Pisabarro
*b and
Jörg
Rademann
*a
aInstitute of Pharmacy – Medicinal Chemistry, Freie Universität Berlin, Königin-Luise-Str. 2+4, 14195 Berlin, Germany. E-mail: joerg.rademann@fu-berlin.de
bStructural Bioinformatics, BIOTEC TU Dresden, Tatzberg 47-51, Dresden 01307, Germany. E-mail: maria_teresa.pisabarro@tu-dresden.de
cInstitute of Medical Physics and Biophysics, University of Leipzig, Härtelstr. 16/18, 04107 Leipzig, Germany
dInstitute of Biochemistry, University of Leipzig, Brüderstr. 34, 04103 Leipzig, Germany
First published on 1st November 2018
Abstract
Binding of sulfated glycosaminoglycans (GAG) to a wide spectrum of extracellular regulatory proteins is crucial for physiological processes such as cell growth, migration, tissue homeostasis and repair. Thus, GAG derivatives exhibit great relevance in the development of innovative biomaterials for tissue regeneration therapies. We present a synthetic strategy for the preparation of libraries of defined sulfated oligohyaluronans as model GAG systematically varied in length, sulfation pattern and anomeric substitution in order to elucidate the effects of these parameters on GAG recognition by regulatory proteins. Through an experimental and computational approach using fluorescence polarization, ITC, docking and molecular dynamics simulations we investigate the binding of these functionalized GAG derivatives to ten representative regulatory proteins including IL-8, IL-10, BMP-2, sclerostin, TIMP-3, CXCL-12, TGF-β, FGF-1, FGF-2, and AT-III, and we establish structure–activity relationships for GAG recognition. Binding is mainly driven by enthalpy with only minor entropic contributions. In several cases binding is determined by GAG length, and in all cases by the position and number of sulfates. Affinities strongly depend on the anomeric modification of the GAG. Highest binding affinities are effected by anomeric functionalization with large fluorophores and by GAG dimerization. Our experimental and theoretical results suggest that the diversity of GAG binding sites and modes is responsible for the observed high affinities and other binding features. The presented new insights into GAG–protein recognition will be of relevance to guide the design of GAG derivatives with customized functions for the engineering of new biomaterials.
Introduction
Glycosaminoglycans (GAG) are complex linear polysaccharides forming the extracellular matrix (ECM) of all vertebrates together with fibrous proteins. Their repeating units, disaccharides with various β-(1 → 4)-linked uronic acids and β-(1 → 3)-linked hexosamines, carry variable N- or O-sulfations, which contribute to the structural and functional diversity of GAG.1 The interaction of GAG with their target proteins in the ECM depends on their sulfation and influences processes such as tissue hemostasis, lipid transport, absorption, cell growth, cell-migration and tissue repair.2 The diverse sulfation patterns of low and highly sulfated GAG including chondroitin-, dermatan-, keratan sulfates, heparin and heparin sulfates have been proposed to provide binding specificity information in a “sulfation code”.3 In addition, it has been reported that GAG recognition specificity can be also gained by H-bond and van der Waals interactions with non-charged amino acids.1,4 In accordance, sulfated GAG are known to sequester and release cytokines, growth factors and mediators to regulate the different stages of the regenerative process in bone and skin.5 Over the recent years, biomaterials derived from sulfated hyaluronic acid (sHA), a natively non-sulfated GAG core structure, have shown to strongly stimulate the process of dermal wound-healing and the regeneration of bone defects on cellular level and in animal models.5c,6 It has been hypothesized that these effects correlate with the binding of proteins to the sulfated hyaluronic acid part of the biomaterials. Evidence for interactions between GAG and sHA were provided at molecular level for several target proteins.5c,7 In addition, there are numerous GAG-binding proteins with diverse and sometimes opposing biological functions. For example, both pro- and anti-inflammatory cytokines have GAG-binding sites.5d,7b,8 Likewise, both proliferative growth factors that support wound healing and some of their anti-proliferative counterparts can bind GAG.9 In addition, other physiological processes such as blood coagulation, angiogenesis and tissue remodeling involve GAG-binding proteins.2c The diversity of GAG–protein interactions and consequent functional effects raise the question whether GAG-based materials can be tailored in such a way that they are able to stimulate desired functions while not inducing unwanted or even detrimental side effects. To address this point, a comprehensive and systematic analysis of the effect of GAG chemical substitutions on a representative panel of functionally relevant target proteins is needed.In this study, we will examine systematically the structure-activity relationships (SAR) of sHA oligosaccharides with different length, sulfation pattern, anomeric substitution and bivalent character with respect to their recognition by ten representative regulatory target proteins including cytokines, chemokines and growth-factors involved in regenerative processes. A fluorescence polarization-assay will be developed based on sHA oligosaccharides labeled with carboxytetramethylrhodamine (TAMRA) as fluorescent probes. Binding of fluorescent ligands to proteins is in direct correlation to increasing fluorescent polarization values and can be used to determine binding constants with standard microtiter plate readers. The displacement of bound fluorescent probes by non-fluorescent competitors enable the collection of binding data for unlabeled compounds. Thermodynamics and stoichiometry of GAG binding will be determined by isothermal calorimetry titration (ITC), and computer-aided molecular modeling and molecular dynamics simulations (MD) will be applied in order to rationalize the experimental data. Our combined experimental and computational approach will identify the relevance of the investigated structural features for GAG–protein recognition on a systematic basis for an extended target protein panel. Contributions of all available binding sites and binding modes to the overall binding will be analyzed, and thermodynamics and stoichiometry of GAG–protein binding will be elucidated. Thus, this approach has the potential to create novel and systematic insights into GAG–protein recognition which can be further exploited for the rational design of smart GAG with customized functional applications for the development of new biomaterials.
Results and discussion
Chemoenzymatic synthesis and anomeric modification of HA: a library of defined sulfated GAG
