
Hydrogel/enzyme dots as adaptable tool for non-compartmentalized multi-enzymatic reactions in microfluidic devices†
David
Simon
ab,
Franziska
Obst
ab,
Sebastian
Haefner
 c,
Toni
Heroldt
a,
Martin
Peiter
a,
Frank
Simon
a,
Andreas
Richter
c,
Brigitte
Voit
*ab and
Dietmar
Appelhans
*a
c,
Toni
Heroldt
a,
Martin
Peiter
a,
Frank
Simon
a,
Andreas
Richter
c,
Brigitte
Voit
*ab and
Dietmar
Appelhans
*a
aLeibniz Institute of Polymer Research Dresden, Hohe Strasse 6, 01069 Dresden, Germany. E-mail: voit@ipfdd.de; applhans@ipfdd.de
bDepartment of Chemistry and Food Chemistry, Faculty of Science, Technische Universität Dresden, 01069 Dresden, Germany
cTechnische Universität Dresden, Faculty of Electrical and Computer Engineering, Institute of Semiconductors and Microsystems, 01062 Dresden, Germany
First published on 14th November 2018
Abstract
This study presents a simple method to integrate hydrogel/enzyme dots for the performance of multi-enzymatic reactions in two microfluidic devices and shows the conversion of enzyme educts under continuous flow as well as the reusability of the hydrogel/enzyme dots in microfluidic devices. For this purpose, five different enzymes were physically entrapped in a hydrogel matrix composed of poly(ethylene glycol) diacrylate, 2-(dimethylamino)ethyl methacrylate, and 2-hydroxyethyl methacrylate. Two separate tri-enzymatic cascade reactions were carried out. In the first cascade the enzymes β-galactosidase, glucose oxidase, and horseradish peroxidase were used and the second cascade consisted of the enzymes phospholipase D, choline oxidase, and again horseradish peroxidase. The (long-term) activity of free and hydrogel-immobilized enzymes was evaluated by UV-vis spectroscopic measurements. Additionally, time-dependent enzyme leakage from the hydrogel was investigated. Following the successful execution of multi-enzymatic reactions in bulk hydrogels, the material was integrated into PDMS-on-glass microfluidic reactors to carry out the enzyme reactions in miniaturized scale and under continuous flow. For that, hydrogel dots with a diameter of 350 μm were covalently attached to planar glass substrates by UV-initiated polymerisation of the enzyme-containing hydrogel precursor. Experiments were carried out both in channel reactors with hydrogel dots arranged in rows and wide chamber reactors with a hexagonal array of hydrogels. Especially the latter one showed a good performance as the flow velocity and thus the shear force on the hydrogel was decreased. Additionally, the residence time of the substrate and consequently the yield were increased. Long-term activity of the tri-enzymatic reactions in microfluidic reactors was proven with an ABTS assay indicating that this approach may be used as a platform for the integration of (multi-)enzymatic hydrogel dot reactions in microfluidic systems without the need of additional modification steps.
Introduction
“Green” chemistry is a great challenge for the chemical industry of the 21th century. One possible way to tackle this problem is the use of enzymes as biocatalysts.1–3 Enzymes have huge potential for chemical synthesis to control the regio-, stereo- and chemo-selectivity of organic and polymeric reactions as well as polymer-analogous reactions and are therefore e.g. applied in large scale in the synthesis of semisynthetic penicillin or enantiopure carboxylic acids in bioreactors.3–11 Moreover, enzymes may reduce the number of required reaction steps, are non-toxic biological materials, work at moderate reaction temperatures (<100 °C), leave no heavy metal contamination and are biodegradable.3,5 The drawback of enzymatic reactions is the instability of enzymes under harsh reaction conditions (e.g. high mechanical stress) and their limited recovery caused by the loss of their activity while separating them from the reaction mixture. In most cases they cannot be reused. In addition, most enzymes are expensive and thus, the process costs of biocatalytic synthesis may be high.To overcome these disadvantages enzymes can be immobilized in inorganic, organic or polymeric materials.4,5,12–15 This step enables the separation of the enzyme from the product in an easy way and thus facilitates the recovery and reusability of enzymes allowing their cost-efficient use in synthesis. Additionally, the immobilization generally increases enzyme stability under storage and operational conditions.16
Immobilization of enzymes can be divided into three general categories: binding to a carrier, cross-linking and entrapment (including encapsulation).16,17 In principle, binding to a carrier is possible by physical interaction, but these interactions are often too weak to prevent enzyme leakage from the carrier, as demonstrated by Bi et al.18 Covalent binding of enzymes to scaffold materials prevents the detaching, but the covalent coupling is partly harmful for the immobilized enzymes due to conformational changes during this method. This can result in the loss of the enzymatic activity. Carrier free immobilization by cross-linking of enzyme aggregates (cross-linked enzyme aggregates, CLEAs) has the advantage to generate highly concentrated biocatalysts. Moreover, this method is cost-efficient due to the exclusion of an additional carrier, but as well bears the risk of conformational changes of the enzymes during the cross-linking.19,20
The integration of enzymes in a hydrogel matrix without chemical linkages provides the opportunity for structuring hydrogel-enzyme-hybrid materials by photolithography on planar substrates. Sheldon et al. carried out enzymatic carrier synthesis in presence of enzymes and defined this as the most promising method for remaining enzyme activity in comparison to (non-)covalently bound enzymes.4,12 For the integration of active enzymes in hydrogels, the network parameters, including cross-linker concentration, cross-linking density, as well as the polymerisation method (radical, ionic, etc.), have to be chosen wisely to minimize the leakage of enzymes. Finally, enzymatic cascades, immobilized in different environments, can be integrated into microfluidic systems to set up a continuously working microreactor.4,5,20–26 Thus, enzymatic cascade reactions can be used for constant production of different chemicals. These advantages have been reviewed by Gruber et al.27
The goal of the herein presented study was the immobilization of two tri-enzymatic cascade reactions (Fig. 1) in photopatterned hydrogel dots on planar glass substrates and their integration in microfluidic chips, an approach which has not yet been reported. The hydrogel was synthesized from poly(ethylene glycol) diacrylate (PEGDA), 2-(dimethyl-amino)ethyl methacrylate (DMAEMA), which provides a positive charge to induce electrostatic interactions with negatively charged enzymes, and 2-hydroxyethyl methacrylate (HEMA) as a neutral monomer to shield the positive charges of DMAEMA. For proof-of-concept studies, the enzymes β-galactosidase (β-Gal), glucose oxidase (GOx), and horseradish peroxidase (HRP) were applied. In a cascade reaction, the substrate lactose is converted to glucose which is then oxidized by GOx whereby hydrogen peroxide is obtained. In the third step, hydrogen peroxide is reduced under catalysis of HRP and the dye ABTS is oxidized to the radical cation [ABTS*]+ which can be detected by UV-vis spectroscopy. After successful integration of this enzymatic cascade into the microfluidic system, a second set of enzymes, namely phospholipase D (PLD), choline oxidase (ChOx), and horseradish peroxidase (HRP) was immobilized in the hydrogel and the activity was characterized as well. Both cascades have a biological relevance and are thus interesting for applications in biosensing. The first cascade allows the detection of lactose and/or glucose, the second can be used for the detection of phosphatidylcholine which is a relevant molecule for cell signaling pathway in liver diseases.28
 | ||
| Fig. 1 Schematic tri-enzymatic cascade reactions carried out with non-immobilized and immobilized enzymes. Cascade 1 (β-Gal, GOx, and HRP; left), cascade 2 (PLD, ChOx, and HRP, right). | ||
PDMS-on-glass microfluidic reactors were used as they can be produced relatively easy by hard and soft photolithography and prototyping works rather rapidly.29 Two different designs of the microfluidic device were developed for the herein presented investigations. At first, a simple geometry was applied were the hydrogel dots were placed in a narrow (900 μm) channel. In order to reduce the shear force on the hydrogel dots and increase the residence time of the substrates on the hydrogels and consequently the yield, a chamber design was developed (Fig. 2).
 | ||
| Fig. 2 Schematic setup for the microfluidic experiments with hydrogel entrapped enzymes (left). Design of the channel and the chamber microfluidic chips with integrated hydrogel dots (red boxes, right). | ||
Even though the immobilization of one or two enzymes is well covered in literature, the realization of trienzymatic cascade reactions in microfluidic channels is rarely discussed.18,26,30–38 The trienzymatic cascade reaction from β-Gal, GOx, and HRP was reported, carried out with a substrate flow of a few nanoliter per minute with surface-bound enzymes,32 which is different from our approach. In most cases several functionalization steps of the surface are necessary to immobilize the corresponding enzymes in the reactor.32–35 In contrast to that, the use of hydrogels allows the immobilization of different enzymes in close proximity which minimizes the diffusion time of educts and intermediates between the catalytic centers (substrate-channeling effect) and thus may increase the conversion.39 Moreover, resilience of immobilized enzymes against storage conditions and mechanical stress within the microfluidic flow needs to be enhanced.5,23 Heo et al. demonstrated that the immobilization of horseradish peroxidase and glucose oxidase in PEGDA hydrogels in larger dimensions can be performed using a photo-induced polymerisation of PEGDA. Furthermore, a concentration-dependent activity of dual-enzymatic cascade reaction was shown, using a simple microfluidic reactor with only three hydrogel dots.40
In contrast, our study focuses on the miniaturization being able to incorporate an array of hydrogel/enzyme dots in microfluidic reactors (Fig. 2), and to achieve stable tri-/multi-enzymatic cascade reactions within several cycles of usage and storage. The enzyme activities for free and immobilized enzymes will be compared and two different designs of microfluidic devices will be tested. In this, the guidelines for reporting of biocatalytic reactions as listed by the STRENDA initiative were followed.41 The herein presented study is the starting point for the future development of serially connected and parallelized enzymatic cascade reactions in microfluidic reactors on one microchip which will allow the optimization of the reaction conditions (e.g. pH value, temperature) according to the enzymes. Additionally, this provides the technical basis to perform multi-enzymatic reactions even with incompatible reaction steps. The need for such systems was recently shown with the three-step synthesis of CMP-N-acetylneuraminic acid using spatially separated enzymes.42
Materials and methods
Materials
Poly(ethylene glycol) diacrylate (PEGDA, Mw 700 g mol−1), 2-(dimethylamino)ethyl methacrylate (DMAEMA, 98%), 2-hydroxyethyl methacrylate (HEMA, ≥99%), sodium phosphate monobasic (anhydrous), sodium phosphate dibasic (anhydrous), 3-(trichlorosilyl)propyl methacrylate (TPM, ≥90%), 2,4,6-trimethylbenzoyl chloride (97%), dimethyl phenylphosphonite (97%), peroxidase from horseradish (HRP, Type I, 50–150 units per mg solid), β-galactosidase from Aspergillus oryzae (β-Gal, 13,4 units per mg solid), glucose oxidase from Aspergillus niger (GOx, Type X-S, 228253 units per g solid), phospholipase D from Streptomyces chromofuscus (PLD, ≥50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units per ml buffered aqueous glycerol solution), 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS, ≥98%), lactose (anhydrous), and lithium bromide (≥99%) were purchased from Sigma-Aldrich. Choline Oxidase from Alcaligenes sp. (ChOx, ≥18.5 units per mg, lyophilized powder) was purchased from Chemos GmbH & Co. KG. D-(+)-Glucose (anhydrous) was purchased from Fluka Bio Chemika and Phosphatidylcholine (PC06, C6 saturated fatty acids) was purchased from Avanti Polar Lipids Inc. Ammonium hydroxide (28–30 wt%) was purchased from Acros Chemicals. Hydrogen peroxide (35%, H2O2) and the glass slides (Menzelglas, Thermo Scientific, 26 × 76 × 1 mm3) were purchased from Merck and D-(+)-glucose (anhydrous) from Fluka Bio Chemika. Polydimethylsiloxane (PDMS, SYLGARD® 184) was purchased from VWR. Negative dry film resist (Ordyl SY355; DFR), developer and rinser was purchased from Elga Europe. Glaswafers (BOROFLOAT 33, thickness: 0.7 mm, diameter: 75 mm, cut edges) were purchased from Schott AG (Mainz, Germany). HEMA and DMAEMA were passed throw neutral aluminum oxide to remove inhibitors. PLD was diluted by addition of PBS buffer to 9523 units per ml. All other chemicals were used as received. Filtered and deionized water (MilliQ) was obtained by Milli-Q® Gradient A10® from Merck Millipore. Phosphate-buffered saline (PBS) buffer (0.1 M, pH 7.4) was prepared by dissolving sodium phosphate dibasic and sodium phosphate monobasic in MilliQ. The photoinitiator lithium phenyl-2,4,6-trimethyl-benzoylphosphinate (LAP) was synthesized as described by Fairbanks et al. and Majima et al.43,44
000 units per ml buffered aqueous glycerol solution), 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS, ≥98%), lactose (anhydrous), and lithium bromide (≥99%) were purchased from Sigma-Aldrich. Choline Oxidase from Alcaligenes sp. (ChOx, ≥18.5 units per mg, lyophilized powder) was purchased from Chemos GmbH & Co. KG. D-(+)-Glucose (anhydrous) was purchased from Fluka Bio Chemika and Phosphatidylcholine (PC06, C6 saturated fatty acids) was purchased from Avanti Polar Lipids Inc. Ammonium hydroxide (28–30 wt%) was purchased from Acros Chemicals. Hydrogen peroxide (35%, H2O2) and the glass slides (Menzelglas, Thermo Scientific, 26 × 76 × 1 mm3) were purchased from Merck and D-(+)-glucose (anhydrous) from Fluka Bio Chemika. Polydimethylsiloxane (PDMS, SYLGARD® 184) was purchased from VWR. Negative dry film resist (Ordyl SY355; DFR), developer and rinser was purchased from Elga Europe. Glaswafers (BOROFLOAT 33, thickness: 0.7 mm, diameter: 75 mm, cut edges) were purchased from Schott AG (Mainz, Germany). HEMA and DMAEMA were passed throw neutral aluminum oxide to remove inhibitors. PLD was diluted by addition of PBS buffer to 9523 units per ml. All other chemicals were used as received. Filtered and deionized water (MilliQ) was obtained by Milli-Q® Gradient A10® from Merck Millipore. Phosphate-buffered saline (PBS) buffer (0.1 M, pH 7.4) was prepared by dissolving sodium phosphate dibasic and sodium phosphate monobasic in MilliQ. The photoinitiator lithium phenyl-2,4,6-trimethyl-benzoylphosphinate (LAP) was synthesized as described by Fairbanks et al. and Majima et al.43,44
Surface activation and TPM modification of glass slides
The glass slides were cleaned and activated following the protocol of the RCA-cleaning.45 Initially they were cleaned by ultrasonic treatment in isopropanol, MilliQ, and ethanol for 10 minutes each. Afterwards, activation was done in H2O2, ammonium hydroxide and MilliQ (1:1:5 vol%) at 70 °C for 10 minutes. After washing with MilliQ and drying in a nitrogen stream, the slides were functionalized with TPM by vacuum deposition in a dry and argon flushed desiccator at 10 mbar and room temperature for two hours. They were stored light protected at 8 °C for maximal two weeks. Analysis of functionalization was done by contact angle measurements (OCA 35xl Drop Shape Analysis-device; DataPhysics GmbH) and XPS studies [AXIS ULTRA spectrometer; Kratos Analytical (ESI†)]. Results from both analytic methods confirmed the expected TPM surface functionalization needed for the attachment of hydrogel dots on glass substrates.
Hydrogel synthesis and enzyme immobilization
The hydrogel precursor solution was prepared by mixing 892.8 μl (1.43 mmol, 60 mol%) PEGDA, 101.4 μl (0.59 mmol, 25 mol%) DMAEMA, 43.4 μl (0.35 mmol, 15 mol%) HEMA and 39.9 mg (38.8 μmol) LAP in 1.727 ml MilliQ. To ensure complete dissolving of the photoinitiator, the mixture was stirred overnight in the dark. Enzymes were added afterwards to the mixture and it was stirred for 1 hour until they were dissolved. In case of the first enzymatic cascade, 8.2 mg HRP, 3.6 mg GOx, and 61.3 mg β-Gal were applied. In case of the second cascade, 83 μL PLD solution (ESI†), 62.4 mg ChOx, and 23.0 mg HRP were added to the hydrogel precursor.Both bulk hydrogels and hydrogel dots covalently attached to glass slides were produced. For the synthesis of bulk hydrogels, 4 μL of enzyme-containing precursor solution were filled into a PDMS mould (4 × 4 × 0.3 mm3) and photopolymerized. For the synthesis of hydrogel dots, a thin layer of the precursor solution was formed on the glass slides. For this, a spacer of two layers of adhesive tape was attached to the edges of the slides. Subsequently, 300 μl of the precursor solution was added into the cavity which was then covered with a matt, black PET sheet (steps i and ii in Fig. 3A). The lithography mask (Fig. ESI-4 or ESI-5†) was placed on the glass slide (step iii in Fig. 3A) prior to the UV irradiation. Corresponding to the two different reactor geometries (Fig. 2, right), hydrogels were either produced in a row (channel reactor) or in an array (chamber reactor). In all cases, the photopolymerisation was performed using a DELOLUX 04 (DELO, Industrie Klebstoffe GmbH, Windach, Germany) as UV light source with an intensity of 8 W cm−2 and an emission spectrum ranging from 315 nm to 500 nm. The irradiation time was set to 7.5 seconds to minimize denaturation of enzymes by exposing to UV irradiation (step iv in Fig. 3A). After the polymerisation, the hydrogels were washed with MilliQ and stored in PBS buffer overnight.
 | ||
| Fig. 3 (I) Synthesis of hydrogel dots covalently attached to a TPM-functionalized glass slide. i) Preparation of glass slide with spacer (2 layers of adhesive tape); ii) filling with hydrogel pre-solution and covering with black poly(ethylene terephthalate) (PET) sheet; iii) covering of glass slide with photomask; iv) photopolymerisation; v) removing photomask, black PET plate, and spacer and rinsing of hydrogel dots with MilliQ. (II) Topographical analysis of the hydrogel dots by confocal microscopy (left: height profile; right: 3D picture); Fig. ESI-6† presenting analysis of hexagonally organized dots. (III) Assembly of the PDMS-on-glass microfluidic chip from glass functionalized with hydrogel dots (A) and PDMS sheet (B) and covering with aluminium plate (C). Fixation of the microfluidic chip in the metal holder and connection to the inlet and outlet tubes (D). | ||
The hydrogel dots were characterized with the confocal 3D-microscope μsurf explorer (Nano Focus, Oberhausen, Germany) after drying in a nitrogen stream using a 10× object lens (Fig. 3B).
UV-vis spectroscopic determination of enzymatic activity and enzyme leakage
The enzymatic activity was measured with an UV-vis-spectroscopic assay by observing the oxidation of the dye ABTS with HRP and H2O2 (see Fig. 1) at 405 nm. The measurement was performed with the SPECORD® 210 PLUS (Analytic Jena AG, Germany) spectrometer and PBS buffer (0.1 M) was used as solvent. The reaction yield was calculated using a calibration curve of [ABTS*]+, (see Fig. ESI-7†) obtained through ABTS conversion by H2O2 and HRP.To measure the enzymatic activity within the enzyme-loaded hydrogel dots, dots containing the enzymes β-Gal, GOx, and HRP or PLD, ChOx, and HRP were synthesized as presented in Fig. 3A. Afterwards, the glass slides were cut into five pieces each possessing one row of 20 hydrogel/enzyme dots. Each sample, composed of a row of 20 hydrogel/enzyme dots, was placed in a single use macro-cuvettes (Carl Roth) and 2633 μl (cascade 1) or 1805 μl (cascade 2) of 0.1 M PBS buffer and 78 μl of the ABTS solution (0.5 mol L−1 in 0.1 M PBS buffer) were added. In case of the first enzymatic cascade, 78 μL of the substrate solution (H2O2, glucose, or lactose, 0.5 mol L−1) were added to the corresponding enzyme solution in the cuvette. In case of the second enzymatic cascade, 750 μl PC06 (0,01 M in PBS buffer at pH 7.4) was added to the hydrogels row sample. After sample preparation, the cuvettes were shaken and immediately placed in the spectrometer. Conversion of the substrates was recorded by measuring the absorption of formed [ABTS*]+ every 30 seconds over a period of 20 minutes and shaking between each measurement point. Experiments were conducted at least three times for the mono-, dual- or tri-enzymatic reaction of cascade 1.
In order to estimate the immobilization efficiency, the activity of non-polymerized hydrogel precursor solution was determined and compared to the enzyme activity within the dots. For this, the volume of the hydrogel dots was calculated based on confocal microscopy and the respective amount of the enzyme-containing hydrogel precursor solution was applied for the activity measurement described above. As 20 hydrogel dots possess a volume of 19.6 nL, the respective amount of the enzyme-containing hydrogel precursor solution was applied. Prior to the measurement, the precursor solution was diluted to ¼ to prevent undesired polymerisation.
Enzyme leakage from hydrogel bulks was investigated by performing activity tests with the PBS storage solution after each storage day and placing the hydrogels into a fresh solution. In detail, 15 hydrogel bulks (4 μL) were stored in 3 mL of PBS buffer at pH 7.4 and 8 °C. Activity measurements were carried out with 673.3 μL of the storage solution, 390 μL ABTS solution (0.1 mol L−1) and 78 μL lactose solution (0.1 mol L−1). An excess of the dye was used to ensure complete conversion of the formed H2O2. The activity was normalized to the initial activity of an equal amount of enzyme-containing hydrogel pre-solution (defined as 100% enzyme activity). Hydrogel bulks were used instead of the small dots to improve the measurement accuracy.
Preparation of photomasks and PDMS sheets
Photomasks for the production of hydrogel dots and microfluidic chips were designed with the CAD software Autodesk Inventor and manufactured by photo plotting (MIVA 26100 ReSolution, MIVA Technologies, Schönaich, Germany) on black-and-white flat films.Polydimethylsiloxane (PDMS) sheets for the formation of PDMS-on-glass microfluidic chips (Fig. 2, Fig. ESI-9†) were fabricated by hard and soft lithography. At first, glass wafers were rinsed with acetone, isopropanol, and water and baked at 150 °C for at least 20 min. Three resist layers (DFR, 50 μm) were laminated on the cooled glass slide and each baked at 85 °C for 3 min. Afterwards, the resist was exposed to UV light through a polymer photomask for 90 s followed by a post-exposure bake for 40 min at 85 °C.42 Subsequently, the resist was evolved in a developer and rinser bath and finally baked for 1 h at 85 °C. According to confocal microscopy, the height of the structure was 150 ± 3 μm. Afterwards, PDMS (10:1 silicone: crosslinker) was poured over the master, degassed, and cured for 3 h at 60 °C. Finally, the PDMS sheets were peeled off from the master, perforated with a biopsy punch (kai industries co., ltd, Ø: 1.5 mm) to form the fluidic inlet and outlet (Fig. 2), and cleaned by ultrasonic treatment in isopropanol.
Fabrication and test of microfluidic reactors
The glass slide with conditioned enzyme-loaded hydrogel dots were rinsed with MilliQ and dried in a nitrogen stream. Afterwards, the corresponding PDMS sheet was placed on the glass to form the microfluidic reactor. The chip was covered with an aluminium frame and placed in the aluminium holder to prevent detachment of the PDMS from the glass. The connection to the pump (Asia Syringe Pump, Syrris, Royston, UK) and to the outlet was done using modified screw terminals with small PDMS flanges (see Fig. 3C, iv). For spectroscopic analysis, the outlet tube was connected to a 50 μL flow cuvette (Helma GmbH, Müllheim, Germany) which was placed in the UV/vis spectrometer (Cary 50, Varian Inc., Palo Alto, CA, USA).For the long-time activity measurement (20 hours measurement time, 5 cycles) of the first cascade reaction, a 0.01 mol L−1 educt solution was used (342.3 mg lactose and 548.7 mg ABTS in 100 mL PBS buffer) and a flow rate of 5 μl min−1 was applied. The absorption at 405 nm was measured every 30 seconds. After each cycle, the chip was disassembled completely and the glass slide with the hydrogels was stored in PBS buffer overnight for reconditioning and elimination of residual reaction products. The experiments were carried out at least in quadruplicates.
For the second cascade reaction only carried out in microfluidic chamber a 100 ml educt solution was prepared by dissolving 90,7 mg PC06 (0.01 M) and 109,7 mg ABTS (0.01 M) in 20 ml PBS buffer. The educt solution was pumped through the microfluidic reactor with a constant flow rate of 5 or 10 μl min−1. The experiments were conducted for at least 15 hours and 5 cycles using four independent microfluidic reactors. The absorption of converted ABTS was measured every 30 seconds. After each flow test the reactor was disassembled completely and the glass slide with enzyme loaded hydrogel dots was stored in PBS buffer overnight for reconditioning to eliminate residual reaction products.
Additionally, the dependency of the first cascade reaction conversion on the reactor geometry, substrate concentration, and flow rate was investigated. For this, substrate concentrations of 5 mmol L−1 and 10 mmol L−1 (channel reactor) and 3 mmol L−1 and 5 mmol L−1 (chamber reactor) and flow rates of 2, 5, and 10 μL min−1 were applied. With a fixed substrate concentration, microfluidic chips were tested at different flow rates. Each flow rate was applied for at least 2 hours to ensure equilibration of the system. For every substrate concentration, freshly prepared microfluidic chips were used to limit the effects of aging. All these experiments were carried out at least in triplicates.
Simulation of microfluidic flow
COMSOL® Multiphysics software was used to carry out stationary 2D microfluidic simulations to explore the velocity field and the shear rate at different flow rates. The hydrogel dots (d = 350 μm) were modelled as cylinders which were transverse to the direction of the flow. Laminar, incompressible flow with no-slip wall boundary conditions at 20 °C was chosen and water was selected as the fluid. The mesh size for the CFD simulations was set to “normal”.Results and discussion
TPM modification of glass surfaces
For the covalent attachment of the hydrogel-enzyme dots to the substrate surface upon polymerisation of the hydrogel monomers (Fig. 3A), TPM modification of the glass slides was carried out. The freshly cleaned glass surfaces were highly hydrophilic and droplets of water spread immediately. After the TPM modification the advancing contact angle was about 70° and the receding contact angle about 36° indicating a moderate decrease of the surface hydrophilicity (Table ESI-1†). These findings correlate with contact angle data measured on a methacrylate-coated glass surface.46–48 Furthermore, the high-resolution C 1s XPS spectrum confirmed TPM modification of glass surfaces (for details see ESI†).Hydrogel synthesis
For the formation of the hydrogel dots on glass slide surface (Fig. 2), a precursor solution containing poly(ethylene glycol) diacrylate (PEGDA), 2-(dimethylamino)ethyl methacrylate (DMAEMA), 2-hydroxyethyl methacrylate (HEMA), and the photoinitiator LAP was photopolymerized in the presence of the enzymes (see exp. part). First experiments with a smaller PEGDA cross-linker (Mn: 575 g mol−1) led to a complete loss of enzymatic activity due to a strongly reduced accessibility of the enzyme active centre caused by the small mesh size of the hydrogel network. Hydrogel formation with the slightly longer PEGDA (Mn: 700 g mol−1) maintained the enzymatic activity. Using the commercially available photoinitiator Irgacure, an exposure time of around 20 seconds was needed for polymerisation. However, applying the photoinitiator LAP the patterning time for the hydrogel dots was reduced to 7.5 seconds which is important to preserve enzymatic activity.49,50DMAEMA was used to receive a positively charged polymeric network allowing ionic interactions with anionic enzymes (ESI†). Thereby the leakage of immobilized enzymes should be reduced. The monomer HEMA was applied to decrease the positive charge density and thus repulsive forces of DMAEMA in the hydrogel.
Initial investigations, using the literature-known method to pattern hydrogels within PDMS-channels,40 led to fuzzy patterned hydrogel dots caused by scattering effects of UV-light through the PDMS layer. Thus, in an alternative strategy without the use of PDMS channel hydrogels dots were obtained by forming a thin layer of the hydrogel precursor solution on the glass slide and photopolymerisation through the photomask (Fig. 3A). The back-scattering was reduced by covering the precursor layer with a black and matt PET sheet. Sharply patterned cylindrical hydrogels with 250 μm diameter and 20 μm height were determined in dry state (Fig. 3B). As desired, the hydrogel dots were covalently attached to the glass surface due to prior functionalization with TPM.
Activity of hydrogel-immobilized enzymes for first cascade reaction without microfluidic device
Using the first enzymatic cascade (Fig. 1), the enzymatic conversions of enzymes immobilized in hydrogel dots and free enzymes were evaluated using the above described UV-vis-spectroscopic ABTS assay: (i) the single enzymatic conversion of ABTS to the UV active product [ABTS*]+ with H2O2 catalysed by HRP (Fig. 4A), (ii) the bi-enzymatic conversion of glucose to gluconolactone and H2O2 and subsequent formation of [ABTS*]+ catalysed by GOx and HRP (Fig. 4B), and (iii) the tri-enzymatic conversion of lactose to glucose and the subsequent formation of H2O2 and [ABTS*]+ catalysed by β-Gal, GOx, and HRP (Fig. 4C).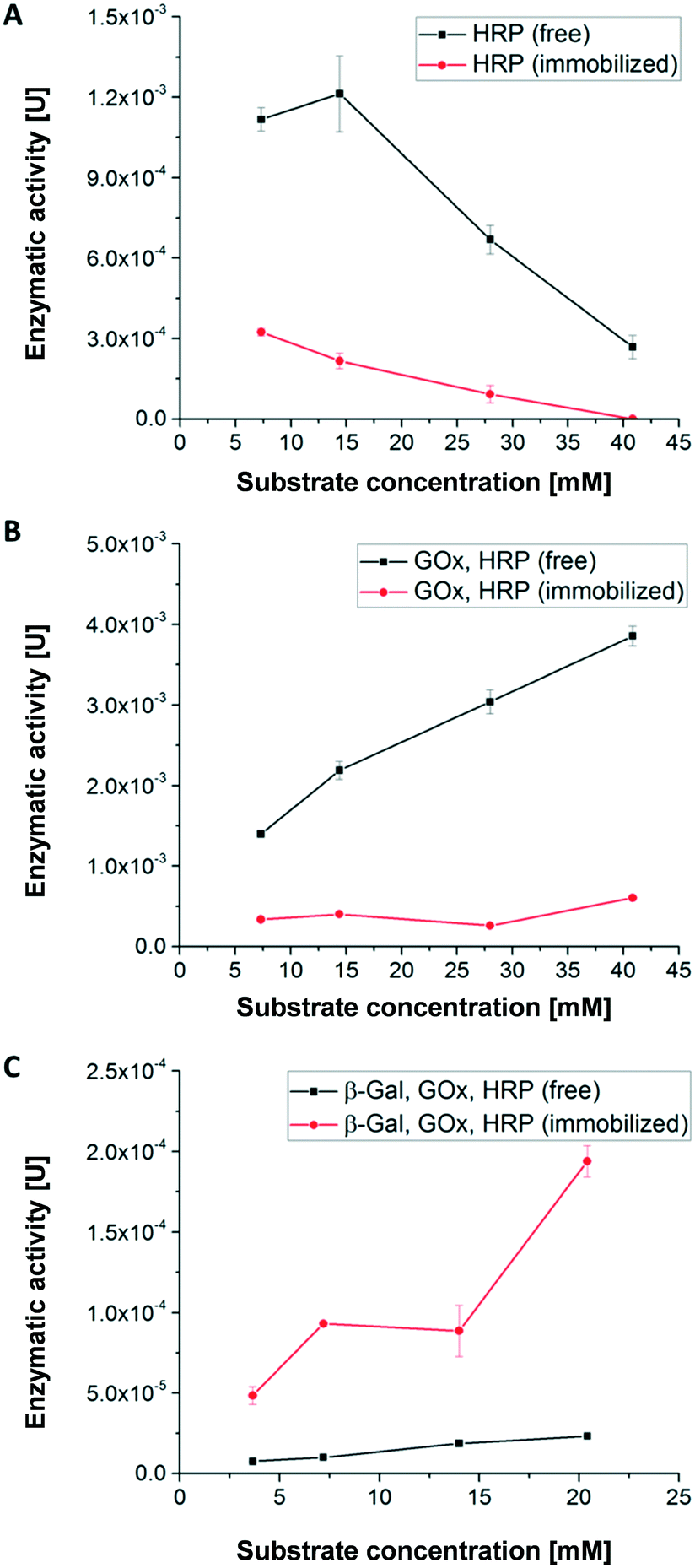 | ||
| Fig. 4 Enzymatic activity of free and immobilized enzymes in dependency of the substrate concentration; enzymes (A) HRP, (B) GOx, HRP, (C) β-Gal, GOx, HRP; substrates (A) H2O2, (B) glucose and (C) lactose. Quantification of [ABTS*]+ was done by UV/vis spectroscopy (λ = 405 nm). | ||
In order to obtain a high enzymatic activity, the influence of the H2O2 concentration on the activity was studied. At low H2O2 concentration (≤14.4 mM) the activity of free HRP is significantly higher than of immobilized HRP (Fig. 4A) which can be attributed to the better accessibility of the catalytic centre of free HRP. Additionally, the polymer network surrounding the enzymes restricts the diffusion of the substrate and thus the substrate conversion per time. At H2O2 concentrations higher than 14 mM, the enzymatic activity of both immobilized and free HRP strongly decreases. This relates to the deactivation of HRP by toxic H2O2 concentration and the formation of reactive oxygen species.51
In case of the bi-enzymatic cascade reaction of GOx and HRP (Fig. 4B) the activity of HRP is triggered by the conversion of glucose and the formation of H2O2 (Fig. 1). Hydrogen peroxide is constantly turned over by HRP for all glucose concentrations. These observations imply that the concentration of hydrogen peroxide remains at a low level and thus the enzymes are not deactivated (Fig. 4B). This effect was described previously.5,12 As in the monoenzymatic reaction of HRP, the activity of the non-immobilized enzymes is higher than that of the immobilized ones.
However, in case of the tri-enzymatic cascade reaction by β-Gal, GOx and HRP (Fig. 4C), the activity of immobilized enzymes is higher than the activity of the free enzymes.
Further experiments were conducted to find an explanation for the superior enzymatic activity of the tri-enzymatic cascade reaction compared to those of mono- and bi-enzymatic (cascade) reactions in hydrogel dots. The isoelectric point of all enzymes plays a minor role due to the presence of anionic surface charges of all enzymes at pH 7.4 (pI: 4.6 for β-Gal, 4.2 for GOx and 3–9 for HRP). Thus, all the enzymes can undergo ionic interactions with the cationic hydrogel network. To investigate the influence of cationic DMAEMA monomer on enzyme activity of the tri-enzymatic cascade reaction within hydrogels, several hydrogels with different DMAEMA and enzyme concentrations were synthesized (further details in ESI†). Results from enzyme activity test (Fig. ESI-6†) outlined that an increasing cationic charge (DMAEMA concentration ≥20%) of the polymeric network slightly increases the activity of tri-enzymatic cascade reaction, but also increases the standard deviation of the enzymatic activity. Zhang et al. demonstrated the activity increase of β-Gal immobilized in hydrogel beads in a medium containing potassium ions.52 These effects are likely to occur in the herein presented hydrogel/enzyme dots as well. But most importantly, the enzymes are kept next to each other by ionic interactions and other non-covalent interactions (e.g. H-bonds) within the hydrogel dots. The spatial proximity of enzymes in the hydrogel network implies short distances for diffusion of educts and intermediates prior to the following conversions, also described as substrate-channeling effect.43 This effect leads to a decreased diffusion time between the different enzymes and thus to an increased activity of the tri-enzymatic cascade reaction.
In summary, the results for the substrate concentration-dependent enzymatic activity imply that a substrate concentration of 5 mM provides good enzymatic conversion and allows reliable detection of the converted ABTS. Thus, this substrate concentration was used for the continuous conversion within a microfluidic channel and chamber to study the first cascade reaction.
Long-term activity of hydrogel-enzyme dots and enzyme leakage for first cascade reaction without microfluidic device
The long-term activity of enzymes in hydrogel-enzyme dots for the first cascade reaction was determined in order to evaluate the enzyme reusability (Fig. 5A). The single immobilized HRP lost its activity after one day caused by the harsh measurement conditions including the high concentration of H2O2. The bi-enzymatic (GOx, HRP) and tri-enzymatic (β-Gal, GOx, HRP) cascade reaction show a constant decrease of their activities to 60% and 40%, respectively, after 21 days. These results reflect the previously obtained results from substrate concentration-dependent activity of various enzyme combinations. However, we could prove that the hydrogel-enzyme dots are long-term stable biohybrid materials suited for the application in microfluidic reactors. | ||
| Fig. 5 (A) Relative long-term activity measured for single- (black), bi- (red) and tri-enzymatic (green) cascade reactions of β-Gal, GOx, and HRP immobilized in hydrogel dots measured over a period of 21 days. (B) Enzymatic activity in storage solution of enzyme containing (β-Gal, GOx, and HRP) hydrogel bulks. 0 d defines the leakage of enzyme from precursor hydrogel solution for preparing requested bulk hydrogel for studying enzyme leakage from hydrogel bulks. | ||
The enzyme leakage from the hydrogel upon conditioning was investigated to determine the immobilization efficiency. Fig. 5B shows the results of 3 storage cycles of enzyme-containing bulk hydrogels in PBS buffer. After the first storage cycle (=1st washing/conditioning step after synthesis) 48% of the immobilized enzymes were released. This indicates that a large amount of the enzymes was not completely immobilized in the hydrogel. The immobilization effectivity has been quantified by the comparison of enzymes' activities for immobilized (Fig. 5B) and non-immobilized (free) enzymes (defined as 100% enzyme activity). For the 2nd and 3rd storage cycle further enzyme leakage became negligible, allowing for keeping about 50% of the original enzyme loading.
Thus, it is demonstrated that after the conditioning (=1st storage cycle) of the hydrogels almost no further release of enzymes occurs. A similar behaviour can be assumed for the hydrogel-enzymes dots even though the higher surface to volume ratio may result in a higher enzyme leakage. However, long-term activity studies for the enzymatic cascade reactions in hydrogel dots confirm the successful and long-lasting enzyme immobilization (Fig. 5A).
Long-term activity of second tri-enzymatic cascade reaction without microfluidic device
The long-term activity of the second enzyme cascade was determined both for free and immobilized enzymes (Fig. 6). Thereby, the reaction steps were not analysed individually, as the general results obtained for the first tri-enzymatic cascade reaction were considered to be transferable. As Fig. 6 shows, the activity of immobilized enzymes is much higher than the activity of free enzymes. While free enzymes lose their activity within three days, there is no loss of activity of immobilized enzymes after 15 days (in the range of scattering of the results), and even after 40 days some activity is maintained. | ||
| Fig. 6 Long-term activity measured for tri-enzymatic cascade reaction PLD, ChOx, and HRP immobilized in hydrogel dots measured over a period of 42 days. | ||
Performance of tri-enzymatic cascade reactions integrated in microfluidic reactors
After the successful enzyme immobilization and activity determination in hydrogel dots, microfluidic reactors containing hydrogel/enzyme dots were produced (Fig. 2) and the enzymatic activity was determined under continuous flow. At first, the reactor was checked for water leakage with various flow rates (5, 10 and up to 250 μl min−1) showing no leakage over 5 days. Subsequently, the lactose and ABTS containing substrate solution for the first tri-enzymatic cascade reaction (Fig. 1) was pumped through the reactor with a flow rate of 5 μl min−1 and the absorption of produced [ABTS*]+ was measured. The usability of microfluidic chamber reactors was tested as described above for the simple microfluidic channel reactors.At first, the enzymatic cascade from the enzymes β-Gal, GOx, and HRP within the channel reactor (Fig. 2) was investigated (Fig. 7A). Within the first two to three hours of the experiment, a low absorption was obtained as measurements were performed offline and some time was needed to pump the educts through the microfluidic chip and to transfer the products into the cuvette. This initial absorption results from impurities of [ABTS*]+ in the substrate ABTS as purchased by the supplier. Additionally, the occurrence of bubbles within the cuvette during the filling process contributes to an absorption value above zero. After this time, the absorption increased and reached values up to 1.4. Throughout the 20 hours of the measurement, this value slightly decreased again. From the average absorption value throughout the measurement, the average yield was calculated and was 2.1% for the first operation cycle of a freshly prepared and conditioned microfluidic chip (Fig. 7B).
 | ||
| Fig. 7 (A) Flow test of 4 microfluidic channel reactors with integrated first tri-enzymatic cascade reaction (Fig. 1). (B) Relative yield of the first tri-enzymatic cascade reaction integrated in microfluidic channel reactor after several cycles of reactor disassembly, storage and reassembly. This experiment series was repeated four times using 6 different microfluidic reactors. Measurement conditions: 5 mM educt concentration (lactose and ABTS) in PBS puffer at pH 7.4 and room temperature and 5 μl min−1 for flow rate. (C) Relative yield of the second tri-enzymatic cascade reaction integrated in microfluidic chamber reactor after 5 cycles of reactor disassembly, storage and reassembly; using 4 different microfluidic chamber reactors. Measurement conditions: PC06 (0,01 M) in PBS buffer at pH 7.4 and room temperature, 5 (black symbols) and 10 (blue symbols) μl min−1 for flow rate. | ||
These results confirm the conversion of the substrates ABTS and lactose within the microfluidic chip in a tri-enzymatic cascade reaction. However, a slight decrease in the enzymatic activity during one cycle of operation has to be noted. This implies that some enzymes leak out of the hydrogels or may be deactivated. To estimate the intensity of this effect and thus the reusability of the microfluidic chips, the reactors were applied in successive usage cycles and the yield was calculated for each run (Fig. 7B).
In detail, the following steps were carried out with four microfluidic reactors for four times: (i) 20 hours run; (ii) disassembly, (iii) conditioning step of hydrogel-enzyme dots overnight in PBS buffer at 4 °C, (iv) reassembly. Fig. 7B and C present the results obtained in the operation cycles for the first (β-Gal, GOx, HRP) and second (PLD, ChOx, HRP) tri-enzymatic cascade reactions. Considering the initial yield of 2.10% for the first cascade reaction, the yield decreases to 1.25% after the first and to 1.11% and 0.85% after the second and third cycle, respectively. In case of the second cascade reaction the initial yield is only 0.65% resulting in lower repeating cycle yields as found for the first cascade reactions.
This decrease of enzymatic activity corresponds to leakage of the enzymes from the hydrogel dots as studied above and is more pronounced using the first cascade reaction. Additionally, it has to be considered that within the microfluidic flow tests the hydrogel dots are exposed to mechanical stress which may result in increased enzyme leakage. It is noted that the highest enzyme activity loss is observed in the first cycle due to enzyme leakage and deactivation especially upon the first flow stress. Moreover, we hypothesize that the remaining enzymes are tighter immobilized within the hydrogel dots and thus better protected from the shear forces. The liver assay also indicates that the first two involved enzymes27 of the second tri-enzymatic cascade reaction are even more prone to deactivation and leakage in microfluidic environments.28 In summary, we can state that hydrogel/enzyme dots are stable enough and reusable under selected testing conditions (e.g. flux rate, temperature, pH value, shear forces during the run) and thus suited for use in continuous microfluidic chamber reactors.
Variation of substrate concentration and flow rate for the first tri-enzymatic cascade reaction in microfluidic reactors
In order to further characterize the microfluidic devices, the conversion of the first tri-enzymatic cascade reaction with β-Gal, GOx, and HRP was determined in dependency of the substrate concentration and educt flow velocity. Additionally, the conversion within channel and chamber reactor was compared and the two reactor geometries were evaluated (Fig. 8). | ||
| Fig. 8 Dependency of the conversion (represented by the absorption of [ABTS*]+) on the substrate concentration and the fluid flux within the channel reactor (A) and the chamber reactor (B) filled with hydrogel-enzyme dots β-Gal, GOx, HRP. | ||
Obviously, the residence time of the substrate on the hydrogel dots is more than twice as high within the microfluidic chamber reactor (165 s instead of 75 s in microfluidic channel reactor as determined by optical observation of the fluid flow at a flow rate of 10 μL min−1). Furthermore, the hydrogel dots, integrated in microfluidic chamber reactor, are placed closer together resulting in a higher amount of hydrogel dots and thus immobilized enzymes. These geometrical effects contribute to a higher substrate conversion. For example, at a fluid flow of 5 μL min−1 and a substrate concentration of 5 mmol L−1, the conversion within the channel reactor is only 0.62%, whereas 2.3% are obtained in the chamber reactor. No dependency of the conversion on the substrate concentrations was found within the scope of measurement accuracy. At a fluid flow of 5 μL min−1 the conversion in the channel reactor is 0.62% at a substrate concentration of 5 mmol L−1 and 0.45% at 10 mmol L−1, respectively (Fig. 8A). In case of the chamber reactor a conversion of 2.3% is obtained with 3 and 5 mmol L−1 substrate concentration. However, a clear dependency of the conversion on the flux is shown. Generally, a reduction of the flux increases the residence time of the educts at the hydrogels and thus the conversion. At low conversions, it can be assumed that halving of the flux doubles the conversion. This correlation is clearly shown for the chamber reactor (Fig. 8B). However, in case of the channel reactor (Fig. 8A) such a correlation cannot be clearly derived from the experimental data due to the experimental error as a result of the low conversions and the difficulty to accurately setup this type of reactor.
In summary, these experiments illustrate the better performance of the chamber reactor compared to the channel reactor as higher conversions can be obtained at similar educt concentration and fluid flux. Additionally the comparable low standard deviations (Fig. 8B; e.g. ≤5% for use of 5 mmol L−1 substrate) for chamber reactors with ≥5 μl min−1 allow an accurate tuning of the conversion and indicate a good reproducibility. Considering in addition the data obtained by theoretical simulation on shear rate, see below, this chamber geometry should be favoured for the application in further investigations.
Simulation of the fluid flow in the microfluidic reactors
Theoretical simulations of the fluid flow were carried out to estimate the mechanical stress onto the hydrogel dots upon operation as a high shear stress may increase enzyme leakage and/or deactivation and cause detachment of the hydrogel dots from the glass slide. Due to the higher flow velocities in the narrow channel reactor (1.4 mm s−1 instead of 0.06 mm s−1), mechanical stress is high (Fig. 9A and B). As expected, the shear stress on hydrogels within the wider reaction chamber is much lower (Fig. 9C and D). This also indicates that a better homogeneous distribution of substrate solution around the hydrogel/enzyme dots (=drained and wetting hydrogel/enzyme dots with substrate solution) is present in chamber reactors than in channel reactors. This result also supports our observation of no detached hydrogel dots in microfluidic chamber reactor (Fig. 7C) after cyclic use applying disassembly, storage and assembly approach.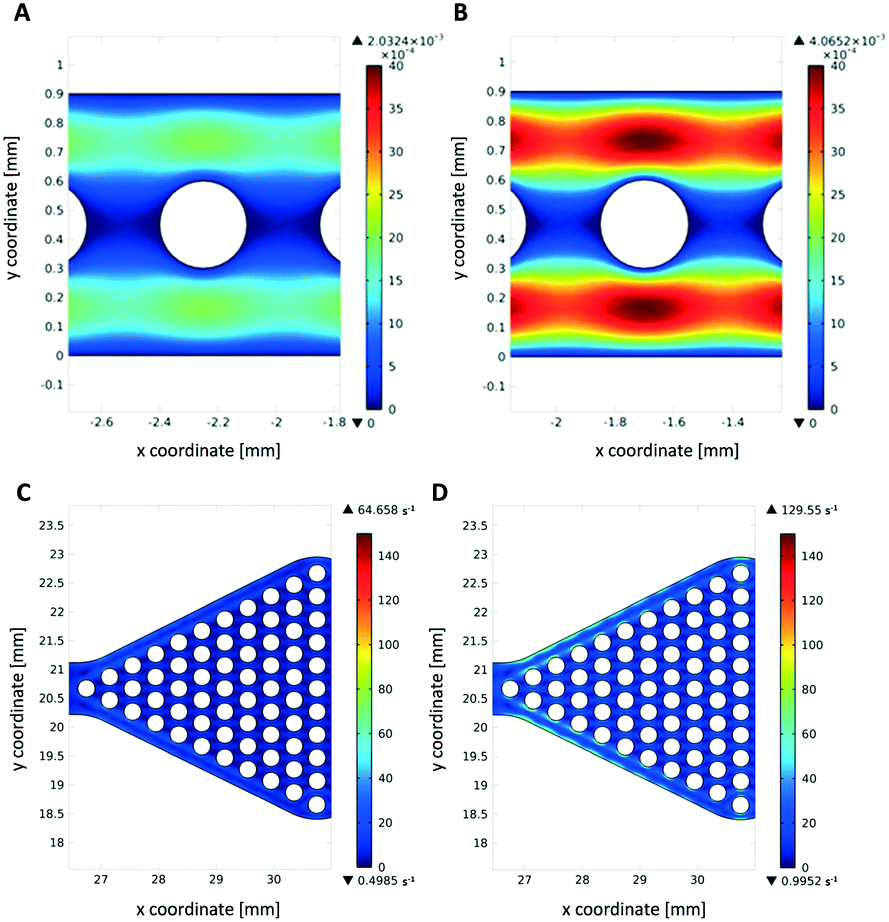 | ||
| Fig. 9 Simulation of shear rate inside microfluidic channel reactors for (A) 5 μl min−1 flow rate (max. shear rate: 92 1/s) and (B) 10 μl min−1 flow rate (max. shear rate: 184 1/s). Simulation of shear rate inside triangular reaction chamber for (C) 5 μl min−1 flow rate (max. shear rate: 65 1/s) and (D) 10 μl min−1 flow rate (max. shear rate: 130 1/s). Size of hydrogel dots used in simulation corresponds to experimentally determined size. Scaling left from each image presents velocity magnitude [m s−1] within a section of each reactor. | ||
Conclusions
We demonstrated a simple and efficient strategy to immobilize two non-compartmentalized tri-enzymatic cascade reactions by the formation of patterned hydrogel/enzyme dots using UV-initiated in situ polymerisation on a glass surface. These hydrogel/enzyme dots were successfully integrated in microfluidic channel and chamber reactors to test their enzymatic conversion ability under continuous flow conditions. This is different to most other approaches where on the other hand enzymes were integrated in microfluidic channels chemically or biologically29–33 or the enzymes were compartmentalized for multienzymatic reactions in microfluidics.50,51Overall, the catalytic activity of two differently robust tri-enzymatic cascade reactions is not only maintained in those hydrogel/enzyme dots over several weeks, but also increases compared to those of free enzymes in solution. After a conditioning cycle, the entrapped enzymes only minimally leak out of the hydrogel dots.
It was also shown that patterned hydrogel/enzyme dots could be miniaturized by photo-patterning developing a suitable preparation method and by that subsequently and smoothly integrated in larger arrays in both microfluidic reactors. For both tri-enzymatic cascade reactions the entrapped enzymes in hydrogel dots withstand the shear forces of the microfluidic flow (5 μl min−1) and generally show a constant reaction rate for a period of at least 15 hours of usage. The reusability of both tri-enzymatic cascade reactions is given for a minimum of at least three cycles. Moreover, the manageability of both microfluidic reactors was also tested by carrying out at least 3 assembly/disassembly/reassembly cycles of each reactor type. Especially, in case of the microfluidic channel reactor (Fig. 7B), a first larger decrease of enzymatic activity is recognizable. Simulation results also confirmed that hydrogel dots in microfluidic channels are more exposed to higher shear rates as found for hexagonally patterned hydrogel dots in reaction chambers.
This promising result will be further improved for non-compartmentalized multi-enzymatic reactions in microfluidic chamber reactors under continuous flow. One key issue will be the design and fabrication of even more diffusible hydrogel/enzyme dots for exchanging educts and products in such reactors. Such integrated enzymes in hydrogel dots may provide the chance of robust sensing of microfluidics with highly activated enzymes over weeks as proven in our long-term studies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding by the German Research Foundation (DFG), Project: GRK 1865/1 “Hydrogel-Based Microsystems” is gratefully acknowledged.References
- K. M. Koeller and C. H. Wong, Nature, 2001, 409, 232–240 CrossRef CAS PubMed.
- R. Wohlgemuth, Curr. Opin. Biotechnol., 2010, 21, 713–724 CrossRef CAS PubMed.
- C. M. Clouthier and J. N. Pelletier, Chem. Soc. Rev., 2012, 41, 1585–1605 RSC.
- N. Miletic, A. Nastasovic and K. Loos, Bioresour. Technol., 2012, 115, 126–135 CrossRef CAS PubMed.
- D. N. Tran and K. J. Balkus, ACS Catal., 2011, 1, 956–968 CrossRef CAS.
- L. Tao, C. Fu and Y. Wei, Polym. Int., 2015, 64, 705–712 CrossRef CAS.
- S. Kobayashi and M. Ohmae, Adv. Polym. Sci., 2006, 194, 159–210 CrossRef CAS.
- S. Puanglek, S. Kimura, Y. Enomoto-Rogers, T. Kabe, M. Yoshida, M. Wada and T. Iwata, Sci. Rep., 2016, 6, 30479 CrossRef CAS PubMed.
- J. Gaitzsch, X. Huang and B. Voit, Chem. Rev., 2016, 116, 1053–1093 CrossRef CAS PubMed.
- Y. Jiang and K. Loos, Polymer, 2016, 8, 243 Search PubMed.
- O. Kirk, T. V. Borchert and C. C. Fuglsang, Curr. Opin. Biotechnol., 2002, 13, 345–351 CrossRef CAS PubMed.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289–1307 CrossRef CAS.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC.
- C. Garcia-Galan, A. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904 CrossRef CAS.
- D. Brady and J. Jordaan, Biotechnol. Lett., 2009, 31, 1639–1650 CrossRef CAS PubMed.
- B. M. Brena and F. Batista-Viera, in Methods in Biotechnology: Immobilization of Enzymes and Cells, Springer, 2nd edn., 2006, ch. 2, pp. 15–30 Search PubMed.
- H. Bi, A. C. Fernandes, S. Cardoso and P. Freitas, Sens. Actuators, B, 2016, 224, 668–675 CrossRef CAS.
- R. Schoevaart, M. W. Wolbers, M. Golubovic, M. Ottens, A. P. G. Kieboom, F. van Rantwijk, L. A. M. van der Wielen and R. A. Sheldon, Biotechnol. Bioeng., 2004, 87, 754–762 CrossRef CAS PubMed.
- C. Roberge, D. Amos, D. Pollard and P. Devine, J. Mol. Catal. B: Enzym., 2009, 56, 41–45 CrossRef CAS.
- J. M. Bolivar and B. Nidetzky, Chim. Oggi, 2013, 31, 50–54 CAS.
- M. Miyazaki and H. Maeda, Trends Biotechnol., 2006, 24, 463–470 CrossRef CAS PubMed.
- J. Lalonde and A. Margolin, in Enzyme Catalysis in Organic Synthesis: A Comprehensive Handbook, Second Edition, Wiley-VCH, Weinheim, 2002, pp. 163–184 Search PubMed.
- M. A. Nunes, M. E. Rosa, P. C. Fernandes and M. H. Ribeiro, Bioresour. Technol., 2014, 164, 362–370 CrossRef CAS PubMed.
- M. Bajić, I. Plazl, R. Stloukal and P. Žnidaršič-Plazl, Process Biochem., 2017, 52, 63–72 CrossRef.
- Y. Zhang and S. Tadigadapa, Biosens. Bioelectron., 2004, 19, 1733–1743 CrossRef CAS PubMed.
- P. Gruber, M. P. C. Marques, B. O'Sullivan, F. Baganz, R. Wohlgemuth and N. Szita, Biotechnol. J., 2017, 12, 1700030 CrossRef PubMed.
- M. Takayama, S. Itoh, T. Nagasaki and I. Tanimizu, Clin. Chim. Acta, 1977, 79, 93–98 CrossRef CAS.
- W. Reschetilowski, Microreactors in Preparative Chemistry, Wiley-VCH, Weinheim, 2013 Search PubMed.
- H. Qu, H. Wang, Y. Huang, W. Zhong, H. Lu, J. Kong, P. Yang and B. Liu, Anal. Chem., 2004, 76, 6426–6433 CrossRef CAS PubMed.
- N. Yanagisawa and D. Dutta, Biosensors, 2011, 1, 58–69 CrossRef CAS PubMed.
- S. Fornera, P. Kuhn, D. Lombardi, A. D. Schlüter, P. S. Dittrich and P. Walde, ChemPlusChem, 2012, 77, 98–101 CrossRef CAS.
- Q. Zhang, J. J. Xu and H. Y. Chen, Electrophoresis, 2006, 27, 4943–4951 CrossRef CAS PubMed.
- F. Costantini, R. Tiggelaar, S. Sennato, F. Mura, S. Schlautmann, F. Bordi, H. Gardeniers and C. Manetti, Analyst, 2013, 138, 5019–5024 RSC.
- Z. Q. Wu, Z. Q. Li, J. Y. Li, J. Gu and X. H. Xia, Phys. Chem. Chem. Phys., 2016, 18, 14460–14465 RSC.
- I. M. Ferrer, H. Valadez, L. Estala and F. A. Gomez, Electrophoresis, 2014, 35, 2417–2419 CrossRef CAS PubMed.
- Y. D. Han, C. Y. Jeong, J. H. Lee, D.-S. Lee and H. C. Yoon, Jpn. J. Appl. Phys., 2012, 51, 06FK01 CrossRef.
- R. Sista, A. E. Eckhardt, T. Wang, M. Sellos-Moura and V. K. Pamula, Clin. Chim. Acta, 2011, 412, 1895–1897 CrossRef CAS PubMed.
- Y. H. Zhang, Biotechnol. Adv., 2011, 29, 715–725 CrossRef CAS PubMed.
- J. Heo and R. M. Crooks, Anal. Chem., 2005, 77, 6843–6851 CrossRef CAS PubMed.
- L. Gardossi, P. B. Poulsen, A. Ballesteros, K. Hult, V. K. Svedas, D. Vasic-Racki, G. Carrea, A. Magnusson, A. Schmid, R. Wohlgemuth and P. J. Halling, Trends Biotechnol., 2010, 28, 171–180 CrossRef CAS PubMed.
- L. Klermund, S. T. Poschenrieder and K. Castiglione, ACS Catal., 2017, 7, 3900–3904 CrossRef CAS.
- B. D. Fairbanks, M. P. Schwartz, C. N. Bowman and K. S. Anseth, Biomaterials, 2009, 30, 6702–6707 CrossRef CAS PubMed.
- T. Majima, W. Schnabel and W. Weber, Makromol. Chem., 1991, 192, 2307–2315 CrossRef CAS.
- W. Kern, J. Electrochem. Soc., 1990, 137, 1887 CrossRef CAS.
- W. G. Koh, A. Revzin, A. Simonian, T. Reeves and M. Pishko, Biomed. Microdevices, 2003, 5, 11–19 CrossRef CAS.
- Y. M. Bae, K.-H. Lee, J. Yang, D. Heo and H. J. Cho, Jpn. J. Appl. Phys., 2014, 53, 067201 CrossRef.
- Q. Zhu and D. Trau, Biosens. Bioelectron., 2015, 66, 370–378 CrossRef CAS PubMed.
- V. Falguera, J. Pagán and A. Ibarz, LWT--Food Sci. Technol., 2011, 44, 115–119 CrossRef CAS.
- Y. A. Vladimirov, D. I. Roshchupkin and E. E. Fesenko, Photochem. Photobiol., 1970, 11, 227–246 CrossRef CAS PubMed.
- Y. Xiao, H. X. Ju and H. Y. Chen, Anal. Chim. Acta, 1999, 391, 299–306 CrossRef CAS.
- Z. P. Zhang, R. J. Zhang, L. Chen and D. J. McClements, Food Chem., 2016, 200, 69–75 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Microfluidic setup, synthesis, characterization, and topographical analysis of hydrogel dots, surface activation, enzyme tests and simulation. See DOI: 10.1039/c8re00180d |
| This journal is © The Royal Society of Chemistry 2019 |