
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceColloidal tectonics for tandem synergistic Pickering interfacial catalysis: oxidative cleavage of cyclohexene oxide into adipic acid†
Bingyu
Yang
a,
Loïc
Leclercq
 *a,
Véronique
Schmitt
b,
Marc
Pera-Titus
*c and
Véronique
Nardello-Rataj
*a
*a,
Véronique
Schmitt
b,
Marc
Pera-Titus
*c and
Véronique
Nardello-Rataj
*a
aUniv. Lille, CNRS, Centrale Lille, ENSCL, Univ. Artois, UMR 8181 - UCCS - Unité de Catalyse et Chimie du Solide, F-59000 Lille, France. E-mail: veronique.rataj@univ-lille1.fr; loic.leclercq@univ-lille.fr
bCentre de Recherche Paul Pascal, 115 Avenue du Dr Albert Schweitzer, F-33600 Pessac, France
cEco-Efficient Products and Processes Laboratory (E2P2L), Xin Zhuang Industrial Zone, UMI 3464 CNRS-Solvay, 3966 Jin Du Road, China. E-mail: marc.pera-titus-ext@solvay.com
First published on 15th October 2018
Abstract
Supramolecular preorganization and interfacial recognition can provide useful architectures for colloidal building. To this aim, a novel approach, based on colloidal tectonics involving two surface-active particles containing both recognition and catalytic sites, has been developed for controlling the formation and the properties of Pickering emulsions. This was illustrated by the combination of dodecyltrimethylammonium phosphotungstate nanoparticles, [C12]3[PW12O40], and silica particles functionalized with alkyl and sulfonic acid groups, [Cn/SO3H]@SiO2. The interfacial self-assembly occurs by the penetration of the alkyl chains of [Cn/SO3H]@SiO2 into the [C12]3[PW12O40] supramolecular porous structure constituted of polar and apolar regions. The emulsions were used as a non-nitric acid route for adipic acid synthesis from the one-pot oxidative cleavage of cyclohexene oxide with aqueous H2O2. The catalytic performance was significantly boosted due to the synergistic interactions between the particles.
Introduction
The recently developed “Pickering Interfacial Catalysis” (PIC) concept, which uses catalytic nanoparticles (NPs) able to stabilize emulsions, appears as a very promising strategy for overcoming diffusion limitations in liquid/liquid reactions.1 Indeed, PIC systems, in which the NPs behave concomitantly as emulsifiers and interfacial catalysts, show several advantages: (i) presence of immiscible interfacial catalytic NPs, (ii) increase of mass transfer between the phases due to an enhanced contact area, (iii) easy separation of the reaction products facilitating catalyst recovery, and (iv) upgrade of the eco-efficiency of the reaction system.1 The potential of PIC has already been demonstrated in oxidation,2 etherification,3 acetalization,4 and transesterification reactions.5PIC systems can be designed with “supramolecular hierarchical” [C12]3[PW12O40] NPs resulting from the ionic attraction between dodecyltrimethylammonium chains and phosphotungstate anions.2 These NPs exhibit an organized internal structure based on [PW12O40] parallel planes separated by interdigitated surfactant chains, generating porous architectures constituted of polar and apolar regions where organic molecules can be accommodated.2,6 In the presence of water and oil, they form very stable Pickering emulsions due to their bridging upon the release of some alkyl chains during oil penetration.6 More broadly, the surface interaction between molecules (tectons) can induce their self-assembly in reversible and predictable colloidal systems.7 This novel approach is here termed as “colloidal tectonics”.8 The formation of [C12]3[PW12O40] NPs and their further use as an emulsifier strongly suggest that colloidal tectonics can be applied to the bottom-up design of emulsions driven by the interfacial self-assembly of NPs.8 To ensure the predictability and stability of the emulsions, “programming” of NPs is essential for controlling recognition events and their iterations (Fig. 1).8
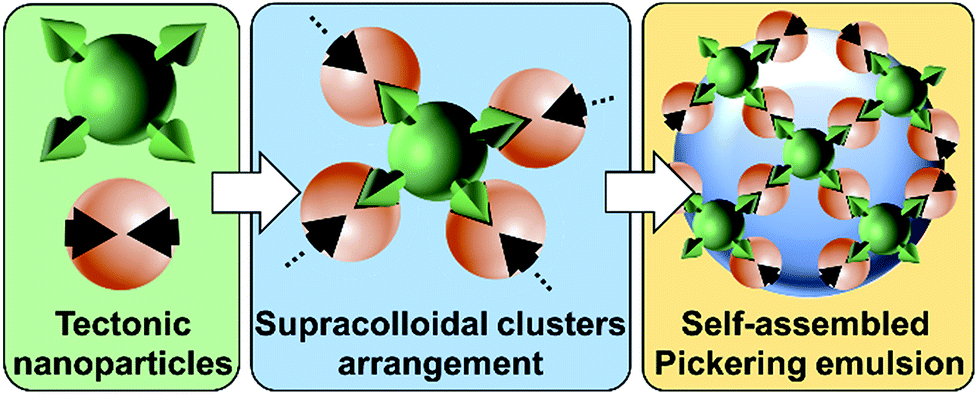 | ||
| Fig. 1 Design of Pickering emulsions driven by the self-assembly of receptors and connectors (tectonic nanoparticles). | ||
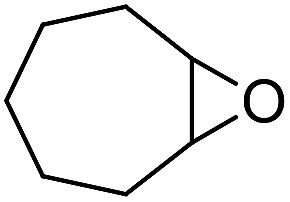
It is well known that NP mixtures can stabilize Pickering emulsions. Unfortunately, this seems to be circumscribed to NPs with opposite charge,9–13 and different polarity14–17 or size.18–20 Alternatively, the colloidal tectonics approach offers a more general platform for building emulsions from [C12]3[PW12O40] NPs. Indeed, their interaction with silica NPs grafted with alkyl and sulfonic acid groups ([Cn/SO3H]@SiO2, n = 3, 8 and 18) should drive the concomitant self-assembly of both NPs driven by partial penetration of the alkyl chains of [Cn/SO3H]@SiO2 into [C12]3[PW12O40] (Fig. 2). To illustrate the potential applications of this strategy, the self-assembled emulsions were evaluated for PIC tandem reactions being only attempted in few studies.21,22 Herein, we explored the synthesis of adipic acid (precursor of Nylon 6,6) from the one-pot oxidative cleavage of cyclohexene oxide with aqueous H2O2 as a non-nitric acid route.23 This eco-friendly synthesis requires the co-adsorption of [Cn/SO3H]@SiO2 and [C12]3[PW12O40] catalyzing the hydrolysis (i and v) and oxidation steps (ii–iv, see Fig. 2) of the overall reaction.24
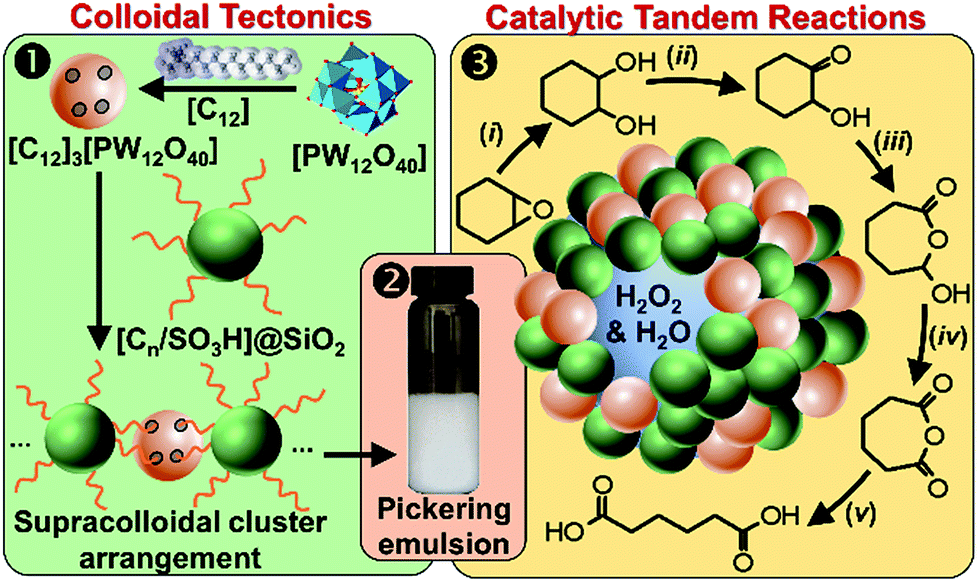 | ||
| Fig. 2 From molecular tectonics to colloidal engineering of catalytic Pickering emulsions for adipic acid synthesis. | ||
Results and discussion
The NPs were prepared using protocols developed in previous studies.2,3 The [C12]3[PW12O40] NPs exhibit a spherical and monodisperse shape (ϕ ≈ 35 nm, see ESI, Fig. S1†), negative ζ-potential (−36 mV) and a water contact angle >90° (Table 1).7 The [Cn/SO3H]@SiO2 NPs also exhibit a spherical and monodisperse shape (ϕ ≈ 12–15 nm, see ESI, Fig. S2–S5†), as well as a gradually negative ζ-potential from −9 to −42 mV with chain lengths from C3 to C18. This behavior is closely related to the grafting degree and the Cn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SO3H molar ratio.5 Increasing the chain length from C3 to C18 results in a lower grafting degree (from 58 to 32%). As all [Cn/SO3H]@SiO2NPs were prepared with the same nominal Cn:SO3H molar ratio (50/50), the Cn:SO3H molar ratio after grafting can be associated with the accessibility of the reactants to the [Cn/SO3H]@SiO2 surface, decreasing due to the steric hindrance and hydrophobicity of the grafted alkyl chains.5 In turn, both the grafting degree and the Cn:SO3H molar ratio are linked to the acidity and hydrophilicity of the modified NPs: [C18/SO3H] < [C8/SO3H] < [C3/SO3H]@SiO2. However, [C8/SO3H]@SiO2 and [C18/SO3H]@SiO2 exhibit a water contact angle >90°, reflecting poor wetting. The emulsifying capacity, defined as the ability of the NPs to emulsify various oils (n-heptane, toluene or di-n-butyl ether) in water, is more prominent for the hydrophobic [C18/SO3H]@SiO2 NPs, generating water-in-oil (W/O) emulsions with all the solvents. Likewise, [C8/SO3H]@SiO2 gave W/O emulsions with toluene, whereas the hydrophilic [C3/SO3H]@SiO2 did not stabilize any emulsion. Self-assembled [C12]3[PW12O40] NPs also stabilized W/O emulsions with all the solvents. As only toluene could generate emulsions with [C8/SO3H]@SiO2, [C18/SO3H]@SiO2 and [C12]3[PW12O40] NPs, we conducted a complete physicochemical investigation on the emulsifying capacity of the different NPs for water/toluene systems.
:SO3H molar ratio.5 Increasing the chain length from C3 to C18 results in a lower grafting degree (from 58 to 32%). As all [Cn/SO3H]@SiO2NPs were prepared with the same nominal Cn:SO3H molar ratio (50/50), the Cn:SO3H molar ratio after grafting can be associated with the accessibility of the reactants to the [Cn/SO3H]@SiO2 surface, decreasing due to the steric hindrance and hydrophobicity of the grafted alkyl chains.5 In turn, both the grafting degree and the Cn:SO3H molar ratio are linked to the acidity and hydrophilicity of the modified NPs: [C18/SO3H] < [C8/SO3H] < [C3/SO3H]@SiO2. However, [C8/SO3H]@SiO2 and [C18/SO3H]@SiO2 exhibit a water contact angle >90°, reflecting poor wetting. The emulsifying capacity, defined as the ability of the NPs to emulsify various oils (n-heptane, toluene or di-n-butyl ether) in water, is more prominent for the hydrophobic [C18/SO3H]@SiO2 NPs, generating water-in-oil (W/O) emulsions with all the solvents. Likewise, [C8/SO3H]@SiO2 gave W/O emulsions with toluene, whereas the hydrophilic [C3/SO3H]@SiO2 did not stabilize any emulsion. Self-assembled [C12]3[PW12O40] NPs also stabilized W/O emulsions with all the solvents. As only toluene could generate emulsions with [C8/SO3H]@SiO2, [C18/SO3H]@SiO2 and [C12]3[PW12O40] NPs, we conducted a complete physicochemical investigation on the emulsifying capacity of the different NPs for water/toluene systems.
| [C12]3[PW12O40] | [Cn/SO3H]@SiO2 | |||
|---|---|---|---|---|
| C3 | C8 | C18 | ||
| a Grafting degree. b Calculated from elemental and TG analyses. c Diameter of NPs based on TEM (S.D. = 15%). d Determined for 1 wt% NPs in water (S.D. = 5%). e Water contact angle measured on compressed NPs (S.D. = 2%). f Calculated from elemental analysis and pH-metric titration. | ||||
| DGa,b (%) | — | 58 | 39 | 32 |
| Cn:SO3H molar ratiob |
— | 64:36 |
73:27 |
82:18 |
| ϕ (nm) | 35 | 15 | 12 | 14 |
| ζ (mV) | −36 | −9 | −38 | −42 |
| θ (°) | 97 | 76 | 96 | 104 |
| Acidityf (mmol H+/g) | 0 | 1.45 | 0.70 | 0.38 |
The water/toluene emulsions (1/1 v/v) were prepared using [Cn/SO3H]/[C12]3[PW12O40] NP mixtures (n = 8 or 18, 3.6 wt%). The emulsions were visualized by optical microscopy to assess the evolution of the mean droplet diameter with the [Cn/SO3H]@SiO2 mass fraction, ϕ (Fig. 3). The mean droplet diameter decreases dramatically with ϕ until a plateau value around ϕ = 0.25, reflecting an attractive (synergistic) interaction between both NPs. As the mean droplet diameter of the NP mixtures is equal to or less than that of single [Cn/SO3H]@SiO2 NPs, the mixed interface formation seems to be governed by the silica NPs, being more “hydrophobic” than [C12]3[PW12O40] (Table 1). Unlike [C8/SO3H]@SiO2 and [C18/SO3H]@SiO2 for which stable W/O emulsions are obtained for all mass fractions, no emulsion was observed with the hydrophilic [C3/SO3H]@SiO2. These results are very surprising because of the negative ζ-potentials of [C12]3[PW12O40] and [Cn/SO3H]@SiO2 NPs (see ζ-potentials in Table 1). Therefore, the repulsion between NPs should inevitably decrease the driving force of the assembly. Besides, the combined NPs exhibit even weaker ζ-potentials compared to that of individual NPs. For instance, [C12]3[PW12O40] and [C18/SO3H]@SiO2 NPs (−36 and −42 mV, respectively) give only −17 mV at ϕ = 0.5 (measured for 1 wt% NPs in water). In parallel, the aqueous conductivity is also greatly affected: [C12]3[PW12O40] and [C18/SO3H]@SiO2 NPs (137 and 153 μS cm−1, respectively) give only 53 μS cm−1 at ϕ = 0.5. As the conductivity is related to the charge and size of the NPs, the formation of larger aggregates causes a decrease in the number of charge carriers and hence the conductivity decreases (i.e. there is self-assembly of [C12]3[PW12O40] and [C18/SO3H]@SiO2 NPs). It is clear that the stability of these aggregates results in the balance between attractive and repulsive forces. When two free negatively charged NPs approach each other, electrostatic repulsion becomes more significant. This repulsive energy can be overcome by van der Waals and depletion attractive forces between the largest [C12]3[PW12O40] and the smallest [C18/SO3H]@SiO2 NPs (35 and 14 nm, respectively), leading to the formation of mixed aggregates. As the interaction energy is mainly governed by van der Waals forces between the alkyl chains of both NPs, this explains the tendency of hydrophobic NPs to agglomerate more easily as long as alkyl chains favour van der Waals interactions. For instance, the self-aggregation of [C12]3[PW12O40] and [C18/SO3H]@SiO2 NPs observed by TEM is shown in Fig. 4. As depicted in Fig. 4, the NPs are arranged into interconnected NP networks where the larger [C12]3[PW12O40] NPs are surrounded by the smallest [C18/SO3H]@SiO2 NPs. This structure may also be regarded as [C18/SO3H]@SiO2 supramolecular host networks in which the introduction of [C12]3[PW12O40] NPs (guests) occurs with the formation of inclusion-type compounds. At this stage, to explain the Pickering stabilization by both NPs, we can suggest a mechanism based on specific interactions between the [C12]3[PW12O40] and [C18/SO3H]@SiO2 NPs leading to the formation of aggregates at the oil/water interface. This assumption is supported by the freeze-fracture cryo-SEM micrographs of the emulsion (Fig. 4).
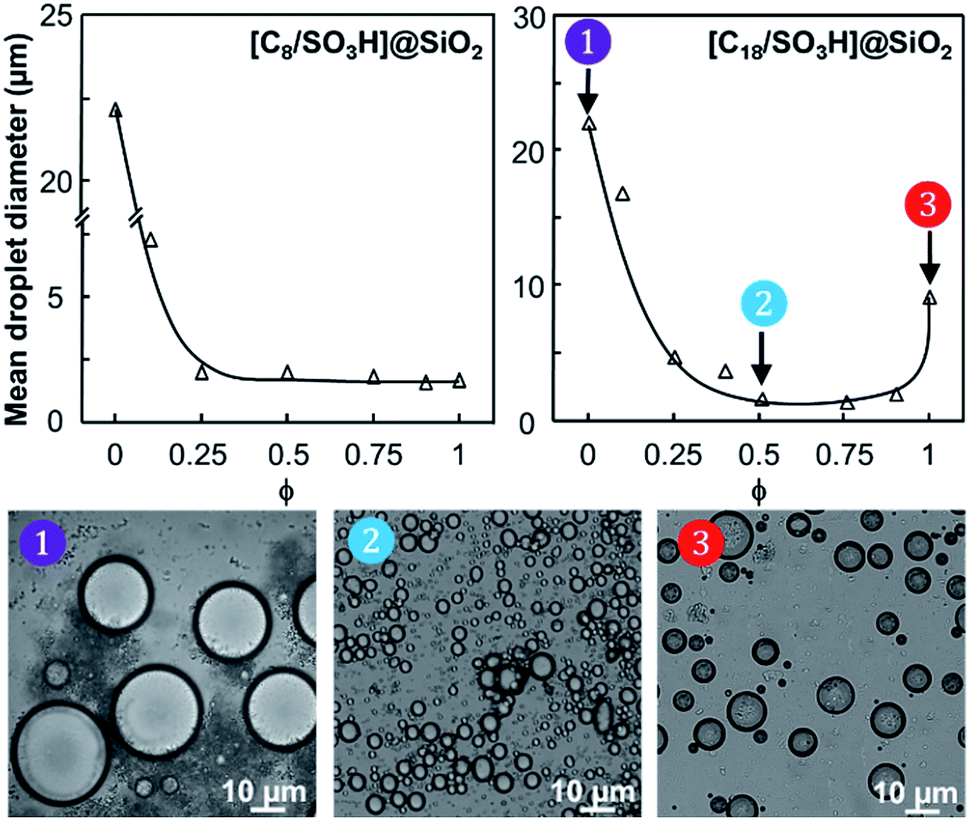 | ||
| Fig. 3 Evolution of the droplet size (S.D. = 10%) as a function of the [Cn/SO3H]@SiO2 mass fraction (ϕ) for water/toluene Pickering emulsions (1/1 v/v) stabilized by [Cn/SO3H]@SiO2 and [C12]3[PW12O40] NPs (3.6 wt%, 11500 rpm). The insets present the micrographs of the W/O emulsions. | ||
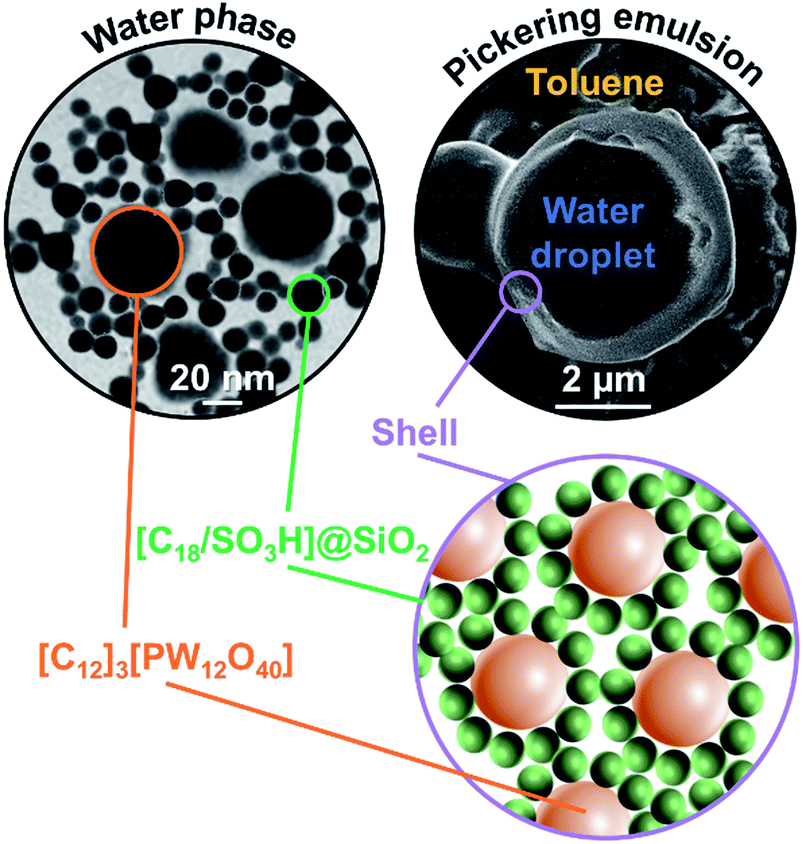 | ||
| Fig. 4 TEM micrograph of [C12]3[PW12O40] and [C18/SO3H]@SiO2 NP mixture (3.6 wt%, ϕ = 0.5) after dispersion in water (left) and cryo-SEM micrograph of water/toluene emulsions (1/1 v/v) of [C18/SO3H]@SiO2/[C12]3[PW12O40] mixtures (3.6 wt%, ϕ = 0.5, 11500 rpm, right). The inset shows a representation of the NPs in water and at the surface droplets. | ||
Next, the emulsion stability was examined as a function of storage time at 60 °C for the [Cn/SO3H]@SiO2 NPs either alone or combined with [C12]3[PW12O40]. As depicted in Fig. 5, the emulsion stability for the NPs alone increases in the order [C8/SO3H]@SiO2 < [C12]3[PW12O40] < [C18/SO3H]@SiO2.
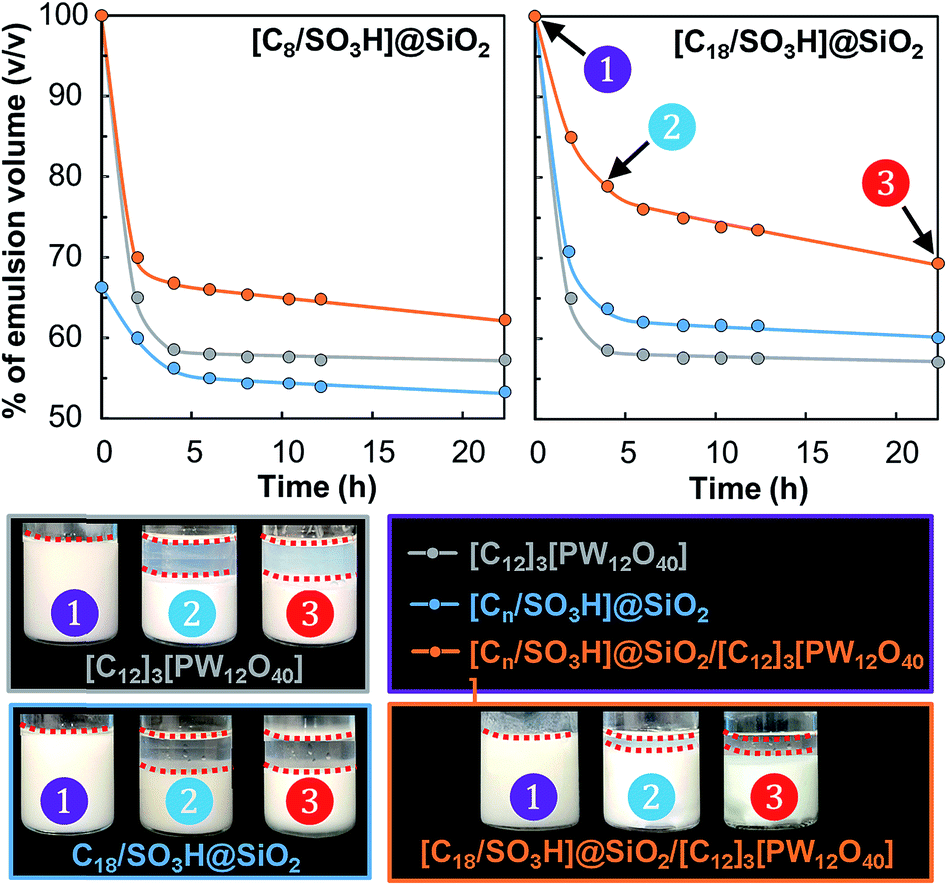 | ||
| Fig. 5 Evolution of the emulsion volume fraction (S.D. = 2%) as a function of the storage time at 60 °C for water/toluene emulsions (1/1 v/v) stabilized by [Cn/SO3H]@SiO2 and [C12]3[PW12O40] NPs or a mixture of both NPs (3.6 wt%, ϕ = 0.5, 11500 rpm). The insets show the W/O emulsions stabilized by [C18/SO3H]@SiO2 and/or [C12]3[PW12O40] NPs. | ||
In order to identify the possible destabilization mechanisms, we also performed a series of multiple light scattering experiments coupled with vertical scanning (Fig. 6).
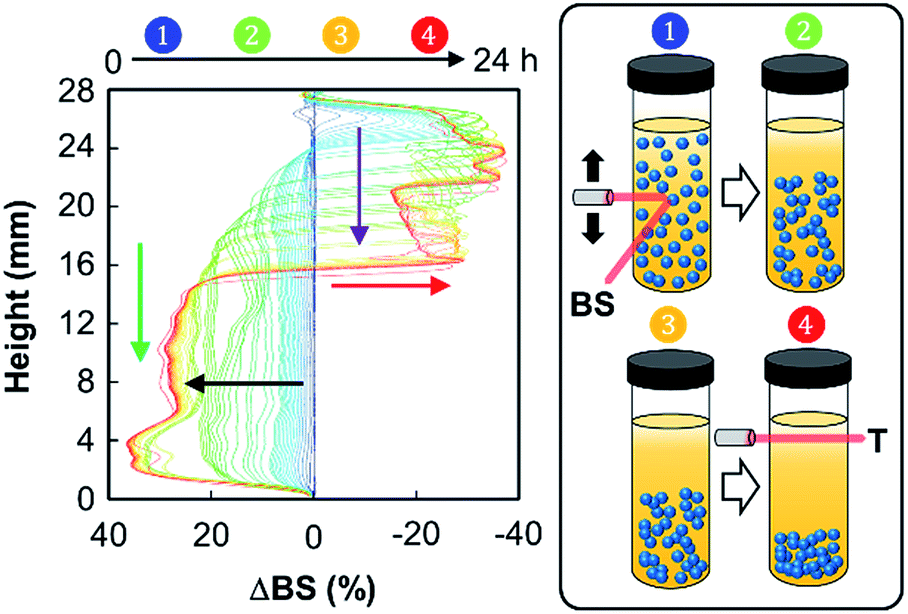 | ||
| Fig. 6 Variation of backscattering at 60 °C (ΔBS) versus sample height and time of [C18/SO3H]@SiO2/[C12]3[PW12O40] mixtures (3.6 wt%, ϕ = 0.5, 11500 rpm) from 0 (blue curve) to 24 h (red curve). The inset shows a representation of the time-evolution of the clarification/sedimentation process (T = transmitted light and BS = backscattered light) of the emulsion. | ||
The time-evolution of backscattering is characteristic of a sedimentation process, leading to the clarification of the continuous phase (toluene). Indeed, the backscattering level increases at the bottom due to a higher droplet concentration in this part of the sample, thus suggesting a sedimentation phenomenon (black arrow). In contrast, when looking at the top of the cell, we observe a decrease of the backscattering, which is due to the clarification (red arrow). Moreover, clarification and sedimentation fronts were clearly devised (purple and green arrows, respectively). This sedimentation process can be explained by the self-aggregation of both NPs. Indeed, van der Waals attraction forces, observed in aqueous solution, also operate between NPs located at the droplet surface, leading the droplets to get closer. As the gravitational force exceeds the thermal motion of the connected droplets (Brownian motion), a concentration gradient builds up in the system. As a result, larger droplets move rapidly to the bottom of the sample (their density is higher than that of the medium). At these conditions, the connected droplets form a close-packed array at the bottom of the system, with the remainder of the volume being occupied by the continuous phase. However, sedimentation does not favor ultimate coalescence because of the NP dense film located at the oil/water interface: the measured droplet size of the emulsion does not vary with time. As depicted in Fig. 5 and 6, droplet sedimentation occurs within the first hours after emulsification. Moreover, sedimentation stops more efficiently when hydrophobic NPs are used, reflecting stronger interactions between the NPs and the oil molecules than those between the NPs, leading to higher droplet stability. The resistance to sedimentation is higher for the mixed NP systems. This confirms the strong attractive interaction between both NPs due to the sponge-like structure of [C12]3[PW12O40] NPs, where some alkyl chains of [Cn/SO3H]@SiO2 can be accommodated.6 This behaviour is more distinct for [C18/SO3H]@SiO2NPs due to the large “hydrophobic” contact area between the alkyl chains after their penetration inside [C12]3[PW12O40] NPs.6 Therefore, these emulsions show stability during the reaction time and can be destabilized on demand by centrifugation (4000 rpm, 20 min). In addition, droplet sedimentation, which is not a destructive and irreversible process, can be readily avoided during the reaction by stirring at 500 rpm (see below).
The water/toluene emulsions stabilized by [C18/SO3H]@SiO2 and [C12]3[PW12O40] NPs were further inspected directly (without use of a replica) by cryo-SEM (Fig. 7). The freeze-fracture of the emulsions varies in appearance depending on the fracture plane and the structural heterogeneity of the droplets. As the frozen droplets are randomly located, freeze-fracture was performed in non-mid planes, resulting in a difficult measure of the droplet. However, cross-fractured droplets (where the plane of the fracture passes through the mid plane) reveal a size compatible with the mean droplet diameter measured by optical microscopy. It is noteworthy that [C12]3[PW12O40] alone can generate crystalline organizations in the interfacial layer (Fig. 7A and A′) driven solely by the self-assembly of [C12]3[PW12O40] NPs at long scale. However, for the single [C12]3[PW12O40] NPs, electrostatic forces and/or steric effects are not relevant due to the negative ζ-potential and monodisperse size distribution (Table 1). The self-assembly of [C12]3[PW12O40] NPs relies on recognition events, leading to a supracolloidal interface governed by hydrophobic forces. Indeed, toluene penetration inside [C12]3[PW12O40] NPs can lead to the release of alkyl chains, allowing their interlocking.6 In the case of [C18/SO3H]@SiO2 NPs, interlocking between NPs probably exists, but without encompassing a large or well-ordered organization. For NP mixtures, the presence of [C18/SO3H]@SiO2, even at a very low mass fraction (ϕ = 0.05), decreases the thickness of the NP layer. Therefore, the attractive interaction between both NPs is larger than the self-assembly of [C12]3[PW12O40] NPs in colloidal crystals.
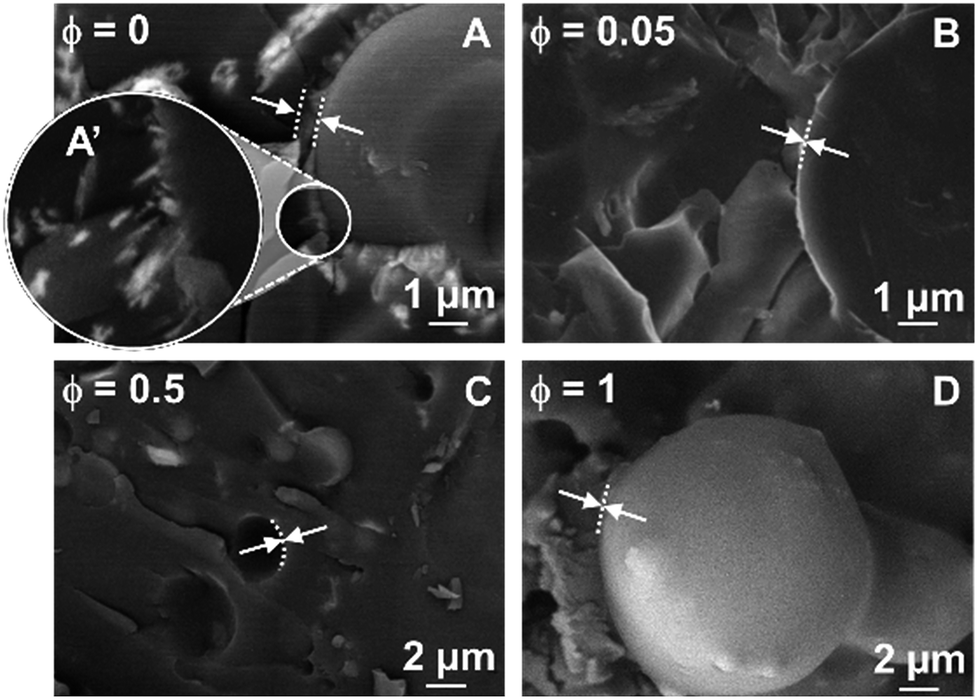 | ||
| Fig. 7 Cryo-SEM micrographs of water/toluene emulsions (1/1 v/v) as a function of the [C18/SO3H]@ SiO2 mass fraction (ϕ) for [C18/SO3H]@SiO2/[C12]3[PW12O40] mixtures (3.6 wt%, 11500 rpm). The white arrows and the dashed lines indicate the thickness of the NPs layer. | ||
With these results in hand, we explored the effect of catalytic properties of [C12]3[PW12O40] and [Cn/SO3H]@SiO2 NPs on the oxidative cleavage of cyclohexene oxide (Table 2). The tests were carried out at 80 °C with a 50/50 mass ratio of [C18/SO3H]@SiO2 and [C12]3[PW12O40] NPs. The diacid/diol ratio was used to assess whether the hydrolysis or oxidation steps are of significance in the mixed emulsions. To assess the influence of each catalyst on the reaction, a first series of experiments was performed either without (entry 1) or with the single NPs (entries 2 and 3). For the catalyst-free reaction, a nearly quantitative cyclohexene conversion (>99%) is observed after 12 h along with a very low adipic acid selectivity (6%) and a diacid/diol ratio of 0.1 (entry 1). These values are typical of uncatalyzed epoxide ring opening in aqueous solution.25 This observation points out that the overall reaction rate is clearly controlled by the rate of the oxidation steps. The above hypothesis is fully supported by a second control experiment using [C18/SO3H]@SiO2 as the catalyst (Table 2, entry 2). A 2-fold increase of the adipic acid selectivity is obtained under the same reaction conditions, which can be attributed to a faster hydrolysis of adipic anhydride into adipic acid. The diacid/diol ratio remains identical to the value measured without catalyst (i.e. 0.1), confirming that the oxidation reactions are not affected by the [C18/SO3H]@SiO2 catalyst. We next performed the oxidative cleavage of cyclohexene oxide with the single [C12]3[PW12O40] NPs (entry 3). The adipic acid selectivity after 12 h is 10-fold higher than without catalyst, while the diacid/diol ratio increases to 2.0. This clearly indicates that [C12]3[PW12O40] sitting at the W/O interface can lower the activation energy of the rate-limiting oxidation steps, resulting in a higher reaction rate. The combination of [C18/SO3H]@SiO2 and [C12]3[PW12O40] NPs acts as an efficient promoter for adipic acid production. Indeed, the selectivity after 12 h is almost 15 times higher than that for the control experiment without catalyst (Table 2, entries 1 and 4), resulting in an isolated yield of 94% and a diacid/diol ratio about 23. This high activity results not only from the microstructured reaction medium, but also from the synergy between both NPs. It is noteworthy that cyclohexene oxide was quantitatively converted into the diol after 30 min, this step being faster than the following oxidation steps (see ESI, Fig. S6†). To discern between mass transfer and intrinsic kinetics, the activation energy was determined from the Arrhenius plot, showing a value of 74.5 kJ mol−1 (see ESI, Fig. S7†), which is typical of diffusion-free tungsten-catalyzed oxidation reactions (range of 50–90 kJ mol−1).26–28 Consequently the reaction rate does not appear to be affected by mass transfer due to the large interfacial surface area generated, the reaction occurring in the interfacial layer. The recycling of the catalytic system was then investigated: the already used emulsified reaction medium was centrifuged (4000 rpm for 20 min). The mixed catalytic NPs were filtered, washed with ethanol (3 × 5 mL), and dried. A new batch of fresh cyclohexene oxide (1.5 mmol) in toluene (1.5 mL), and H2O2 (5.25 mmol) in water (1.5 mL) were added to the NPs. The reaction proceeds without any loss of activity (i.e. conversion) and selectivity after five consecutive recycles. Therefore, catalytic and amphiphilic NPs capable of stabilizing Pickering emulsions improve: (i) the performance of the catalytic systems due to the presence of NPs at the interface leading to a huge interfacial area and promoting the mass transfer, (ii) phase separation, and (iii) the ecological aspect of the process.
| n° | Oil | [X]3[PW12O40] | [Cn/SO3H]@SiO2 | Sb% | Diacid/diolc |
|---|---|---|---|---|---|
| a [Cn/SO3H]@SiO2 = 50 mg, [X]3[PW12O40] = 50 mg, toluene = 1.5 mL, water = 1.5 mL, cyclohexene oxide = 1.5 mmol, H2O2 = 5.25 mmol, 80 °C, 500 rpm, 12 h. Conv. of substrate >99%. b (Mol. of adipic acid)/(mol. of converted cyclohexene oxide) × 100. The side products were mainly glutaric acid, succinic acid, 2-hydroxycyclohexanone and 7-hydroxyoxepan-2-one. c Ratio of adipic acid to 1,2-cyclohexanediol. d [Na]3[PW12O40] = 15 μmol. e H2SO4 = 9.5 × 10−3 mmol. f [C18/SO3H]@SiO2 = 25 mg, [C12]3[PW12O40] = 25 mg. | |||||
| 1 | T | — | — | 6 | 0.1 |
| 2 | T | — | [C18/SO3H]@SiO2 | 12 | 0.1 |
| 3 | T | [C12]3[PW12O40] | — | 63 | 2.0 |
| 4 | T | [C12]3[PW12O40] | [C18/SO3H]@SiO2 | 94 | 23.3 |
| 5d | T | [Na]3[PW12O40] | [C18/SO3H]@SiO2 | 25 | 0.7 |
| 6e | T | [C12]3[PW12O40] | H2SO4 | 75 | 3.9 |
| 7f | T | [C12]3[PW12O40] | [C18/SO3H]@SiO2 | 48 | 1.2 |
| 8 | DBE | [C12]3[PW12O40] | [C18/SO3H]@SiO2 | 52 | 1.0 |
| 9 | H | [C12]3[PW12O40] | [C18/SO3H]@SiO2 | 19 | 0.3 |
| 10 | T | [C12]3[PW12O40] | [C8/SO3H]@SiO2 | 92 | 18.2 |
To provide further evidence of the synergy between the different catalytic NPs, a series of control experiments were performed under the same reaction conditions by replacing the [C18/SO3H]@SiO2 and [C12]3[PW12O40] NPs by [C18/SO3H]@SiO2 and the water-soluble [Na]3[PW12O40], or by [C12]3[PW12O40] NPs and homogeneous H2SO4 (entries 5 and 6). For both catalytic systems, the presence of a homogeneous catalyst ([Na]3[PW12O40] or H2SO4) instead of the two surface-active NPs does not affect the substrate conversion (>99%), but discourages diacid formation (compare entries 4, 5 and 6). These results clearly indicate that the presence of [C18/SO3H]@SiO2 and [C12]3[PW12O40] NPs at the W/O interface is essential to achieve high adipic acid selectivity. As both NPs interact to provide a nanostructured catalytic Pickering interface, in which the water/oil (W/O) interfacial area hugely favors mass transfer and catalyzes the various hydrolysis and oxidation steps (see Fig. 2), the overall reaction rate is enhanced, allowing the formation of the desired product (diacid) after 12 h. Next, the effect of the catalyst amount on the catalytic activity was investigated (entry 7). As the catalyst amount was divided by 2, an unstable emulsion was obtained. At these conditions, the surface-active NPs provide a high substrate conversion after 12 h, but the diacid selectivity is affected. All these observations support an interfacial catalytic mechanism, where the co-adsorption of both catalysts at the W/O interface is essential for producing adipic acid. As a consequence, there is a close relation between the structure of both NPs (i.e. the presence and spatial distance of the hydrophobic binding sites) and the catalytic performance of the resulting emulsions.
Additional oxidative cleavage experiments were performed using either n-heptane or di-n-butyl ether as solvents. Unlike toluene, the activity, selectivity and diacid/diol ratio decrease for both solvents (entries 8 and 9). We can reasonably assume that these linear solvents act as competitive inhibitors of the porous [C12]3[PW12O40] NPs. The binding of n-heptane and di-n-butyl ether with [C12]3[PW12O40] NPs is expected to reduce the [C18/SO3H]@SiO2/[C12]3[PW12O40] interaction. The results indicate that the most hydrophobic solvent (n-heptane) provides a stronger inhibitory effect due to easier penetration into the [C12]3[PW12O40] NPs in agreement with the eluotropic series.29 To gain more insight into the attractive interactions between the NPs, an experiment was performed for the [C8/SO3H]@SiO2/[C12]3[PW12O40] system (entry 10). Even if the conversion remains almost quantitative, the adipic acid selectivity is slightly lower than for the [C18/SO3H]@SiO2/[C12]3[PW12O40] system. Concomitantly, the diacid/diol ratio drops from 23 to 18 (entries 4 and 10). Again, we can reasonably argue that this effect is correlated with the chain length of the [Cn/SO3H]@SiO2NPs. Indeed, the strength of London forces is directly proportional to the “hydrophobic” contact area after penetration, increasing the cohesion of the catalytic interface.
On the basis of literature data, to obtain selectively adipic acid (>70%) from the oxidative cleavage of cyclohexene and/or its derivatives with W-based catalysts under mild reaction conditions (<100 °C and >600 rpm) and acceptable time (<24 h), several elegant approaches have been explored in the last two decades including organic acids,30 phase transfer catalysts,31–33 catalytic surfactants,34 catalytic ionic liquids35 and supported catalysts.36,37 Although all these systems solve the phase incompatibility issue, they suffer from one or more of the following drawbacks: (i) catalyst leaching, (ii) complex phase separation, especially in the presence of surfactants or organic acids, (iii) requirement of washing and neutralization of the reaction medium (systems using homogeneous acids), which generates waste water treatment problems, and/or (iv) sometimes there is a need for a co-solvent especially with supported catalysts (e.g., acetonitrile). All these issues can be solved here as the PIC system allows reaction completion (after 12 h) under mild conditions (80 °C and 500 rpm) without co-solvents. Products and catalysts are readily separated by simple centrifugation. Even if silica NPs require synthetic surface modification, the catalytic [Cn/SO3H]@SiO2/[C12]3[PW12O40] system is efficient and globally eco-friendly because it minimizes the use of organic solvents (or co-solvents) and can be easily re-used without waste generation.
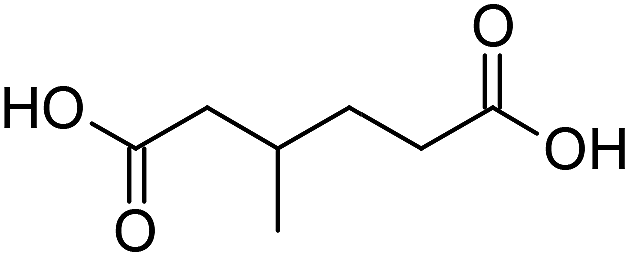
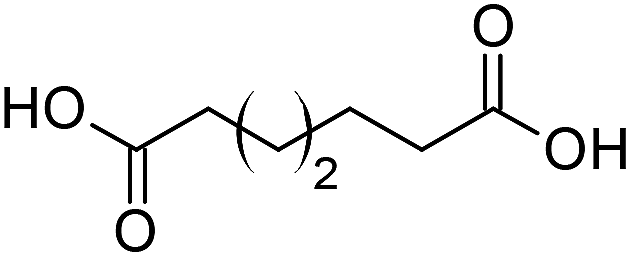
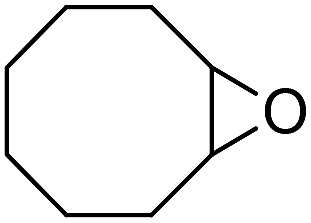
Finally, to extend the scope of the system, the oxidative cleavage of other cycloalkene oxides (i.e. cycloheptene, cyclooctene, 1-methylcyclohexene, and 4-methylcyclohexene) was carried out at the same reaction conditions (Table 3). To allow a proper comparison of the catalytic performance of the different substrates, the reaction time was set at 12 h. As depicted in Table 3, all cycloalkene oxides were quasi-quantitatively converted (conv. ≥99%) to their corresponding carboxylic acids with an isolated yield ≥69%. As the oxidation of alkene oxides to carboxylic acids occurs via the formation of diol intermediates, which constitute the major side product based on 1H NMR spectroscopic analysis of the crude reaction mixture, the overall reaction rate is clearly controlled by the rate of the oxidation steps (see Fig. 2). Consequently, the susceptibility of the cycloalkene oxides to oxidation steps is as follows: 1-methylcyclohexene < 4-methylcyclohexene < cyclooctene < cycloheptene < cyclohexene oxides. However, it is noteworthy that all cycloalkene oxides can be oxidized to their corresponding acids with a yield ≥90% after 24 h. Therefore, the combination of [C18/SO3H]@SiO2 and [C12]3[PW12O40] NPs acts as an efficient promoter for acid production at acceptable reaction times and facilitates the recycling of the catalytic system.
Conclusions
Pickering emulsions could be designed and stabilized by two catalytic amphiphilic NPs using the colloidal tectonics approach. The porous [C12]3[PW12O40] NPs allow the accommodation of C18 chains belonging to [C18/SO3H]@SiO2, resulting in their interlocking. In the presence of a W/O biphasic system, these attractive interactions act as elastic “springs” in the interfacial layer, conditioning the droplet size and the emulsion stability. At optimal conditions, such systems are efficient for the conversion of cycloalkene oxides into diacids combining the benefits of supramolecular chemistry (flexible, versatile and highly dynamic self-assembly systems) with homogeneous (high activity and selectivity) and heterogeneous catalysis (simple phase separation and catalyst reuse). This novel approach opens interesting and unprecedented perspectives and foreshadows the advent of predictable colloidal self-assembly methodologies for catalysis. Work is under way to extend these systems to other tandem reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by Solvay via a CIFRE grant. Cryo-SEM pictures have been taken at Placamat. Chevreul Institute (FR 2638), Ministère de l’Enseignement Supérieur et de la Recherche, Région Nord - Pas de Calais and Fonds Européen de Développement Régional (FEDER) are acknowledged for supporting and funding partially this work.References
- M. Pera-Titus, L. Leclercq, J.-M. Clacens, F. De Campo and V. Nardello-Rataj, Angew. Chem., Int. Ed., 2015, 54, 2006 CrossRef CAS PubMed.
- L. Leclercq, A. Mouret, A. Proust, V. Schmitt, P. Bauduin, J.-M. Aubry and V. Nardello-Rataj, Chem.–Eur. J., 2012, 18, 14352 CrossRef CAS PubMed.
- Z. Fan, A. Tay, M. Pera-Titus, W.-J. Zhou, S. Benhabbari, X. Feng, G. Malcouronne, L. Bonneviot, F. De Campo, L. Wang and J.-M. Clacens, J. Colloid Interface Sci., 2014, 427, 80 CrossRef CAS PubMed.
- Y. Xie, M. Sun, Y. Shen, H. Li, G. Lv, Z. Cai, C. Yang, G. A. A. Ali, F. Wang and X. Zhang, RSC Adv., 2016, 6, 101808 RSC.
- B. Yang, L. Leclercq, J.-M. Clacens and V. Nardello-Rataj, Green Chem., 2017, 19, 4552 RSC.
- L. Leclercq, A. Mouret, P. Bauduin and V. Nardello-Rataj, Langmuir, 2014, 30, 5386 CrossRef CAS PubMed.
- L. Leclercq, A. Mouret, S. Renaudineau, V. Schmitt, A. Proust and V. Nardello-Rataj, J. Phys. Chem. B, 2015, 119, 6326 CrossRef CAS PubMed.
- L. Leclercq, Front. Chem., 2018, 6, 168 CrossRef PubMed.
- H. Ma and L. L. Dai, Langmuir, 2011, 27, 508 CrossRef CAS PubMed.
- L. L. Dai, S. Tarimala, C.-Y. Wu, S. Guttula and J. Wu, Scanning, 2008, 30, 87 CrossRef CAS PubMed.
- T. Nallamilli, E. Mani and M. G. Basavaraj, Langmuir, 2014, 30, 9336 CrossRef CAS PubMed.
- T. R. Nallamilli, B. P. Binks, E. Mani and M. G. Basavaraj, Langmuir, 2015, 31, 11200 CrossRef CAS PubMed.
- S. D. C. Pushpam, M. G. Basavaraj and E. Mani, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2015, 92, 052314 CrossRef PubMed.
- S. Frasch-Melnik, I. N. Norton and F. Spyropoulos, J. Food Eng., 2010, 98, 437 CrossRef CAS.
- D. Miller, E.-M. Wiener, A. Turowski, C. Thunig and H. Hoffmann, Colloids Surf., A, 1999, 152, 155 CrossRef CAS.
- R. C. Pasquali, M. P. Taurozzi, N. Sacco and C. Bregni, Lat. Am. J. Pharm., 2008, 27, 839 CAS.
- F. C. Wang, F. Peyrone and A. G. Marangoni, Cryst. Growth Des., 2016, 16, 297 CrossRef CAS.
- S. Barg, B. P. Binks, H. Wang, D. Koch and G. Grathwohl, J. Porous Mater., 2012, 19, 859 CrossRef CAS.
- S. Wang, Y. He and Y. Zou, Particuology, 2010, 8, 390 CrossRef CAS.
- H. Xu, M. Lask, J. Kirkwood and G. Fuller, Langmuir, 2007, 23, 4837 CrossRef CAS PubMed.
- S. Crossley, J. Faria, M. Shen and D. E. Resasco, Science, 2010, 327, 68 CrossRef CAS PubMed.
- P. A. Zapata, J. Faria, M. P. Ruiz and D. E. Resasco, Top. Catal., 2012, 55, 38 CrossRef CAS.
- H. Ulrich, Raw Materials for Industrial Polymers, Hanser Publishers, Munich, 1988 Search PubMed.
- R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977 RSC.
- Z. Wang, Y.-T. Cui, Z.-B. Xu and J. Qu, J. Org. Chem., 2008, 73, 2270 CrossRef CAS PubMed.
- G. G. Allan and A. N. Neogi, J. Catal., 1976, 16, 197 CrossRef.
- X. Jian and A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 547 CrossRef CAS.
- K. Kamata, K. Yamaguchi and N. Mizuno, Chem.–Eur. J., 2004, 10, 4728 CrossRef CAS PubMed.
- S. H. Hansen, P. Helboe, U. Lund, Adsorption and partition chromatography in Separation methods, ed. Z.Deyl, Elsevier, Amsterdam, 1984, p. 169 Search PubMed.
- Y. Deng, Z. Ma, K. Wang and J. Chen, Green Chem., 1999, 1, 275 RSC.
- K. Sato, M. Aoki and R. Noyori, Science, 1998, 281, 1646 CrossRef CAS PubMed.
- I. Vural-Gürsel, Q. Wang, T. Noël, V. Hessel and J. T. Tinge, Ind. Eng. Chem. Res., 2013, 52, 7827 CrossRef.
- M. Shang, T. Noël, Y. I. Su and V. Hessel, Ind. Eng. Chem. Res., 2016, 55, 2669 CrossRef CAS.
- W. Zhu, H. Li, X. He, Q. Zhang, H. Shu and Y. Yan, Catal. Commun., 2008, 9, 551 CrossRef CAS.
- M. Vafaeezadeh and M. M. Hashemi, Chem. Eng. J., 2013, 221, 254 CrossRef CAS.
- M. Vafaeezadeh and M. M. Hashemi, Catal. Commun., 2014, 43, 169 CrossRef CAS.
- Z. Bohström, I. Rico-Lattes and K. Holmberg, Green Chem., 2010, 12, 1861 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data, procedure characterization methods and additional figures. See DOI: 10.1039/c8sc03345e |
| This journal is © The Royal Society of Chemistry 2019 |