
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA low-valent dinuclear ruthenium diazadiene complex catalyzes the oxidation of dihydrogen and reversible hydrogenation of quinones†
Xiuxiu
Yang
a,
Thomas L.
Gianetti
 *ab,
Michael D.
Wörle
a,
Nicolaas P.
van Leest
c,
Bas
de Bruin
c and
Hansjörg
Grützmacher
*a
*ab,
Michael D.
Wörle
a,
Nicolaas P.
van Leest
c,
Bas
de Bruin
c and
Hansjörg
Grützmacher
*a
aDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1, 8093 Zürich, Switzerland. E-mail: hgruetzmacher@ethz.ch
bDepartment of Chemistry and Biochemistry, The University of Arizona, Tucson, Arizona 85721, USA. E-mail: tgianetti@email.arizona.edu
cVan't Hoff Institute for Molecular Sciences (HIMS), University of Amsterdam (UvA), Science Park 904, 1098 XH Amsterdam, The Netherlands
First published on 15th November 2018
Abstract
The dinuclear ruthenium complex [Ru2H(μ-H)(Me2dad)(dbcot)2] contains a 1,4-dimethyl-diazabuta-1,3-diene (Me2dad) as a non-innocent bridging ligand between the metal centers to give a [Ru2(Me2dad)] core. In addition, each ruthenium is bound to one dibenzo[a,e]cyclooctatetraene (dbcot) ligand. This Ru dimer converts H2 to protons and electrons. It also catalyzes reversibly under mild conditions the selective hydrogenation of vitamins K2 and K3 to their corresponding hydroquinone equivalents without affecting the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds. Mechanistic studies suggest that the [Ru2(Me2dad)] moiety, like hydrogenases, reacts with H2 and releases electrons and protons stepwise.
C double bonds. Mechanistic studies suggest that the [Ru2(Me2dad)] moiety, like hydrogenases, reacts with H2 and releases electrons and protons stepwise.
Hydrogenase enzymes are the most efficient biological catalysts for the mutual interconversion of hydrogen to protons and electrons, H2 ⇆ H+ + H−.1–4 A detailed understanding of the mechanisms of these reactions is necessary for the development of efficient artificial catalytic systems for the use of H2 as a renewable energy source.5 Currently, there are three classes of hydrogenases known which either contain a bimetallic core, [Fe,Fe] or [Fe,Ni], or a single Fe center as active sites.3,4 Intensive spectroscopic investigations, including the determination of the structures of several hydrogenases by single-crystal X-ray diffraction methods,6–8 allowed extraction of the essential features needed for activity: (i) redox active metal centers; (ii) an electron reservoir; (iii) a cooperating ligand9–11 participating reversibly in the heterolytic cleavage/formation of H2; and (iv) a free coordination site for substrate binding (see a simplified sketch of the active [Fe,Fe] core at the top of Fig. 1).
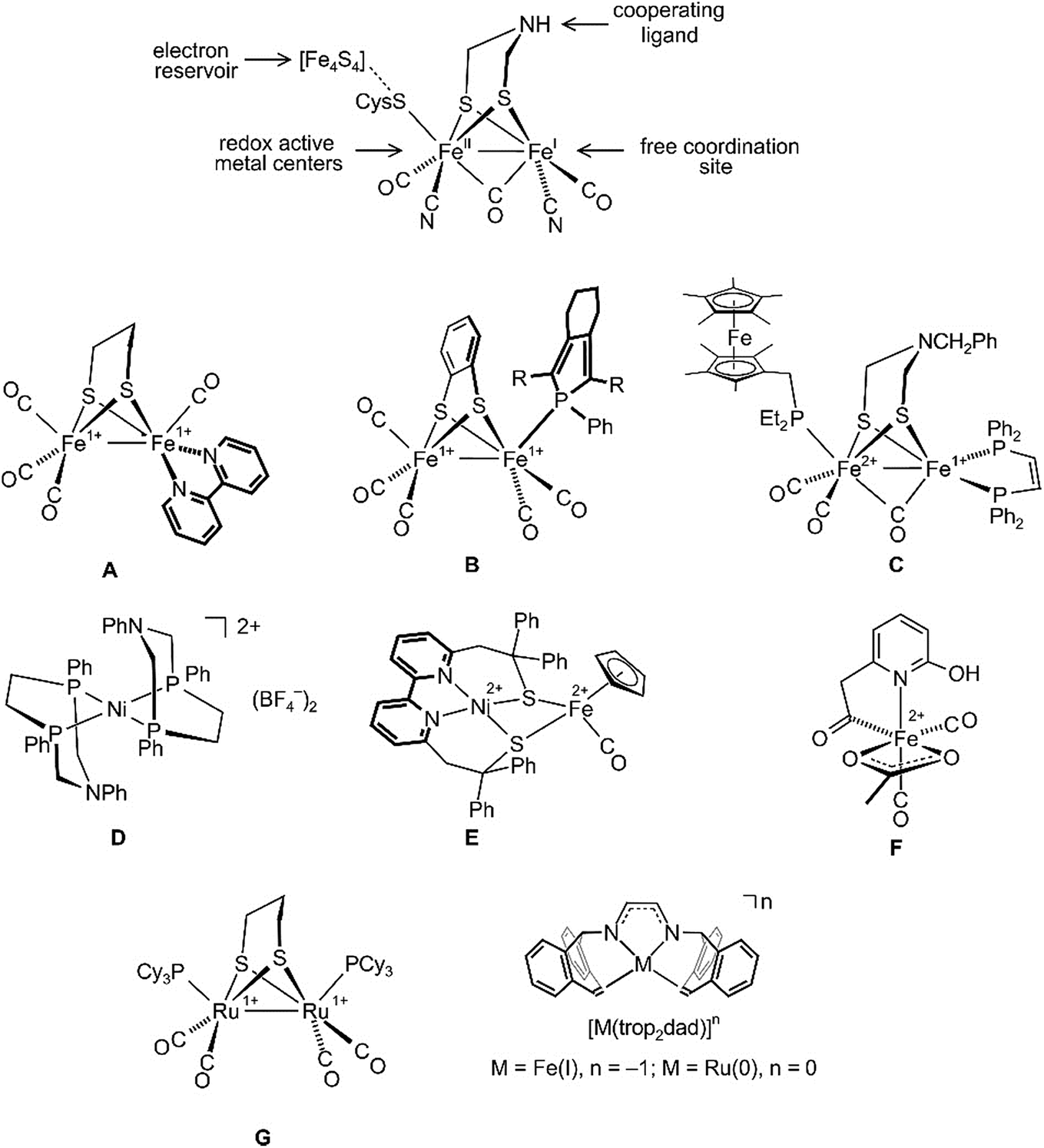 | ||
| Fig. 1 Structure of the [Fe,Fe] hydrogenase active site (top) and some hydrogenase mimics (A–G), as well as the illustration of diazadiene olefin complexes related to this work (bottom). | ||
All hydrogenases contain CO and some CN as archetypical σ-donor/π-acceptor ligands that keep iron in a low spin state and link these enzymes to classical organometallic chemistry. Consequently, the synthesis of hydrogenase model complexes is an intensively investigated topic of organometallic chemistry and some recent relevant examples A–G are shown in Fig. 1. Complexes A and B employ redox non-innocent ligands like bipyridine12 or phosphole13 in order to mimic Fe4S4 ferredoxines as ubiquitous electron reservoirs in enzymes.14,15 Both complexes, A and B, are active electrocatalysts for the production of H2 from acidic media. To date, the closest model to natural [Fe,Fe] hydrogenases is complex C, which can catalyze the oxidation of H2 to H+ in the presence of an oxidant and a base.16 The discovery that basic sites in a chelating diphosphane ligand greatly enhance the efficiency of heterolytic H2 splitting and the electrochemical oxidation of H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17,18 has led to the development of iron or nickel complexes like D as functional hydrogenase models which achieve truly impressive turnover frequencies (TOFs) of up to 100000 s−1 for the electrocatalytic production of H2.19–23 When tethered to conducting support materials, derivatives of D allow fabrication of membrane-electrode assemblies at which H2 is produced at a very low overpotential.18 The structural and functional model E for [Ni,Fe] hydrogenase was reported, which likewise generates H2 from mildly acidic solutions with high rates.24 The mononuclear iron model complex F in combination with an apoenzyme was used to prepare an [Fe] hydrogenase model.25 Remarkably, this semiartificial enzyme, like its natural counterpart, is able to reversibly hydrogenate methylene tetrahydromethanopterin. Due to the enhanced stability of Ru hydrides, the replacement of Fe with Ru in artificial enzymes has also been investigated.26–29 Several dinuclear ruthenium complexes were proposed as hydrogenase mimics.30–32 For example, Rauchfuss et al. achieved the photochemical addition of H2 across the Ru–Ru bond in complex G and moreover could demonstrate that the terminal hydride ligand in the resulting diruthenium dihydride complex is more easily protonated than the bridging hydride.30
17,18 has led to the development of iron or nickel complexes like D as functional hydrogenase models which achieve truly impressive turnover frequencies (TOFs) of up to 100000 s−1 for the electrocatalytic production of H2.19–23 When tethered to conducting support materials, derivatives of D allow fabrication of membrane-electrode assemblies at which H2 is produced at a very low overpotential.18 The structural and functional model E for [Ni,Fe] hydrogenase was reported, which likewise generates H2 from mildly acidic solutions with high rates.24 The mononuclear iron model complex F in combination with an apoenzyme was used to prepare an [Fe] hydrogenase model.25 Remarkably, this semiartificial enzyme, like its natural counterpart, is able to reversibly hydrogenate methylene tetrahydromethanopterin. Due to the enhanced stability of Ru hydrides, the replacement of Fe with Ru in artificial enzymes has also been investigated.26–29 Several dinuclear ruthenium complexes were proposed as hydrogenase mimics.30–32 For example, Rauchfuss et al. achieved the photochemical addition of H2 across the Ru–Ru bond in complex G and moreover could demonstrate that the terminal hydride ligand in the resulting diruthenium dihydride complex is more easily protonated than the bridging hydride.30
Recently, diazadiene olefin complexes [M(trop2dad)]n with low-valent iron or ruthenium centers became accessible (Fig. 1).33,34 In these complexes, the trop2dad ligand combines the well-established chemical and redox non-innocence of diazadienes (dads) and related ligands35–37 with the σ-donor/π-acceptor properties of olefins.30 Some of these low-valent metal complexes have remarkable properties. For example, [Ru0(trop2dad)] was found to be an efficient catalyst for the clean conversion of aqueous basic methanol or formaldehyde solutions into H2 and carbonate.34,38
Herein we report the synthesis of the dinuclear complex [Ru2(Me2dad)(dbcot)2] (3) which under 1 bar of H2 is converted to [Ru2H(μ-H)(Me2dad)(dbcot)2] (3(μ-H)H) as a fully artificial but functional [Fe,Fe] hydrogenase model (Fig. 2).39 This complex catalyzes the oxidation of hydrogen to protons and electrons as well as the reversible and selective hydrogenation of vitamin K3 (VK3) or vitamin K2 (VK2) forming dihydrovitamin K3 (VK3H2) or VK2 hydroquinone (VK2H2) without affecting the CC double bonds of these vitamins.
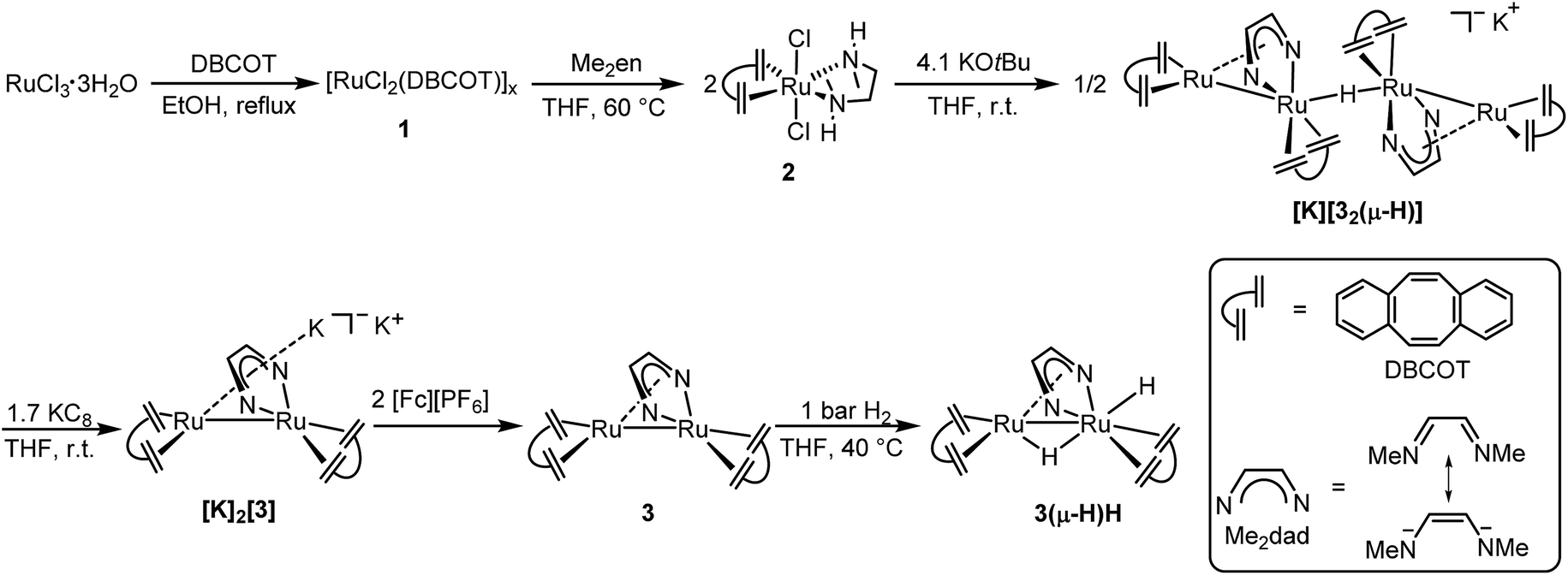 | ||
| Fig. 2 Preparation of [Ru2H(μ-H)(Me2dad)(dbcot)2] (3(μ-H)H). Illustration of the dbcot and Me2dad ligand, and six-step synthesis of binuclear complex 3(μ-H)H. | ||
Refluxing dbcot with ruthenium(III) chloride trihydrate in ethanol and THF quantitatively produces the brown coordination polymer ([RuCl2(dbcot)]x) 1, which reacts further with N,N′-dimethyldiaminoethane (Me2en) in THF and forms the mononuclear complex [RuCl2(Me2en)(dbcot)] 2 in 92% yield (Fig. 2). The deprotonation of 2 by 2.05 eq. of KOtBu produced a tetranuclear complex, [K][Ru4(μ-H)(Me2dad)2(dbcot)4] ([K][32(μ-H)]), in 40% yield as the major species (for detailed spectroscopic data of all isolated complexes reported in this work see the ESI†).30
Reducing the tetranuclear [K][32(μ-H)] with 3.4 equivalents of KC8 generates the dianionic dinuclear [K]2[Ru2(Me2dad)(dbcot)2] ([K]2[3]) in 85% yield. The 2:1 ratio of dbcot to Me2dad and lack of hydride signals in the 1H NMR spectrum suggests the presence of a Ru dimer complex with no hydrides. The low-frequency shift of the Me2dad and olefinic protons in [K]2[3] compared to [K][32(μ-H)] is consistent with a more electron rich complex (see ESI Table 2†). The oxidation of [K]2[3] with 2 eq. of [Fc][PF6] gives a neutral dinuclear complex Ru2(Me2dad)(dbcot)2 (3) in 77% yield. The 1H NMR spectra of 3 show sharp resonances at 0 °C, indicating the structure shown in Fig. 2, in which one Ru center is coordinated in a κ2N,N fashion, and the other in an η4 fashion by the bridging Me2dad ligand. But broad signals for the aromatic and olefinic protons are observed at room temperature, indicating molecular dynamics phenomena. Various NMR experiments show that two dynamic processes occur: (i) exchange of the κ2N,N/η4 coordination mode of the Me2dad ligand between the two Ru centers and (ii) rotation of the dbcot ligands (see ESI Fig. 3 for details†). Finally, exposure of 3 to 1 bar of H2 quickly forms a dinuclear dihydride complex, Ru2H(μ-H)(Me2dad)(dbcot)2 (3(μ-H)H), in quantitative yield. The two Ru–H resonances at −0.74 ppm and −2.74 ppm in THF-d8 show a coupling of 2JHH = 8.7 Hz, which suggests that both hydrides coordinate to the same Ru atom.40 The NOESY spectrum allows us to propose the structure of 3(μ-H)H as shown in Fig. 2 with a bridging hydride (δ = −0.74 ppm) and a terminal hydride (δ = −2.74 ppm)(see ESI Fig. 5†).
Single crystals of [K][32(μ-H)] and [K]2[3] were obtained from a THF/DME/hexane mixture in the presence of 18-crown-6. Crystals of 3(μ-H)H were grown by slow evaporation of a saturated benzene solution. All structures were investigated by X-ray diffraction methods and plots are shown in Fig. 3. [K][32(μ-H)] is a tetranuclear complex which contains two bimetallic ruthenium moieties bridged by a hydride.41 The complex [K]2[3] is best described as an ion pair [K]+{K[3]}− in which the {K[3]}− anion consists of a sandwich complex with a (dbcot)RuN2C2 unit as the central deck to which a Ru(dbcot) fragment binds in an η5-fashion to one side and a K(18-crown-6) fragment to the other side. The dinuclear dihydride 3(μ-H)H contains a bridging and a terminal hydride located on a plane of symmetry including also the two Ru centers, similar to the complex Ru2(S2C3H6)(μ-H)(H)(CO)3(PCy3)2G reported by Rauchfuss et al.30 Note that in the Ru2 complex fragments, the κ2N,N/η4 coordination mode of the diazadiene ligand centers is retained in all complexes. This structural motif is known for related dimeric [Ru2(CO)5(R2dad)] complexes.42
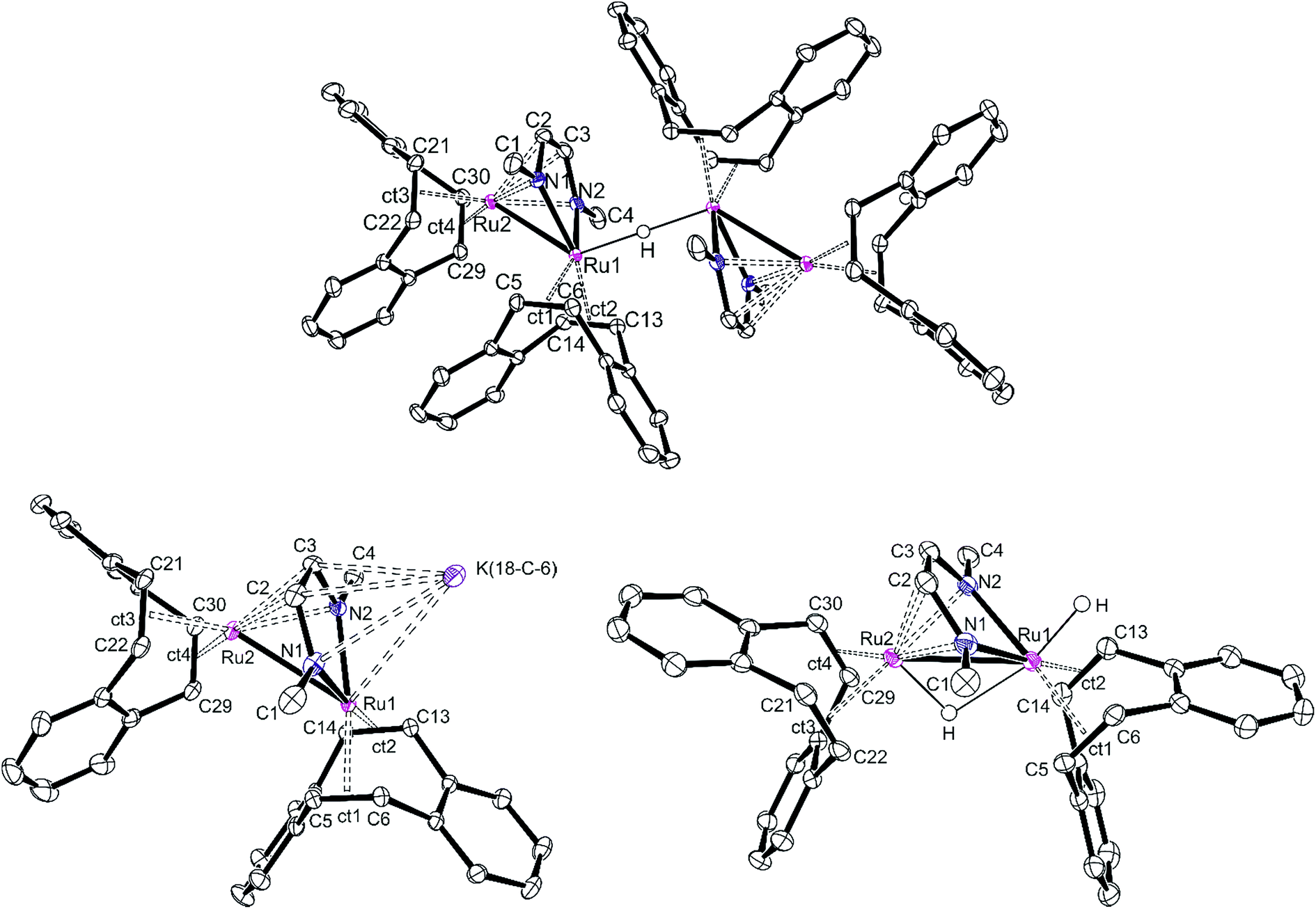 | ||
| Fig. 3 Structures (ORTEP plots) of [K][32(μ-H)] (top), [K]2[3] (bottom left) and 3(μ-H)H (bottom right). [K] represents a K+ cation either coordinated to 18-crown-6 or THF molecules. Thermal ellipsoids are drawn at the 50% probability level. Non-relevant hydrogen atoms, solvent molecules, crown ethers, and potassium cations, which are not part of the anionic Ru2 complex, have been removed for clarity. Ct1−4 are the centroids of the coordinated C5C6, C13C14, C21C22, and C19C20 bonds, respectively. Selected bond distances (Å). [K][32(μ-H)], Ru1–Ru2 2.6947(5), Ru1–Nav 2.107(2), Ru1–ctav 2.014(2), Ru2–Nav 2.173(2), Ru2–ctav 1.991(3), N1–C2 1.381(3), C2–C3 1.382(4), C3–N2 1.379(2), C–Colefin av 1.440(3); [K]2[3], Ru1–Ru2 2.8239(8), Ru1–Nav 2.066(2), Ru1–ctav 1.985(2), Ru2–N2 2.476(2), Ru2–N2 2.293(2), Ru2–ctav 1.970(3), N1–C2 1.399(4), C2–C3 1.406(4), C3–N2 1.390(3), C–Colefin av 1.452(4); 3(μ-H)H (average values from two molecules in one cell), Ru1–Ru2 2.7300(3), Ru1–Nav 2.093(2), Ru1–ctav 2.025, Ru2-Nav 2.180(2), Ru2–ctav 2.018, N1–C2 1.378(3), C2–C3 1.398(3), C3–N2 1.371(3), C–Colefin av 1.425(3). | ||
The Ru–Ru distances in [K][32(μ-H)], [K]2[3] and 3(μ-H)H are 2.6947(5), 2.8239(8) and 2.7300(3) Å, respectively, similar to the ones observed in the Ru(I) dimer complexes (2.632–2.937 Å),24,42,43 suggesting the presence of Ru–Ru bonds. The longer Ru–Ru distance in [K]2[3] (2.8239(8) Å) reflects the highly reduced state of this species. Also, the olefinic bonds in [K]2[3] (average of 1.450(3) Å) are longer than the ones in [K][32(μ-H)] (average of 1.433(3) Å) and 3(μ-H)H (average of 1.424(3) Å), indicating strong back donation from the Ru–Ru unit into the π*-orbitals of the coordinated CCdbcot bonds. This effect increases with increasing anionic charge of the complex (in free dbcot, the average olefin bond length is 1.321 Å). Note that the CCdbcot bond lengths coordinated to Ru2 are longer than those bound to Ru1, indicating a higher electron density at Ru2. Of special interest are the C–N and C–C bond lengths of the diazadiene ligand because they reflect the oxidation state of the ligand and consequently also of the metal. The neutral diimine form, RNCH–CHNR, binds to a low-valent metal center, Mn, and is characterized by short C–N bonds (≈1.29 Å) and a long C–C bond (≈1.46 Å). With increasing shift of electron density from the metal center to the ligand, the C–N bonds are lengthened while the C–C bond shortens: in the diazadiene radical anion [RNCH–CHNR]˙− coordinated to Mn+1, C–N ≈ 1.33 Å and C–C ≈ 1.39 Å; in the dianionic bisamido olefin form [RN–CHCH–NR]2− coordinated to Mn+2, C–N ≈ 1.38 Å and C–C ≈ 1.35 Å.36,44–46 The C–N and C–C bonds of the Me2dad in [K][32(μ-H)], [K]2[3] and 3(μ-H)H are approx. 1.38 Å and 1.39 Å, respectively, indicating a reduced form of the ligand. Consequently, the oxidation states at the Ru centers vary between 0 and +1. These data illustrate the redox non-innocent behavior of the Me2dad ligand in these complexes.36,46 The bridging coordination modes of 2e− reduced dad and the closely related pyridine-diimine ligands have been reported before.47,48
The ability to split H2 into protons and electrons like that of hydrogenases was investigated using the “Rauchfuss test”.16 In the presence of ten equivalents of PPh3 and [Fc][PF6], 3(μ-H)H catalytically splits hydrogen into protons and electrons under 1 bar H2 at 40 °C, forming protonated triphenylphosphane [Ph3PH][PF6], and Cp2Fe in THF within 1.5 hours (Fig. 4a). More compellingly, 3(μ-H)H is able to catalyze reversibly the hydrogenation of the biologically relevant vitamins VK3 and VK2 (Fig. 4b).49,50 Hydrogenation of VK3 was investigated under 1 bar or 15 bar H2 pressure, at 40 °C or 70 °C (ESI Table 3, entries 1–3†). With 1 bar H2, a TON of 252 was achieved. Remarkably, 3(μ-H)H remained active even after 2 months. At 70 °C and 15 bar H2, 0.13 mol% 3(μ-H)H converts 65% of VK3 to VK3H2 to give a TON of 220. Under 15 bar H2, the TOF values at 40 °C and 70 °C are 2.2 and 10 h−1, respectively. VK2 is a more delicate substrate because it tautomerizes.51 At room temperature, the hydrogenation of VK2 is more selectively achieved (ESI Table 3, entry 4†). In addition, the CC double bonds of VK2 remain intact and the hydrogenation occurs selectively at the quinone moiety of the substrate. These catalytic reactions can be reversed. The dehydrogenation of VK3H2 and VK2H2 was tested at 40 °C under Ar (ESI Table 3, entries 5–7†). The TON values of VK3H2 and VK2H2 are 70 (in 48 h) and 24 (in 40 h), respectively. The TOFs for hydrogenation and dehydrogenation reactions are comparable. A kinetic isotope effect of kH/kD = 1.9(2) was measured experimentally for the hydrogenation of VK3 by measuring kH and kD in separate experiments (ESI, part 7.1†).
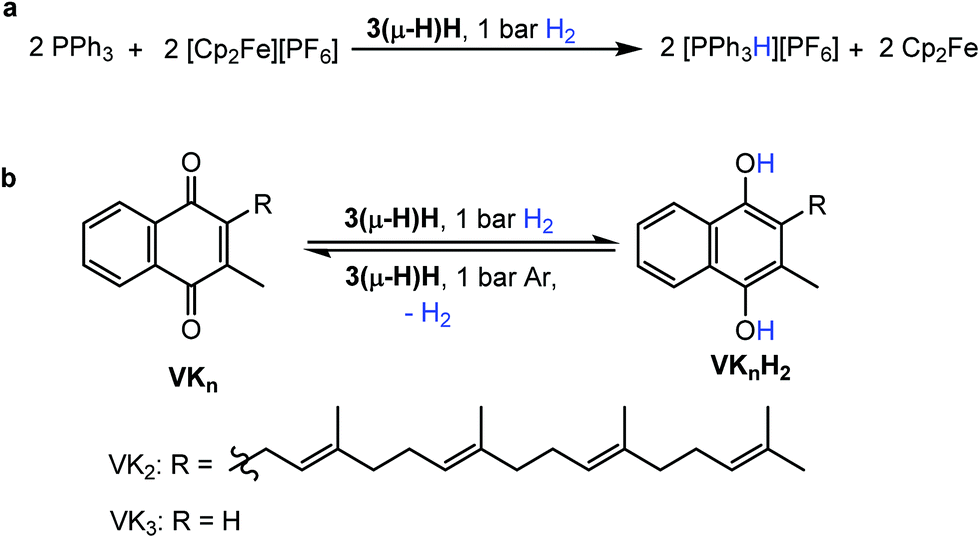 | ||
| Fig. 4 Reactivity of complex 3(μ-H)H. (a) Catalytic splitting of H2 by 3(μ-H)H using PPh3 and [Fc][PF6] as the proton and electron acceptors, respectively. (b) Hydrogenation of VK3 and VK2 to dihydrovitamin K3 (VK3H2) and VK2 hydroquinone (VK2H2) and the reverse dehydrogenations both promoted by 3(μ-H)H as the catalyst. | ||
Stoichiometric reactions were performed and monitored by NMR spectroscopy in order to gain some insights into possible reaction mechanisms. 3(μ-H)H can be rapidly and quantitatively oxidized at room temperature by ferrocenium salts, [Fc][X] (X = PF6 or OTf), to give the complex [Ru2(μ-H)(Me2dad)(dbcot)2]+[PF6]− or [Ru2(μ-H)(OTf)(Me2dad)(dbcot)2] with a bridging hydride (Fig. 5a) in good yield. The complex [Ru2(μ-H)(Me2dad)(dbcot)2][PF6] [3(μ-H)][PF6] (Ru–H, δ = −10.34 ppm) was characterized by NMR in THF-d8, while[Ru2(μ-H)(OTf)(Me2dad)(dbcot)2] [3(μ-H)(OTf)] was isolated in crystalline form. Characterization of this complex by single crystal X-ray diffraction reveals the presence of a structure closely related to 3(μ-H)H with a triflate anion bound to Ru1 instead of the terminal hydride (see Fig. 2 and ESI Fig. 2†). In THF solution, partial dissociation occurs to give [3(μ-H)][OTf] (Ru–H, δ = −10.34 ppm) and [3(μ-H)(OTf)] (Ru–H, δ = −10.18 ppm) in a 1:9 ratio. These results indicate that the radical cation salt [3(μ-H)H]˙+[X]− as a primary oxidation product rapidly loses hydrogen to give [3(μ-H)][X] (Fig. 6). The cyclic voltammogram of 3(μ-H)H shows only one irreversible oxidation peak in THF (ESI Fig. 14†). Our attempts to characterize [3(μ-H)H]˙+[X]− by EPR spectroscopy failed so far and oxidation of 3(μ-H)H with [Fc][OTf] or reduction of [3(μ-H)(OTf)] with Cp2Co at 20 K in MeTHF glass afforded only very weak EPR signals characteristic of (a mixture of) metal-centered radical species with g-values in the range of 2.6–1.8 and 2.5–1.7, respectively, which are unlikely to stem from any of the paramagnetic on-cycle catalytic intermediates (such as [3(μ-H)H]˙+ or [3(μ-H)]˙; vide infra). Note that complex [3(μ-H)(OTf)] is catalytically active in both reactions (a) and (b) shown in Fig. 4.
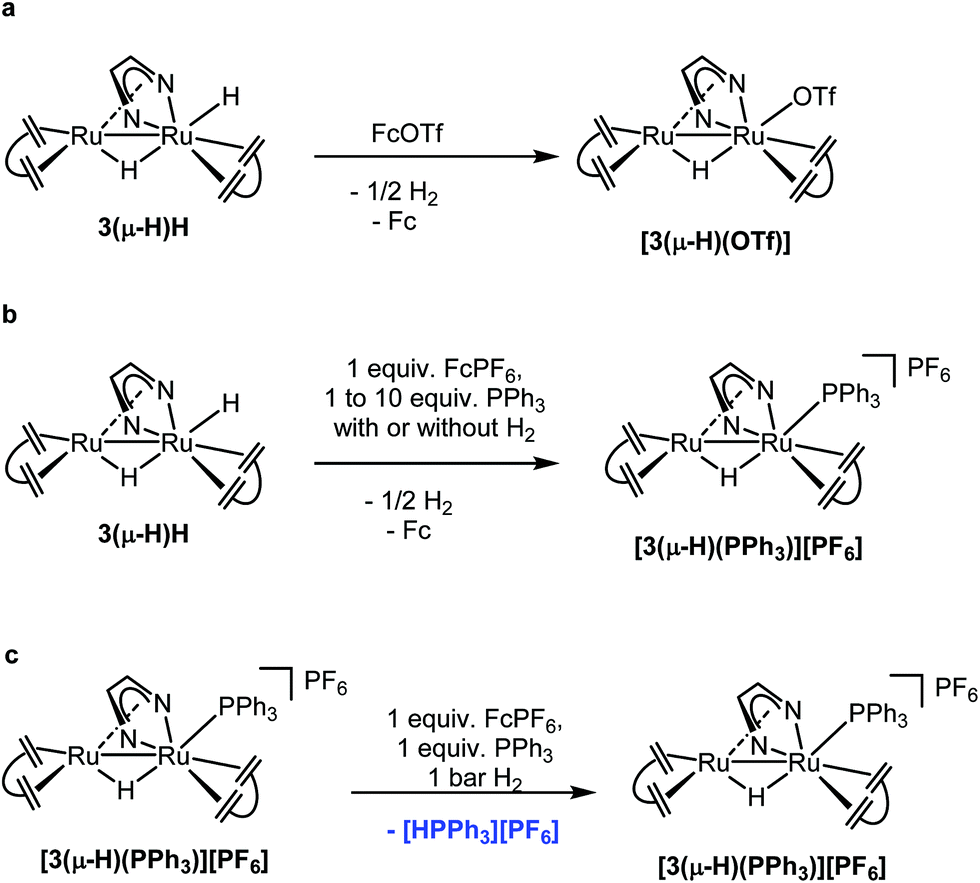 | ||
| Fig. 5 Stoichiometric reactions performed to support the proposed mechanisms. | ||
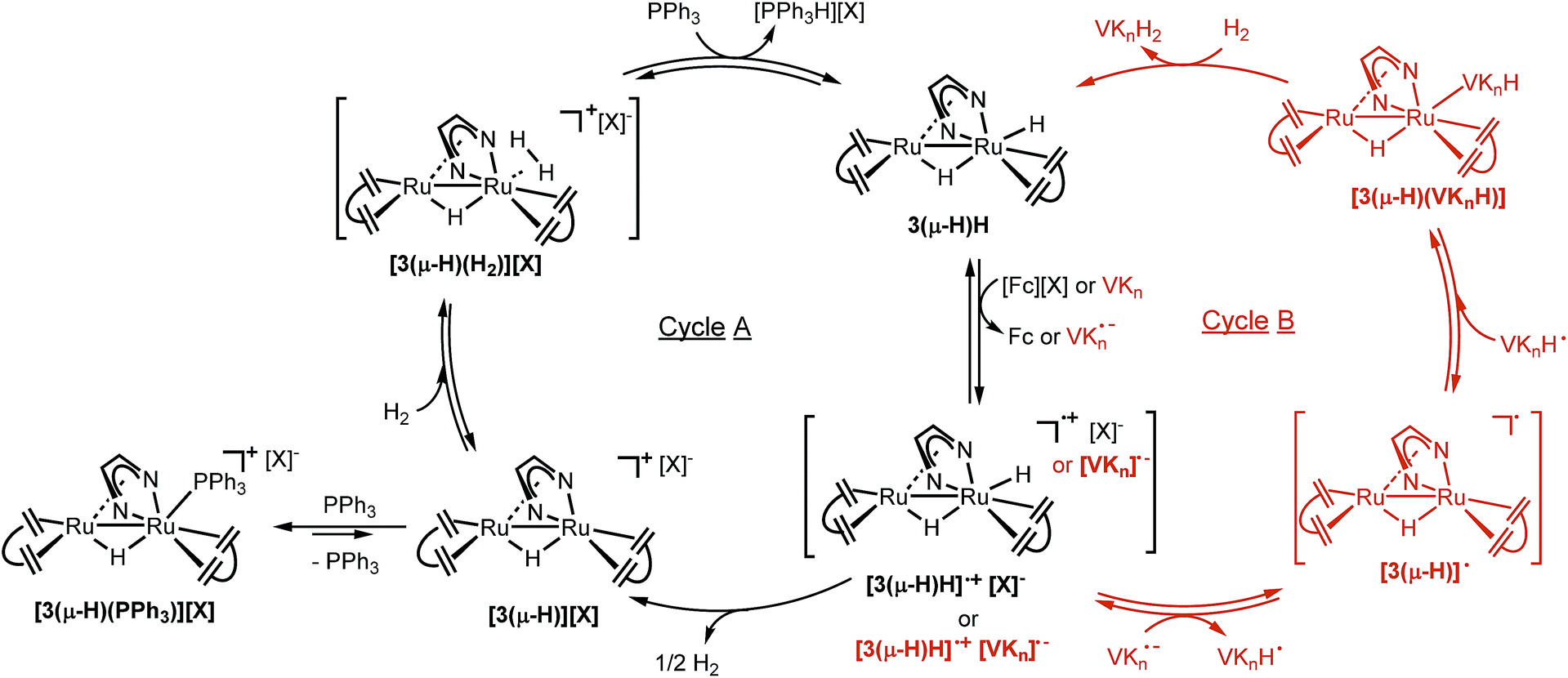 | ||
| Fig. 6 Proposed mechanisms of H2 splitting by 3(μ-H)H using PPh3 and [Fc][PF6] as the proton and electron acceptors (Cycle A, left), and hydrogenation of vitamin Kn (n = 2 or 3) to their hydroquinone analogs by 3(μ-H)H (Cycle B, right). | ||
In the next experiment, 3(μ-H)H was oxidized with one equivalent of [Fc][PF6] in the presence of one equivalent of PPh3. Exclusively, [Ru2(μ-H)(Me2dad)(dbcot)2(PPh3)][PF6], [3(μ-H)(PPh3)][PF6], 31P NMR: δ = 32.8 ppm, was obtained and no phosphonium salt, [Ph3PH][PF6], was formed (Fig. 5b and ESI, Fig. 6†). The compound [3(μ-H)(PPh3)]+ was also obtained from the reaction of [3(μ-H)(OTf)] and PPh3. Furthermore, the PPh3 ligand in [3(μ-H)(PPh3)]+ can be replaced by a stronger ligand such as P(OMe)3 (ESI, Fig. 7†). When 3(μ-H)H was exposed to 1 bar of D2, either at room temperature or under catalytic conditions, the complex [Ru2D(μ-H)(Me2dad)(dbcot)2] was observed (ESI, Fig. 8 and 9†), along with deuterium incorporation into the substrate, VK3D2, when present (ESI, Fig. 9†). In combination, these experiments show that (i) only the terminal hydride in 3(μ-H)H participates in the reactions, and that (ii) the ligand at the terminal site of Ru1 can be exchanged. When experiments were performed under 1 bar of H2 in the absence of an oxidant, no reaction took place and 3(μ-H)H remained intact. When 3(μ-H)H was reacted in the presence of a large excess of PPh3 (10 equivalents) but only 1 equivalent of [Fc][PF6] under 1 bar of H2, the cationic PPh3 complex [3(μ-H)(PPh3)]+ was observed but no [Ph3PH]+ (Fig. 5b and ESI, Fig. 10†). However, the formation of [PPh3H]+ from a mixture of [3(μ-H)(PPh3)]+, PPh3 and H2 was observed in the presence of 1 equiv. of Fc+ (Fig. 5c and ESI, Fig. 11†). These experiments suggest that two equivalents of ferrocenium are needed to observe turnover with 3(μ-H)H as the catalyst, and that the second equivalent is needed to rapidly convert 3(μ-H)H into [3(μ-H)]+ in order to prevent the reaction between the [Ph3PH]+ and the hydridic complex 3(μ-H)H, according to [Ph3PH+] + 3(μ-H)H → [3(μ-H)(PPh3)]+ + H2, which was experimentally found to be exergonic (see ESI Fig. 12†).
Taken together, these results support the proposed catalytic cycle A shown in Fig. 6. 3(μ-H)H is first oxidized with loss of one electron to give the radical cation [3(μ-H)H]˙+, which rapidly loses half an equivalent of H2 to yield [3(μ-H)]+. This complex with a labile coordination site at Ru1 is intercepted by PPh3 to give [3(μ-H)(PPh3)]+ which is the resting state within the catalytic cycle. The complex [3(μ-H)(PPh3)]+ is in equilibrium with [3(μ-H)]+ (likely as a solvated adduct [3(μ-H)(solv)]+ and present at low concentration), which may coordinate with H2 to give [3(μ-H)(H2)]+. Deprotonation of [3(μ-H)(H2)]+ by PPh3 (vide infra) gives the phosphonium salt [Ph3PH]+ and regenerates 3(μ-H)H. A second equivalent of oxidant is needed to turn over the catalytic cycle to give back [3(μ-H)(PPh3)]+ as the resting state via the reaction sequence given above.
A slightly different reaction path was observed for the reaction of 3(μ-H)H with quinones. The reaction between 3(μ-H)H and VK3, in the absence of H2, formed a thick suspension, which prevents further characterization by NMR spectroscopy. However, upon addition of a large excess of NaOTf, a signal attributed to complex [3(μ-H)(OTf)] was detected by 1H NMR spectroscopy. No reaction between NaOTf and 3(μ-H)H was observed in the absence of VK3. These data suggest that the terminal hydride of 3(μ-H)H was transferred to VK3, which then formed a protonated semiquinone oxygen bound species [3(μ-H) (VK3H)] (see the proposed structure based on DFT calculations in Fig. 8), and that VK3H− is labile enough to be partially displaced by OTf−. When followed by EPR spectroscopy at room temperature, the reaction between 3(μ-H)H and VK3 led to the formation of one organic radical species, which is detected by EPR spectroscopy at room temperature when 3(μ-H)H is reacted with VK3. The signal disappears with time resulting in the formation of the EPR silent and sparsely soluble [3(μ-H) (VK3H)] as the only product. With the symmetrical benzoquinone 2,5-di-tert-butyl-p-benzoquinone as the model substrate, in situ monitoring of the reaction with 3(μ-H)H afforded immediately the paramagnetic HSQ˙ (protonated semi-quinone) species (doublet, giso = 2.005, AHiso = 10.0 Hz; see ESI part 7.4†).
These observations support the catalytic cycle B shown in red in Fig. 6, in which the neutral semiquinone radical VKH˙ and the neutral organometallic Ru2 radical [3(μ-H)]˙ were formed. It is still unclear if these radical species are formed via direct hydrogen transfer from 3(μ-H)H to VKn, or via initial electron transfer to form the oxidized complex [3(μ-H)H]˙+ and the reduced quinone [VKn]˙−, followed by proton transfer to form [3(μ-H)]˙ and [VKnH]˙. Subsequent radical recombination with net reduction of the semiquinone radical forms the observed complex [3(μ-H)(VKnH)]. This complex is subsequently hydrogenated by H2 to give the product VKnH2 and 3(μ-H)H, which is the resting state in this cycle as observed during a single turnover experiment (ESI, Fig. 13†). In this mechanism, the observation of 3(μ-H)H as the only species present after a single turnover experiment is consistent with a less acidic product VKnH2 compared to [Ph3PH]+.
The activation steps of H2 by the Ru2(Me2dad) core, in both catalytic reactions, were investigated by DFT calculations without using structural simplification (ΔG values are given here, see Fig. 7 and ESI part 8†). In the absence of other substrates, H2 adds to [3(μ-H)]+ to give [3(μ-H)(H2)]+ in a weakly exergonic reaction. This complex can react with a basic substrate like PPh3 to give the products 3(μ-H)H and [Ph3PH]+ in an overall exergonic reaction (−11.3 kcal mol−1) although we did not succeed in finding any transition states for this bimolecular reaction. Alternatively, heterolytic cleavage across one Ru–N bond is energetically accessible viaTS-1 at 15.8 kcal mol−1 to give [3(μ-H)(H)2]a+. This complex can either rearrange into complex [3(μ-H)(H)2]b+ to give a hydrogenated CN bond of the Me2dad ligand or the acidic NH group is deprotonated to give the products 3(μ-H)H and [Ph3PH]+. Note that the direct heterolytic cleavage of H2 across a Ru C vector viaTS-2 and the hydrogenation of one of the CCdbcot bonds viaTS-3 are both highly unfavorable processes. With [3(μ-H)]+, PPh3 forms a stable complex, [3(μ-H)(PPh3)]+ (−22.3 kcal mol−1), which agrees with our assumption that this is the resting state in the catalytic splitting of H2 with PPh3 and [Fc][X]. The DFT calculations, for the reaction with VK3 (Fig. 8), confirm that VK3H− forms a stable complex, [3(μ-H)(VK3H)] (−38.9 kcal mol−1), in the reaction with [3(μ-H)]+. But in this case, the final products VK3H2 and 3(μ-H)H (−45.0 kcal mol−1) are more stable. That is, the adduct [3(μ-H)(VK3H)] is not the resting state in this cycle but 3(μ-H)H. The cleavage of H2 involves a classical sigma-bond metathesis transition state TS-4 at 32 kcal mol−1 in which hydrogen is activated across the oxygen atom of the coordinated VK3H and the ruthenium metal center. Subsequently, TS-4 collapses in a strongly exergonic reaction to give the products VK3H2 and 3(μ-H)H. This finding agrees with the measured kinetic isotope effect (1.89), which indicates that the cleavage of H2 is likely involved in the rate determining step. The activation of H2 may also occur across a Ru–N bond and involves the second Ru center situated far from the Ru-VK3H unit but this activation barrier is significantly higher (Ea = 42.5 kcal mol−1).
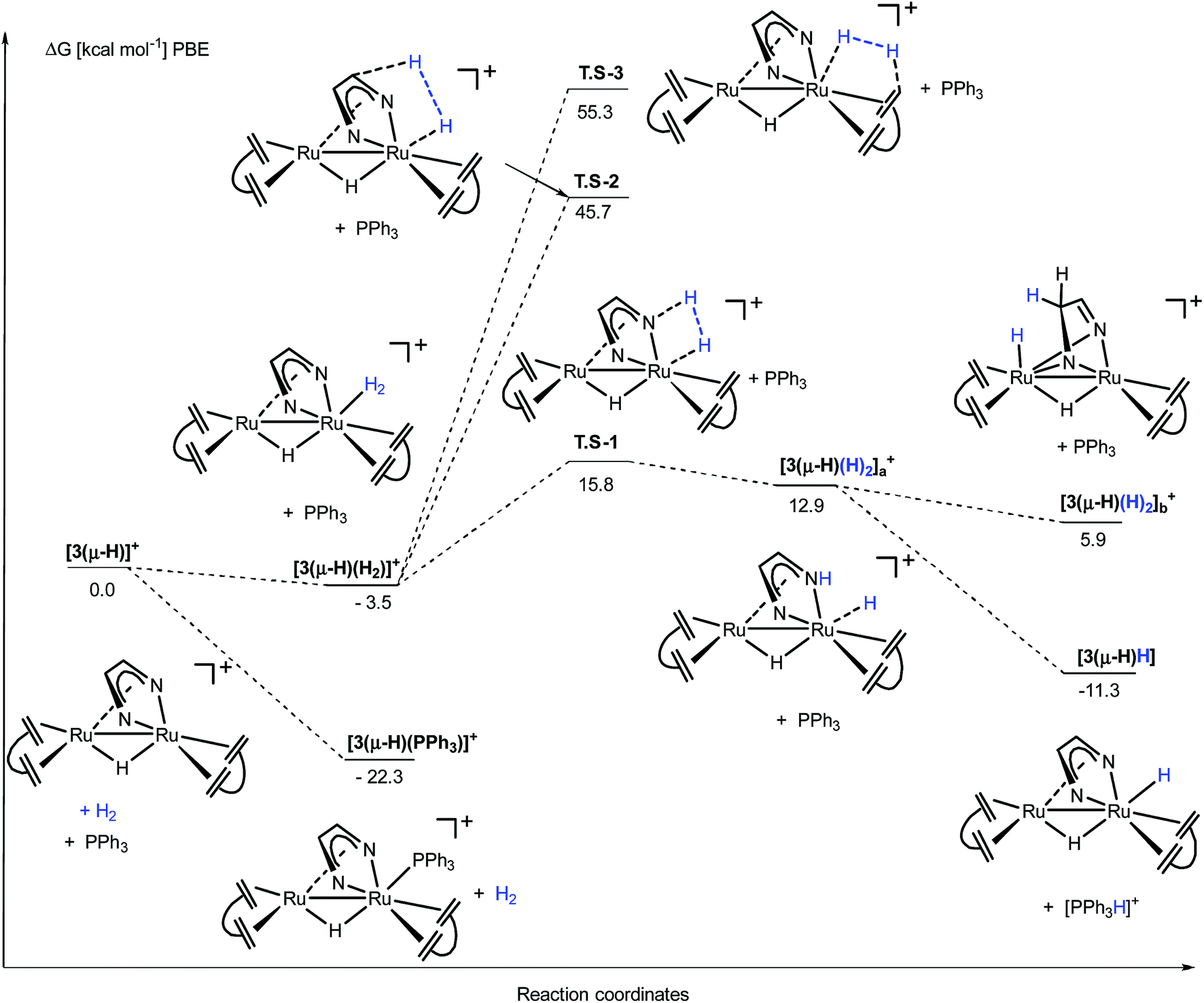 | ||
| Fig. 7 DFT calculation for the activation of H2 from [3(μ-H)]+ in the presence of PPh3. The transition state for the deprotonation of the H2 complex by PPh3 was not found; however, the direct reaction path from [3(μ-H)(H2)]+ (−3.5 kcal mol−1) to 3(μ-H)H + [Ph3PH]+ (−11.3 kcal mol−1) cannot be excluded. | ||
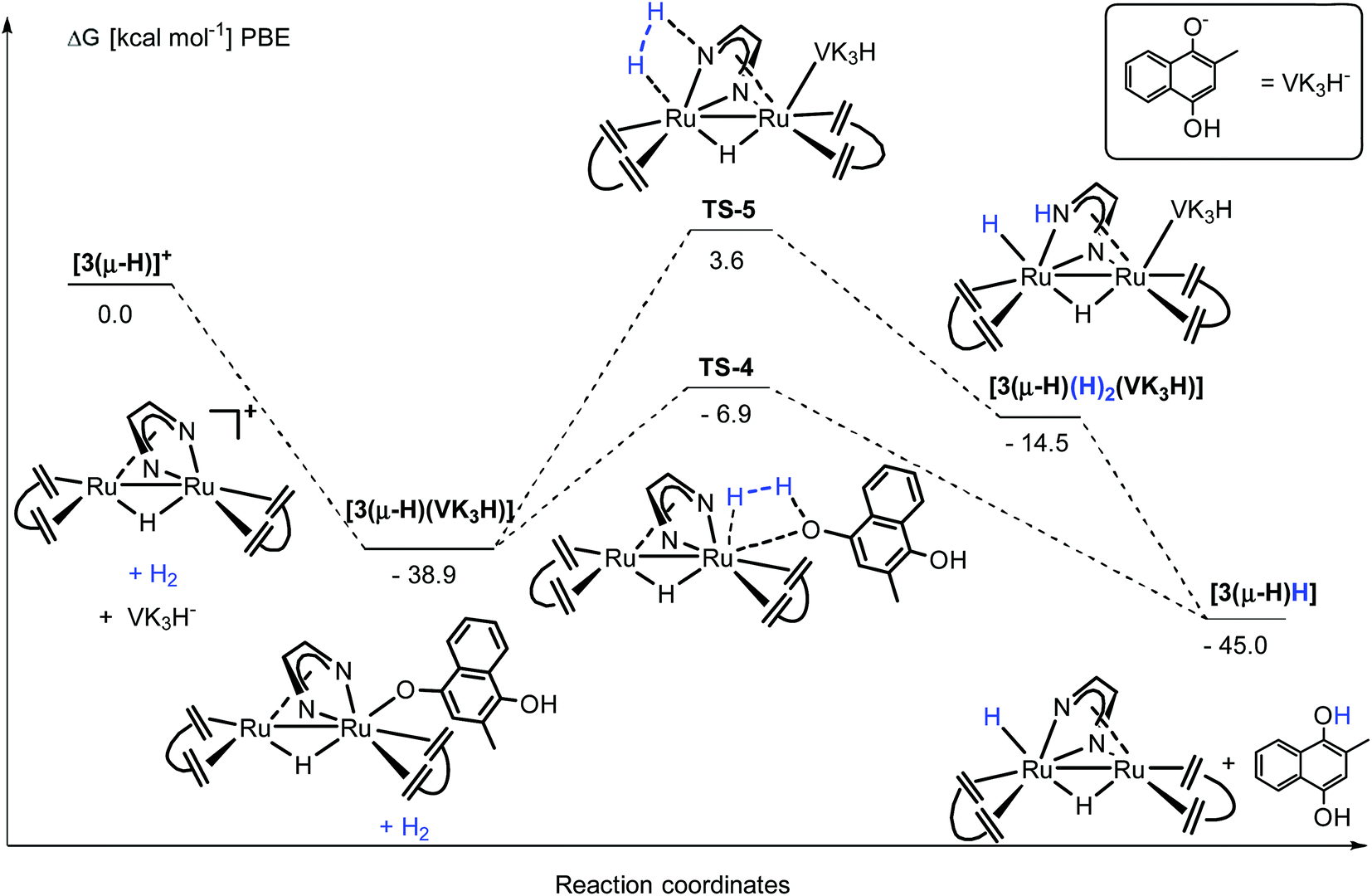 | ||
| Fig. 8 DFT calculation for the activation of H2 from [3(μ-H)]+ in the presence of VK3H−. | ||
Conclusions
The complex [Ru2H(μ-H)(Me2dad)(dbcot)2], 3(μ-H)H, can be regarded as a fully artificial mimic of binuclear [Fe,Fe] hydrogenases. The Ru2(Me2dad) core serves as an electron reservoir and [K]2[Ru2(Me2dad)(dbcot)2], [K]2[3], is the most reduced species and [Ru2(Me2dad)(dbcot)2], 3, is the most oxidized species. The Me2dad ligand binds in an N,N-κ2-fashion to one Ru center, resulting in a five-membered RuN2C2 heterocycle with a conjugated π-electron system. Formally, this RuN2C2 heterocycle coordinates to a second Ru center as a heteroatom analogue of archetypical cyclopentadienyl ligands, a structural feature known for dinuclear Ru2(dad) complexes. However, the Ru2(dad) complexes reported in the literature carry CO as additional ligands and are likely too unstable to serve as efficient catalysts. In this study, the very rigid and concave shaped dibenzocyclooctatriene, dbcot, was used as a neutral four electron π-donor and π*-acceptor ligand, which stabilizes all complexes. A Ru–Ru interaction of about 2.7 Å is observed in all complexes, structurally characterized by single crystal X-ray diffraction methods, which is remarkably invariant. Furthermore, the CCdbcot units bound to the Ru centers do not differ much in length (by 0.05 Å between [K]2[3] (longest) and [3(μ-H)][PF6] (shortest)), indicating that the electron densities at the Ru centers in the various complexes are rather similar. Small structural and electronic variances at the metal centers are also a feature of [Fe,Fe] and [Ni,Fe] hydrogenases.3,24,52–58 The complex 3(μ-H)H is a catalyst that splits H2 into protons and electrons in the presence of PPh3 and [Fc][PF6], forming [Ph3PH][PF6] and Cp2Fe (Rauchfuss test for hydrogenase activity), and reversibly and selectively hydrogenates vitamins VK3 or VK2, which are natural substrates for the enzyme hydrogen:quinone oxidoreductase. Spectroscopic data strongly suggest that like in hydrogenases, multiple coupled electron and proton transfer steps might be involved in these reactions. Clearly, the observed activities and efficiencies must be significantly improved. But this investigation demonstrates that redox and chemically non-innocent ligands may be key components and their variation may allow further improvements and uncovering of new bearings in synthesizing small molecular hydrogenase mimics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Reinhard O. Kissner and Jean-Pierre H. Oudsen for their kind help with EPR measurements. This work was supported by the Swiss National Science Foundation and the ETH Zürich. X. Y. is grateful for financial support from the China Scholarship Council. T. L. G. was supported by the ETH Zürich Postdoctoral Fellowship Program, co-funded by the ETH Zurich-Marie Curie action for people (FEL-14 15-1).Notes and references
- M. Senger, K. Laun, F. Wittkamp, J. Duan, M. Haumann, T. Happe, M. Winkler, U.-P. Apfel and S. T. Stripp, Angew. Chem., Int. Ed., 2017, 56, 16503–16506 CrossRef CAS PubMed.
- M. Winkler, M. Senger, J. Duan, J. Esselborn, F. Wittkamp, E. Hofmann, U.-P. Apfel, S. T. Stripp and T. Happe, Nat. Commun., 2017, 8, 16115 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rudiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed.
- R. Cammack, M. Frey and R. Robson, Hydrogen as a fuel: Learning from nature, CRC Press, London and New York, 2002 Search PubMed.
- A. Volbeda, M.-H. Charon, C. Piras, E. C. Hatchikian, M. Frey and J. C. Fontecilla-Camps, Nature, 1995, 373, 580–587 CrossRef CAS PubMed.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS PubMed.
- S. Shima, O. Pilak, S. Vogt, M. Schick, M. S. Stagni, W. Meyer-Klaucke, E. Warkentin, R. K. Thauer and U. Ermler, Science, 2008, 321, 572–575 CrossRef CAS PubMed.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273–4303 CrossRef CAS PubMed.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- M. Trincado and H. Grützmacher, Cooperative Catalysis: Designing Efficient Catalysts for Synthesis, Wiley-VCH Verlag GmbH & Co., Weinheim, 2015, ch. 3, pp. 67–105 Search PubMed.
- S. Roy, T. L. Groy and A. K. Jones, Dalton Trans., 2013, 42, 3843–3853 RSC.
- R. Becker, S. Amirjalayer, P. Li, S. Woutersen and J. N. H. Reek, Sci. Adv., 2016, 2, e1501014 CrossRef PubMed.
- R. Lill, Nature, 2009, 460, 831–838 CrossRef CAS PubMed.
- S. C. Lee, W. Lo and R. H. Holm, Chem. Rev., 2014, 114, 3579–3600 CrossRef CAS PubMed.
- J. M. Camara and T. B. Rauchfuss, Nat. Chem., 2012, 4, 26–30 CrossRef CAS PubMed.
- C. J. Curtis, A. Miedaner, R. Ciancanelli, W. W. Ellis, B. C. Noll, M. Rakowski DuBois and D. L. DuBois, Inorg. Chem., 2003, 42, 216–227 CrossRef CAS PubMed.
- A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Métayé, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384–1387 CrossRef CAS PubMed.
- T. B. Liu, D. L. DuBois and R. M. Bullock, Nat. Chem., 2013, 5, 228–233 CrossRef CAS PubMed.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
- S. E. Smith, J. Y. Yang, D. L. DuBois and R. M. Bullock, Angew. Chem., Int. Ed., 2012, 51, 3152–3155 CrossRef CAS PubMed.
- A. Dutta, D. L. DuBois, J. A. S. Roberts and W. J. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16286–16291 CrossRef CAS PubMed.
- N. Priyadarshani, A. Dutta, B. Ginovska, G. W. Buchko, M. O'Hagan, S. Raugei and W. J. Shaw, ACS Catal., 2016, 6, 6037–6049 CrossRef CAS.
- D. Brazzolotto, M. Gennari, N. Queyriaux, T. R. Simmons, J. Pecaut, S. Demeshko, F. Meyer, M. Orio, V. Artero and C. Duboc, Nat. Chem., 2016, 8, 1054–1060 CrossRef CAS PubMed.
- S. Shima, D. F. Chen, T. Xu, M. D. Wodrich, T. Fujishiro, K. M. Schultz, J. Kahnt, K. Ataka and X. L. Hu, Nat. Chem., 2015, 7, 995–1002 CrossRef CAS PubMed.
- T. Liu, M. R. DuBois, D. L. DuBois and R. M. Bullock, Energy Environ. Sci., 2014, 7, 3630–3639 RSC.
- G. Gezer, S. Verbeek, M. A. Siegler and E. Bouwman, Dalton Trans., 2017, 46, 13590–13596 RSC.
- T. Matsumoto, Y. Nakaya, N. Itakura and K. Tatsumi, J. Am. Chem. Soc., 2008, 130, 2458–2459 CrossRef CAS PubMed.
- G. M. Chambers, R. Angamuthu, D. L. Gray and T. B. Rauchfuss, Organometallics, 2013, 32, 6324–6329 CrossRef CAS.
- A. K. Justice, R. C. Linck, T. B. Rauchfuss and S. R. Wilson, J. Am. Chem. Soc., 2004, 126, 13214–13215 CrossRef CAS PubMed.
- C. Sommer, C. P. Richers, W. Lubitz, T. B. Rauchfuss and E. J. Reijerse, Angew. Chem., Int. Ed., 2018, 57, 5429–5432 CrossRef CAS PubMed.
- M. Yuki, K. Sakata, Y. Hirao, N. Nonoyama, K. Nakajima and Y. Nishibayashi, J. Am. Chem. Soc., 2015, 137, 4173–4182 CrossRef CAS PubMed.
- C. Lichtenberg, L. Viciu, M. Vogt, R. E. Rodriguez-Lugo, M. Adelhardt, J. Sutter, M. M. Khusniyarov, K. Meyer, B. de Bruin, E. Bill and H. Grutzmacher, Chem. Commun., 2015, 51, 13890–13893 RSC.
- R. E. Rodríguez-Lugo, M. Trincado, M. Vogt, F. Tewes, G. Santiso-Quinones and H. Grützmacher, Nat. Chem., 2013, 5, 342–347 CrossRef PubMed.
- F. Richard Keene, Coord. Chem. Rev., 1999, 187, 121–149 CrossRef.
- K. G. Caulton, Eur. J. Inorg. Chem., 2012, 435–443 CrossRef CAS.
- D. L. J. Broere, R. Plessius and J. I. van der Vlugt, Chem. Soc. Rev., 2015, 44, 6886–6915 RSC.
- M. Trincado, V. Sinha, R. E. Rodriguez-Lugo, B. Pribanic, B. de Bruin and H. Grützmacher, Nat. Commun., 2017, 8, 14990 CrossRef CAS PubMed.
- F. Wittkamp, M. Senger, S. T. Stripp and U. P. Apfel, Chem. Commun., 2018, 54, 5934–5942 RSC.
- D. Schott, C. J. Sleigh, J. P. Lowe, S. B. Duckett, R. J. Mawby and M. G. Partridge, Inorg. Chem., 2002, 41, 2960–2970 CrossRef CAS PubMed.
- D. A. Vicic, T. J. Anderson, J. A. Cowan and A. J. Schultz, J. Am. Chem. Soc., 2004, 126, 8132–8133 CrossRef CAS PubMed.
- K. Vrieze, J. Organomet. Chem., 1986, 300, 307–326 CrossRef CAS.
- E. A. Seddon and K. R. Seddon, The chemistry of ruthenium, Elsevier, 1984 Search PubMed.
- A. Mederos, S. Domínguez, R. Hernández-Molina, J. n. Sanchiz and F. Brito, Coord. Chem. Rev., 1999, 193–195, 913–939 CrossRef CAS.
- C. Mealli, A. Ienco, A. D. Phillips and A. Galindo, Eur. J. Inorg. Chem., 2007, 2007, 2556–2568 CrossRef.
- K. P. Butin, E. K. Beloglazkina and N. V. Zyk, Russ. Chem. Rev., 2005, 74, 531–553 CrossRef CAS.
- C. Tejel, M. A. Ciriano, M. P. del Río, F. J. van den Bruele, D. G. H. Hetterscheid, N. Tsichlis i Spithas and B. de Bruin, J. Am. Chem. Soc., 2008, 130, 5844–5845 CrossRef CAS PubMed.
- G. V. Koten and K. Vrieze, in Advances in Organometallic Chemistry, ed. F. G. A. Stone and R. West, Academic Press, 1982, vol. 21, pp. 151–239 Search PubMed.
- S. Jiang, T. Y. Zhang, X. Zhang, G. H. Zhang and B. Li, Dalton Trans., 2015, 44, 16708–16712 RSC.
- T. Matsumoto, H.-C. Chang, M. Wakizaka, S. Ueno, A. Kobayashi, A. Nakayama, T. Taketsugu and M. Kato, J. Am. Chem. Soc., 2013, 135, 8646–8654 CrossRef CAS PubMed.
- A. M. Swartz, M. Barra and D. Kuntz, J. Org. Chem., 2004, 69, 3198–3201 CrossRef CAS PubMed.
- Y. Higuchi, H. Ogata, K. Miki, N. Yasuoka and T. Yagi, Structure, 1999, 7, 549–556 CrossRef CAS PubMed.
- T. Krämer, M. Kampa, W. Lubitz, M. van Gastel and F. Neese, ChemBioChem, 2013, 14, 1898–1905 CrossRef PubMed.
- M.-E. Pandelia, H. Ogata and W. Lubitz, ChemPhysChem, 2010, 11, 1127–1140 CrossRef CAS PubMed.
- P. Amara, A. Volbeda, J. C. Fontecilla-Camps and M. J. Field, J. Am. Chem. Soc., 1999, 121, 4468–4477 CrossRef CAS.
- Y. Nicolet, A. L. de Lacey, X. Vernède, V. M. Fernandez, E. C. Hatchikian and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 2001, 123, 1596–1601 CrossRef CAS PubMed.
- S. Stripp, O. Sanganas, T. Happe and M. Haumann, Biochemistry, 2009, 48, 5042–5049 CrossRef CAS PubMed.
- P. E. M. Siegbahn, J. W. Tye and M. B. Hall, Chem. Rev., 2007, 107, 4414–4435 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1535510, 1535543, 1535559, 1535561 and 1852336. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02864h |
| This journal is © The Royal Society of Chemistry 2019 |