Oligohyaluronans were synthesized as di-, tetra- and hexasaccharides (HA-2 1, HA-4 2, and HA-6 3, Scheme 1) using a recently published chemoenzymatic approach.7d,10 The anomeric centers of the reducing and unprotected HA-oligosaccharides were fixed in the form of β-glycosyl azides 4–6 in good yields using an optimized method employing the reagent dimethyl-imidazolium chloride (DMC)11 in water.7d Azides 5 and 6 were coupled with the 5(6)-carboxy-tetramethylrhodamine (TAMRA)-labeled propargyl amide 7 using Cu-catalyzed azide–alkyne coupling reaction.12 The respective 5′- and 6′-isomers were separated on HPLC to furnish the fluorescently labeled compounds 8 and 9 in 93% (for both isomers) and 79% yield (for both isomers), respectively. In order to dissect the effect of the TAMRA fluorophore from that of the triazole, the tetrahyaluronan azide 5 was coupled with ethynyltrimethylsilane, and the intermediary 4-trimethyl-silyl-triazol was deprotected with tetrabutylammonium-fluoride (TBAF) buffered with acetic acid in aqueous THF. The triazole-substituted tetrasaccharide 10 was obtained as a control compound in 76% yield over two steps. In an alternative and versatile route for anomeric functionalization, oligohyaluronans were converted to thioglycosides. First, HA-4 was activated with DMC and base.10 The intermediary 1,2-oxazoline was reacted with 1-thiobenzoic acid furnishing the HA-4 1-thiobenzoate 11. Treatment of 11 with base released the 1-β-thioglycoside, which was first alkylated with methyl iodide furnishing the HA-4 methyl thioglycoside 12 in 63% yield. Alternatively, 11 was deprotected with base, and the obtained thiol was desulfurized by a free radical-mediated reduction with an equimolar amount of tris-(2-carboxyethyl)-phosphine (TCEP).13 The obtained 1-deoxy-hyaluronan tetrasaccharide 13 contains an 1-desoxy-sugar, β-D-2-acetamido-1-deoxy-glucosamine. As reported before, after the basic release of thiobenzoate 11, the in situ generated glycosyl-thiolate was further alkylated with 2-[2-[2-[(triphenyl-methyl)-thio]-ethoxy]-ethyl]-bromide to furnish a hyaluronan tetrasaccharide attached to a terminal S-trityl-protected polyethylene glycol spacer.10 Subsequent acidic deprotection of the S-trityl group to the free thiol followed by oxidation with air in 0.1 M NaOH provided disulfide 14 in 91% yield over two steps. Finally, compounds 8–10 and 12–14 were fully O-sulfated using SO3-pyridine-complex in DMF and purified by dialysis to provide the nona-sulfo-tetrameric fluorescent probe 9s-HA-4-TAMRA 15 in 61–85% yield and, the anomeric triazol-, methyl thioglycoside- and 1-desoxy-derivatives 16, 17 and 18 in 61%, 47% and 72% yield, respectively. The sulfations were accomplished to give the trideca-sulfo-fluorescent tracer 13s-HA-6-TAMRA 19 and the bivalent octadeca-sulfo-disulfide (9s-HA-4-S-PEG-S-)220 in 61–76% and 74% yield, respectively.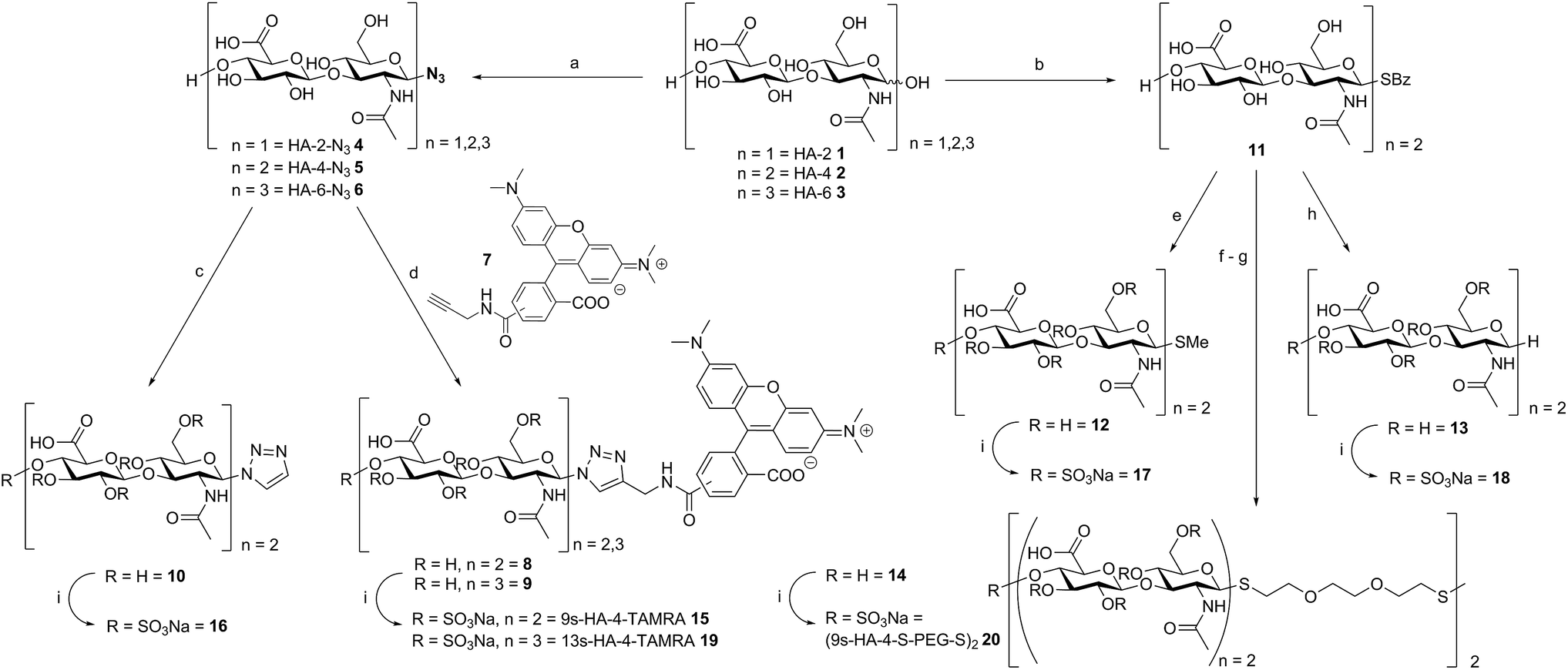 | ||
Scheme 1 Synthesis of polysulfated hyaluronan oligosaccharides with a variety of anomeric substituents. Reaction conditions: (a) DMC, N-methylmorpholine, NaN3, H2O, 0 °C to RT, 30 h, 59% (4), 72% (5) and 48% (6); (b) (i) DMC, DIPEA, D2O/MeCN 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 0 °C to RT, 90 min; (ii) thiobenzoic acid in MeCN, 5 min; 74%; (c) (i) ethynyltrimethylsilane, TBTA, CuSO4·5H2O, Na-ascorbate in MeOH/H2O 3:1, 2 h; (ii) TBAF in THF/AcOH/H2O 12:1:4, 16 h, 76%; (d) TBTA, CuSO4·5H2O, Na-ascorbate in MeOH/H2O 3:1, 2 h, 93% (8) and 79% (9); (e) MeI, NaOMe in MeOH, 16 h, 63%; (f) 2-[2-[2-[(triphenylmethyl)-thio]ethoxy]ethyl]-bromide, NaOMe, TCEP in MeOH, 36 h, 50 °C, 93%; (g) (i) TFA/DCM/TES/H2O 10:10:1:1, 5 min; (ii) 0.1 M NaOH, O2, 35 °C, 4 h, 91%; (h) NaOMe in MeOH, DOWEX-H+, then TCEP, 2-hydroxy-1-[4-(2-hydroxyethoxy)phenyl]-2-methyl-1-propanone in MeOH/H2O 4:1, hν, 1 h, 71%; (i) SO3·Py, DMF, 6 h, 61–82% (15), 61% (16), 47% (17), 72% (18), 61–76% (19), 74% (20); DMC = 2-chloro-1,3-dimethyl-imidazolium chloride, DIPEA = N,N-diisopropylethylamine, TBTA = tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine, TCEP = tris(2-carboxyethyl)phosphine, TFA = trifluoroacetic acid, TES = triethylsilane. :1, 0 °C to RT, 90 min; (ii) thiobenzoic acid in MeCN, 5 min; 74%; (c) (i) ethynyltrimethylsilane, TBTA, CuSO4·5H2O, Na-ascorbate in MeOH/H2O 3:1, 2 h; (ii) TBAF in THF/AcOH/H2O 12:1:4, 16 h, 76%; (d) TBTA, CuSO4·5H2O, Na-ascorbate in MeOH/H2O 3:1, 2 h, 93% (8) and 79% (9); (e) MeI, NaOMe in MeOH, 16 h, 63%; (f) 2-[2-[2-[(triphenylmethyl)-thio]ethoxy]ethyl]-bromide, NaOMe, TCEP in MeOH, 36 h, 50 °C, 93%; (g) (i) TFA/DCM/TES/H2O 10:10:1:1, 5 min; (ii) 0.1 M NaOH, O2, 35 °C, 4 h, 91%; (h) NaOMe in MeOH, DOWEX-H+, then TCEP, 2-hydroxy-1-[4-(2-hydroxyethoxy)phenyl]-2-methyl-1-propanone in MeOH/H2O 4:1, hν, 1 h, 71%; (i) SO3·Py, DMF, 6 h, 61–82% (15), 61% (16), 47% (17), 72% (18), 61–76% (19), 74% (20); DMC = 2-chloro-1,3-dimethyl-imidazolium chloride, DIPEA = N,N-diisopropylethylamine, TBTA = tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine, TCEP = tris(2-carboxyethyl)phosphine, TFA = trifluoroacetic acid, TES = triethylsilane. | ||
Anomerically modified derivatives were isolated as uniform compounds with one anomeric signal in 1H-NMR (4.9 ppm, J = 6.4 Hz for thiol-substitution, 6.0 ppm, J = 9.6 Hz for triazole-substituted C-1) and 13C-NMR (around 84.7 ppm), and a distinct molar mass as detected by MS-analysis (for details see ESI†).
Site-selective partial sulfation
The library synthesis of oligohyaluronans with defined and distinct sulfation patterns posed a considerable challenge as the use of many protecting group schemes is excluded for selective sulfation due to the lability of sulfates at both basic and acidic pH. We have developed methods for the synthesis of the 6,6′′-di-sulfo 2s-HA-4-N321, the penta-sulfo 5s-HA-4-N322, the hepta-sulfo 7s-HA-4-N323, the penta-sulfo 5s-HA-2-N324, the nona-sulfo 9s-HA-4-N325 and the trideca-sulfo 13s-HA-6-N326 (Scheme 2). HA-4-N35 was treated with 6 equivalents of sulfur trioxide pyridine complex in DMF in order to exploit the higher reactivity of primary alcohols as nucleophiles for site-selective sulfation. Elution of the reaction mixture over a weak anion exchanger with NaCl furnished the di-O-6-sulfo-tetrahyaluronan azide 21 in 35% yield after desalting. Mono- and tri-sulfated by-products were formed in low amounts and baseline-separated. Products were isolated as pure compounds with sulfates at defined positions. Screening several protecting groups for the 4,6-positions of HA-4 revealed that only phenyl boronate diesters were stable enough to endure sulfation of the protected intermediate and were labile enough to be cleaved without sulfate loss. Treatment of HA-4-N35 with phenyl boronic acid under exclusion of water at 80 °C in DMF led the quantitative formation of the bis-4,6-O-protected compound. The reaction progress was monitored by 13C-NMR in DMF-d6 (chemical shift of C-6 from 61.6 ppm to 63.8 ppm, see SFig. 1 in the ESI†). The protected intermediate was penta-sulfated with SO3-pyridine, and the cyclic boronates were cleaved during aqueous work-up14 furnishing 5s-HA-4-N322 in 45% yield. Finally, the 7s-HA-4-N323 was synthesized by the quantitative and selective 6,6′′-bis-dimethoxytritylation with an excess of 4,4′-dimethoxytrityl-chloride in pyridine-d5 and activation with 4-dimethylamino pyridine (DMAP) at 85 °C. Progress of the reaction was monitored by 13C-NMR considering the quantitative shift of the CH2-group at the primary alcohol from 61.8 ppm to 63.7 ppm (SFig. 2†). The bis-tritylated hyaluronan was hepta-sulfated and the dimethoxytrityl group was cleaved with 1,1,1,3,3,3-hexafluoro-2-propanol (pKa = 9.3)15 without sulfate loss furnishing heptasulfate 23 in 38% yield. The hydrogenolytical cleavage of trityl groups from the HA-4-N3 was not possible due to side reduction of the azide function to the corresponding amine. In the general per-sulfation protocol for oligohyaluronan-azides, six equivalents of SO3-pyridine complex per OH-group were applied in DMF, and fully sulfated sugars were precipitated as sodium salts. This protocol enabled the synthesis of high density charged oligohyaluronans and delivered the per-sulfated di-, tetra-, and hexahyaluronan azides with five (24), nine (25) and 13 (26) sulfates in 72%, 74% and 83% yield, respectively.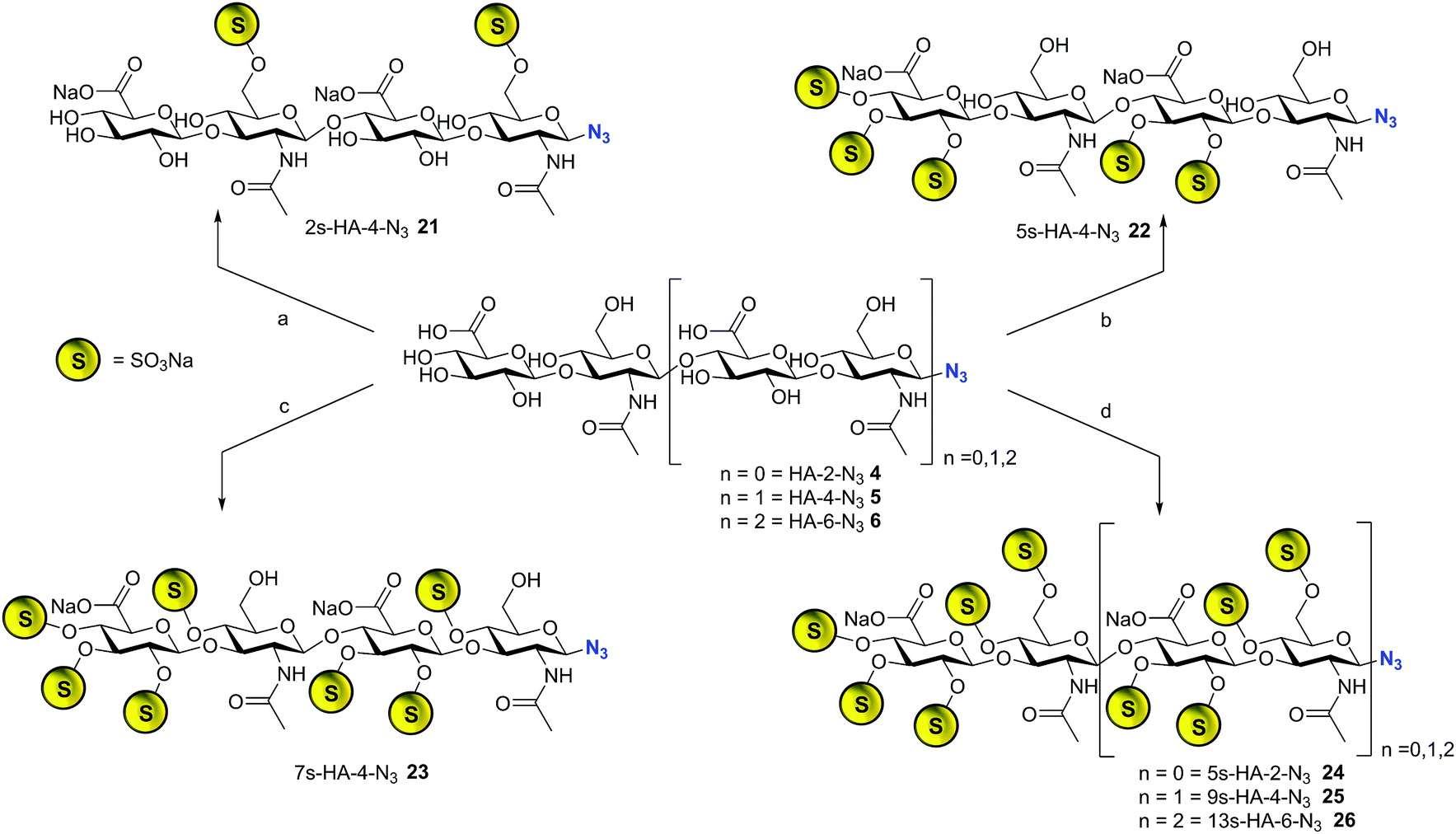 | ||
| Scheme 2 Synthesis of selectively sulfated oligohyaluronan azides. Reaction conditions (a) SO3·Py, DMF, 0 °C, 1 h, 35%; (b) (i) PhB(OH)2, DMF, 80 °C, 4 h; (ii) SO3·Py, 60 °C, 1 h, 45%; (c) (i) DMTrt-Cl, Py, 85 °C, 48 h; (ii) SO3·Py, 40 °C, 4 h, (iii) HFIP, water/CHCl3, 38%; (d) SO3·Py, DMF, 6 h, 72% (24) 6 h, 74% (25) 6 h, 83% (26); DMTrt-Cl = 4,4′-dimethoxytriphenylmethyl chloride, HFIP = 1,1,1,3,3,3-hexafluoro-2-propanol. | ||
Selection of representative regulatory proteins
Ten extracellular proteins involved in the regulation of tissue and bone healing, which are accumulated or released by interactions with GAG, were selected as representative examples. As cytokines interleukin 8 (IL-8/CXCL-8), responsible for neutrophil recruitment to guide inflammation,16 the pro- and antiinflammatory interleukin 10 (IL-10),5d stromal cell-derived factor 1 (SDF-1/CXCL-12), responsible for MSC attraction,17 and transforming growth factor β1 (TGF-β1) as key mediator in several phases of the healing process were chosen.18 As examples of growth factors served the strongly mito- and angiogenic fibroblast growth factors 1 and 2 (FGF-1, FGF-2)19 and the osteoinductive bone morphogenetic protein-2 (BMP-2).20 Sclerostin was selected as an endogenous inhibitor of Wnt-signaling, one of the fundamental signaling pathways for osteoblast differentiation and bone formation.21 Last but not least, the tissue inhibitor of metalloproteinase 3 (TIMP-3)22 and the blood coagulation inhibitor antithrombine-III (AT-III)23 were included in the study.Protein binding assays
Binding of all sulfated hyaluronans to the selected proteins was measured using fluorescence polarization (FP) assays. First, dissociation constants of tetra- and hexameric fluorescent probes 15 and 19 were determined by FP saturation experiments titrating 10 nM solutions of the probe molecules with increasing concentrations of the proteins (Table 1). The recorded FP values were converted into binding isotherms by considering the differences in the fluorescence quantum yields between the bound and the free FP probe (see ESI,† FP section). The fluorescent probes 15 and 19 bound to all the targeted proteins with high affinities. KD values were all in the nanomolar range, except for compound 15 with IL-10 and AT-III, which had values in the low micromolar range (IL-10: KD = 4.0 μM, AT-3: KD = 1.16 μM; Table 1). This demonstrates the general applicability of compounds 15 and 19 as fluorescent probes in GAG–protein binding assays. In most cases, the hexameric fluorescent ligand 19 showed higher binding affinities compared to the tetrameric probe 15 (see Scheme 3). Binding of all unlabeled oligohyaluronan sulfates (25, 26) was determined in FP-competition assays measuring the replacement of the bound FP probe 15. The protein concentration employed in this approach corresponded to the EC50 value of the bound FP probe to ensure sufficiently strong polarization signals and high assay sensitivity. In few exceptional cases higher protein concentrations were applied (for details see the ESI†). The equation of Nikolovska-Coleska and Wang24 was used to calculate KD values from FP-competition (see ESI,† FP section).| Proteina | Type of protein | K D [μM] | ||||
|---|---|---|---|---|---|---|
| 5s-HA-2-R | 9s-HA-4-R | 13s-HA-6-R | ||||
| R = -N3 (24)c | -TAMRA (15)b | -N3 (25)c | -TAMRA (19)b | -N3 (26)c | ||
| a IL-8 = interleukin 8; IL-10 = interleukin 10; CXCL-12 = stromal cell derived factor 1; TGF-β1 = transforming growth factor β1; FGF-1/-2 = fibroblast growth factor -1/-2; BMP-2 = bone morphogenetic protein 2; TIMP-3 = tissue inhibitor of metalloproteinase 3; AT-III = antithrombine III. n.d. = not determined. b Determined by FP titration. c Determined by FP competition with probe 15. | ||||||
| IL-8 | Chemokine | 117.4 ± 14.0 | 0.214 ± 0.013 | 5.0 ± 0.6 | 0.029 ± 0.003 | 1.51 ± 0.44 |
| IL-10 | Cytokine | 65.9 ± 10.0 | 4.0 ± 0.5 | 3.2 ± 1.3 | 0.422 ± 0.028 | 0.812 ± 0.12 |
| BMP-2 | Growth factor | 110.1 ± 31.1 | 0.075 ± 0.006 | 2.3 ± 0.3 | 0.107 ± 0.008 | 0.214 ± 0.069 |
| Sclerostin | Wnt signaling inhibitor | 266.3 ± 97.0 | 0.035 ± 0.001 | 2.8 ± 0.03 | 0.021 ± 0.002 | 0.23 ± 0.05 |
| TIMP-3 | MMP-inhibitor | n.d. | 0.03 ± 0.001 | 0.27 ± 0.062 | 0.053 ± 0.002 | 0.67 ± 0.05 |
| CXCL-12 | Chemokine | n.d. | 0.069 ± 0.001 | 3.4 ± 0.7 | 0.030 ± 0.002 | 0.75 ± 0.024 |
| TGF-β1 | Cytokine | n.d. | 0.047 ± 0.002 | 4.8 ± 1.7 | 0.056 ± 0.004 | 1.01 ± 0.64 |
| FGF-1 | Growth factor | n.d. | 0.1 ± 0.007 | 0.051 ± 0.027 | 0.058 ± 0.004 | 0.003 ± 0.002 |
| FGF-2 | Growth factor | n.d. | 0.023 ± 0.001 | 0.294 ± 0.095 | 0.019 ± 0.001 | 0.093 ± 0.003 |
| AT-III | Serine protease inhibitor | n.d. | 1.16 ± 0.15 | 3.76 ± 0.74 | 0.48 ± 0.048 | 0.65 ± 0.51 |
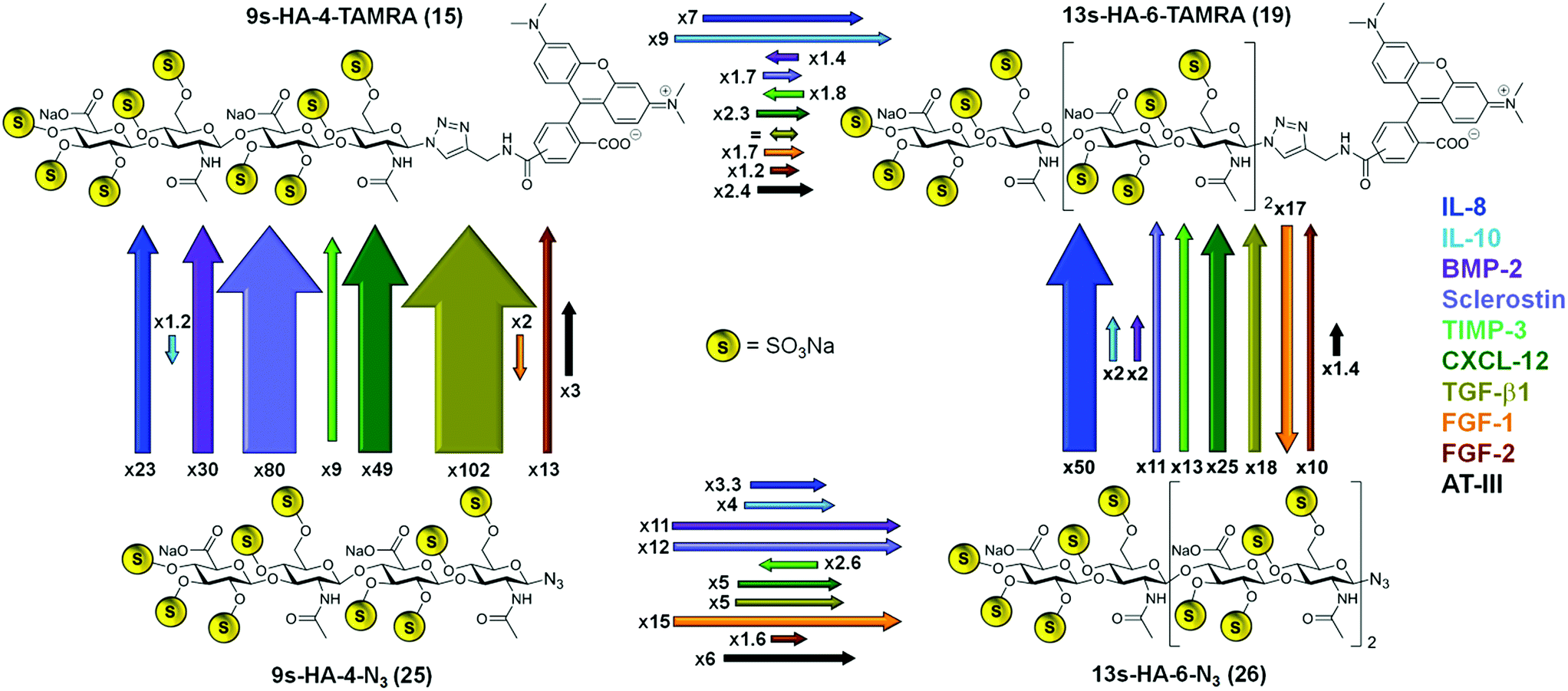 | ||
| Scheme 3 Summary of the recognition of four defined sulfated hyaluronanes by ten regulatory proteins obtained experimentally by fluorescence polarization. Affinity ratios of fluorophor-labeled tetra- and hexahyaluronans 15 and 19 (top) versus the azide-functionalized tetra- and hexahyaluoronans 25 and 26 (bottom) are represented schematically by arrows. Each of the ten regulatory proteins analyzed is labeled with a different color, which is also used for the arrows representing its affinity ratios. The length and thickness of each arrow correspond to the ratio of the experimentally determined KD values for this case. A ratio of < = 10 is illustrated by the length of the arrow and a ratio >10 is represented by the thickness of the arrow. Each arrow is labeled with a number representing the determined affinity ratio for this case. | ||
In most cases, the binding of the fluorescent probes 15 and 19 was considerably enhanced compared to the unlabeled azides 25 and 26 (Scheme 3). The effect of fluorophore addition was especially pronounced for the tetrahyaluronans. The 9s-HA-4-TAMRA 15 bound stronger than the azide analog 25 in eight cases of the ten target proteins. The strongest effects were observed for TGF-β (102-fold) and sclerostin (80-fold affinity). In the case of the hexa-saccharides 19 and 26, TAMRA addition was associated with an increase in binding affinity toward nine of the ten target proteins, having the maximum effect on IL-8 (50-fold). In three cases the introduction of the TAMRA fluorophore correlated with a decrease in binding affinity, namely for FGF-1 with compounds 15 and 19 and, almost negligible, for IL-10 with 15. The sugar length of our oligohyaluronans had also a strong impact on binding affinities (Scheme 3). Elongation of 9s-HA-4-TAMRA 15 by two sugar units resulted in a general increase in binding affinity, being more pronounced for IL-10 (9-fold) and IL-8 (7-fold). In the case of the corresponding azide analogs, elongation had the strongest effects for FGF-1 (15-fold), sclerostin (12-fold) and BMP-2 (11-fold). Solely, TIMP-3 showed reverse effects for the labeled and unlabeled compounds regardless of anomeric modifications, which could indicate of a smaller, confined GAG-binding region.25 In addition, the 5s-HA-2-N324 was tested in competition experiments with a selection of the target proteins (i.e. the first five listed in Table 1). Binding affinities were considerably decreased for all proteins compared to 9s-HA-4-N325 by factors between 21 (for IL-10) and 95 (for sclerostin) suggesting that a tetramer represents the minimum length required for GAG–protein interaction. Binding affinities of the HA-probes determined by FP were considerable higher than those measured in earlier studies by NMR. For example, the affinity of 25 for IL-10 determined by HSQC-titration was 1.07 ± 0.13 mM,7d a value much less favorable (334 times) than the value measured in the FP competition assay. Lower affinities in NMR titrations have been reported earlier26 and can been attributed in part to the effects of the very high protein concentrations employed in these experiments (>0.5 mM).
For an additional explanation, it should be noted that NMR-titrations record the chemical shift of one defined N–H crosspeak of the protein and thus report binding very specifically for one protein site. If several alternative binding modes are recorded in one experiment, local binding can result in a significantly higher overall binding affinity (for an example see ESI†).
Next, the effects of a variety of anomeric substitutions were tested with a selection of five target proteins (Table 2, SScheme 1†). Differences in the binding affinities between the anomeric azide 25, 1,2,3-triazole 16, methyl-thioglycoside 17, and the desoxy-derivative 18 were low for IL-8, IL-10 and TIMP-3, encouraging further chemical variations in this apparently variable anomeric position. On the contrary, sclerostin exhibited a significantly stronger decrease in the binding of the four sulfated tetrahyaluronans 16, 17, 18 compared to the azide 25 (16 → 25, 7-fold; 17 → 25, 13-fold), which was remarkable considering the minor structural differences. Most surprisingly, the binding affinities of the bivalent (9s-HA-4-S-PEG-S-)220 toward several proteins were increased drastically compared to the monomeric 9s-HA-4-N325. For example, binding to IL-8 was improved by a factor of 417, constituting the best binder for this protein so far (KD = 0.012 μM), while binding to IL-10 and BMP-2 was enhanced by a factor of 71 and 59, respectively. The high affinity of the bivalent sHA 20 suggested that the dimeric ligand can bind to more than one GAG-binding site of one or of several proteins. In order to investigate these possibilities, isothermal calorimetry (ITC) of IL-8 and IL-10 was conducted with monovalent 9s-HA-4-N325, bivalent (9s-HA-4-S-PEG-S-)220 and 9s-HA-4-TAMRA 15 (Fig. 1, STables 1, 2, SScheme 2, 3†). IL-8 was used at 10 μM (monomer) concentration and ligands were added dropwise at 50 and 100 μM in the same buffer. Monovalent ligand 25 and TAMRA-labeled ligand 15 bound IL-8 with a stoichiometry of 0.37 and 0.42, respectively (average of three independent experiments), suggesting that IL-8 was bound predominantly as a protein dimer.27 Bivalent ligand 20 bound IL-8 with a stoichiometry of 0.20 (ca. two times less than the monomeric ligand). This finding indicated that indeed both monovalent parts of 20 were involved in binding, and that most of the bivalent ligands bound more than two protein dimers. The affinity of the monovalent azide 25 was 34 nM. Binding was most favorable for the TAMRA-labeled ligand 15 (KD = 9 nM), suggesting the participation of the TAMRA residue in binding to IL-8. The binding affinity of the bivalent ligand 20 was 16 nM. In all cases the binding free energy was composed of a large enthalpic and a small entropic contribution. The bivalent compound 20 showed a considerable rise of entropy in agreement with the loss of conformational freedom at a flexible linker. The observed higher affinities compared to the FP experiments can have several causes. First, KD values determined by FP were related to the concentrations of the protein monomer. If all IL-8 is in dimeric form, the effective protein concentrations at the binding isotherms must be divided by two. Secondly, deviations might reflect the diversity of binding sites and modes. If e.g. the complex of IL-8 with the fluorescent probe 15 is able to bind other sulfated GAG as ligands, this would result in reduced binding affinities in the competition assay. Similar results were already reported for GAG binding to TIMP3 using H/D exchange NMR experiments.25a
| Proteina | K D [μM]b | |||||
|---|---|---|---|---|---|---|
| 9s-HA-4-R | (9s-HA-4-R)2 | Fondaparinux | ||||
| R = -N3 (25) | -Triazol (16) | -SMe (17) | -H,H (18) | -S-PEG-S- (20) | (27) | |
| a IL-8 = interleukin 8; IL-10 = interleukin 10; BMP-2 = bone morphogenetic protein 2; TIMP-3 = tissue inhibitor of metalloproteinase 3. b Determined by FP competition with probe 15. c EC50FSB values. n.d. = not determined. | ||||||
| IL-8 | 5.0 ± 0.6 | 10.6 ± 2.0 | 7.87 ± 0.84 | 6.5 ± 0.79 | 0.012 ± 0.003 | 19.0 ± 2.5 |
| IL-10 | 3.2 ± 1.3 | 7.54 ± 0.71 | 9.13 ± 1.23 | 6.2 ± 1.4 | 0.045 ± 0.014 | 10.8 ± 1.7 |
| BMP-2 | 2.3 ± 0.3 | 2.81 ± 0.3 | 2.36 ± 0.4 | 3.4 ± 1.0 | 0.039 ± 0.009 | 80.9 ± 8.5 |
| Sclerostin | 2.8 ± 0.03 | 20.4 ± 7.1 | 37.3 ± 8.8 | 27.8 ± 14.3 | 0.045 ± 0.007c | 629.0 ± 335 |
| TIMP-3 | 0.27 ± 0.06 | 0.35 ± 0.01 | 0.86 ± 0.1 | 0.44 ± 0.04 | 0.02 ± 0.001c | n.d. |
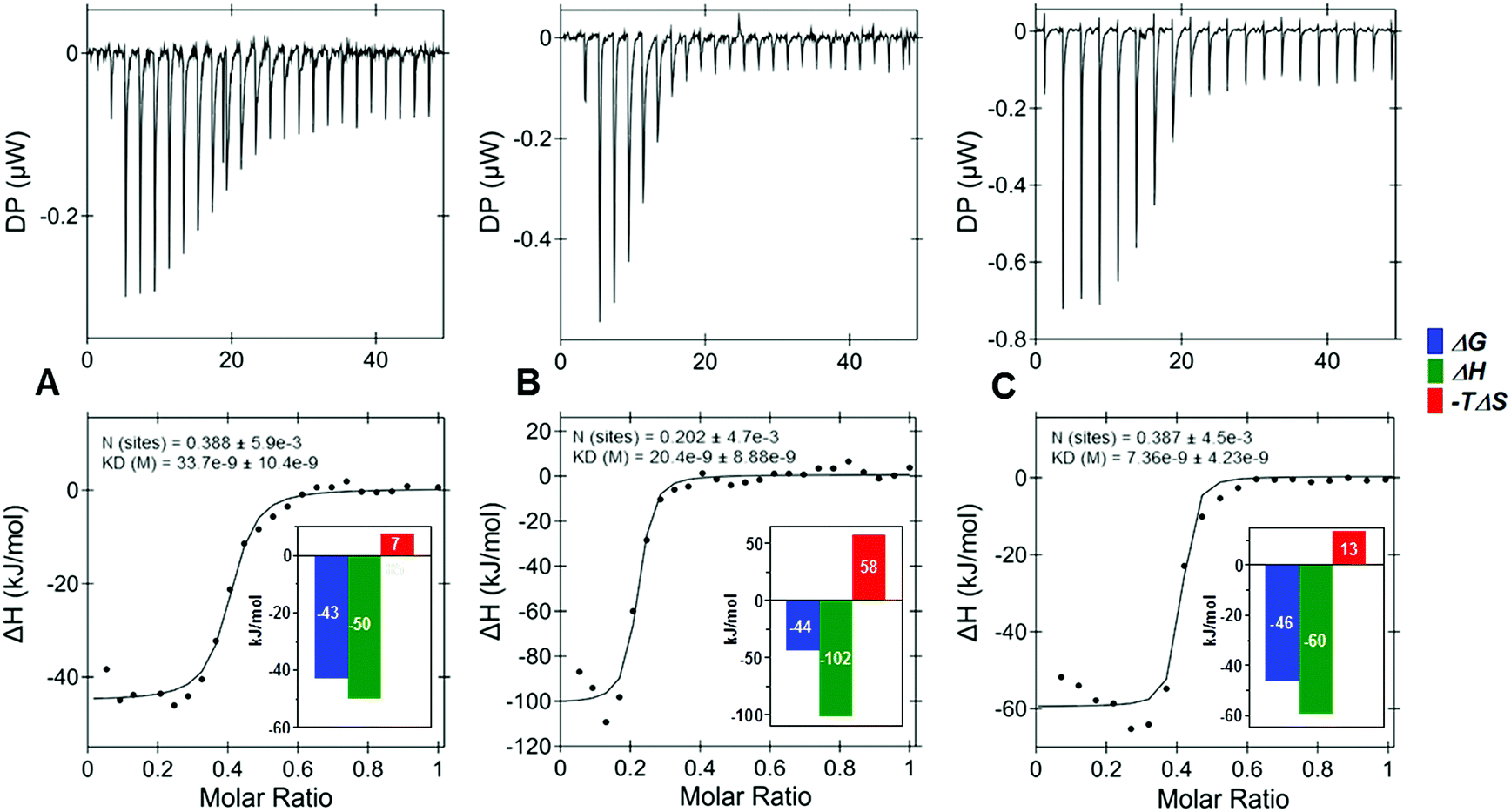 | ||
| Fig. 1 Isothermal titration calorimetry of a 50 μM solution of (A) 9s-HA-4-N3 (25), (B) (9s-HA-4-S-PEG-S-)2 (20), and a 100 μM solution of (C) 9s-HA-4-TAMRA (15) titrated to IL-8 (10 μM) yielded dissociation constants (KD) of 34 ± 10 nM, 16 ± 10 and 9 ± 4 nM, respectively, determined as the average of three independent experiments. | ||
The high affinities of 9s-HA-4 derivatives 15, 25, and (9s-HA-4-S-PEG-S)220 were also compared with the octa-sulfo-pentaheparan fondaparinux 27, a clinically used anticoagulant (Table 2).28 Fondaparinux is a nanomolar ligand of antithrombine III (AT-III) effecting the inhibition of factor Xa.23 Despite its high degree of sulfation, fondaparinux was significantly less active toward the four selected proteins, albeit with some differences. A remarkable low binding for fondaparinux was observed with BMP-2 (81 μM) and sclerostin (630 μM), suggesting that these proteins recognize highly sulfated oligohyaluronans with more efficiency.5c,7c,29
Finally, the binding of partially sulfated oligohyaluronans was investigated with IL-8, IL-10, BMP-2 and sclerostin (Table 3). Non-sulfated HA-4-N35 showed no binding to any of the tested proteins at 10 mM concentration, suggesting that electrostatic interactions are essential to promote GAG binding.6 2s-HA-4-N321, sulfated exclusively at O-6 position, showed weak binding to all four proteins tested (between 1.3 mM for IL-8 and ca. 100 μM for sclerostin, Table 3). 5s-HA-4-N322, non-sulfated at O-4,6 positions, displayed a strongly enhanced binding in comparison to 21 and the shorter disaccharide 5s-HA-2-N324, which was most pronounced for IL-8 (45- and 4-fold) and sclerostin (7- and 19-fold). These findings suggest that sulfation degree and sulfate distribution in a GAG sequence could affect protein recognition resulting in some binding specificity.3a,b,e–h,5c,7a–c,25,29,30 The additional presence of sulfate groups at O-4 position in 7s-HA-4-N323 with respect to 22 had a small effect on the binding to IL-8, BMP-2 and sclerostin (1.2–1.9-fold), whereas binding to IL-10 was amplified by a factor of eight with a KD of 6.4 μM. The fully sulfated 9s-HA-4-N325 showed the strongest increase in binding with BMP-2 (10-fold) and sclerostin (5-fold). While binding increased with the number of sulfates in the case of all tetrahyaluronans, it should be noted again that also the length of the saccharides had a significant impact on the affinities.1 For instance, the 5s-HA-4-N322 bound much stronger than the 5s-HA-2-N324 (Tables 3 and 1). Our binding data confirm that highly sulfated oligohyaluronans are indeed quite potent ligands of all tested regulatory proteins. The differences observed in the KD values obtained for IL-8 and IL-10 with different binding assays (NMR, FP, ITC) seem to reflect diversity of binding sites and modes. In order to evaluate such GAG binding diversity toward extracellular regulatory proteins at atomic detail, we applied molecular modeling and dynamics simulation (MD) techniques.
| Proteina | K D [μM]b | ||||
|---|---|---|---|---|---|
| HA-4-N3 (5) | 2s-HA-4-N3 (21) | 5s-HA-4-N3 (22) | 7s-HA-4-N3 (23) | 9s-HA-4-N3 (25) | |
| a IL-8 = interleukin 8; IL-10 = interleukin 10; BMP-2 = bone morphogenetic protein 2. b Determined by FP competition with probe 15. | |||||
| IL-8 | >10000 |
1328.0 ± 164 | 29.4 ± 7.8 | 15.6 ± 5.0 | 5.0 ± 0.6 |
| IL-10 | >10000 |
246.6 ± 77.2 | 50.0 ± 8.1 | 6.4 ± 2.1 | 3.2 ± 1.3 |
| BMP-2 | >10000 |
185.4 ± 25.6 | 33.5 ± 6.6 | 23.0 ± 1.9 | 2.3 ± 0.3 |
| Sclerostin | >10000 |
98.7 ± 11.5 | 14.0 ± 3.8 | 11.2 ± 0.8 | 2.8 ± 0.3 |
Molecular modeling
Blind docking was applied in order to predict potential recognition sites for the sulfated oligohyaluronans 15, 16 and 19–26 on the surface of the target proteins IL-8, IL-10, BMP-2 and sclerostin. MD simulations were used to refine the resulting GAG–protein complexes and to obtain binding energies (including per residue and pairwise interactions) and atomic-detailed information about the established interactions in order to rationalize the mechanisms involved in GAG–protein recognition.5c,7b,c,8,17,29 The results obtained for IL-8 and Sclerostin are discussed below. For IL-10 and BMP-2 details are given in the ESI (Discussion S1†).IL-8
As IL-8 can exist as a monomer and dimer, the binding analysis was carried out for both forms. Blind docking on the monomer predicted two distinct binding sites depending on GAG sulfation and length (Fig. 2, SFig. 1†). The disulfate 2s-HA-4-N321 was predicted to bind in different orientations along the IL-8 β1 strand having residue R31 as common anchor. The higher sulfated derivatives 15, 19, 22–23 and 25–26 were predicted to bind with different orientations along the IL-8 C-terminal α-helix (Fig. 2, SFig. 1†), which has been previously described as GAG recognition site.7b,31 5s-HA-4-N322 was oriented parallel to the helix from the non-reducing to the reducing end, and an antiparallel orientation was preferred for the higher sulfated azide derivatives (23, 25 and 26). Binding poses for the TAMRA derivatives 9s-HA-4-TAMRA 15 and 13s-HA-6-TAMRA 19, and for (9s-HA-4-S-PEG-S-)220 (half molecule, see ESI†) were predicted with the respective TAMRA and S-PEG groups positioned on the β1 strand and in proximity to the C-terminal α-helix. Refinement of the docking poses by MD simulations allowed us to investigate the determinants for complex formation in terms of receptor protein flexibility, residues involved in recognition and their contributions to binding free energy (both pairwise and per residue interactions). | ||
| Fig. 2 Predicted diversity in binding sites and modes of highly sulfated hyaluronan derivatives on IL-8 (PDB ID: 1IL8). IL-8 is depicted in gray cartoon (light and dark representing each monomer unit). In outlined boxes, schematic representation of secondary structure of IL-8 (grey; left), and summary of all binding sites and modes predicted for TAMRA (green; right), for azide (light brown) and for the half-bivalent molecule (dark brown). In full color boxes, representative snapshots from 100 ns MD simulations of most favorable binding sites and modes of 15 (in green) on IL-8 monomer (m, left), and of 15 and 19 on IL-8 dimer (d, middle and right), 25 (in light brown) on IL-8 monomer (m, left), and of 25 and 26 on IL-8 dimer (d, middle and right) and the bivalent 20 (in dark brown) on IL-8 dimer (d). sHA molecules are shown in sticks and colored by atom type. IL-8 interacting residues are highlighted in yellow and numbered (indicating those belonging to the second monomer unit with a comma). Length and thickness of the blue arrows correspond to the increase on binding obtained computationally. | ||
The binding energies computed for sHA derivatives 15, 19–23 and 25–26 with MM-GBSA32 are shown in Table 4 and STable 3.† It can be observed that binding of sulfated tetrahyaluronan azides to IL-8 monomer increases with the number of sulfates, which is in agreement with our experimental binding data (Table 3). Binding of the fully sulfated hexahyaluronans 13s-HA-6-TAMRA 19 and 13s-HA-6-N326, however, was not predicted to be stronger than that of the tetramer analogs (15 and 25). In the case of TAMRA derivatives (15 and 19) the energetically most favored binding poses were those with the sugar moiety oriented along the C-terminal α-helix (Table 4, STable 3†). Here, the calculated increased binding of 15 compared to 25 was only small (4–5 kcal mol−1) due to additional interactions of the fluorophore with IL-8 residues T42, K59, N61 and Q64 (Fig. 2). Thus, the free binding energies calculated with the IL-8 monomer did not explain the experimentally observed 23-fold increase in binding affinity of 15vs.25 and the 7-fold of hexahyaluronan 19vs. tetrahyaluronan 15 in FP assays. The same computational protocol was applied to the IL-8 dimer and the GAG ligands 15, 19–23 and 25–26. Several binding sites and modes were predicted (Table 4, SFig. 2, STables 4 and 5†), which were not observed in previous studies with other GAG derivatives and IL-8 dimer.7b For 2s-HA-4-N321, binding poses were located between the β1 strand of one monomer and the N-loop of the other, in contrast to the results obtained for IL-8 monomer in which only binding poses along β1 strand were predicted. For 5s-HA-4-N322, poses were found between the β1 and β3′ strands and the N-terminal loop, which differ from the binding poses found for the monomer at the C-terminal α-helix. For 7s-HA-4-N323 and 9s-HA-4-N325 the most populated binding poses were found along the two C-terminal α-helices of the dimer imitating the pose found for the monomer protein. In the case of 9s-HA-4-TAMRA 15, a great diversity of binding poses was predicted in which, surprisingly, the TAMRA moiety was disposed along the IL-8 dimerization axis (Fig. 2, SFig. 2†). The energetically most favored binding poses of 15 were more favorable than those predicted for its azide analog 9s-HA-4-N325 and recognized a different protein region (N-loop and β1 to β3 of each monomer) (Table 4, Fig. 2, STable 4†). The binding energy calculated per residue and pairwise for 9s-HA-4-TAMRA 15 indicated that the TAMRA group contributed strongly to the binding (SFig. 3†), making contacts with IL-8 residues R11 from one monomer and K8, R11, Q13 and, to a lesser extent, I15 of the second monomer (Fig. 2, STable 5†). Furthermore, our energy calculations indicated that 15 stabilizes the IL-8 dimer (STable 4†). Thus, our theoretical models suggested plausible GAG–protein interactions that can explain the 23-fold increase of binding experimentally determined for 15 with respect to 25 in FP assays. A very good linear correlation was observed between the energies obtained for the most favorable predicted binding sites and modes of the investigated GAG and the experimental KD values (SFig. 4†). The observed differences in binding between ITC and FP measurements could be explained by the contributions of the predicted multiple binding sites and modes33 (Fig. 2). In the case of two binding sites being predicted, we observed for both sites a high correlation between calculated and experimentally obtained binding data. The best correlations were obtained for the site with most favorable predicted energies. Besides, the predicted alternative binding sites (significantly populated and with good interaction energies) also showed good correlations (SFig. 5†), which makes conceivable the idea of binding diversity as we have previously reported for similar systems.8,25a Our findings revealed that, indeed, certain GAG substitutions could strongly modify recognition by directing the molecule toward other regions of the protein, which might have strong implications for binding specificity. In terms of the different GAG lengths, the results indicated a strong increase of the free binding energies of the hexa- vs. the tetrahyaluronans (19, 26vs.15, 25, respectively, Table 4, STable 4†) with a stronger effect for the azides than for the TAMRA derivatives.
| Protein | ΔG [kcal mol−1] | |||||||
|---|---|---|---|---|---|---|---|---|
| 2s-HA-4-N3 (21) | 5s-HA-4-N3 (22) | 7s-HA-4-N3 (23) | 9s-HA-4-N3 (25) | 9s-HA-4-TAMRA (15) | 13s-HA-6-N3 (26) | 13s-HA-6-TAMRA (19) | (9s-HA-4-S-PEG-S-)2 (20) | |
| a Shown here for the energetically most favored binding modes. b Shown for the lowest energy structure of the NMR ensemble. N- and C-termini were not considered. c Only determined for half of the molecule 9s-HA-4-S-PEG-SMe (−37.8 ± 4.4, see ESI and STable 3 for further details). d Also determined for 9s-HA-4-S-PEG-SMe (−68.3 ± 8.2, see ESI and STable 4 for further details). n.d. = not determined. | ||||||||
| IL-8monomer | −21.8 ± 5.0 | −31.6 ± 4.7 | −41.0 ± 4.7 | −45.6 ± 3.2 | −51.4 ± 5.5 | −45.2 ± 3.1 | −49.6 ± 1.9 | n.d.c |
| IL-8dimer | −33.4 ± 4.7 | −34.1 ± 6.4 | −50.4 ± 5.7 | −42.9 ± 5.6 | −72.5 ± 4.8 | −61.5 ± 7.0 | −85.1 ± 20.0 | −87.1 ± 7.1d |
| Sclerostinb | −45.3 ± 4.2 | −55.7 ± 10.5 | −78.1 ± 7.5 | −102.3 ± 10.0 | −102.5 ± 3.7 | −104.6 ± 13.2 | −127.0 ± 9.2 | n.d. |
The binding region found for the tetra- and hexa-TAMRA (15 and 19) was similar (along the IL-8 dimerization axis, Fig. 2). For the 9s-HA-4-N3 and 13s-HA-6-N3 (25 and 26), we observed that the longer molecule establishes more interactions with the protein, which translates into a large increase in binding energy (Table 4 and STable 4†) and correlates highly with the experimental FP binding data. Indeed, the hexamer contacts both units of the dimer: one site along the IL-8 dimerization axis and a second one involving the C-terminal α-helix of one monomer and the N-loop of both monomers (Fig. 2, SFig. 2, 5†). A similar recognition scenario was also found for half of the bivalent derivative 20 in which the most energetically favorable binding modes were located at the dimerization axis (Fig. 2, SFig. 2 and STable 4†). This allowed the modeling of the full bivalent molecule 20 by joining its two halves. The protein residues interacting with 20 were similar to those found to recognize 13s-HA-6-TAMRA 19, and our MD-based energy calculations showed small differences in binding free energy, predicting slightly stronger binding of 20vs.19 (Table 4, STable 4 and SFig. 6†). These results offer a plausible molecular mechanism of recognition explaining the experimentally recorded by FP and remarkably strong assessed binding of 20.
Indeed, our ITC results point toward this direction with a stoichiometry of 0.20 referring to a binding ratio of ca. 1:5 of 20 to the IL-8 monomers (Fig. 1 and SScheme 2†), meaning that 20 not only could be positioned along the dimerization axis bringing two IL-8 dimers together and, therefore, acting as a molecular glue, but it could additionally establish interactions with the C-terminal α-helix, which we find to be less energetically favorable (STable 4 and SFig. 4†).
Sclerostin
Using the lowest energy structure from the NMR ensemble of sclerostin,34 two distinguishable GAG binding regions were predicted for differentially sulfated GAG at the twisted anti-parallel β-strands and at the flexible loop (SFig. 7†). Low sulfated tetrahyaluronans 2s-HA-4-N3 21 and 5s-HA-4-N3 22 as well as dihyaluronan 5s-HA-2-N3 24 were predicted to bind to part of the twisted anti-parallel β-strands of sclerostin. For higher sulfated GAG additional binding poses were obtained at the flexible loop and also involving a few residues from the β-strands region, which showed more favorable binding energies (Table 4, STable 6 and SFig. 7†). The co-existence of multiple binding poses33 from both regions agrees with the conclusions obtained from experimental data. For 7s-HA-4-N3 23 most poses were predicted at the β-strands region and additional poses were found at the loop region resulting in a small increase in binding with respect to 22, which correlated well with our experimental data (Table 3, STable 6 and SFig. 8, 9†). A similar scenario was found for the highly sulfated tetrahyaluronans 9s-HA-4-N3 25 and 9s-HA-4-TAMRA 15 (SFig. 7–9†). For the azide derivatives, we found the best correlations of experimental and predicted binding energies for the most populated binding sites at the sclerostin β-strands region (SFig. 9B). Addition of the TAMRA group resulted in better binding due to additional hydrophobic and electrostatic interactions with sclerostin (STables 6 and 9†). Remarkable in the case of sclerostin was the strongly reduced binding affinity of the anomeric 1H-triazole 16 compared to azide 25 and TAMRA derivative 15 (Tables 1 and 2). Docking of the triazole 16 predicted binding poses exclusively at the sclerostin β-strands region, and the obtained energies were lower than for the TAMRA derivative (STable 6†). On the contrary, azide 25 bound to the β-strands region and, additionally and more favorably, it formed a cluster of binding poses distributed at the loop. These predicted multiple binding poses33 could offer a plausible explanation for the stronger binding of the azide 25 in comparison to the triazole 16 assessed experimentally. GAG elongation to 13s-HA-6-N326 resulted in binding poses at the β-strands region of sclerostin. For 13s-HA-6-TAMRA 19 a well-defined cluster of binding poses was predicted covering the β-strands and also the flexible loop. The poses found for this long molecule exhibited two different binding modes with similarly favorable binding energies: one with the TAMRA moiety oriented along the β-strands and the sugar part positioned toward the flexible loop, and a second pose at the same position oriented in an antiparallel fashion. The long cluster overlaps with the two clusters observed for the tetra-TAMRA 15. The highest binding free energy was calculated for the 13s-HA-6-TAMRA 19 followed by its azide analog 26, which is in good agreement with the order of binding strength from our experimental data (Tables 1 and 4, STable 6,†Scheme 3). The increment in GAG length also correlated with an increase in the binding energies as observed experimentally (Tables 1 and 4, Scheme 3, SFig. 9†), although the difference observed between the computational values was smaller than the difference appreciated experimentally. This may be due to the extraordinary inherent flexibility of sclerostin and the fact that the docking was performed only with the lowest energy conformation of the NMR ensemble. Therefore, two additional sclerostin conformations from the NMR ensemble (models 10 and 16) were selected in order to further investigate the recognition of tetra- (15 and 25) and hexahyaluronan (19 and 26) (STables 7 and 8†). For the three sclerostin NMR structures investigated the tetra-azide 25 yielded binding poses either at the β-strands (common interacting residues R114, R116, R119, K134 and R136) or loop region (common interacting residues R89, R97, K99, R102 and R145). In the case of the 13s-HA-6-N326, binding modes were predicted involving simultaneous interactions with the flexible loop and β-strands (residues 114–148) regions. Interestingly, these binding modes had been observed with the initial lowest energy NMR structure for the larger 13s-HA-6-TAMRA 19, but not for the smaller azide analog 26. This is attributed to the fact that in the two additionally considered NMR ensemble structures, the loop is disposed closer in space to the β-strand regions (SFig. 7, 10 and 11†). Similar predictions were obtained for the 9s- HA-4- and 13s-HA-6-TAMRA, in which the fluorophore was either oriented toward the β-strands and the GAG moiety positioned toward the loop, or vice versa (SFig. 10, 11, STables 7–9†). Nevertheless, the TAMRA moieties exhibited a major dispersion when disposed at the loop, mainly due to the intrinsic high conformational plasticity of this region and also because of the flexibility of the linker between the saccharide part of the molecule and the fluorophore group. In this binding region, the TAMRA established interactions with basic and hydrophobic residues of the protein. In contrast, when positioned at the β-strands, the disposition of the TAMRA group was more defined and with more hydrophilic protein residues involved in its recognition. In summary, for the three studied NMR ensemble conformations of sclerostin the obtained GAG binding energies were in the same range although with certain differences (Table 4, STables 6–8†). Thus, the values obtained for the 9s-HA-4-TAMRA 15 and the 13s-HA-6-N326 showed more variation for binding poses distributed along the sclerostin loop region. Interestingly, NMR ensemble models 10 and 16 revealed much stronger differences in binding for the 13s-HA-6-N3 derivative in comparison to its 9s-HA-4-N3 analog, while the TAMRA derivatives exhibited similar binding free energies (STables 7 and 8†). The use of representative structures of the NMR ensemble of sclerostin allowed us to better explore the conformational space available, particularly of its highly flexible loop, and to better appreciate the diversity in binding sites and modes between the sulfated tetra- and hexahyaluronan azides and TAMRA derivatives. In summary, our computer-aided studies exploiting the chemical space of the protein complexes of defined sulfated hyaluronans predicted a diversity of binding sites and binding modes for the studied regulatory target proteins and could explain the experimentally observed high affinities of defined sulfated GAG. The established theoretical models provide insights in atomic detail on how the introduction of defined functionalities in GAG may influence their recognition by proteins, which may be of high relevance for future engineering of new customized biomaterials.Conclusion
A systematic analysis of GAG–protein recognition was performed using a model GAG collection of defined sulfated oligohyaluronans, a set of ten representative regulatory proteins of the extracellular matrix, and a combined experimental and computational approach. A library of defined sulfated and anomerically modified oligohyaluronans was synthesized by chemoenzymatic protocols and compounds were varied with respect to the parameters oligosaccharide length, number and position of sulfates and the anomeric modification. Anomeric modifications were introduced into the azides and thioglycosides of di-, tetra-, and hexahyaluronans. Tetra- and hexahyaluronans were labeled with the fluorescent dye carboxytetramethylrhodamine (TAMRA). Tetrahyaluronan was dimerized using a PEG-thiol-linker in order to provide a bivalent ligand. Oligohyaluronans with defined alternating numbers and positions of sulfates including di-, penta-, hepta-, nona-, trideca, and, finally, octadeca-sulfo oligohyaluronanes were prepared exploiting the differential reactivity of sugar hydroxyl groups or by developing intermediary protecting group strategies. All these highly complex structures were fully characterized by NMR spectroscopy and high-resolution MS. The fluorescently labeled nonasulfo-tetrahyaluronan and the respective tridecasulfo-hexahyaluronans were used to develop a fluorescence polarization (FP) assay. This assay enabled the rapid and systematic determination of binding constants and structure–activity relationships for all labeled- and non-labeled defined sulfated oligohyaluronans and ten representative regulatory target proteins. Binding affinities obtained by FP and FP-competition experiments were confirmed for selected examples (IL-8, IL-10) by isothermal titration calorimetry (ITC) and were found to be significantly higher than earlier published affinities determined by NMR. In addition, ITC provided the thermodynamic analysis and stoichiometry of GAG–protein interactions.The comprehensive experimental and computational study presented here provided three main new insights into GAG–protein recognition:
Firstly, binding of sulfated GAG to regulatory proteins is determined by a combination of three parameters: sugar length, number and position of sulfates, and by the anomeric modification of the GAG. The relative contributions of these parameters have been quantified by recording affinity ratios for pairs of compounds on 10 proteins, thereby creating a systematic data set. With this, we have been able to elucidate structure–activity relationships and to analyze, how variations of these three parameters contribute to ligand affinity and selectivity. Sulfated oligohyaluronans consistently displayed high binding affinities in comparison to previously studied GAG-oligosaccharides such as heparin.7b,35 In most cases, stronger binding was observed with increased sulfation and elongation of the oligohyaluronans. While the effect of sulfation on selectivity was limited, there are few examples in which saccharide length affected selectivity. Anomeric modifications showed the strongest effects on ligand affinity and selectivity. Addition of large amphiphilic fluorophores to the anomeric center and the dimerization of the sHA ligands led to the highest binding affinities. Presumably, anomeric variations can address secondary binding sites, which differ from protein to protein.
Secondly, blind docking and extended MD simulations revealed that the binding of sulfated GAG is characterized by a hitherto unreported diversity of binding sites and modes for the studied proteins. The theoretical models provided insights in atomic detail on how defined structural variations introduced in our GAG can influence their recognition by proteins. In particular, the obtained molecular models showed that GAG binding affinity is not only increased by certain anomeric modifications through additional interactions with the target protein, but also by altering recognition toward additional protein sites. For instance, the anomeric substitution with the fluorophore TAMRA was predicted to redirect and enforce GAG binding with increased affinity toward a completely different recognition region on IL-8. Moreover, a bivalent sHA-compound constructed of two GAG connected by a PEG-linker was predicted to be able to recruit additional binding regions that could explained the striking increase in binding in comparison to the monovalent azide analogs found in our FP-experiments. The binding diversity predicted by our theoretical models could explain the high affinities determined by FP and ITC experiments and the differences to binding data obtained by FP-competition and NMR.
Last but not least, we found that GAG-binding is driven primarily by enthalpic contributions. Only the introduction of a flexible linker in a bivalent GAG ligand led to a significant loss of entropy upon binding. These findings correlate well with our modelling results also showing that the binding enthalpy and the free energy of binding rises proportionally with the number of binding interactions between GAG and the protein.
In summary, our insights into GAG–protein recognition suggest that defined chemical modifications of GAG can be exploited for the rational design of non-native GAG derivatives with customized functional properties and for the development of new biomaterials.36 Thus, the next generation of biomaterials designed for tissue regeneration should be able to scavenge or release regulatory proteins as required for the different stages of the healing process and, thus, could be tailored to the stage-specific needs of the patient.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work has been financially supported by the DFG German Research Council (SFB-TRR 67: A4, A7, A8). We thank Georg Künze and Anja Penk for protein production, and Yoandy Sánchez Najarro for technical support.Notes and references
- N. S. Gandhi and R. L. Mancera, Chem. Biol. Drug Des., 2008, 72, 455–482 CrossRef CAS.
- (a) R. Raman, V. Sasisekharan and R. Sasisekharan, Chem. Biol., 2005, 12, 267–277 CrossRef CAS; (b) S. Mizumoto, S. Yamada and K. Sugahara, Curr. Opin. Struct. Biol., 2015, 34, 35–42 CrossRef CAS; (c) J. D. Esko and R. J. Linhardt, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Cold Spring Harbor Laboratory Press The Consortium of Glycobiology Editors, La Jolla, California, Cold Spring Harbor, NY, 2009 Search PubMed.
- (a) H. Habuchi, O. Habuchi and K. Kimata, Glycoconjugate J., 2004, 21, 47–52 CrossRef CAS; (b) P. Martinez, A. Denys, M. Delos, A. S. Sikora, M. Carpentier, S. Julien, J. Pestel and F. Allain, Glycobiology, 2015, 25, 502–513 CrossRef CAS; (c) C. Zong, A. Venot, X. Li, W. Lu, W. Xiao, J. L. Wilkes, C. L. Salanga, T. M. Handel, L. Wang, M. A. Wolfert and G. J. Boons, J. Am. Chem. Soc., 2017, 139, 9534–9543 CrossRef CAS; (d) C. Zong, R. Huang, E. Condac, Y. Chiu, W. Xiao, X. Li, W. Lu, M. Ishihara, S. Wang, A. Ramiah, M. Stickney, P. Azadi, I. J. Amster, K. W. Moremen, L. Wang, J. S. Sharp and G. J. Boons, J. Am. Chem. Soc., 2016, 138, 13059–13067 CrossRef CAS; (e) C. I. Gama, S. E. Tully, N. Sotogaku, P. M. Clark, M. Rawat, N. Vaidehi, W. A. Goddard III, A. Nishi and L. C. Hsieh-Wilson, Nat. Chem. Biol., 2006, 2, 467–473 CrossRef CAS PubMed; (f) J. Angulo, R. Ojeda, J. L. de Paz, R. Lucas, P. M. Nieto, R. M. Lozano, M. Redondo-Horcajo, G. Gimenez-Gallego and M. Martin-Lomas, ChemBioChem, 2004, 5, 55–61 CrossRef CAS PubMed; (g) K. Raman, C. Mencio, U. R. Desai and B. Kuberan, Mol. Pharm., 2013, 10, 1442–1449 CrossRef CAS PubMed; (h) N. V. Sankarayanarayanan, T. R. Strebel, R. S. Boothello, K. Sheerin, A. Raghuraman, F. Sallas, P. D. Mosier, N. D. Watermeyer, S. Oscarson and U. R. Desai, Angew. Chem., Int. Ed., 2017, 56, 2312–2317 CrossRef CAS PubMed.
- (a) N. S. Gandhi and R. L. Mancera, Glycobiology, 2009, 19, 1103–1115 CrossRef CAS; (b) S. A. Samsonov, J. Teyra and M. T. Pisabarro, J. Comput.-Aided Mol. Des., 2011, 25, 477–489 CrossRef CAS; (c) A. Sarkar and U. R. Desai, PLoS One, 2015, 10, e0141127 CrossRef; (d) A. Sarkar, W. Yu, U. R. Desai, A. D. MacKerell and P. D. Mosier, Glycobiology, 2016, 26, 1041–1047 CrossRef CAS.
- (a) J. Salbach, T. D. Rachner, M. Rauner, U. Hempel, U. Anderegg, S. Franz, J. C. Simon and L. C. Hofbauer, J. Mol. Med., 2012, 90, 625–635 CrossRef; (b) P. Martin, Science, 1997, 276, 75–81 CrossRef CAS; (c) A. K. Picke, J. Salbach-Hirsch, V. Hintze, S. Rother, M. Rauner, C. Kascholke, S. Moller, R. Bernhardt, S. Rammelt, M. T. Pisabarro, G. Ruiz-Gomez, M. Schnabelrauch, M. Schulz-Siegmund, M. C. Hacker, D. Scharnweber, C. Hofbauer and L. C. Hofbauer, Biomaterials, 2016, 96, 11–23 CrossRef CAS; (d) G. Kuenze, S. Koehling, A. Vogel, J. Rademann and D. Huster, J. Biol. Chem., 2016, 291, 3100–3113 CrossRef CAS.
- (a) R. Elia, P. W. Fuegy, A. VanDelden, M. A. Firpo, G. D. Prestwich and R. A. Peattie, Biomaterials, 2010, 31, 4630–4638 CrossRef CAS; (b) J. Salbach, S. Kliemt, M. Rauner, T. D. Rachner, C. Goettsch, S. Kalkhof, M. von Bergen, S. Moeller, M. Schnabelrauch, V. Hintze, D. Scharnweber and L. C. Hofbauer, Biomaterials, 2012, 33, 8418–8429 CrossRef CAS; (c) R. Barbucci, A. Magnani, R. Rappuoli, S. Lamponi and M. Consumi, J. Inorg. Biochem., 2000, 79, 119–125 CrossRef CAS; (d) A. van der Smissen, V. Hintze, D. Scharnweber, S. Moeller, M. Schnabelrauch, A. Majok, J. C. Simon and U. Anderegg, Biomaterials, 2011, 32, 8938–8946 CrossRef CAS; (e) A. van der Smissen, S. Samsonov, V. Hintze, D. Scharnweber, S. Moeller, M. Schnabelrauch, M. T. Pisabarro and U. Anderegg, Acta Biomater., 2013, 9, 7775–7786 CrossRef CAS; (f) S. Kliemt, C. Lange, W. Otto, V. Hintze, S. Moller, M. von Bergen, U. Hempel and S. Kalkhof, J. Proteome Res., 2013, 12, 378–389 CrossRef CAS; (g) J. Salbach-Hirsch, N. Ziegler, S. Thiele, S. Moeller, M. Schnabelrauch, V. Hintze, D. Scharnweber, M. Rauner and L. C. Hofbauer, J. Cell. Biochem., 2014, 115, 1101–1111 CrossRef CAS; (h) M. C. Schulz, P. Korn, B. Stadlinger, U. Range, S. Moller, J. Becher, M. Schnabelrauch, R. Mai, D. Scharnweber, U. Eckelt and V. Hintze, J. Mater. Sci.: Mater. Med., 2014, 25, 247–258 CrossRef CAS.
- (a) V. Hintze, A. Miron, S. Moeller, M. Schnabelrauch, H. P. Wiesmann, H. Worch and D. Scharnweber, Acta Biomater., 2012, 8, 2144–2152 CrossRef CAS; (b) A. Pichert, S. A. Samsonov, S. Theisgen, L. Thomas, L. Baumann, J. Schiller, A. G. Beck-Sickinger, D. Huster and M. T. Pisabarro, Glycobiology, 2012, 22, 134–145 CrossRef CAS; (c) V. Hintze, S. A. Samsonov, M. Anselmi, S. Moeller, J. Becher, M. Schnabelrauch, D. Scharnweber and M. T. Pisabarro, Biomacromolecules, 2014, 15, 3083–3092 CrossRef CAS; (d) S. Koehling, G. Kuenze, K. Lemmnitzer, M. Bermudez, G. Wolber, J. Schiller, D. Huster and J. Rademann, Chem.–Eur. J., 2016, 22, 5563–5574 CrossRef CAS.
- J. P. Gehrcke and M. T. Pisabarro, J. Mol. Graphics Modell., 2015, 62, 97–104 CrossRef CAS PubMed.
- S. Faham, R. E. Hileman, J. R. Fromm, R. J. Linhardt and D. C. Rees, Science, 1996, 271, 1116–1120 CrossRef CAS.
- S. Koehling, M. P. Exner, S. Nojoumi, J. Schiller, N. Budisa and J. Rademann, Angew. Chem., Int. Ed., 2016, 55, 15510–15514 CrossRef CAS.
- (a) T. Isobe and T. Iskikawa, J. Org. Chem., 1999, 64, 6984–6988 CrossRef CAS; (b) T. Tanaka, H. Nagai, M. Noguchi, A. Kobayashi and S. Shoda, Chem. Commun., 2009, 3378–3379, 10.1039/b905761g.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (b) C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS.
- C. Walling and R. Rabinowitz, J. Am. Chem. Soc., 1957, 79, 5326 CrossRef CAS.
- P. J. Duggan and E. M. Tyndall, J. Chem. Soc., Perkin Trans. 1, 2002, 1325–1339, 10.1039/b006767i.
- A. Apffel, J. A. Chakel, S. Fischer, K. Lichtenwalter and W. S. Hancock, Anal. Chem., 1997, 69, 1320–1325 CrossRef CAS PubMed.
- C. G. Larsen, A. O. Anderson, E. Appella, J. J. Oppenheim and K. Matsushima, Science, 1989, 243, 1464–1466 CrossRef CAS.
- N. Panitz, S. Theisgen, S. A. Samsonov, J. P. Gehrcke, L. Baumann, K. Bellmann-Sickert, S. Koehling, M. T. Pisabarro, J. Rademann, D. Huster and A. G. Beck-Sickinger, Glycobiology, 2016, 26, 1209–1221 CAS.
- (a) A. Desmouliere, A. Geinoz, F. Gabbiani and G. Gabbiani, J. Cell Biol., 1993, 122, 103–111 CrossRef CAS; (b) A. B. Roberts, M. B. Sporn, R. K. Assoian, J. M. Smith, N. S. Roche, L. M. Wakefield, U. I. Heine, L. A. Liotta, V. Falanga and J. H. Kehrl, et al. , Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 4167–4171 CrossRef CAS.
- W. H. Burgess and T. Maciag, Annu. Rev. Biochem., 1989, 58, 575–606 CrossRef CAS.
- (a) B. Zhao, T. Katagiri, H. Toyoda, T. Takada, T. Yanai, T. Fukuda, U. I. Chung, T. Koike, K. Takaoka and R. Kamijo, J. Biol. Chem., 2006, 281, 23246–23253 CrossRef CAS; (b) A. H. Reddi, Nat. Biotechnol., 1998, 16, 247–252 CrossRef CAS.
- X. Li, Y. Zhang, H. Kang, W. Liu, P. Liu, J. Zhang, S. E. Harris and D. Wu, J. Biol. Chem., 2005, 280, 19883–19887 CrossRef CAS PubMed.
- W. H. Yu, S. Yu, Q. Meng, K. Brew and J. F. Woessner Jr, J. Biol. Chem., 2000, 275, 31226–31232 CrossRef CAS PubMed.
- S. T. Olson, I. Bjork, R. Sheffer, P. A. Craig, J. D. Shore and J. Choay, J. Biol. Chem., 1992, 267, 12528–12538 CAS.
- (a) Z. Nikolovska-Coleska, R. Wang, X. Fang, H. Pan, Y. Tomita, P. Li, P. P. Roller, K. Krajewski, N. G. Saito, J. A. Stuckey and S. Wang, Anal. Biochem., 2004, 332, 261–273 CrossRef CAS; (b) X. Huang, J. Biomol. Screening, 2003, 8, 34–38 CrossRef CAS.
- (a) S. Rother, S. A. Samsonov, T. Hofmann, J. Blaszkiewicz, S. Koehling, S. Moeller, M. Schnabelrauch, J. Rademann, S. Kalkhof, M. von Bergen, M. T. Pisabarro, D. Scharnweber and V. Hintze, Acta Biomater., 2016, 45, 143–154 CrossRef CAS; (b) S. Rother, S. A. Samsonov, U. Hempel, S. Vogel, S. Moeller, J. Blaszkiewicz, S. Koehling, M. Schnabelrauch, J. Rademann, M. T. Pisabarro, V. Hintze and D. Scharnweber, Biomacromolecules, 2016, 17, 3252–3261 CrossRef CAS PubMed.
- L. Fielding, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 51, 219–242 CrossRef CAS.
- H. Fernando, C. Chin, J. Rosgen and K. Rajarathnam, J. Biol. Chem., 2004, 279, 36175–36178 CrossRef CAS PubMed.
- M. Suflita, L. Fu, W. He, M. Koffas and R. J. Linhardt, Appl. Microbiol. Biotechnol., 2015, 99, 7465–7479 CrossRef CAS PubMed.
- J. Salbach-Hirsch, S. A. Samsonov, V. Hintze, C. Hofbauer, A. K. Picke, M. Rauner, J. P. Gehrcke, S. Moeller, M. Schnabelrauch, D. Scharnweber, M. T. Pisabarro and L. C. Hofbauer, Biomaterials, 2015, 67, 335–345 CrossRef CAS.
- S. Rother, S. A. Samsonov, S. Moeller, M. Schnabelrauch, J. Rademann, J. Blaszkiewicz, S. Koehling, J. Waltenberger, M. T. Pisabarro, D. Scharnweber and V. Hintze, ACS Appl. Mater. Interfaces, 2017, 9, 9539–9550 CrossRef CAS.
- G. S. Kuschert, A. J. Hoogewerf, A. E. Proudfoot, C. W. Chung, R. M. Cooke, R. E. Hubbard, T. N. Wells and P. N. Sanderson, Biochemistry, 1998, 37, 11193–11201 CrossRef CAS.
- (a) J. M. Wang, P. Morin, W. Wang and P. A. Kollman, J. Am. Chem. Soc., 2001, 123, 5221–5230 CrossRef CAS; (b) B. R. Miller III, T. D. McGee Jr, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef.
- K. Atkovska, S. A. Samsonov, M. Paszkowski-Rogacz and M. T. Pisabarro, Int. J. Mol. Sci., 2014, 15, 2622–2645 CrossRef.
- V. Veverka, A. J. Henry, P. M. Slocombe, A. Ventom, B. Mulloy, F. W. Muskett, M. Muzylak, K. Greenslade, A. Moore, L. Zhang, J. Gong, X. Qian, C. Paszty, R. J. Taylor, M. K. Robinson and M. D. Carr, J. Biol. Chem., 2009, 284, 10890–10900 CrossRef CAS.
- S. Maza, M. Mar Kayser, G. Macchione, J. Lopez-Prados, J. Angulo, J. L. de Paz and P. M. Nieto, Org. Biomol. Chem., 2013, 11, 3510–3525 RSC.
- (a) M. Calderón, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 22, 190–218 CrossRef PubMed; (b) N. Lohmann, L. Schirmer, P. Atallah, E. Wandel, R. A. Ferrer, C. Werner, J. C. Simon, S. Franz and U. Freudenberg, Sci. Transl. Med., 2017, 9, eaai9044 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03649g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |