
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploiting enhanced paramagnetic NMR relaxation for monitoring catalyst preparation using T1–T2 NMR correlation maps
Carmine
D'Agostino
 *a and
Pierre
Bräuer
b
*a and
Pierre
Bräuer
b
aSchool of Chemical Engineering and Analytical Science, The University of Manchester, The Mill, Sackville Street, Manchester, M13 9PL, UK. E-mail: carmine.dagostino@manchester.ac.uk
bDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Philippa Fawcett Drive, West Cambridge Site, Cambridge, CB3 0AS, UK. E-mail: pb584@cam.ac.uk
First published on 15th October 2018
Abstract
A new method to characterise the evolution of surface sites during metal-supported catalyst preparation has been developed, which exploits NMR relaxation times and their sensitivity to paramagnetic ions. This method opens up new possibilities in terms of monitoring surface species during catalyst preparation.
 Carmine D'Agostino | Carmine D'Agostino received his BEng and MEng in Chemical Engineering at the Universita' di Napoli “Federico II” and a PhD in Chemical Engineering at the University of Cambridge. He is currently a Lecturer in Chemical Engineering at The University of Manchester, working in the Catalysis & Porous Materials group. His research interests focus on investigating diffusion, dynamics and adsorption of fluids within porous structures and catalysts using spectroscopic techniques, including high-field and low-field NMR. He received several awards, including the Young Scientist Award at the International Conference on Catalysis and a prestigious Junior Research Fellowship from Wolfson College, University of Cambridge. |
Once catalysts have been prepared, their characterisation is a key step in both understanding catalyst performance in reactions as well as optimising and improving preparation procedures. A variety of tools are nowadays routinely used for catalyst characterisation,6 each of them aiming at elucidating different physico-chemical aspects. Textural properties such as surface area, pore volume and pore size distribution are usually characterised with gas adsorption isotherms7 or mercury porosimetry,8 the former usually suited for micro and mesoporous catalysts, the latter more suitable for macroporous catalysts. Information on crystal structure are often obtained using X-ray diffraction (XRD),9 which can give insights into crystallinity, unit cell dimensions, crystal size and lattice parameters. Microscopy techniques such as transmission and scanning microscopy are very useful in order to understand surface morphology of both support and metal deposition.10 Other techniques have been developed including temperature-programmed studies,11 which can be useful to quantify metal oxidation state as well as adsorbed and/or deposited species such as coke, and a variety of spectroscopic techniques, including Raman, infrared (IR),12 and NMR.13 Most of the NMR methods used to characterise solid catalysts are based on solid-state magic angle spinning (SS MAS) NMR of 1H, 13C, 29Si, 27Al and 129Xe nuclei, which can be very useful in zeolite studies for example, as it is able to elucidate Al and Si distribution, acidity and in some cases porosity.6,14 Magnetic resonance imaging (MRI) techniques have also been proposed to study heterogeneous catalysts and their preparation. For example, MRI with T1 and T2 contrast has been used to study the impregnation step during the preparation of Ni/γ-Al2O3 hydrogenation catalyst pellets and understand transport and interactions of Ni precursors as well as metal distribution across single catalyst pellets.15
We have recently shown that NMR relaxation time measurements can be used as a non-invasive, rapid tool to characterise adsorption and molecular dynamics of species inside catalysts pores, which can be related to surface characteristics of solid catalysts.16–19 With this methodology we have been able to assess a variety of important aspects, including solvent affinity,20 water-tolerance18 and effect of mechanical treatments.17 Prompted by our initial work, we had the idea to see if the technique can be exploited to develop new protocols to monitor deposition of metal precursors over solid supports during catalyst preparation.
In the work reported here, we have used two-dimensional T1–T2 NMR relaxation measurements to characterise the evolution of the various surface adsorption sites on catalysts prepared by wet impregnation. In particular, we investigate wet impregnation of copper sulphate (CuSO4) over alumina (Al2O3) and probe the evolution of the observed surface adsorption sites by probing changes in NMR T1–T2 maps of 1-octene, the probe molecule used for this study. Choosing this system, we show that we are able to distinguish different adsorption sites over the catalyst surface and their evolution as the concentration of salt used for the preparation increases. The use of short-chain liquid hydrocarbons has shown to give rise to narrow T1–T2 peaks17 as compared with other probe molecules, particular compared to those with higher viscosities such as higher alcohols and polyols.21 This, together with the paramagnetic nature of CuSO4, which is expected to shift and decrease significantly T1 and T2 values of molecules in very close proximity of paramagnetic sites,22 relative to those interacting with the Al2O3 surface, is able to produce T1–T2 maps with clearly distinguishable and well-separated peaks associated to different adsorption environments, which can therefore be unambiguously analysed and quantified to provide new insights into adsorption site evolution as a function of metal precursor loading.
The γ-Al2O3 used for the experiments was supplied by Johnson Matthey. BET and BJH analysis were carried out in order to obtain the textural properties of the porous oxide, which has an average pore size of 12 nm, a pore volume of 0.52 cm3 g−1 and a surface area 90 m2 g−1. Samples of γ-Al2O3 doped with CuSO4 were prepared with a procedure similar to what has been reported in the literature for similar catalysts.23 In more details, alumina particles were dried in the oven at 105 °C for 3 hours and then added to aqueous solutions of copper(II) sulphate pentahydrate at known composition. The particles were stirred and left within the solutions for at least 24 hours. The moist solids were removed from the solution after impregnation and then dried in an oven at 70 °C for 2 hours and at 150 °C for further 2 hours, stirring several times during the drying process in order to ensure a more homogeneous drying. The actual content of paramagnetic CuSO4 salt inside the γ-Al2O3 particles was estimated by knowing the CuSO4 concentration of the aqueous bulk solution used for the impregnation and the pore volume of a known amount of solid used for the sample preparation. The deposition of CuSO4 and associated increase in the paramagnetic nature of the samples due to the presence of paramagnetic Cu2+ ions has been confirmed in magnetic susceptibility measurements previously reported.24
NMR relaxation experiments were performed on a Bruker DMX 300 operating at a 1H frequency of 300.13 MHz using a T1–T2 saturation recovery pulse sequence, which comprises a saturation recovery part to encode T1 (using a comb of 90° pulses) followed by a Carr–Purcell Meiboom–Gill (CPMG) echo train of 180° pulses to encode T2. The sequence is schematically shown in Fig. 1. The T1 recovery interval, tdelay, was varied logarithmically between 1 ms and 10 s in 32 steps. The echo spacing between the 180° pulses of the CPMG was set to 250 μs.
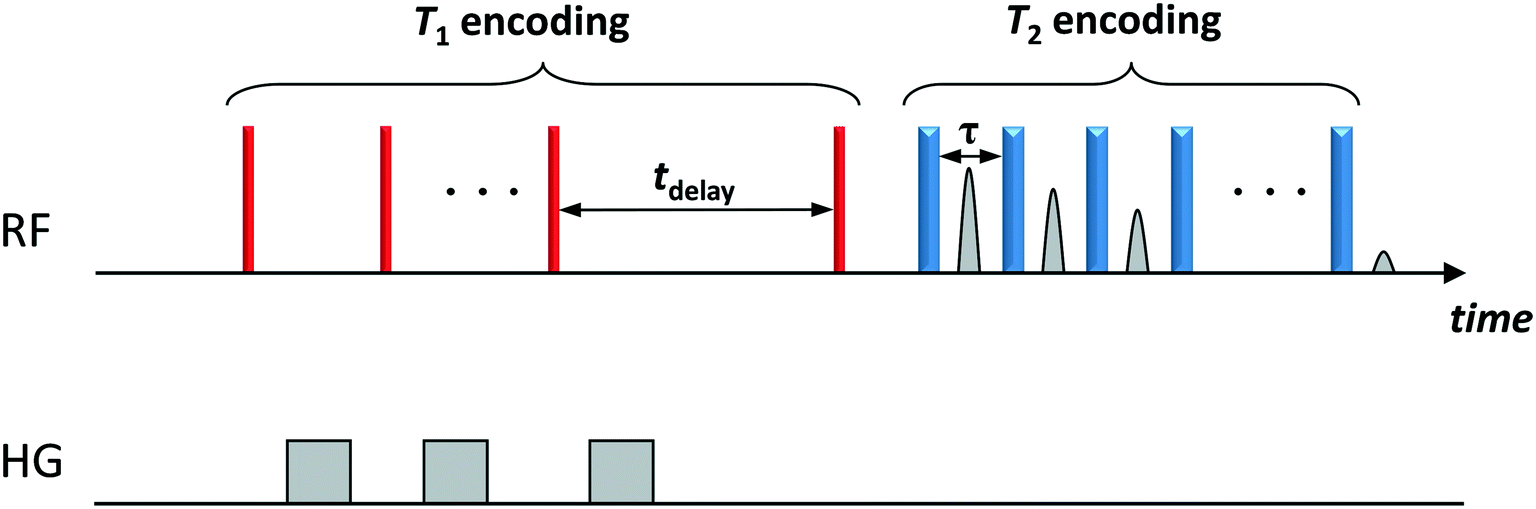 | ||
| Fig. 1 Two-dimensional T1–T2 NMR pulse sequence. The thin (red) and thick (blue) vertical bars represent 90° and 180° radiofrequency (RF) pulses, respectively. T1 relaxation is encoded in the variable time tdelay. T2 relaxation is encoded in the train of n 180° pulses. A single data point is acquired at the centre of each echo time, τ. The grey bars (HG) represent homospoil magnetic field gradients. | ||
Two-dimensional T1–T2 maps for bare Al2O3 and wet impregnated CuSO4/Al2O3 are reported in Fig. 2 for samples with different amount of CuSO4, reported as concentration of paramagnetic ions [Cu2+]. In the case of pure Al2O3, [Cu2+] = 0 ppm, a single peak, denoted as peak I, can be observed, which is attributed to the 1-octene interacting with the surface of the Al2O3 support. Typical values of T1 and T2 for this peak, shown in Fig. 3, are similar to those reported for other hydrocarbons on mesoporous oxide supports.17 With the introduction of small amount of CuSO4, a new peak, denoted as peak II, appears in addition to peak I, the latter attributed to Al2O3 and clearly distinguishable. This is clearly visible when [Cu2+] = 15 ppm. As the CuSO4 concentration increases, the relative intensity of peak II, compared to that of peak I, increases significantly and, for concentrations of [Cu2+] = 184 ppm and above, peak I disappears and only peak II remains visible. It is clear from the results that the new peak II appearing upon wet impregnation with the salt can be attributed to 1-octene in close proximity to new adsorption sites being formed, the latter being created upon CuSO4 wet impregnation, and clearly distinguishable from the peak of 1-octene interacting with the Al2O3, peak I. This assumption is strongly supported by the T1 and T2 values of peak II, which are reported in Fig. 3 and compared with values for peak I. Paramagnetic species are well-known to be strong relaxation sinks, which enhance significantly relaxation rate of probe molecules, hence decreasing values of relaxation times;25 therefore, it is expected that T1 and T2 values of molecules interacting with CuSO4 relaxation sinks will have much shorter T1 and T2 values compared to the same molecules interacting with the Al2O3 surface, which is indeed the case when comparing T1 and T2 values in Fig. 3.
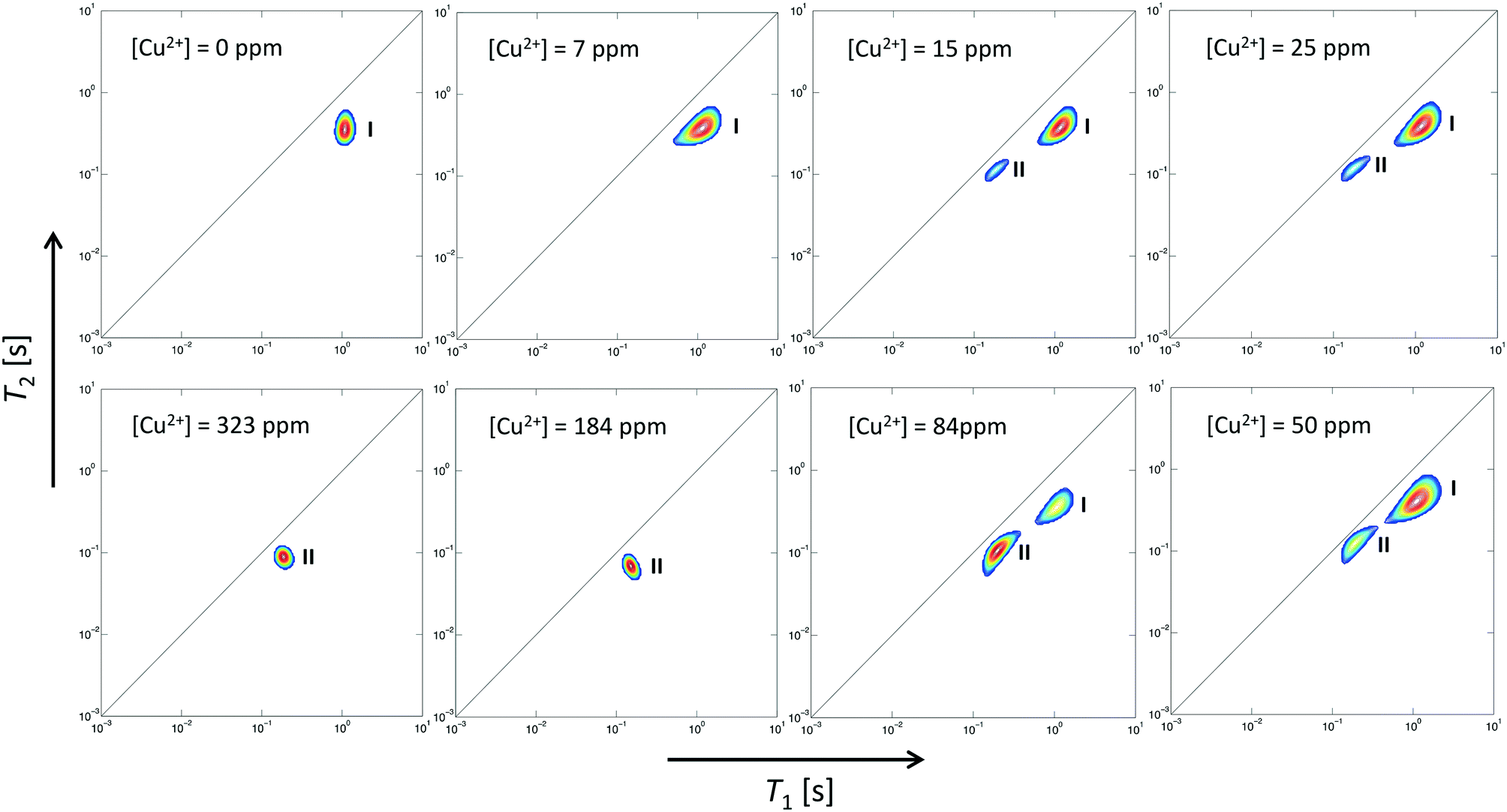 | ||
| Fig. 2 Two-dimensional T1–T2 NMR correlation maps for 1-octene in CuSO4/Al2O3 samples at different concentration of CuSO4, expressed as [Cu2+]. The deposition of CuSO4 gives rise to a new peak, peak II, associated with 1-octene interacting with CuSO4 sites, in addition to that of 1-octene interacting with Al2O3, peak I, which becomes the dominant peak at high CuSO4 concentrations. | ||
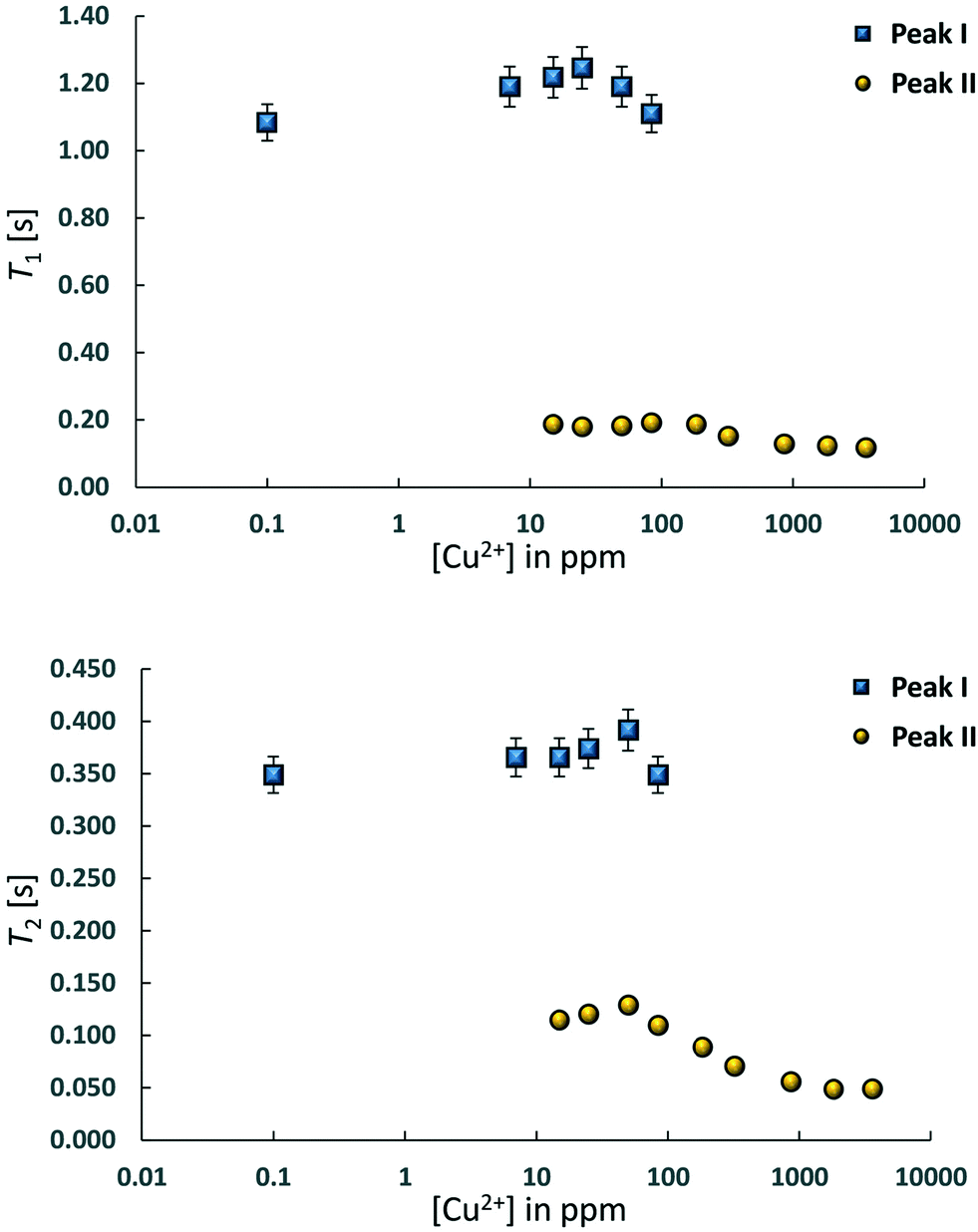 | ||
| Fig. 3 T 1 and T2 values for 1-octene in CuSO4/Al2O3 samples at different concentration of CuSO4, expressed as [Cu2+]. Peak I is associated to 1-octene interacting with Al2O3 whilst peak II is that of 1-octene interacting with CuSO4. The paramagnetic nature of Cu2+ ions leads to much smaller values of T1 and T2 NMR relaxation times of peak II. | ||
Further evidence that peak II is associated to CuSO4 sites can be obtained by analysing the single values of T1 and T2 of this peak as a function of Cu2+ concentration, see Table 1. As the Cu2+ concentration increases, single values of T1 and T2 of peak I, associated to Al2O3 sites, remain approximately constant whereas for peak II, associated to CuSO4 sites, such values experience a significant drop above [Cu2+] = 84 ppm, which can be associated to enhanced relaxation due to an increasing amount of paramagnetic Cu2+ sites, as expected from the theory of relaxation in the presence of paramagnetic sinks.26 Interestingly, this concentration corresponds to the value above which peak I, associated to Al2O3 sites, becomes negligible compared to peak II, associated to the presence of paramagnetic Cu2+ ions as CuSO4 species.
| [Cu2+] in ppm | Peak I (Al2O3) | Peak II (CuSO4) | ||
|---|---|---|---|---|
| T 1 [ms] | T 2 [ms] | T 1 [ms] | T 2 [ms] | |
| 0 | 1084 | 350 | — | — |
| 7 | 1190 | 366 | — | — |
| 15 | 1218 | 366 | 187 | 115 |
| 25 | 1246 | 374 | 180 | 120 |
| 50 | 1190 | 392 | 183 | 129 |
| 84 | 1110 | 349 | 190 | 110 |
| 184 | — | — | 187 | 89 |
| 323 | — | — | 152 | 71 |
| 858 | — | — | 129 | 56 |
| 1815 | — | — | 120 | 49 |
| 3605 | — | — | 118 | 48 |
The simultaneous presence of these two peaks in the concentration range [Cu2+] = 15–84 ppm also suggests that for CuSO4/Al2O3 samples in this range of concentration there is a heterogeneous distribution of adsorption sites over the surface, those attributed to Al2O3 and those due to CuSO4, in the form of a “patchy” catalyst surface with two clearly distinguishable regions, quantitatively comparable, as can be shown in Fig. 4, which reports the area ratio of peak I, associated to Al2O3, over peak II, associated to CuSO4, as a function of [Cu2+]. The ratio decreases with the Cu2+ concentration in a linear fashion, which is a further confirmation of the peak assignment made. This information can clearly be related to site accessibility, hence to the relative amount of molecules interacting with each site. From values of 184 ppm and higher, the peak associated to the Al2O3 support, peak I, disappears completely and this suggests that at this point the CuSO4 salt has covered most of alumina surface, hence the hydrocarbon has a limited access to purely Al2O3 surface sites and will mostly feel the influence adsorption sites due to CuSO4, or at the least that the amount of probe molecules interacting with the Al2O3 sites becomes negligible relatively to those interacting with the CuSO4 sites. Indeed, at higher concentration, only peak II remains prominent.
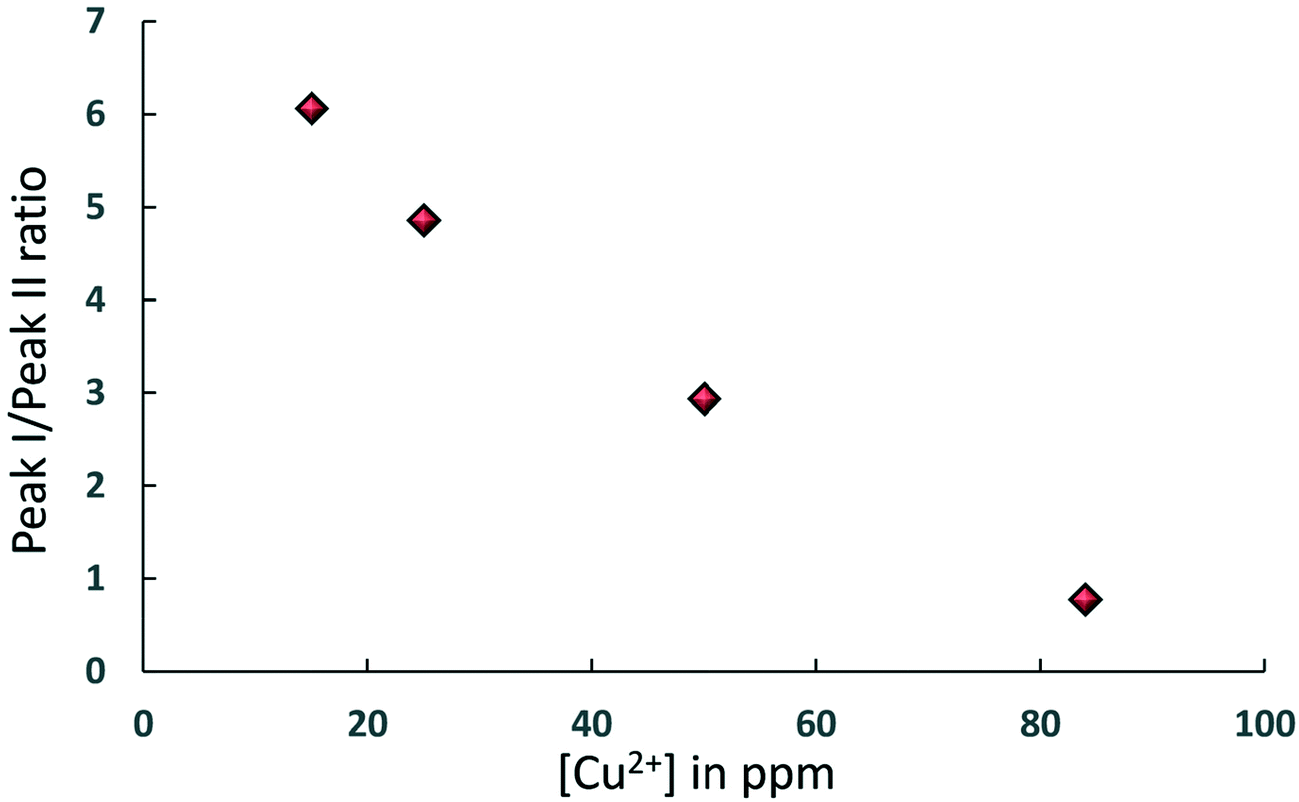 | ||
| Fig. 4 Area ratio of peak I, associated to Al2O3, over peak II, associated to CuSO4, as a function of [Cu2+] for samples where both peaks are present in the T1–T2 NMR correlation maps. | ||
One other point to note is on the values of the T1/T2 ratio, which can be related to an adsorbate/adsorbent surface affinity, for 1-octene on both Al2O3 and CuSO4 sites. Across the whole concentration range T1/T2 ∼ 3 for Al2O3 and T1/T2 ∼ 2 for CuSO4 sites. This suggests that the affinity of 1-octene towards CuSO4 is slightly lower compared to that towards Al2O3 and this could be due to the fact that CuSO4 is likely to be present in the form of hydrate, hence having a more hydrophilic character and a lower affinity with the hydrocarbon.
In summary, the finding reported here shows for the first time the ability of NMR relaxation methods to monitor surface evolution during catalyst preparation, particularly catalysts obtained by deposition of paramagnetic metal salts. We believe that these results open up new possibilities in characterisation and understanding of catalyst preparation, in particular evolution of surface sites and quantification of surface interactions of chemical species with the different surface sites. Future work on this topic will focus on quantitative aspects of this approach.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Prof. Lynn Gladden (University of Cambridge) for the access to experimental facilities and Dr Andy York (Johnson Matthey) for providing catalytic materials. Pierre Bräuer would also like to acknowledge Johnson Matthey for providing financial support for his scholarship.References
- R. Cano, A. F. Schmidt and G. P. McGlacken, Direct arylation and heterogeneous catalysis; ever the twain shall meet, Chem. Sci., 2015, 6, 5338–5346 RSC.
- S. A. Kondrat, P. J. Miedziak, M. Douthwaite, G. L. Brett, T. E. Davies, D. J. Morgan, J. K. Edwards, D. W. Knight, C. J. Kiely, S. H. Taylor and G. J. Hutchings, Base-Free Oxidation of Glycerol Using Titania-Supported Trimetallic Au-Pd-Pt Nanoparticles, ChemSusChem, 2014, 7, 1326–1334 CrossRef CAS PubMed.
- K. Wilson, A. F. Lee and J.-P. Dacquin, in Catalysis for Alternative Energy Generation, ed. L. Guczi and A. Erdôhelyi, Springer New York, New York, NY, 2012, pp. 263–304 Search PubMed.
- M. Schilling, R. Bal, C. Görl and H. G. Alt, Heterogeneous catalyst mixtures for the polymerization of ethylene, Polymer, 2007, 48, 7461–7475 CrossRef CAS.
- F. Pinna, Supported metal catalysts preparation, Catal. Today, 1998, 41, 129–137 CrossRef CAS.
- G. Leofanti, G. Tozzola, M. Padovan, G. Petrini, S. Bordiga and A. Zecchina, Catalyst characterization: characterization techniques, Catal. Today, 1997, 34, 307–327 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, Adsorption of Gases in Multimolecular Layers, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- H. Giesche, Mercury Porosimetry: A General (Practical) Overview, Part. Part. Syst. Charact., 2006, 23, 9–19 CrossRef.
- G. Perego, Characterization of heterogeneous catalysts by X-ray diffraction techniques, Catal. Today, 1998, 41, 251–259 CrossRef CAS.
- J. M. Thomas, Reflections on the value of electron microscopy in the study of heterogeneous catalysts, Proc. R. Soc. A, 2017, 473, 1–27 CrossRef PubMed.
- A. M. Efstathiou, Temperature-programmed desorption (TPD), reaction (TPR) and oxidation (TPO) of species formed on Rh/MgO after interaction with H2 and CO, J. Mol. Catal., 1991, 69, 41–60 CrossRef CAS.
- J. A. Lercher, V. Veefkind and K. Fajerwerg, In situ IR spectroscopy for developing catalysts and catalytic processes, Vib. Spectrosc., 1999, 19, 107–121 CrossRef CAS.
- W. Zhang, S. Xu, X. Han and X. Bao, In situ solid-state NMR for heterogeneous catalysis: a joint experimental and theoretical approach, Chem. Soc. Rev., 2012, 41, 192–210 RSC.
- M. Müller, G. Harvey and R. Prins, Quantitative multinuclear MAS NMR studies of zeolites, Microporous Mesoporous Mater., 2000, 34, 281–290 CrossRef.
- L. Espinosa-Alonso, A. A. Lysova, P. Peinder, K. P. de Jong, I. V. Koptyug and B. M. Weckhuysen, Magnetic Resonance Imaging Studies on Catalyst Impregnation Processes: Discriminating Metal Ion Complexes within Millimeter-Sized gamma-Al2O3 Catalyst Bodies, J. Am. Chem. Soc., 2009, 131, 6525–6534 CrossRef CAS PubMed.
- C. D'Agostino, J. Mitchell, M. D. Mantle and L. F. Gladden, Interpretation of NMR relaxation as a tool for characterising the adsorption strength of liquids inside porous materials, Chem. – Eur. J., 2014, 20, 13009–13015 CrossRef PubMed.
- K. Ralphs, C. D'Agostino, R. Burch, S. Chansai, L. F. Gladden, C. Hardacre, S. L. James, J. Mitchell and S. F. R. Taylor, Assessing the surface modifications following the mechanochemical preparation of a Ag/Al2O3 selective catalytic reduction catalyst, Catal. Sci. Technol., 2014, 4, 531–539 RSC.
- C. D'Agostino, S. Chansai, I. Bush, C. Gao, M. D. Mantle, C. Hardacre, S. L. James and L. F. Gladden, Assessing the effect of reducing agents on the selective catalytic reduction of NOx over Ag/Al2O3 catalysts, Catal. Sci. Technol., 2016, 6, 1661–1666 RSC.
- C. D'Agostino, M. R. Feaviour, G. L. Brett, J. Mitchell, A. P. E. York, G. J. Hutchings, M. D. Mantle and L. F. Gladden, Solvent inhibition in the liquid-phase catalytic oxidation of 1,4-butanediol: understanding the catalyst behaviour from NMR relaxation time measurements, Catal. Sci. Technol., 2016, 6, 7896–7901 RSC.
- C. D'Agostino, T. Kotionova, J. Mitchell, P. J. Miedziak, D. W. Knight, S. H. Taylor, G. J. Hutchings, L. F. Gladden and M. D. Mantle, Solvent effect and reactivity trend in the aerobic oxidation of 1,3-propanediols over gold supported on titania: NMR diffusion and relaxation studies, Chem. – Eur. J., 2013, 19, 11725–11732 CrossRef PubMed.
- C. D'Agostino, G. Brett, G. Divitini, C. Ducati, G. J. Hutchings, M. D. Mantle and L. F. Gladden, Increased affinity of small gold particles for glycerol oxidation over Au/TiO2 probed by NMR relaxation methods, ACS Catal., 2017, 7, 4235–4241 CrossRef.
- I. Foley, S. A. Farooqui and R. L. Kleinberg, Effect of paramagnetic ions on NMR relaxation of fluids at solid surfaces, J. Magn. Reson., Ser. A, 1996, 123, 95–104 CrossRef CAS PubMed.
- T. Sakamoto, H. Yonehara and C. J. Pac, Efficient oxidative coupling of 2-naphthols catalyzed by alumina-supported copper(II) sulfate using dioxygen as oxidant, J. Org. Chem., 1994, 59, 6859–6861 CrossRef CAS.
- C. D'Agostino, P. Brauer, P. Charoen-Rajapark, M. D. Crouch and L. F. Gladden, Effect of paramagnetic species on T1, T2 and T1/T2 NMR relaxation times of liquids in porous CuSO4/Al2O3, RSC Adv., 2017, 7, 36163–36167 RSC.
- T. R. Bryar, C. J. Daughney and R. J. Knight, Paramagnetic Effects of Iron(III) Species on Nuclear Magnetic Relaxation of Fluid Protons in Porous Media, J. Magn. Reson., 2000, 142, 74–85 CrossRef CAS PubMed.
- J. Korringa, D. O. Seevers and H. C. Torrey, Theory of spin pumping and relaxation in systems with a low concentration of electron spin resonance centers, Phys. Rev., 1962, 127, 1143–1150 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |