
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct decarboxylative Giese reactions
David M.
Kitcatt
a,
Simon
Nicolle
b and
Ai-Lan
Lee
 *a
*a
aInstitute of Chemical Sciences, School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK. E-mail: a.lee@hw.ac.uk
bGlaxoSmithKline, Gunnels Wood Rd, Stevenage SG1 2NY, UK
First published on 31st January 2022
Abstract
The quest to find milder and more sustainable methods to generate highly reactive, carbon-centred intermediates has led to a resurgence of interest in radical chemistry. In particular, carboxylic acids are seen as attractive radical precursors due their availability, low cost, diversity, and sustainability. Moreover, the corresponding nucleophilic carbon-radical can be easily accessed through a favourable radical decarboxylation process, extruding CO2 as a traceless by-product. This review summarizes the recent progress on using carboxylic acids directly as convenient radical precursors for the formation of carbon–carbon bonds via the 1,4-radical conjugate addition (Giese) reaction.
 David M. Kitcatt | David M. Kitcatt received his MChem (Hons) at the University of Bath in 2020, during which he also completed his year in industry at the Francis Crick Institute. Since then, he is currently pursuing his doctoral studies at Heriot-Watt University under the supervision of Dr Ai-Lan Lee (HWU) and Dr Simon Nicolle (GSK), funded by an EPSRC/GSK industrial CASE Award. His research interests focus on the development of mild radical decarboxylation methodologies to achieve synthetically useful chemical transformations. |
 Simon Nicolle | Simon Nicolle obtained his PhD in Organic Chemistry from Nottingham University (2016) where he worked on the development of new methodologies for the preparation of diazo compounds and exploring new processes featuring reactive metallocarbene intermediates, under the supervision of Prof. Chris Moody and Prof. Chris Hayes. He started his industrial career in 2016 by joining the Fibrosis Discovery Unit at GlaxoSmithKline (GSK), Stevenage, as a postdoctoral researcher and later as a medicinal chemist. Beyond the contribution to the research of new therapeutics, his role also involves the application of modern methodologies to accelerate drug discovery. |
 Ai-Lan Lee | Ai-Lan obtained her MSci (Hons) (2000) and PhD (2004) from University of Cambridge, working under the supervision of Prof. Steven V. Ley. She was subsequently awarded a Lindemann Trust Fellowship (2004–2005) to work at Boston College with Prof. Amir Hoveyda. In 2006, Ai-Lan was appointed as a fixed-term Lecturer at the University of Edinburgh, carrying out research with Prof. David Leigh. She started as a Lecturer at Heriot-Watt University in 2007 and is currently Associate Professor and Reader. Her research interests include decarboxylative radical reactions and development of new gold-, palladium- and photoredox-catalysed reactions. |
1. Introduction
The past few decades have seen a flourishing of innovative radical chemistries to overcome unique synthetic challenges. Chemists have realised the old perception that radicals are overly reactive and uncontrollable was ill-conceived,1 and many research groups have played their part in the development of novel and elegant synthetic transformations for the construction of complex organic motifs. Radical chemistry provides access to valuable products using simple radical precursors (RPs), achieving good chemo- and regioselectivity without the need for harsh reaction conditions and protecting groups.1Within radical chemistry, a concentrated area of research has focused upon developing new synthetic methodologies to form carbon–carbon bonds via addition to alkenes. Traditional methods involved organometallic, two-electron, conjugate addition reactions onto electron-deficient olefins (Michael acceptors), but these organometallic reagents were often toxic, difficult to prepare and/or unstable, thus limiting their applicability.2 Recent decades have witnessed the extensive utility of Giese-type3 reactions involving additions of radical intermediates onto versatile Michael acceptors, allowing scope expansion to secondary, tertiary, or heteroatom-stabilised substrates as suitable coupling partners.
The Giese reaction3 comprises the generation of a nucleophilic carbon-centred radical I from a radical precursor 1, which adds to a π-bond in conjugation with an electron-withdrawing group (EWG) (e.g., 2) through a regioselective 1,4-addition pathway, establishing a new carbon–carbon bond4 and producing an electrophilic radical intermediate II which is subsequently either trapped by a hydrogen atom-transfer (HAT) process to form a second σ-bond giving the Giese adduct 3, or alternatively, II could undergo single electron transfer (SET) followed by protonation to form 3 (Scheme 1A). Nucleophilic carbon-centred radicals I proceed to add to electron-deficient olefins at rates on the order of 106 M−1 s−1.3b,5 Conveniently, this rate is faster than that of the common counter-productive reactions, including atom-transfer reactions with sensitive functional groups such as alcohols and amines, as well as carbonyl additions, which are on the order of 102 M−1 s−1.6 This difference in rates underscores the chemoselective advantage of radical conjugate additions, which can proceed in the presence of unprotected functional groups.7
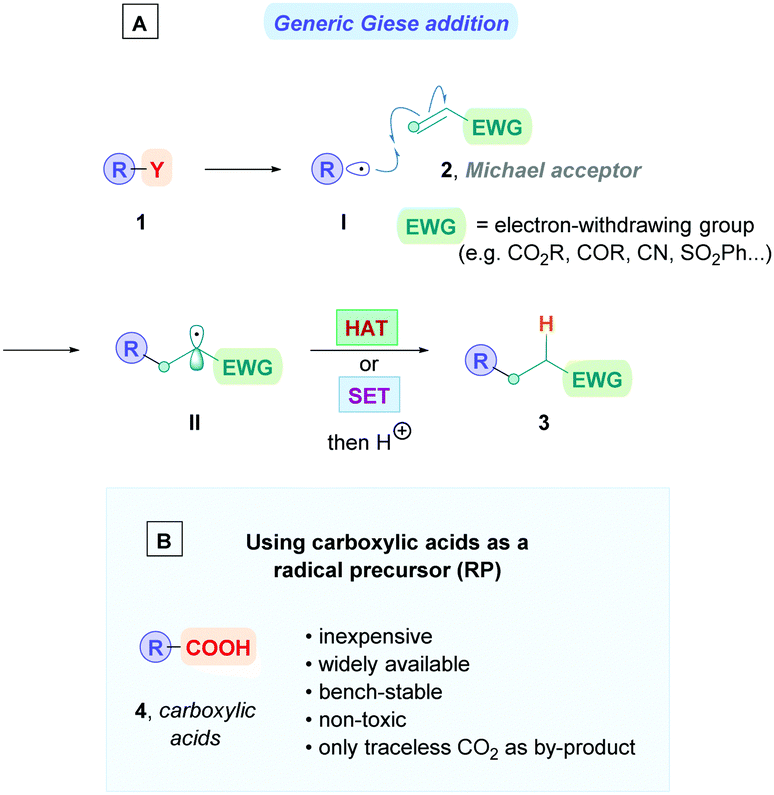 | ||
| Scheme 1 (A) Classic mechanism for the radical Giese reaction. (B) The advantages of using carboxylic acids as radical precursors. | ||
Traditionally, Giese reactions depended upon toxic tin hydride reagents (e.g., 5 in Scheme 2A) with alkyl halides or selenides as radical precursors (6), together with an initiation source in the form of light, heat or a radical initiator (e.g., AIBN) (Scheme 2A).3 Upon initiation, a tin radical (III) is generated which undergoes X-atom transfer with the radical precursor 6 to produce the alkyl radical I. Subsequent conjugate addition onto Michael acceptor 2 and HAT with another equivalent of tin hydride 5 furnishes the Giese adduct 3. These reactions suffered from competitive side-reactions, most noticeably where the tin radical can add to the Michael acceptor (hydrostannylation) (Scheme 2B), oligomerisation of the Michael acceptor (Scheme 2C), or premature H-abstraction of the generated alkyl radical (Scheme 2D), though these limitations can be suppressed by employing appropriate reaction conditions. The restricted scope of possible radical precursors has further added to the limitations due to availability and costs. To circumvent these issues, there has been growing endeavours towards innovative methodologies that are environmentally benign and utilise more sustainable radical precursors.
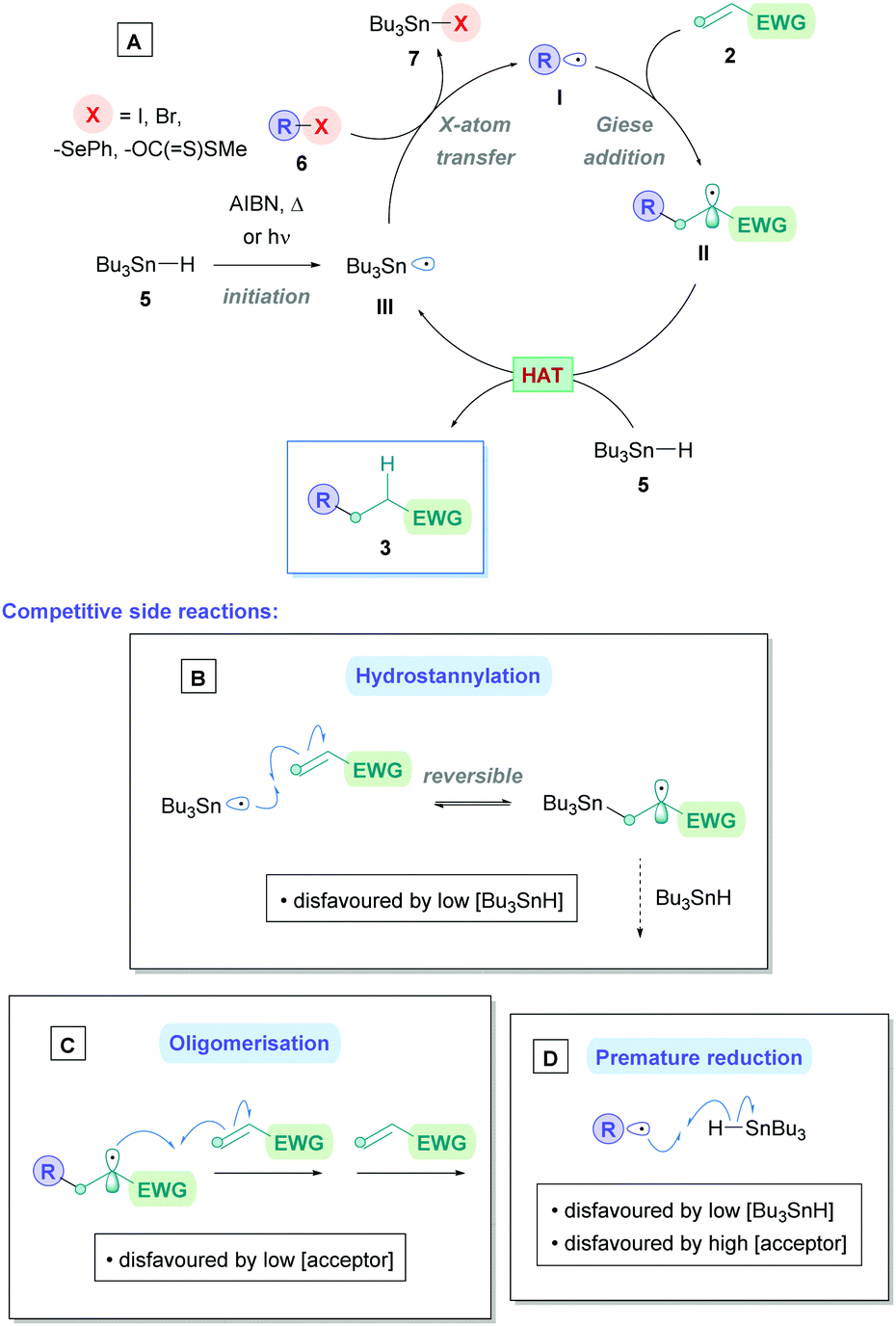 | ||
| Scheme 2 (A) Traditional Giese reaction mechanism using tin hydride reagent.7 Competitive reactions include (B) hydrostannylation of acceptor, (C) oligomerisation of acceptor, and (D) premature reduction of alkyl radical. | ||
It is worth noting that the Giese reaction can be interrupted, whereby radical II has been exploited to participate in other pathways allowing the final HAT process to be bypassed.8 These examples will not be discussed as they are beyond the scope of this review.
Within this context, carboxylic acids have become a versatile radical precursor over recent decades owing to their non-toxicity, low costs, bench-stability, and wide availability from renewable biomass sources.9 Exhibiting extensive structural diversity, carboxylic acids can be found in amino acids, fatty acids, and sugar acids, making them ideal substrates for synthesising high-value compounds such as pharmaceuticals. Moreover, the desired radicals are generated by a radical decarboxylation mechanism with the extruded CO2 as the traceless by-product, providing an entropic driving force for radical formation. It is unsurprising, therefore, that carboxylic acids have found widespread exploitation for decarboxylative Giese reactions.
Radical decarboxylation reactions have been known for a long time, with the likes of Kolbe,10 Hunsdiecker,11 and Barton12 being amongst the earliest pioneers, yet their protocols suffered from narrow substrate scope and quite forcing conditions such as high temperatures and ultraviolet (UV) irradiation. It is only within the last 30 years that more benign decarboxylation methods have been developed that can accommodate a broader range of substrates.
Decarboxylative Giese reactions (DGRs) can proceed using either the indirect method or the direct method. The indirect method involves pre-activation of the carboxylic acid to a redox-active ester designed to induce facile decarboxylation. Historically, the Barton decarboxylation13 (Scheme 3A) was the popular protocol, but the affiliated Barton esters (8) were often unstable and require toxic reducing agents. More modern advancements have given rise to new redox-active esters, the most popular being N-(acyloxy)phthalimide (NHPI) esters (9)14 which are more stable and undergo facile reductive single-electron transfer (SET) with visible light, or inexpensive transition metal catalysis with nickel or iron catalysts (Scheme 3B).9a,15 Perhaps the most notable example of using NHPI esters was elegantly demonstrated by the Baran group, providing a convenient means to access Giese products using nickel catalysis (Scheme 3B).15e Their protocol bypassed several shortcomings of conventional Barton's chemistry, providing a cheap and operational simple method for a broad scope of substrates. Further experimentation uncovered that indirect DGR could be achieved via a one-pot strategy.15e Despite the commendable progress, the indirect method has the disadvantage of producing undesirable by-products which must be separated from the product, on top of the extra step(s) needed for acid pre-functionalisation. Conversely, the direct method simply involves decarboxylation of the unactivated acid (4) through oxidative electron transfer (Scheme 3C), thus minimising waste and negating pre-functionalisation. For these reasons, the direct method is the more attractive approach and will be the subject of this review.
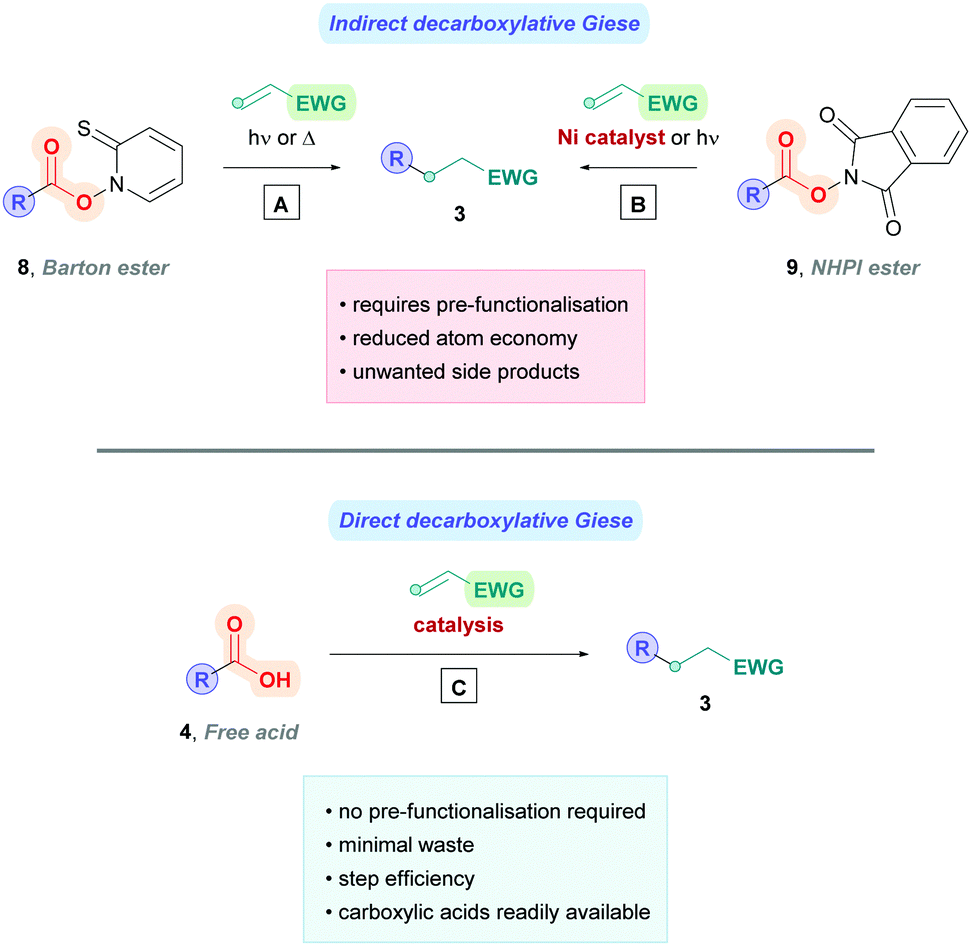 | ||
| Scheme 3 Differences between (A and B) indirect and (C) direct decarboxylative Giese reactions. | ||
There are two potential main pathways associated with which the direct method can proceed. The carboxylic acid 4 could be subjected to hydrogen atom abstraction (HAT), which generates the carboxyl radical V, followed by subsequent decarboxylation to alkyl radical I (Scheme 4A), though this pathway is rarely used owing to costly energetic requirements to homolytically cleave the strong O–H bond (468.6 ± 12.6 kJ mol−1 for acetic acid).16 Alternatively, a more common approach is deprotonation of the carboxylic acid 4 with a base to form the corresponding carboxylate IV (Scheme 4B), which is then oxidised through SET to V (Ered1/2 = +1.25 to +1.31 V vs. the saturated calomel electrode (SCE))17 followed by subsequent decarboxylation to I.
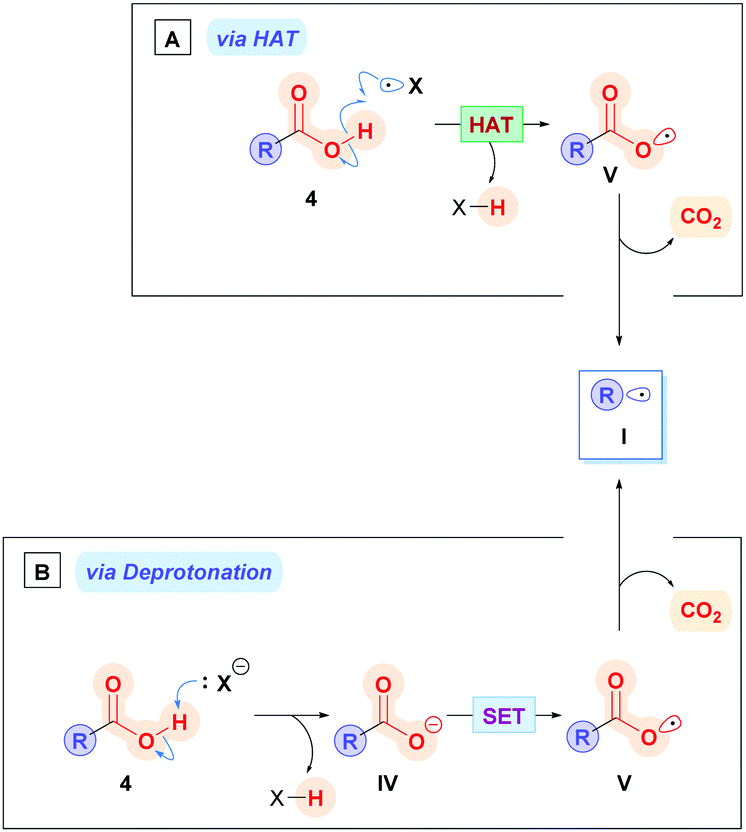 | ||
| Scheme 4 Two modes of radical decarboxylation: (A) via HAT or (B) via deprotonation. | ||
While a number of reviews3b,7,18 have been published on the Giese reaction, it should be noted that the last general review on radical conjugate additions was published in 2005 by Srikanth and Castle.18,19 An updated review is apt due to extensive progress made in this field in recent years. Indeed, a review on photomediated Giese reactions was very recently published by Kanegusuku and Roisen during the preparation of this review, covering selected high impact examples of photomediated Giese reactions from various radical precursors using homogenous catalysis.7 Our review is distinct and complementary as it will focus more comprehensively on the development and latest applications of direct decarboxylative Giese additions over the last few decades, which will not be limited to photomediated reactions. Carboxylic acids radical precursors covered in this review will also include α-oxo acids, oxamic acids and oxalic acid monoesters. Discussions will first begin with photocatalysis, showing applications of homogenous (involving transition metal and organocatalysts, and synergistic dual catalysis) and heterogenous photocatalysis. Examples mediated by silver catalysis under Kochi conditions will be discussed next, followed by discussions on the application of organic electrochemistry to this transformation. This review will also seek to highlight the successes and limitations of the different examples, as well as identifying areas where further progress would be beneficial.
2. Photocatalysis
Currently, the most popular method to achieve direct DGRs is through visible-light photoredox catalysis. The advent of visible-light photosensitisers (or photocatalysts) to act as electron-transfer ‘reagents’ have provided the means to generate highly reactive intermediates under mild conditions, beneficially serving a plethora of valuable transformation.20 Photocatalytic DGRs typically proceed via a reductive quenching pathway, and the various proposed mechanistic details can be summarised by the catalytic cycle shown in Scheme 5. Visible-light photoexcitation of the photocatalyst (PC) generates an oxidising species (*PCox) to initiate decarboxylation. As discussed previously, the carboxylic acid (4) is usually deprotonated to the more easily oxidised carboxylate (IV) (Ered1/2 = +1.25 to +1.31 V vs. the saturated calomel electrode (SCE))17 before decarboxylation, thus the presence of stoichiometric base is commonplace. If no base is present, then the acid would be more difficult to oxidise (Ered1/2 = +2.51 V vs. SCE for 2-phenylacetic acid).21 Oxidative single electron transfer (SET) between *PCox and the carboxylate IV generates the reducing species (PCred) and the corresponding carboxyl radical. Facile decarboxylation subsequently affords the carbon-centred radical I which undergoes conjugate addition with a Michael acceptor 2. The resulting secondary radical intermediate II is reduced to the carbanion species VIvia SET with PCred, which regenerates PC to close the cycle. Subsequent protonation of the carbanion affords the desired Giese product 3.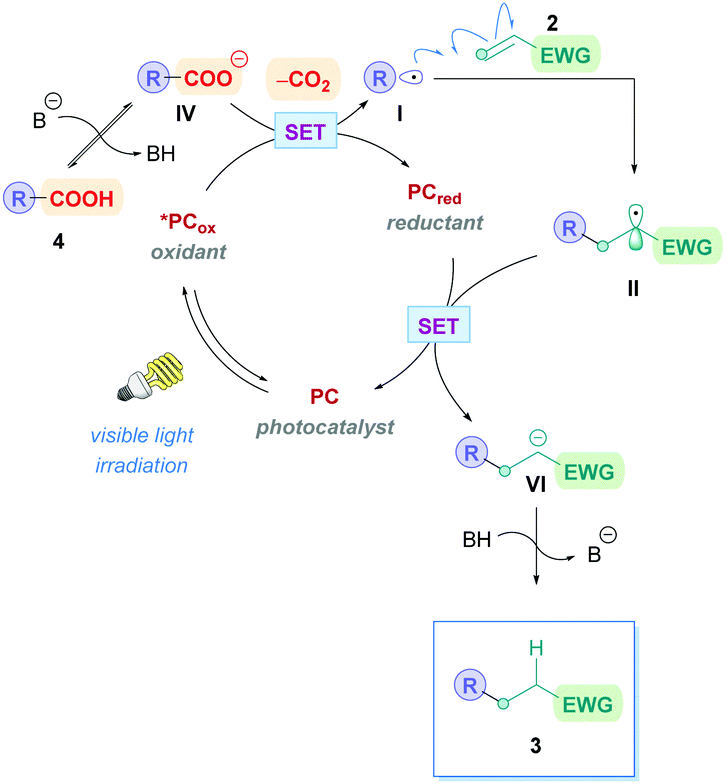 | ||
| Scheme 5 General mechanism for direct decarboxylative Giese additions via photocatalysis. Mechanisms are consistent with a reductive quenching pathway. | ||
The use of iridium-based photocatalysts in DGRs will initially be discussed, followed by photocatalytic systems involving titania (TiO2) and iron, before progressing to the more extensively exploited organo-based photocatalysts. Since the majority of the photocatalysed examples covered follow the pathway of Scheme 5, mechanistic explanations will be kept to a minimum. Processes that deviate from this mechanism, however, will be highlighted.
2.1 Transition metal-based photoredox catalysis
By far the most prevalent photocatalysts used for direct DGRs have been iridium-based, with some of the most common photoactive complexes shown in Scheme 6A. To facilitate direct DGR, these Ir-PCs proceed via Ir(III)/Ir(II) catalytic cycle (Scheme 6B) – referring to the mechanism depicted in Scheme 5 as a template, PCox would correspond to *IrIII and PCred would correspond to the IrII complex.
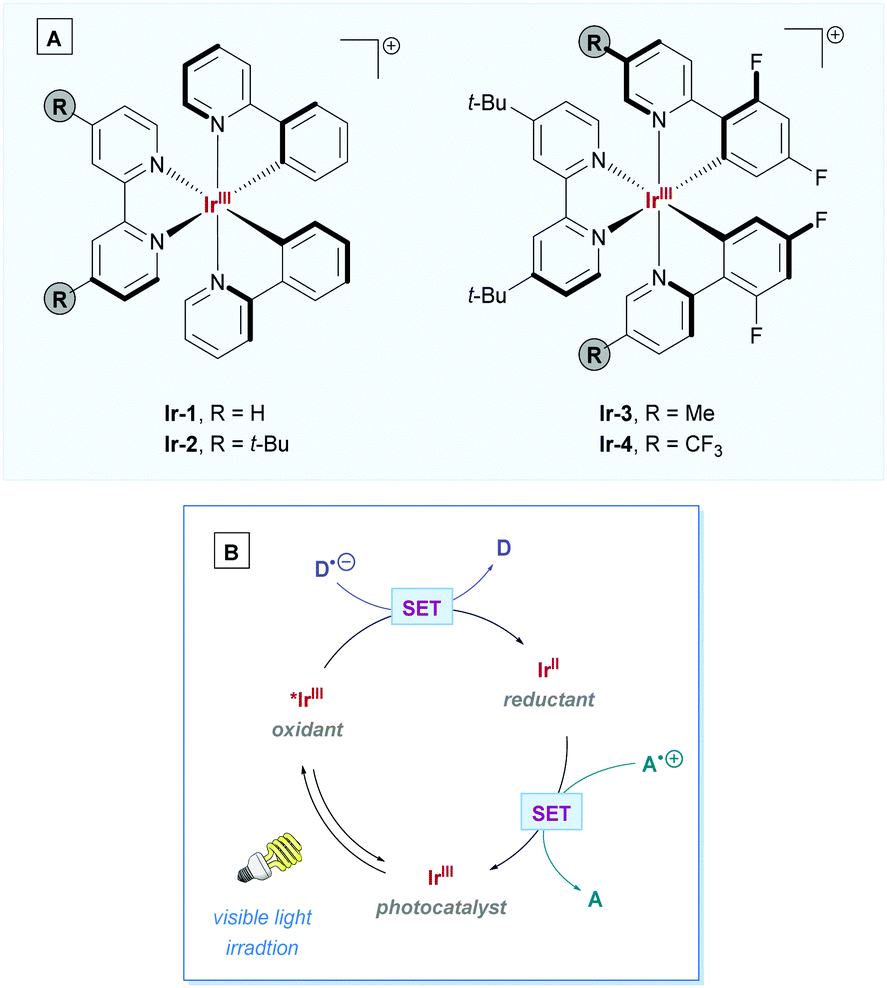 | ||
| Scheme 6 (A) Commonly employed Ir(III) photocatalysts: [Ir(ppy)2(bpy)]+ (Ir-1); [Ir(ppy)2(dtbbpy)]+ (Ir-2); [Ir(dF(Me)ppy)2(dtbbpy)]+ (Ir-3); and [Ir(dF(CF3)ppy)2(dtbbpy)]+ (Ir-4). (B) General Ir(III) photocatalytic cycle following the reductive quenching pathway of Scheme 5. Details are omitted to highlight the change in oxidation states of the iridium centre. D = electron donator, A = electron acceptor. | ||
The first example of iridium-based photocatalysis for direct DGR was reported in 2013 by Miyake, Nishibayashi and co-workers, who looked into the conjugate addition of arylacetic acids bearing amino groups at the para-position (Scheme 7).23 Using 1.1 equiv. of Michael acceptor, Giese addition was achieved using [Ir-1][BF4] as the chosen iridium photocatalyst in the presence of 14 W white LED illumination, affording benzylated adducts (12) containing tertiary, secondary and primary amino groups from low to high yields (16–98%). During optimisation, they reported no homocoupling of the benzyl radicals, indicating the benefits of employing photocatalysis to add benzyl radicals across olefins. The transformation was reported with a reaction time of 18 h, under a mild temperature of 25 °C. This protocol was only limited to para-substituted anilines because, interestingly, when testing the benzylation of phenylacetic acids bearing a morpholino group at the para-, meta- and ortho-positions (12h–j), a high yield (96%) was obtained for 12h but no yields were observed for 12i and j. Stern–Volmer studies with 12i suggested that oxidation of 12i did occur but with no decarboxylation. Based on other studies,24 it was postulated that the radical character (or spin population) of the carbon atom at the β-position with respect to the carboxyl group of the oxidised 12j was not sufficient to induce efficient decarboxylation. In contrast, oxidation of 12j was scarce probably due to intramolecular hydrogen bonding between the amino group and the carboxylic acid that inhibits oxidative SET.25
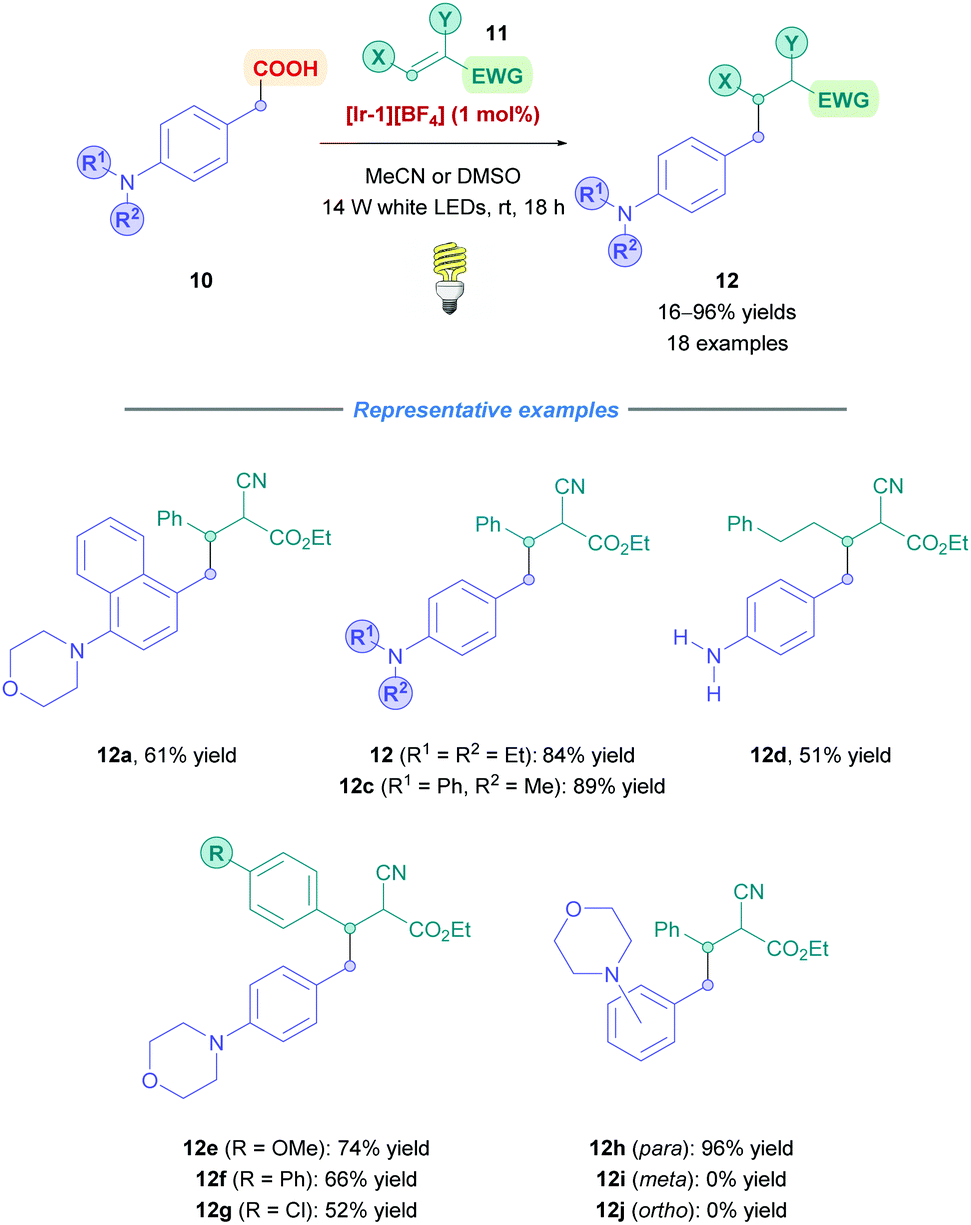 | ||
| Scheme 7 Ir-Catalysed direct DGR of arylacetic acids. | ||
The MacMillan group later made significant improvements to light-assisted DGRs by providing a more generalised protocol for a wide variety of cyclic and acyclic carboxylic acid substrates (Scheme 8).26 In the presence of stoichiometric inorganic base (organic bases gave poorer results), photocatalyst [Ir-4][PF6] was irradiated with a 26 W fluorescent light bulb at room temperature to access conjugate adducts (14) using hydrocarbon-substituted (14d), α-oxy, and α-amino (14a–c, e–f) carboxylic acids as starting materials with moderate to excellent yields (53–97%) across the board. The reaction conditions were also compatible with a broad range of Michael acceptors including olefins bearing ketones (14a), aryl bromides (14b), aldehydes, esters (14b and c) and carboxylic acids. Furthermore, the electronic nature of aromatic systems at the α-position of the olefin (14b) had little impact on the reaction efficiency. The group further demonstrated the applicability of this protocol by providing a three-step racemic synthesis of the anticonvulsant drug pregabalin (15, commercialised by Pfizer under the trade name Lyrica).
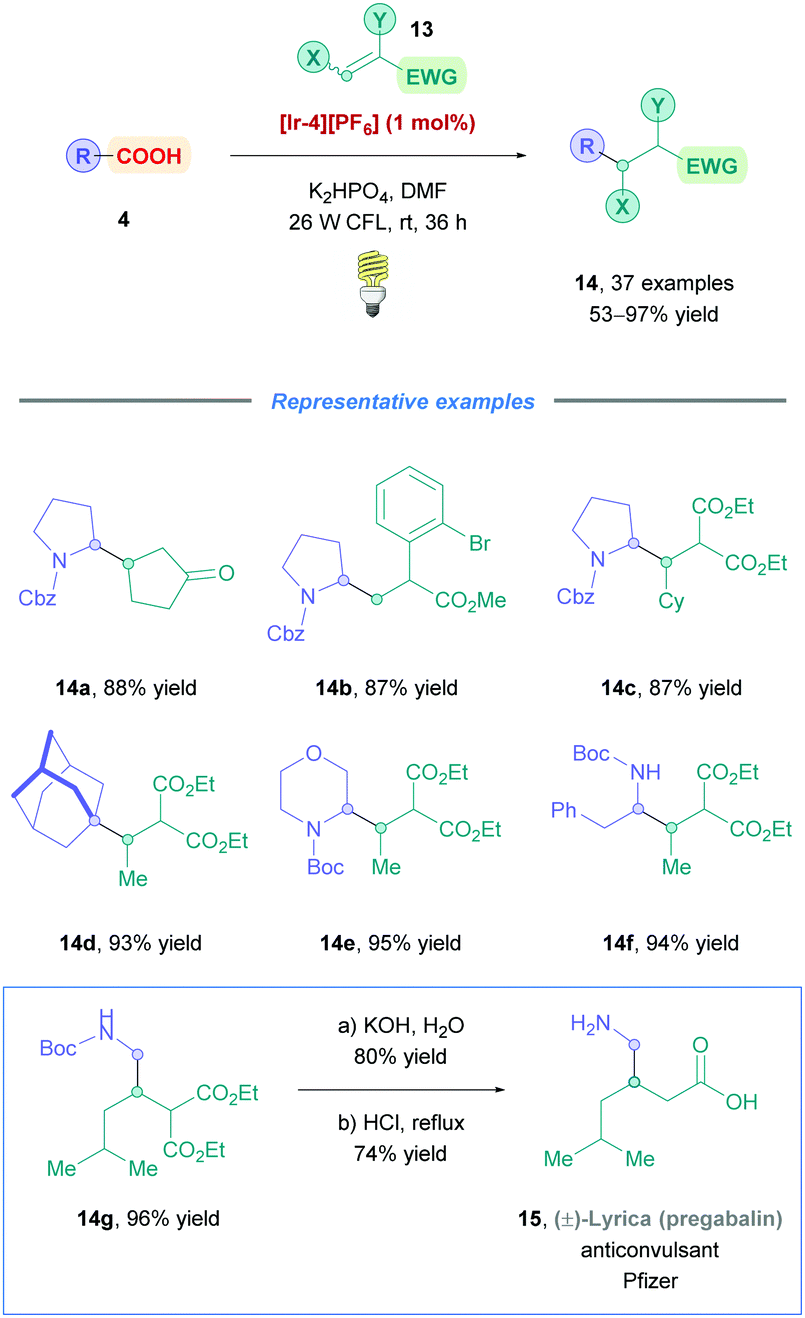 | ||
| Scheme 8 MacMillan's general Ir-catalysed direct DGR protocol. | ||
These seminal works later helped inspire other research groups to exploit Ir-based photoredox catalysis on a range of different carboxylic acid (Section 2.1.1.1) and Michael acceptor substrates (Section 2.1.1.2).
2.1.1.1 Exploration of carboxylic acid substrates. Fu and co-workers successfully applied Ir photoredox catalysis to develop a new type of acyl conjugate addition using α-keto acids (16) as nucleophiles (Scheme 9).27 Due to their thermal-instability,28 Michael acceptors are rendered incompatible for the harsh conditions often seen in transition-metal-catalysed decarboxylation coupling reactions.29 However, the characteristic mild conditions of Ir photoredox catalysis provided an alternative means to access these acyl conjugate products (17). Employing [Ir(dF(CF3)ppy)2(phen)][PF6] ([Ir-5][PF6]) as the PC, Fu assessed a variety of 2-oxo-2-(hetero)arylacetic acids with a series of Michael acceptors bearing nitriles (17a), sulfones (17b), esters (17d), ketones (17c, e–h), amides, and aldehydes, accomplishing moderate to excellent yields (45–94%). Phenylacetic acids with both EWGs and EDGs, including halogens, were shown to be well tolerated, with lower yields observed for those with electron-withdrawing substituents, presumably due to corresponding lower nucleophilicity (17evs.17f). Moderate yields were reported for 2-heteroaryl-substituted glyoxylic acids (e.g., 17h). Substituents on both the α- and β-positions of the Michael acceptor were well tolerated, including the useful CF3 group at the β-position (17d). Some acceptors, such as simple acrylic acid, were unsuccessful as substrates, possibly due to competitive reductive quenching with the 2-oxocarboxylate. Michael acceptors that give rise to tertiary and benzylic radicals (cf. radical II in Scheme 5) were also incompatible, due to their lower oxidation potentials being insufficient to re-oxidise the corresponding Ir(II) species (cf.Scheme 6B).
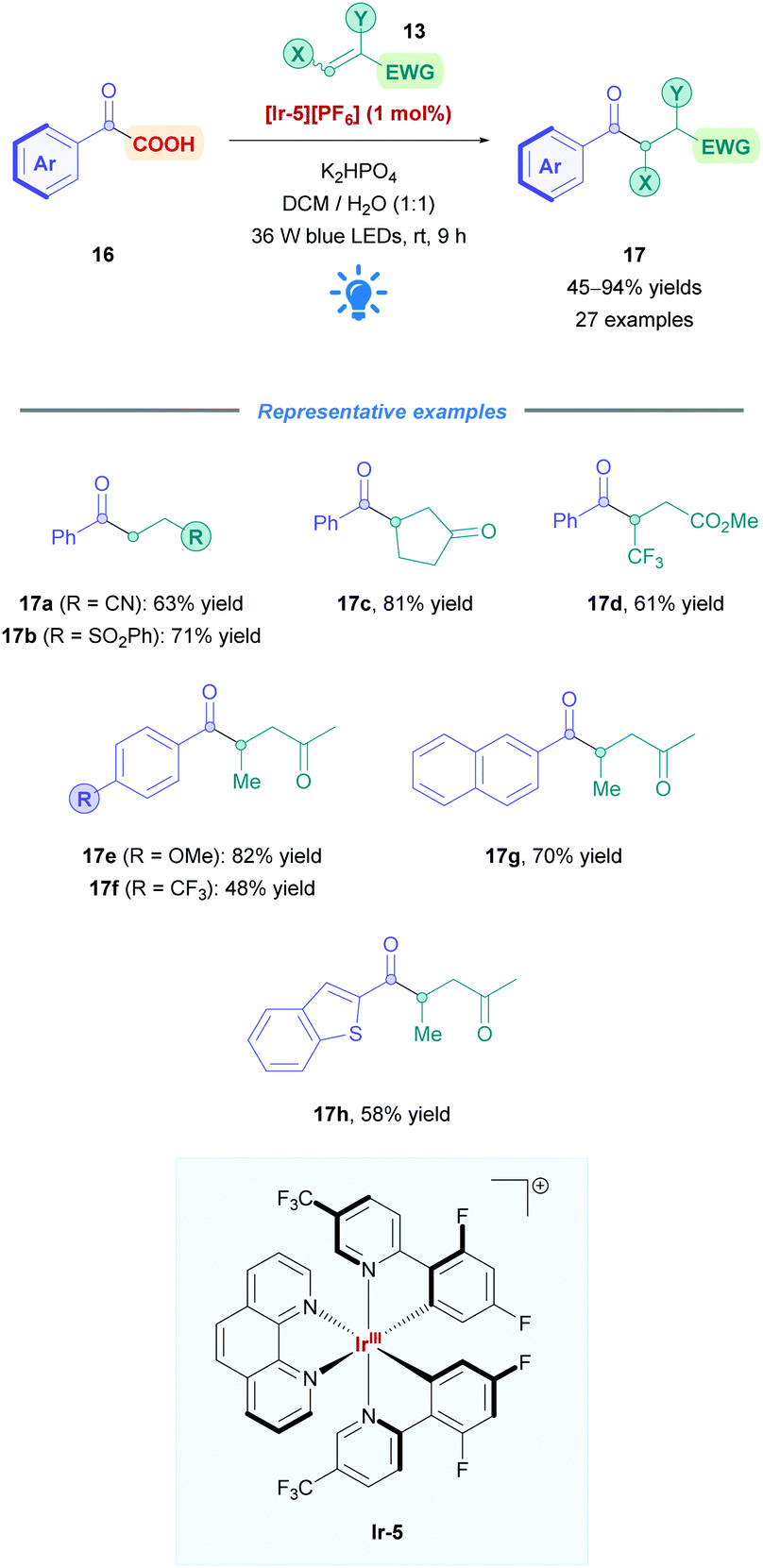 | ||
| Scheme 9 Fu's Ir-catalysed direct DGR of α-oxocarboxylic acids. | ||
α-Keto acids have also been used to achieve Giese alkylations. Upon decarboxylation of radical VII (Scheme 10A), the resulting acyl radical VIII can undergo further decarbonylation to give the corresponding alkyl radical IX, whilst extruding traceless CO in the process. The propensity for the acyl radical to decarbonylate is dependent upon the stability of the resulting alkyl radical. Consequently, decarbonylations that give more stable tertiary radicals are favoured, while primary and secondary α-keto acids tend to give their acylated Giese adducts. This trend can be seen in the work by Xu and co-workers, where they reported the photocatalytic decarboxylation/decarbonylation of α-keto acids (18) using [Ir-4][PF6], achieving Giese alkylation of tertiary radicals in excellent yields (20, 75–97%) and acylation when using secondary and primary α-keto acids.30 In a similar fashion, oxalic acid monoesters can also be used as radical precursors (Scheme 10B). Upon initial decarboxylation of X to give radical XI, a second decarbonylation event can occur to give the desired alkyl radical IX. Using photocatalysis (with [Ir-4][PF6]), MacMillan and co-workers employed caesium oxalates (21) as activated alcohols to conjugate alkyl radicals to electron-deficient olefins (19) with moderate to excellent yields of 20 (54–99%).31 Because of the same trends reasoned above, this protocol is most efficient to install tertiary alkyl radical, though two examples of secondary oxalates were reported. Overman and co-workers have also used caesium oxalates to engage in selective 1,6-conjugate additions,32 which has successfully been employed for the synthesis of the diterpenoid trans-clerodane.33
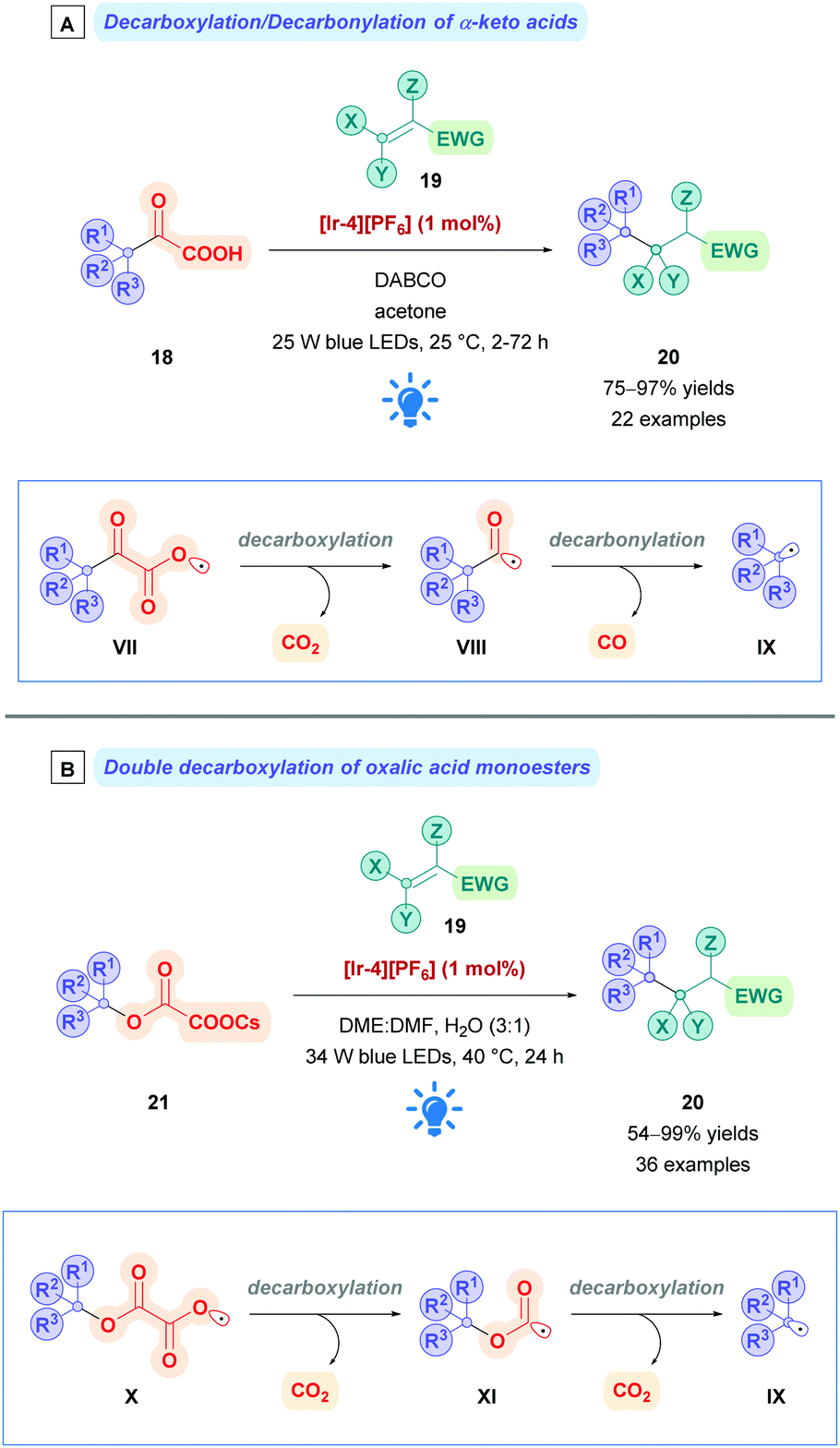 | ||
| Scheme 10 Photocatalysed Giese alkylations using (A) α-keto acids and (B) oxalates. | ||
Rueping and co-workers expanded the utility of amino acid derived α-amino radicals for both primary and secondary amines (Scheme 11).34 Employing PC [Ir-2][PF6], N-aryl glycine derivatives (24a–d, h–m) were of primary focus, but other amino acids such as N-phenyl alanine (24e), valine (24f), and serine (24g) were also investigated, overall achieving moderate to good yields (42–86%). Halogenated aryl rings were tolerated (24a–c), and poor reactivity was observed with EDGs in the para position; yields were improved when these groups occupied the ortho position (24d). Accessing a quaternary centre (24i) was also successful in appreciable yields. The group also attempted to further the scope by applying the reaction conditions to amino acids bearing N-unsaturated heterocycles (23). Though using PC [Ir-2][PF6] did not give the desired transformation, switching to the more oxidising [Ir-4][PF6] furnished the conjugate products (e.g., 25a–b) in good yields (60% and 73% yields respectively). The group's methodology has the added benefit of using catalytic amounts of basic Li2CO3, which was shown to be essential for successful coupling. The reaction time was also noticeably shorter (9 h) compared to most photocatalytic reactions, however, the presence of an inert Ar atmosphere was a necessity.
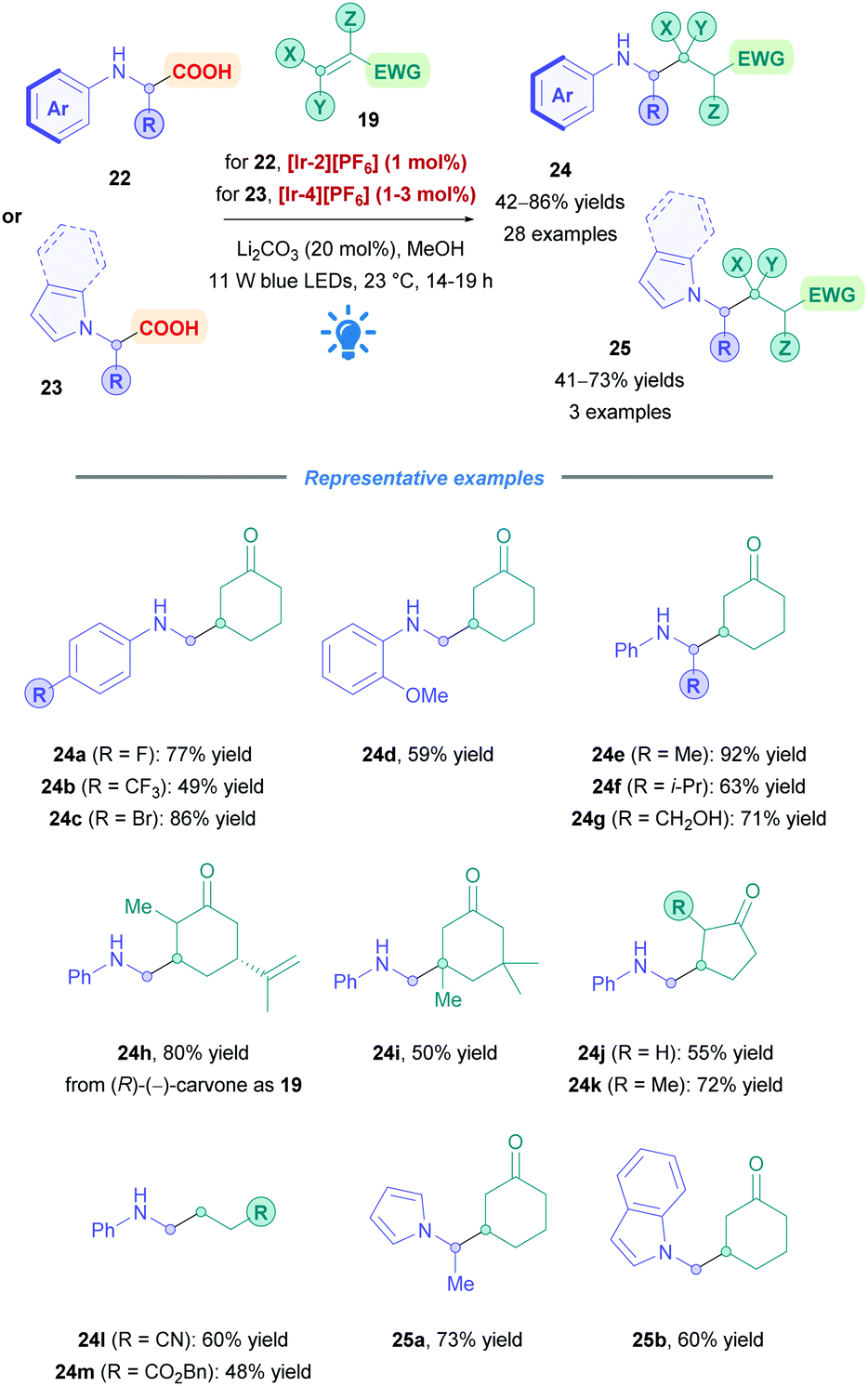 | ||
| Scheme 11 Rueping's Ir-catalysed direct DGR of N-aryl-α-amino and N-heterocycles. | ||
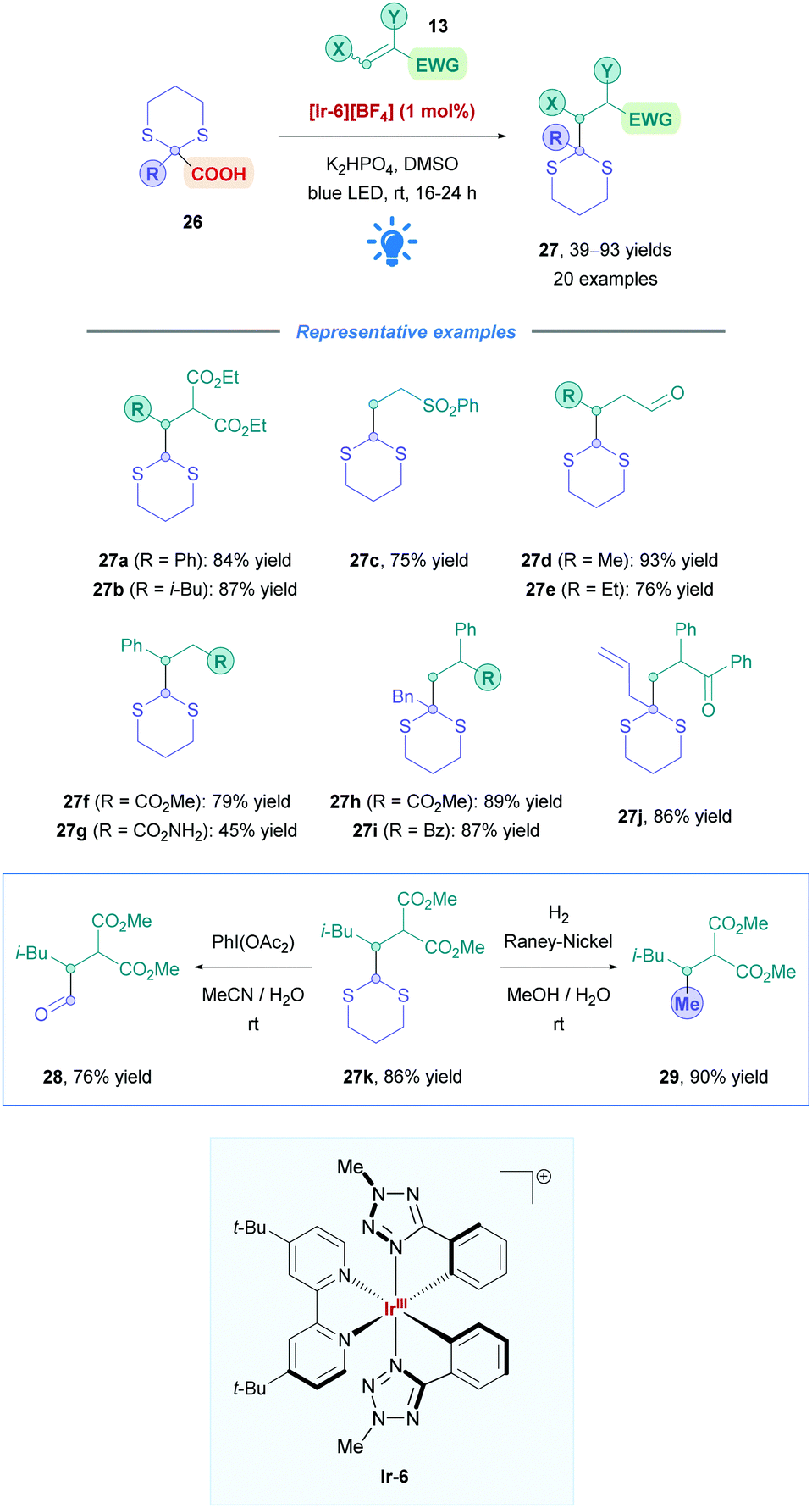
The most popular and effective Ir photocatalyst for DGRs is [Ir-4][PF6], but it is expensive and requires additional ligand synthesis step(s) for its preparation.35 To overcome these limitations, Cozzi and co-workers employed an alternative Ir complex [Ir-6][BF6] which possesses phenyl tetrazole ligands. This catalyst has the advantage of being more easily prepared, and the efficiency is comparable to photocatalyst [Ir-4][PF6].36 They applied catalyst [Ir-6][BF6] to the DGR of dithiane radical precursors (26), acting as a potential surrogate for a methyl  radical (Scheme 12).37 Due to their inherent instability, the generation of primary radicals, especially methyl radicals, via decarboxylation continues to be a challenge within photocatalysis. Dithianes also act as useful functional handles for the installation of carbonyl-bearing groups (27k into 28) and alkylation (27k into 29). The reaction has a broad substrate scope with excellent yields (up to 93%) for a variety of Michael acceptors (e.g., unsaturated esters, amides, and aldehydes) (27a–g), as well as 2-substituted dithiane-2-carboxylic acids (27h–j). Due to the lower reduction potential of its photoexcited state (*EIII/II1/2 = +0.4 V vs. ferrocene/ferrocenium couple), catalyst [Ir-6][BF6] has a greater selectivity for the oxidation of α-functionalised acids than catalyst [Ir-4][PF6]. Scale-up from 0.2 mmol to 2.4 mmol was also successful without any issues.
radical (Scheme 12).37 Due to their inherent instability, the generation of primary radicals, especially methyl radicals, via decarboxylation continues to be a challenge within photocatalysis. Dithianes also act as useful functional handles for the installation of carbonyl-bearing groups (27k into 28) and alkylation (27k into 29). The reaction has a broad substrate scope with excellent yields (up to 93%) for a variety of Michael acceptors (e.g., unsaturated esters, amides, and aldehydes) (27a–g), as well as 2-substituted dithiane-2-carboxylic acids (27h–j). Due to the lower reduction potential of its photoexcited state (*EIII/II1/2 = +0.4 V vs. ferrocene/ferrocenium couple), catalyst [Ir-6][BF6] has a greater selectivity for the oxidation of α-functionalised acids than catalyst [Ir-4][PF6]. Scale-up from 0.2 mmol to 2.4 mmol was also successful without any issues.
 | ||
| Scheme 12 Ir-Catalysed direct DGR of dithiane. | ||
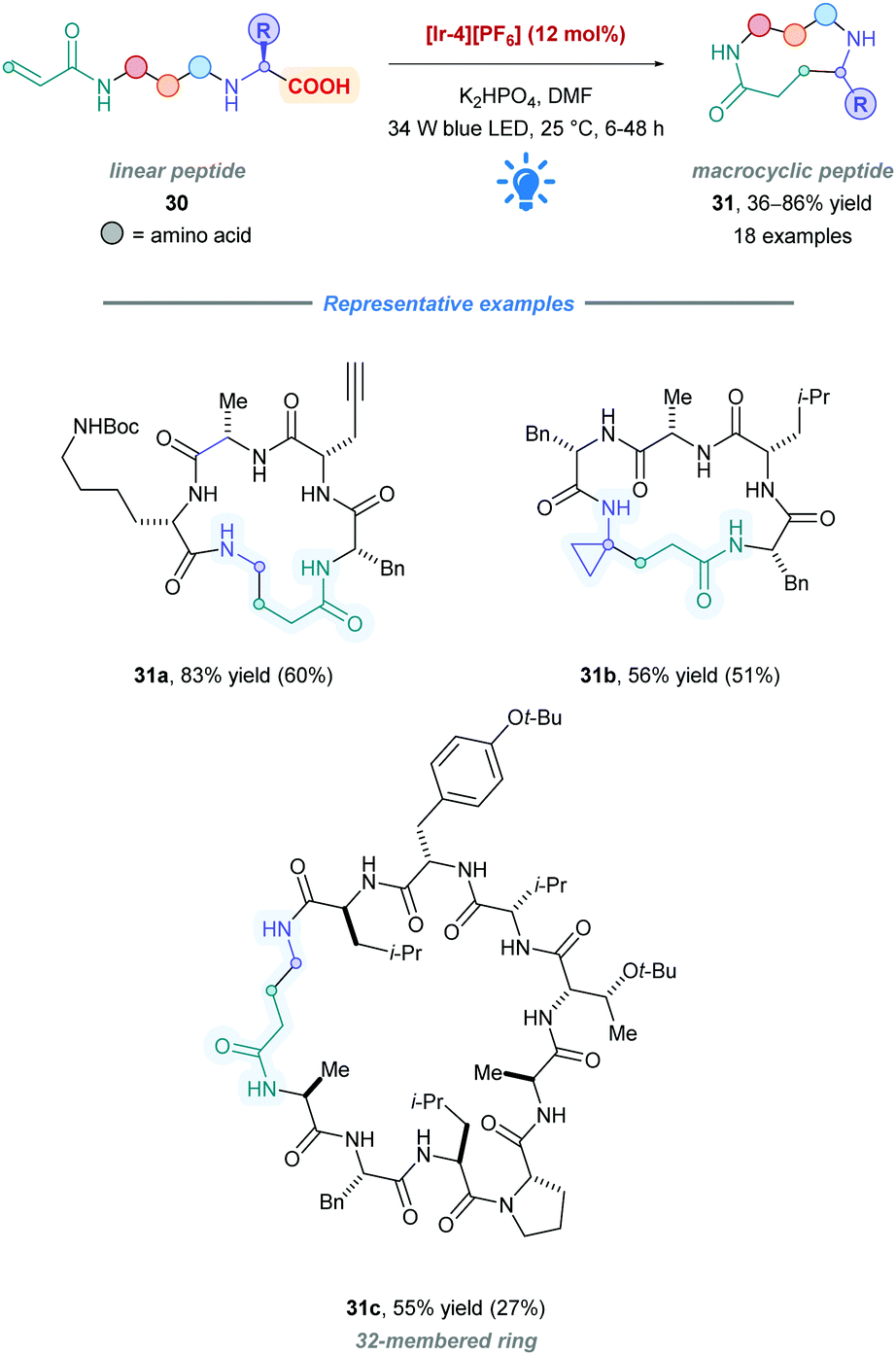
Following their seminal 2014 work,26 the MacMillan group applied their previous protocol to access macrocyclic peptides (31) (Scheme 13).38 They sought to utilize the C-terminal carboxylate to generate an α-amino radical, which subsequently undergoes intramolecular conjugate addition with a pendant Michael acceptor in the form of an acrylamide. High catalytic loadings (12 mol% of [Ir-4][PF6]) accommodated the high dilution necessary to diminish oligomerisation pathways. Peptide lengths ranging from 3 to 15 amino acids (31a–c) with many functional side chains were all shown to be successfully cyclised with reasonable yields. Owing to their lower pKa and oxidation potential, high selectivity was observed for decarboxylation of the α-amino acid C-terminal residue in preference to any side-chain carboxylic acids (Ered1/2 = +1.2 V vs. SCE for Boc-Gly-CO2K).38 In many of the cases, purification of the cyclic peptides, unfortunately, resulted in significantly reduced yields when compared to crude yields.
 | ||
| Scheme 13 Ir-Catalysed direct DGR for peptide macrocyclization. Yield of isolated product obtained by preparative HPLC in parentheses. | ||
Very recently, Ruf and co-workers expanded upon the work of MacMillan and co-workers to show successful C-terminal derivatisation of small peptide substrates.39 They used a more extensive array of Michael acceptors including α,β-unsaturated ketones, as well as less electrophilic olefins such as cinnamic acid ester and vinyl-substituted (hetero)aromatic motifs.
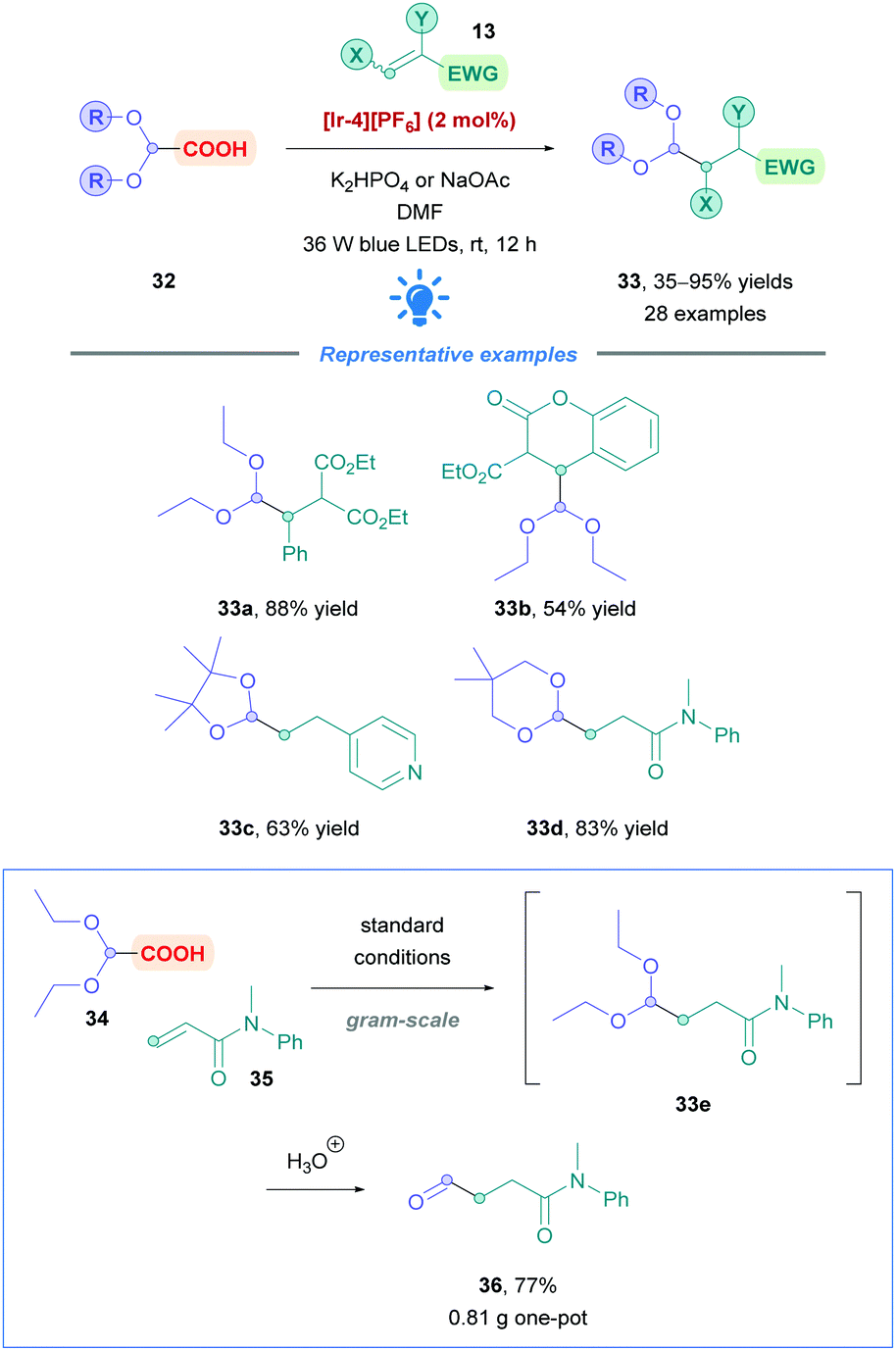
Xu and co-workers sought to introduce a formyl group across Michael acceptors using glyoxylic acid derivatives (32) as synthetic equivalents.40 Upon Giese addition of the glyoxylic acid derivative 32 simple treatment with acid furnishes the desired aldehyde (Scheme 14). The group paid particular attention to Michael acceptors that are generally less effective substrates for transition-metal-catalysed hydroformylation using CO and H2 as syngas.41 A broad scope of Michael acceptors showed moderate to excellent yields (35–95%), including α,β-unsaturated esters (33a–b), amides (33d), aldehydes, ketones, and nitriles. It is worth mentioning also that vinylpyridine and α-aryl styrene were successful substrates (e.g.33c). Good functional group tolerance was observed such as with aryl chlorides, aryl fluorides and boronic esters. Giese addition onto styrene, or styrenes possessing EWGs, proved to be fruitless, possibly due to oligomerisation, or an energy transfer event between the Ir PC and styrene. In addition to glyoxylic acid diethylacetals as the primary substrate, cyclic acetals 4,4,5,5-tetramethyl-1,3-dioxolane-2-carboxylic acid and 5,5-dimethyl-1,3-dioxane-2-carboxylic acid underwent efficient Giese addition (33c and d, respectively). The value of this protocol was demonstrated through the gram-scale, one-pot synthesis of aldehyde 36, among others, achieving yields comparable to that of the small-scale reactions.
 | ||
| Scheme 14 Ir-Catalysed direct DGR of glyoxylic acids. | ||
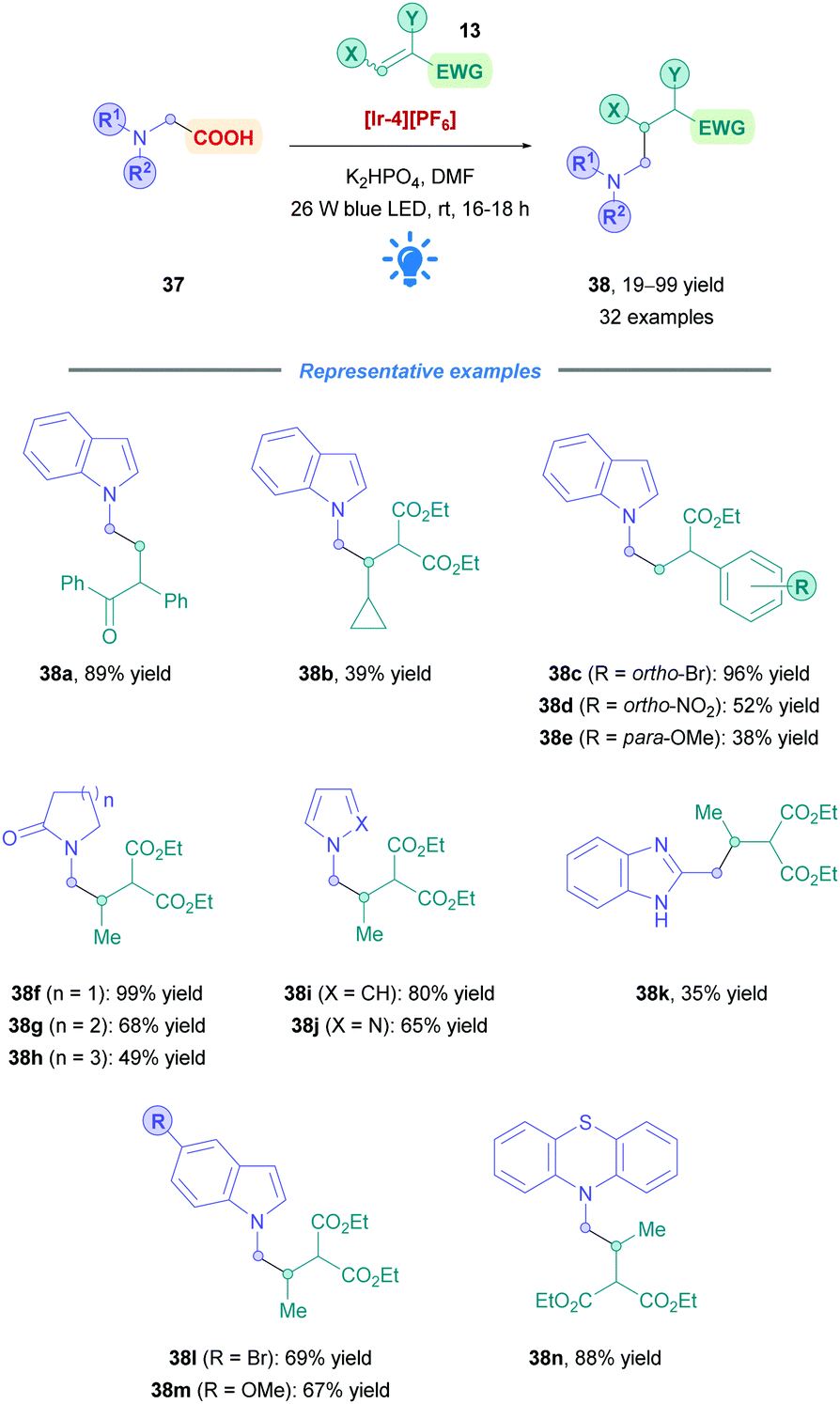
The next advancement was driven by the fact that N-substituted heterocycles are important scaffolds for the pharmaceutical industry. Seeking to expand access to this motif, AstraZeneca reported the DGR of N-methyl radicals, using N-substituted acetic acids (37) as easily accessible radical precursors (Scheme 15).42 They employed [Ir-4][PF6] to install weakly nucleophilic heterocyclic N-substituted acetic acids with low to excellent yields (19–99%), with substrates including indoles (38a–e, l and m), indazoles, imidazoles, benzimidazoles (38k), pyrroles (38i), and pyrazoles (38j). Efficient Giese addition was observed for olefins bearing halogens, EWGs and EDGs with good to excellent yields (38c–e, l and m). Moreover, cyclic amides of different ring sizes (38f–h) and fused phenyl substituted cyclic amides showed smooth DGR. Giese addition appeared not to be significantly hindered when increasing the steric bulk on the olefin. Indeed, it was shown that unsubstituted maleates and acrylates underwent limited conjugate addition. Prolonged irradiation of up to 16–18 h was required for all substrates, possibly due to the weak nucleophilicity of the generated N-methyl radicals.
 | ||
| Scheme 15 Ir-Catalysed direct DGR of α-amino acids. | ||
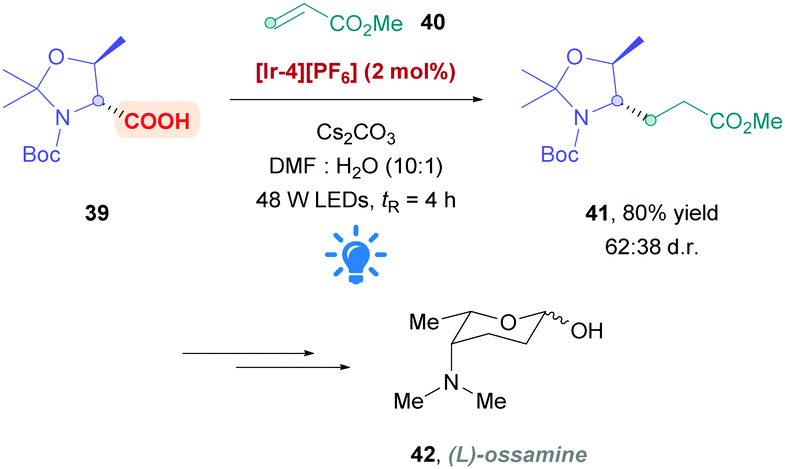
MacMillan's original protocol26 has also been applied by the Fujimoto group to the formal synthesis of L-ossamine (42), a deoxyaminosugar motif found in the polyketide natural product ossamycin (Scheme 16).43 Using Ir complex [Ir-4][PF6] as the PC, light-promoted DGR of the protected D-threonine onto methyl acrylate was performed in flow with a residence time of 4 h to give conjugate addition product 41 in 80% yield and moderate diastereoselectivity (62![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :38 d.r.).
:38 d.r.).
 | ||
| Scheme 16 Use Ir-catalysed direct DGR for the total synthesis of (L)-ossamine. | ||
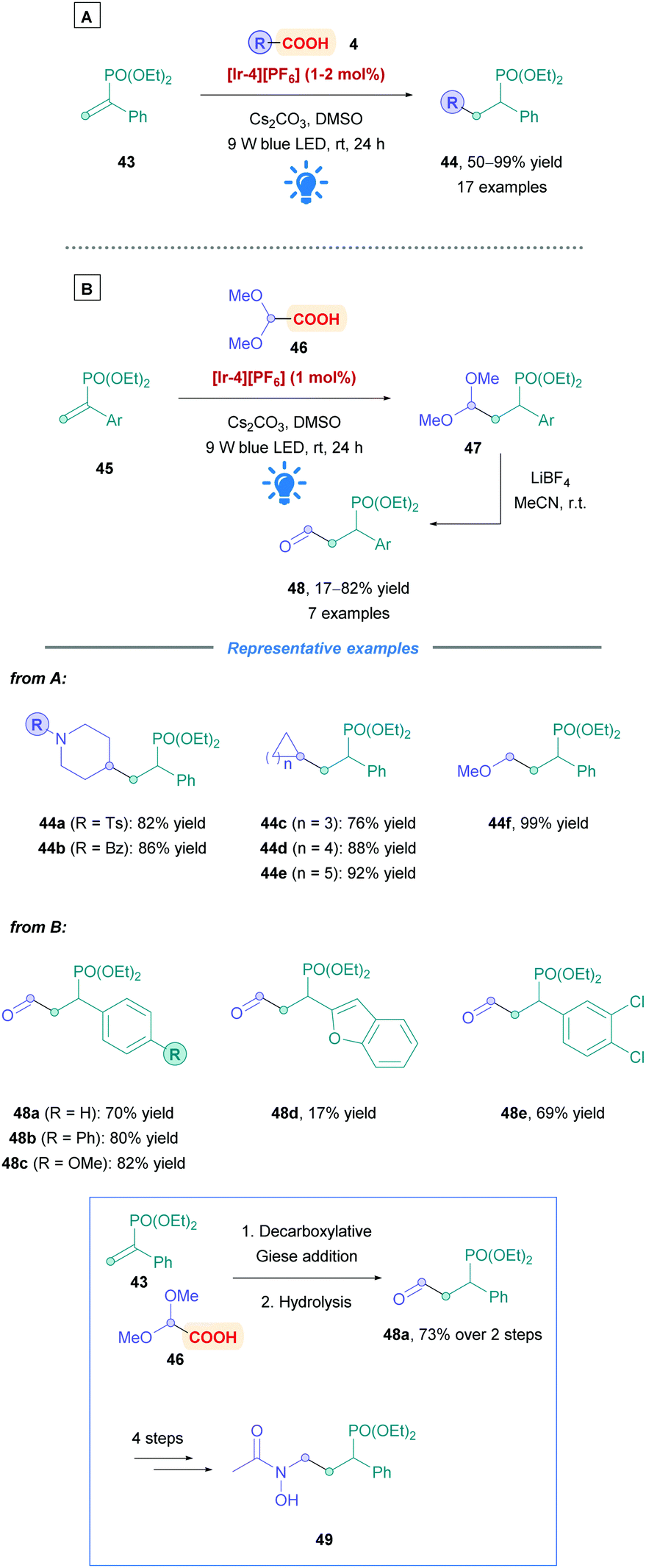
2.1.1.2 Further studies on other Michael acceptor substrates. In addition to exploring the possible carboxylic acid substrates that could be used, some work has been carried out to identify Michael acceptors that offer unique functional handles. For example, Tian and co-workers used α-aryl vinylphosphonates (43) to generate valuable α-aryl alkylphosphonates (44) with moderate to excellent yields (50–99%) (Scheme 17A).44 α-Aryl phosphonates have stirred an interest within medicinal chemistry because of their attractive biological properties.44,45 Furthermore, vinylphosphonates are less prone to undergo oligomerisation in comparison to their conjugated carbonyl counterparts. The group reported a broad substrate scope using Ir PC [Ir-4][PF6], with good efficiency and functional group tolerance (44a–f and 48a–e). Addition of benzyl radicals was shown to be limited, but using potassium benzyltrifluoroborate as a radical precursor, instead of phenylacetic acid, significantly improved yields. Hydroformylation, similarly to the work of Xu,40 could be achieved through sequential Giese addition using glyoxylic acid acetal, followed by LiBF4-mediated hydrolysis, affording a variety of diethyl (1-aryl-3-oxopropyl)phosphonates (45) (Scheme 17B). They also showcased the utility of their protocol by providing a synthesis of the fosmidomycin derivative 49. Competitive decarboxylative alkylation experiments gave evidence that unsaturated carbonyls are better radical acceptors than vinylphosphonates.
 | ||
| Scheme 17 Ir-Catalysed direct DGR onto vinylphosphonates with (A) alkyl acids and (B) glyoxylic acids. | ||
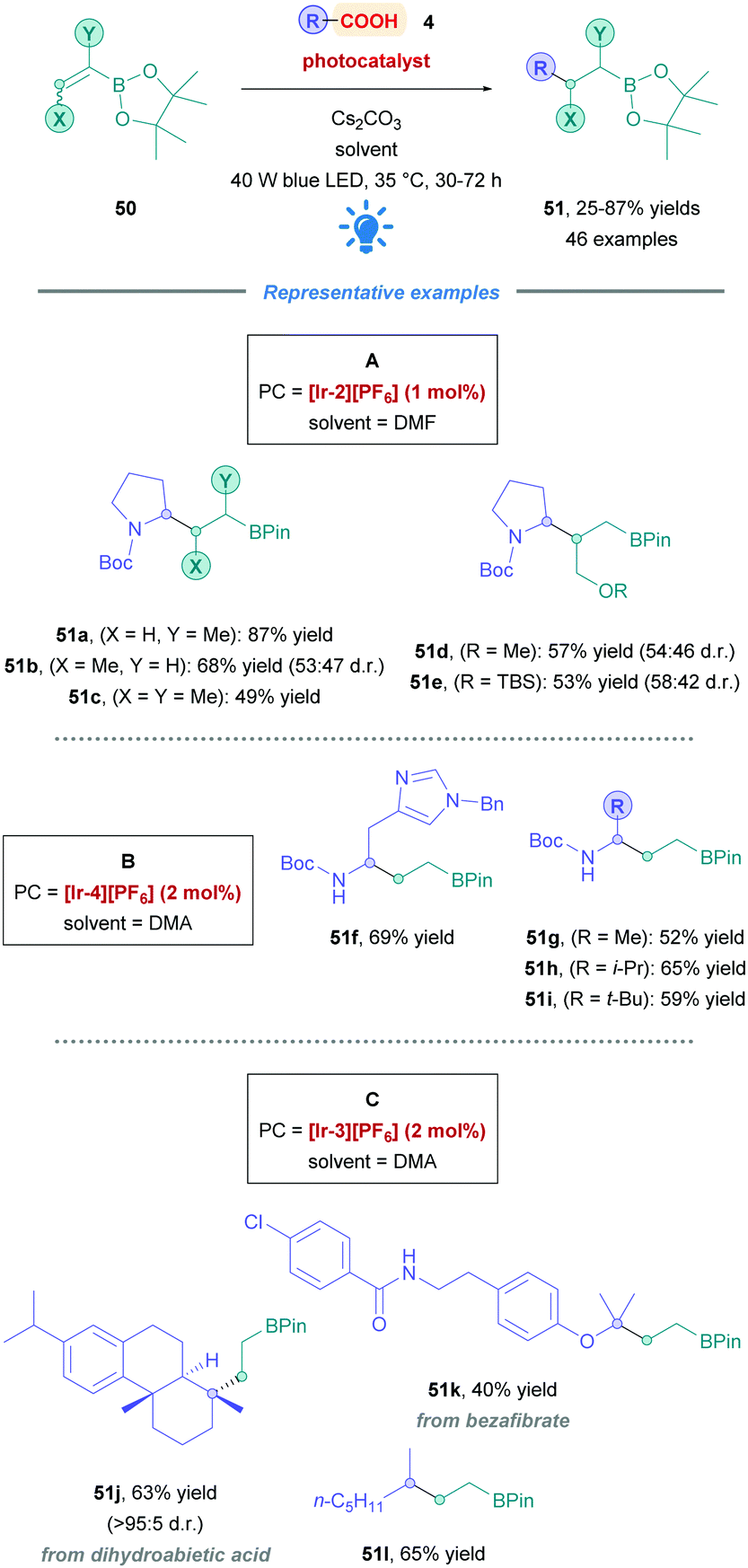
The Aggarwal group described a novel DGR of amino acid derivatives onto vinyl boronic esters (50), accessing a myriad of boronic esters (Scheme 18) (51).46 The proposed mechanism surprisingly involved a unique reductive SET of an α-boryl radical by the reduced-state PCred, which was further supported by deuterium-labelling studies and DFT (density functional theory) calculations. Various cyclic and acyclic amino acids proceeded with good yields (51a–i). Catalyst [Ir-2][PF6] was most effective for amino acids possessing tertiary N-protected amines (Conditions A). However, amino acids bearing free NH groups required minor modifications to the reaction conditions, changing the PC to [Ir-4][PF6] and solvent to DMA (Conditions B). Various vinyl boronic ester derivatives were examined, obtaining products with moderate to good yields (51a–e, 53–87%). Unfortunately, α-styrenyl boronic esters was rendered incompatible because of the rapid protodeboronation under the reaction conditions. The group further investigated the substrate scope by applying the protocol to simple alkyl carboxylic acids (51j–l). Again, reaction conditions were further tuned by employing the more reducing PC [Ir-3][PF6] (Condition C). Natural product or marketed drug carboxylic acids (e.g., 51j–k) underwent Giese addition with moderate yields (40–63%), demonstrating the potential for application to late-stage functionalisations.
 | ||
| Scheme 18 Ir-Catalysed direct DGR onto vinyl boronic esters (A) cyclic and (B) acyclic amino acids, and (C) alkyl carboxylic acids. | ||
Identifying new peptide-based drug candidates remains of interest within the pharmaceutical industry.47 Peptides solely consisting of natural amino acids tend to have poor pharmacokinetic properties primarily due to their low membrane permeability and metabolic instability.47 Thus, much effort has focused on the synthesis and installation of unnatural amino acids (UAAs) to overcome some of these limitations. Recently, Gómez-Suárez and co-workers developed a convenient preparation of diverse range of UAAs through direct DGR onto chiral dehydroalanine derivative (Dha, 52)48 (Scheme 19).49 This versatile Michael acceptor can be accessibly synthesised in a 3-step procedure from benzyl cysteine on multigram scales.49 Employing [Ir-4][PF6] as the PC, a whole host of acid substrates where tested to give either acylated or alkylated syn oxazolidinones (53) with excellent diastereoselectivity (d.r. >20:1). Broad substrate scope with high functional group tolerance was observed for aliphatic α-keto acids (53a and c), as well as primary, secondary and tertiary aliphatic acids (53b, d, and e). Low to excellent yields were obtained throughout (10–98%). Having accessed the oxazolidinone core, and treatment with acid gives rise to the desired (L)-amino acid (e.g., 54). Subsequent N-Fmoc protection then renders the UAA suitable for solid phase peptide synthesis. Alternatively, further enolate-assisted alkylation methodologies could be employed to further increase complexity and stereochemical information, such as the formation of 55.
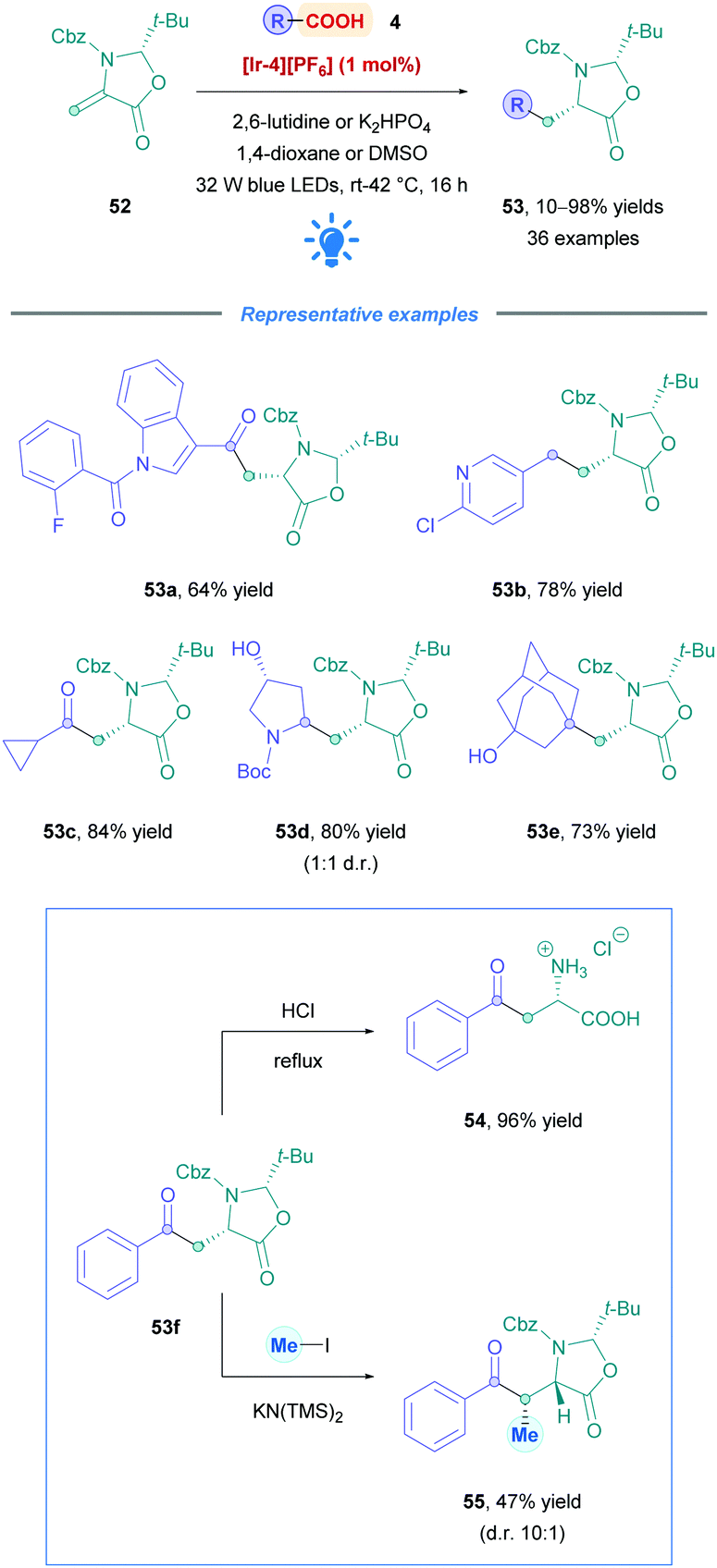 | ||
| Scheme 19 Diastereoselective Ir-catalysed direct DGR onto Dha. | ||
During their total synthesis of (+)-fastigiatine 59, the Rychnovsky group employed a MacMillan-type protocol26 to install the protected glycine derivative 57 onto the bicyclic α,β-unsaturated ketone 56 to afford the Giese product 58 (Scheme 20).50 Using PC [Ir-4][PF6], they achieved a very good yield of 59 (87%) with the desired diastereomer favoured by a 1:5.2 ratio. Interestingly, a previous approach using aminomethyl cuprate addition resulted in poorer diastereoselectivity.
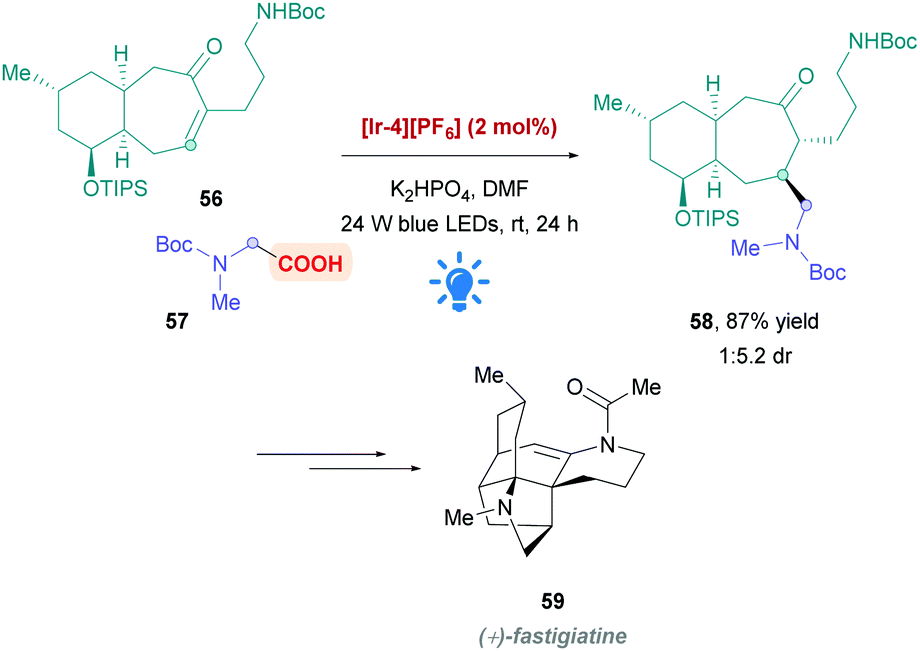 | ||
| Scheme 20 Use Ir-catalysed direct DGR for the total synthesis of (+)-fastigiatine. | ||
Another intriguing mode for Fe-based photocatalysis is through intramolecular charge-transfer, whereby the organic substrate is directly bound to the metal centre. This approach negates the need for long-lived transition states and takes advantage of photo-labile metal–ligand bonds. In 1953, Parker and co-workers reported the photolysis of ferrioxalate which gave rise to decarboxylation,54 and the decades proceeding has seen extensive mechanistic studies of this process.55 A recent review by Reiser and co-workers discusses some of the latest advances in visible-light induced homolysis of earth-abundant organometallic complexes.56 Recently, Jin and co-workers took advantage of the photoredox-active nature of Fe(III) carboxylates to achieve direct DGR onto vinylidene cyanide derivatives (60). Fe(III) carboxylates were generated from catalytic amounts of Fe2SO4, and gave low to excellent yields (17–99%) of the corresponding Giese adducts (61).57 They postulated a mechanism (Scheme 21) whereby the carboxylic acid 4 initially coordinates to the Fe(III) species L-FeIII, forming the Fe(III)–carboxylate complex XIV. Visible-light irradiation (427 nm) promotes excitation of XIV, triggering an intramolecular ligand-to-metal-charge-transfer (LMCT) event which induces decarboxylation to generate the alkyl radical I and the reduced Fe(II) complex L-FeII. Radical addition of I onto Michael acceptor 60 generates radical intermediate XII, which engages in SET with L-FeII to form the anion XIII whilst the Fe(II) is re-oxidised to Fe(III) (L-FeIII) to close the catalytic cycle. Final protonation of XIII furnishes the desired Giese adduct 61. The entire process benefits from being redox neutral, avoiding the use of any additional oxidant.
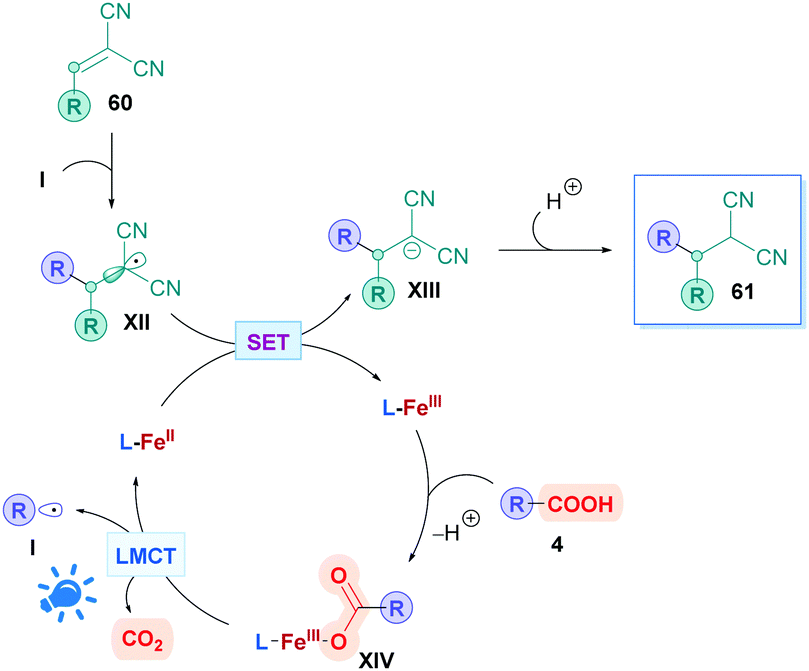 | ||
| Scheme 21 Proposed mechanism for Fe-catalysed direct DGR. | ||
Reaction optimisation studies unveiled that the di-(2-picolyl)amine 62 served as the best ligand, and no additional base was required for acid deprotonation (Scheme 22). The Michael acceptor scope was primarily restricted to 2-benzylidenemalononitrile derivatives 60, with aryl motifs bearing electron-withdrawing and -donating groups showing to be well tolerated (e.g., 61a–c). 2-Alkylidenemalononitrile (61d) and malonate-type Michael acceptors (61e) could also be employed with synthetically useful yields (69%). Regarding the acid scope, a wide variety of different acid types could be employed. Stabilised radical precursors including α-oxy (61f), -amino (61g), and benzyl acids (61h and i) gave generally good overall yields (30–97%) of their corresponding adducts. Acids that form unstabilised primary (61j), secondary (61k), and tertiary (61l–m) radicals also gave low to good yields (29–68%).
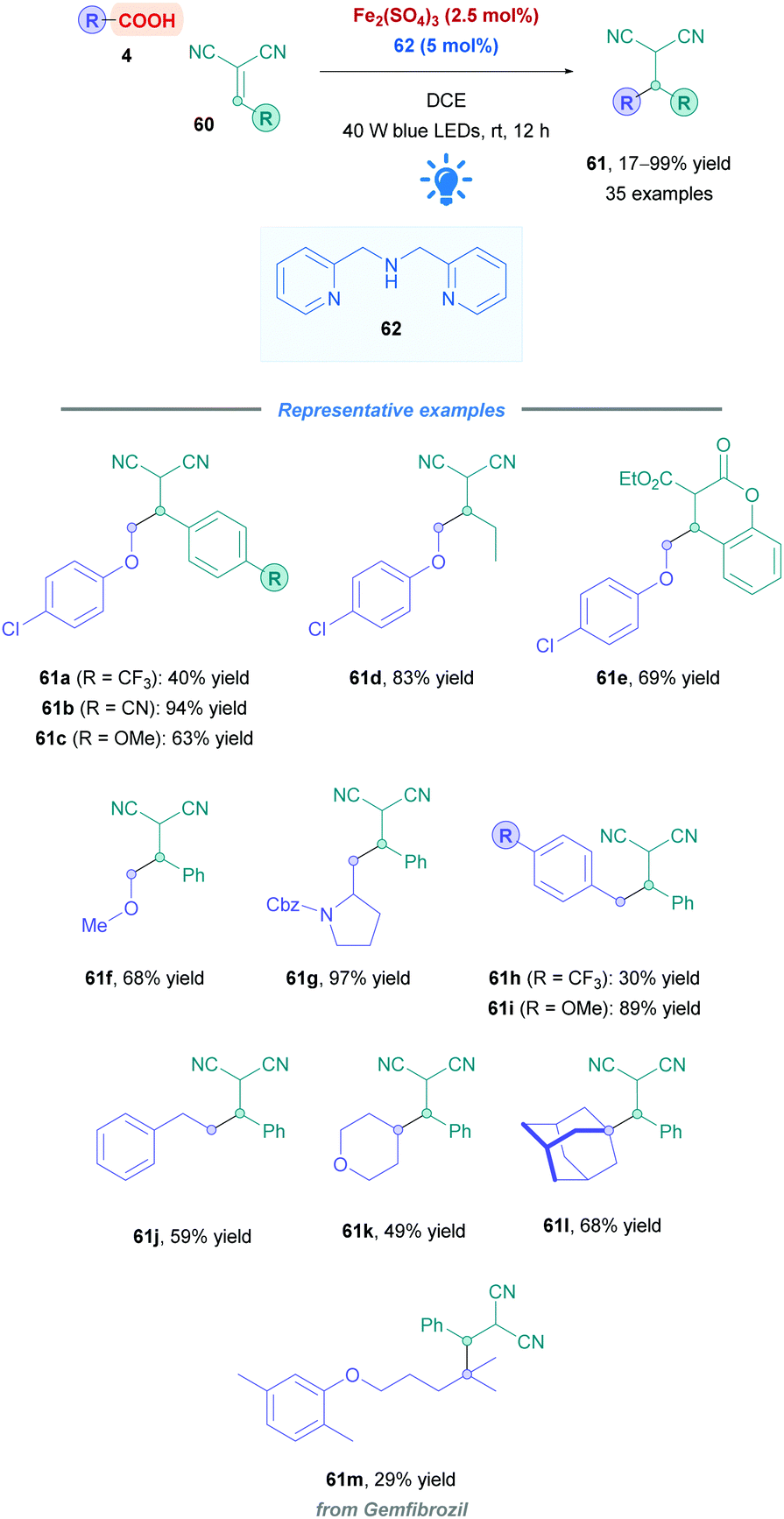 | ||
| Scheme 22 Fe-Catalysed direct DGR. | ||
Although photocatalysis using 3d transition metals does not yet recapitulate the transformations accessible using heavier transition metals, important advances are continuously made in this exciting research area. One possible development in this field would be to establish a broader substrate scope for direct DGRs.
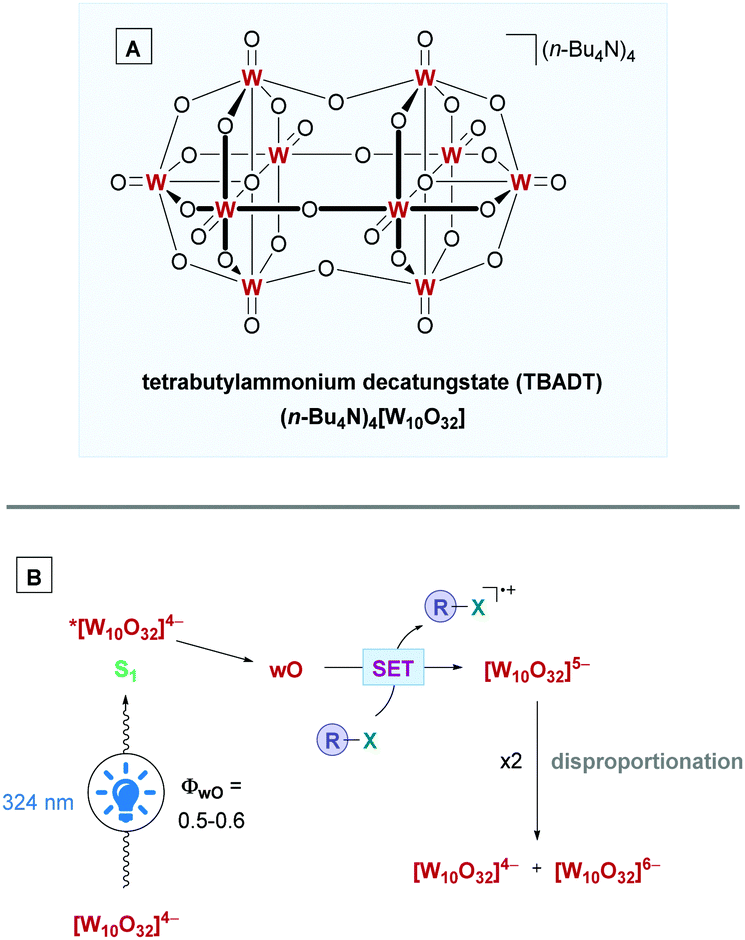 | ||
| Scheme 23 (A) Structure of TBADT. (B) Proposed mechanism for oxidation using TBADT. | ||
The mechanism of action for TBADT is depicted in Scheme 23B. Irradiation of [W10O32]4− with light (324 nm)61 initially forms the singlet excited state *[W10O32]4− (S1). Due to its short lifetime, in the order of a few tens of picoseconds, it does not interact with organic substrates and rapidly decays to the relaxed, more reactive excited state. This excited state is an unknown species and is frequently labelled as wO.58a,61wO is formed with a quantum yield (ΦwO) of around 0.5–0.6.62 The organic substrate can then interact with wO through oxidative SET, although, depending on the redox properties of the substrate, a HAT process is also possible. The reduction potential of wO, E(wO/[W10O32]5−) has been estimated to range from +2.26 to +2.61V vs. SCE,63 with a new study predicting a potential of +2.44 V vs. SCE.64 Reduction of wO gives the [W10O32]5− anion, which disproportionates over a longer time scale to the reduced form [W10O32]6− and the oxidised form [W10O32]4−. All the above decatungstate species mentioned, except for [W10O32]4−, give rise to the blue colouration.
Because of its high oxidation potential, Ravelli and co-workers sought to exploit TBADT for the direct DGR of benzyl radicals from aryl acetic acids.21 In contrast to secondary, tertiary, and α-heteroatom radicals, benzyl radicals exhibit a lack of reactivity towards electron-deficient olefins and have a tendency to dimerise.65 To overcome these issues, the group envisaged that the deactivated catalyst [W10O32]5− could serve to activate simple olefins through reduction, regenerating the photocatalyst [W10O32]4− and affording the corresponding radical anion of the olefin. This activated olefin would then act as a more efficient radical trap for the benzyl radical. The reduction potential of the reduced photocatalyst ([W10O32]5−) is estimated to be within the range of −0.9 and −1.4 V vs. SCE,21,66 whilst the formal reduction potentials (E0′) of simple olefins, e.g., maleic anhydride, maleimides, fumarates, and fumaronitrile, are within the range −1.09 and −1.47 V vs. SCE,63b thus the [W10O32]5− species can be sufficiently reducing for these olefins.
Reaction optimisation using phenylacetic acid and fumaronitrile with TBADT (4 mol%) in aq. MeCN solution, under UV radiation (310 nm), revealed that NaHCO3, NaClO4, and biphenyl were essential additives for an efficient Giese addition (83% optimised yield). The NaHCO3 served as a base to deprotonate the acid to the more reducible carboxylate, and the combination NaClO4/biphenyl is postulated to facilitate the electron transfer process.63b,67
The optimised conditions showed moderate to excellent yields for a variety of arylacetic acids (63) with Michael acceptors (19) (Scheme 24). Phenylacetic acids bearing the electron-donating methoxy group in the ortho- and para-positions were well tolerated (64c, 70% and 64a, 81%), and the electron-withdrawing meta-methoxy derivative unsurprisingly gave the lowest yield (64b, 54%), presumably due to lower radical nucleophilicity.68 Aryl moieties bearing deactivating groups in the form of halogens (64d–e) were similarly well tolerated but gave lower yields (44–56%). Good yields were also obtained for thiopheneacetic acid (64m, 52%) and N-Boc-protected aminophenylacetic acid (64f, 79%,) – the unprotected aminophenylacetic acid was insoluble under the reaction conditions. Interestingly, selective decarboxylation was observed when using (3,4-methylenedioxy)phenylacetic acid (giving 64l), despite the presence of the methylene hydrogen atoms which can be subjected to HAT by TBADT.69 2-Phenylpropionic and mandelic acid derivatives also proved to be successful substrates, however, mandelic acid itself was unsuccessful, giving rise to benzaldehyde instead. Other Michael acceptors including N-phenyl maleimide and benzylidenemalononitirile could also serve as efficient radical traps for benzyl radicals. Dimethyl fumarate (giving 64j) required an excess of acid and biphenyl to be omitted to improve yields, and its cis isomer dimethyl maleate (giving 64k) proved to be sluggish due to its higher reduction potential (−1.65 V vs. SCE).63b
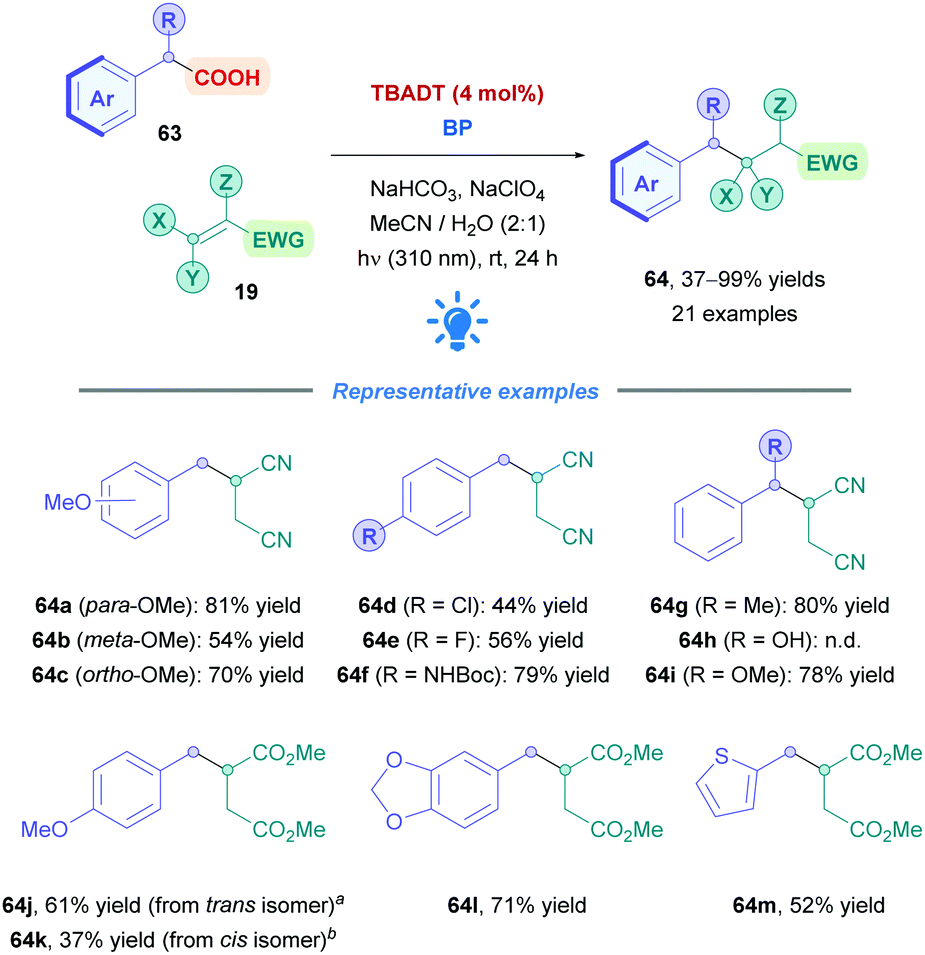 | ||
| Scheme 24 TBADT-catalysed direct DGR of benzylcarboxylic acids. | ||
The proposed mechanism is presented in Scheme 25, where, firstly, photoexcitation of [W10O32]4− populates the highly oxidising wO state. wO could undergo oxidative SET with carboxylate XV to the corresponding carboxyl radical XVI. This process, however, is slow as evidenced by low yields obtained from when performing the reaction without biphenyl. Two possible explanations could account for this behaviour. There may be electrostatic repulsion effects between wO and XV since they are both negatively charged. Alternatively, back-electron transfer (BET) may be involved that prevents decarboxylation. This slow reactivity is bypassed by the presence of biphenyl (BP), which is readily oxidised by wO (Ered1/2(BP˙+/BP) = +1.95 V vs. SCE)70 to the long lived radical cation BP˙+ together with [W10O32]5−. As well as being positively charged to overcome the electrostatic repulsions, BP˙+ can oxidise XV to trigger decarboxylation, giving the benzyl radical XVII and the regenerated BP. Meanwhile, the [W10O32]5− species can oxidise the olefin 19 to its activated radical anion XVIII whilst closing the catalytic cycle by reforming [W10O32]4−. Subsequent conjugate addition of XVII with XVIII, followed by protonation (either from the aqueous solvent or protonated base), affords the final product 64.
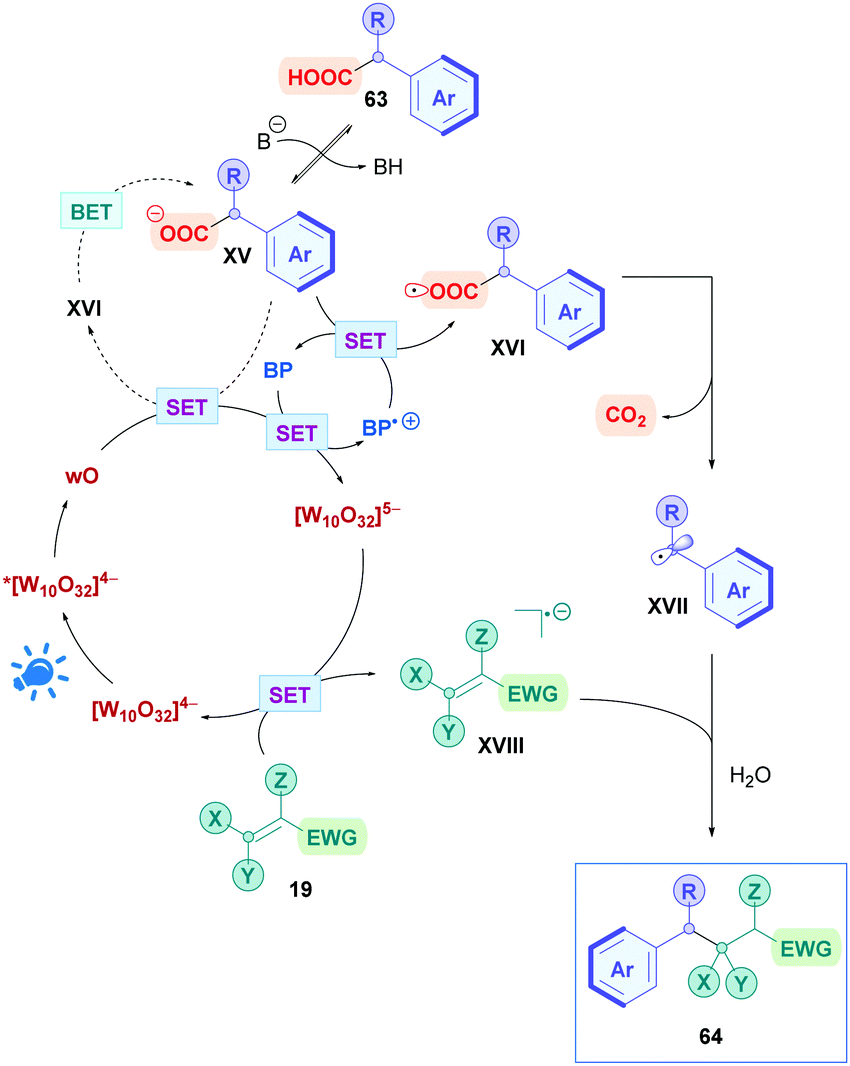 | ||
| Scheme 25 Proposed mechanism for TBADT-catalysed direct DGR of benzylcarboxylic acids. | ||
2.2 Organophotocatalysis
Despite the impressive utility of Ir-based photocatalysis, unsurprisingly, the major drawback for these catalysts is the scarcity of the metals used, leading to high financial costs and sustainability issues. Consequently, the field of organophotocatalysis has greatly expanded in recent years to find cheaper alternatives. In addition to lower costs, organic PCs are more sustainable and offer a myriad of modular scaffolds with characteristic and tunable physiochemical properties.71 Organo-PCs tend to achieve higher oxidation powers than their TM-PC counterparts, allowing access to thermodynamically challenging chemical transformations.71aIt is of no surprise that organophotoredox catalysis has found application within the realm of direct DGRs. This section will discuss some of the different families of organo-PCs that has been employed, again identifying any significant advantages and limitations for each system.
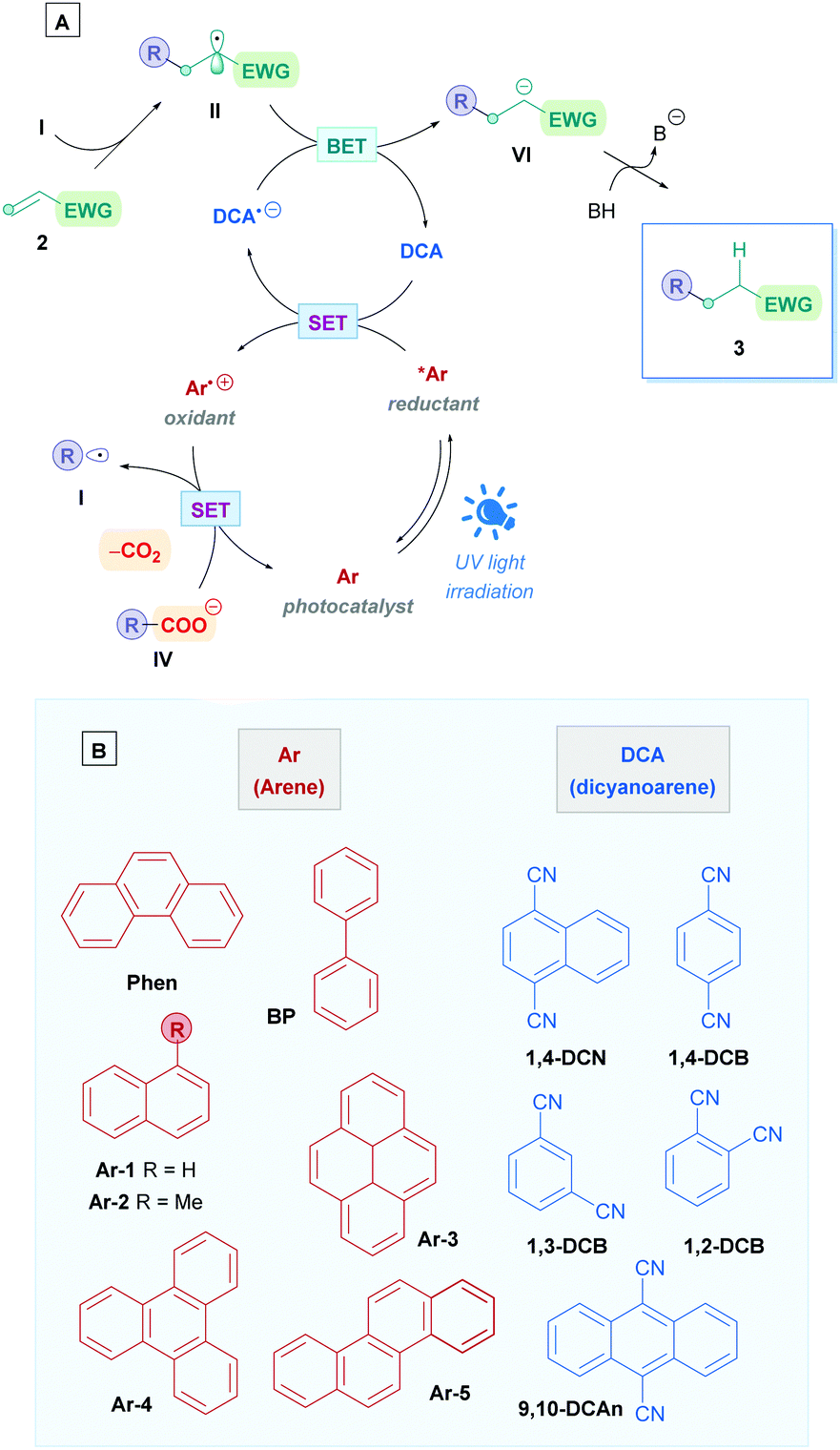 | ||
| Scheme 26 (A) Proposed mechanism for the direct DGR of carboxylic acids using Ar/DCA-coupled photoelectron transfer. (B) Some commonly employed arenes and dicyanoarenes for photocatalysis. DCN = dicyanonaphthalene, DCB = dicyanobenzene, DCAn = dicyanoanthracene. | ||
Yoshimi, Hatanaka and co-workers applied this catalytic process to promote Giese addition for a variety of different chemical substrates, which is summarised in Scheme 31. In their seminal work, they used phenanthrene (Phen) as the PC and 1,4-dicyanobenzene (1,4-DCB) as the co-catalyst to install N-protected amino acids onto simple Michael acceptors to generate N-Boc-γ-amino acids (Scheme 31A).72a Only one equivalent of olefin 2 was required to obtain moderate to excellent yields (36–88%) without the need of a base. In addition to single amino acids, N-protected dipeptides and tripeptides (66) were also successful substrates (Scheme 28A). Interestingly, acrylic acid also participated smoothly as a radical acceptor, suggesting that the difference between pKa of the starting α-amino acid and the γ-amino acid product is sufficiently large to prevent further decarboxylation, which would otherwise diminish the yield of 66. The group also carried out an assessment of this decarboxylative coupling for primary, secondary and tertiary carboxylic acids with acrylonitrile.72d Employing catalytic amounts of Phen and 1,4-dicyanonaphthalene (1,4-DCN) as catalytic partners, they achieved moderate yields (33–66%) for the respective Giese adducts. Long reaction times (18 h) were required due to the relatively higher pKa of the starting carboxylic acid, however, the time could be decreased with the addition of base such as tetra-n-butyl ammonium hydroxide. Sodium hydroxide was avoided due to the insolubility of the carboxylate sodium salts in acetonitrile. DMF can be used as a solvent to overcome this, but only with chrysene (Ar-5) and 1,4-DCB as catalysis partners. Moreover, they also showed that 1,4-DCN could be used as a solo catalyst to install N-Boc-amino acids onto acrylonitrile.72c However, the high oxidation potential of 1,4-DCN (1 equiv.) renders amino acids possessing aromatic rings incompatible, and longer reaction times or a more powerful light source (UV) were required. The proposed mechanism is depicted in Scheme 27, which is comparable to most of the mechanisms already shown in this review.
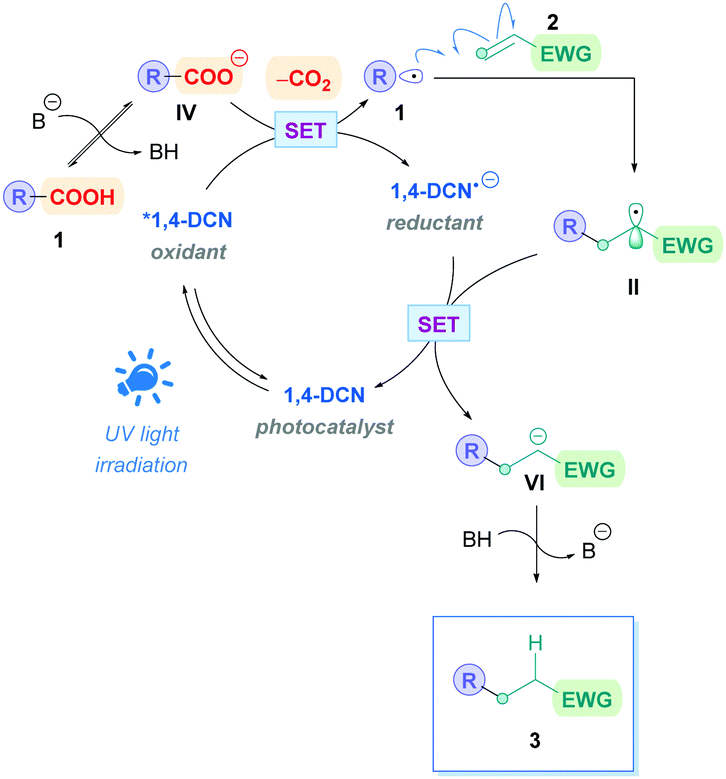 | ||
| Scheme 27 Proposed mechanism for DCN-catalysed direct DGR. | ||
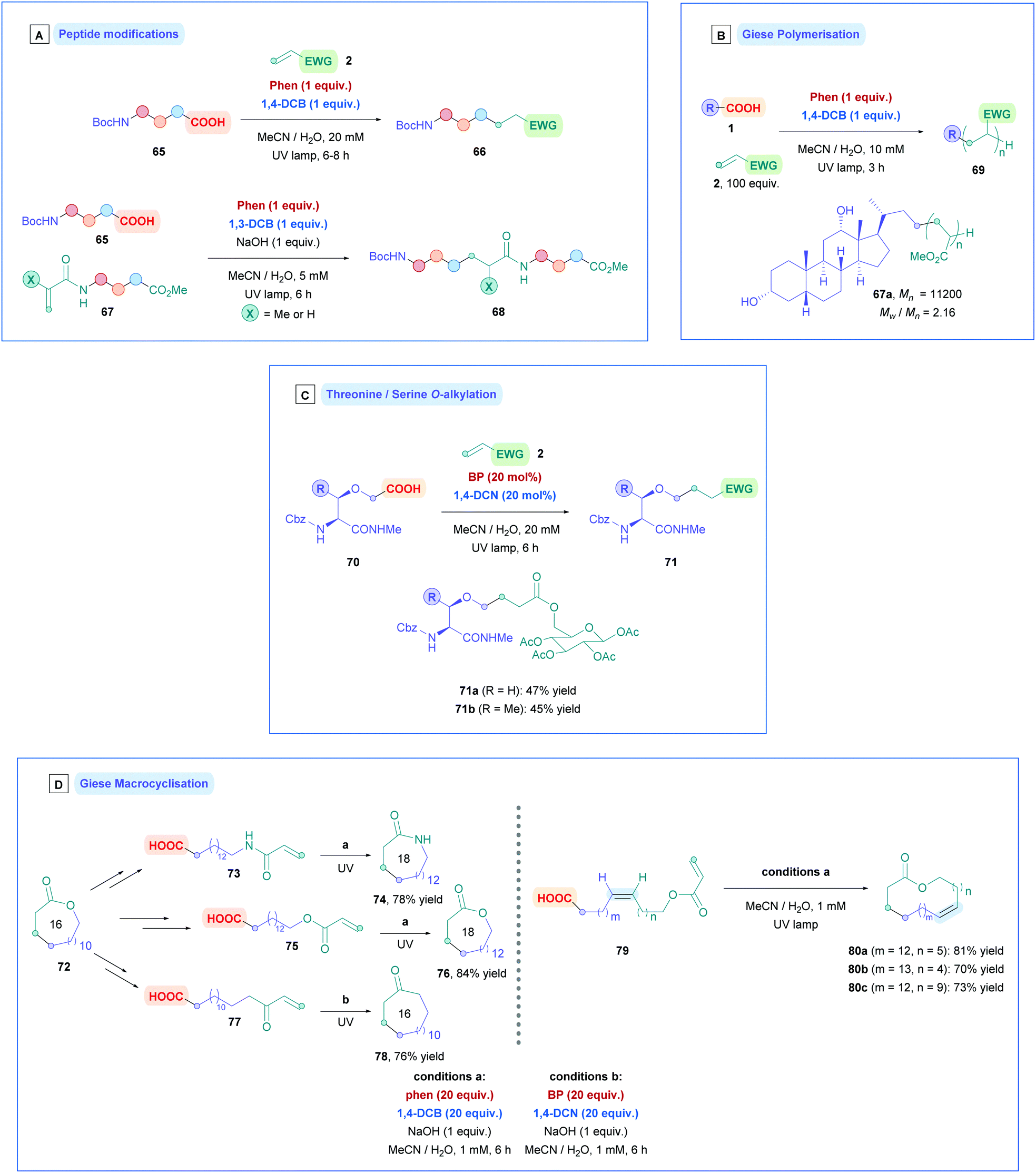 | ||
| Scheme 28 (A) Ar/DCA-catalysed direct DGR of amino acids and peptides. (B) Ar/DCA-catalysed direct decarboxylative Giese polymerisation. (C) Ar/DCA-catalysed direct DGR of threonine and serine derivatives. (D) Ar/DCA-catalysed direct DGR for macrocyclisation. | ||
Further studies to assess the influence of amino acid side chains for tripeptide systems were later conducted.72e Amino acids possessing electron rich substituents such as phenol and indole impeded the efficiency of the Giese addition, possibly due to these moieties quenching the photogenerated Phen radical cation (Ar·+). However, yields can be improved by appropriately installing a Boc protecting group onto these moieties. In contrast, the central amino acids possessing alkyl, phenyl, thioether, hydroxy and amide-containing side chains where all able to participate efficiently.
Installation of α-amino acids and peptides (65), along with a small collection of aliphatic carboxylic acids, onto N-acryloyl amino acid esters and peptides (67) was also successful using the photocatalytic combination of Phen and 1,3-DCB (Scheme 28A), synthesising N-alkylcarbonyl peptides (68).72f Lower yields (25–58%) were observed due to the slower addition of the alkyl radicals onto the less electron-deficient N-acryloyl moiety. As a further consequence of the slower radical addition, unwanted photosubstitution processes meant that 1,4-DCB could not be used, whereby formation of by-product alkylcyanobenzene via decyanation was apparent.
Recently, N-acryloyl amino acids and peptides have seen implementation in the sequential inter- and intramolecular radical cascade reactions involving the aromatic rings of tyrosine and phenylalanine derivatives (Scheme 29A).72k Preliminary studies using O-Boc protected tyrosine (81) with Phen and 1,3-DCB revealed that three products could be obtained (82–84). This can be rationalised using the proposed mechanism in Scheme 29B. Photoinduced decarboxylation generates the α-amino radical which can undergo Giese addition to the acrylamide partner 2, forming intermediate XIX. Radical XIX could either be reduced by BET with 1,3-DCB−˙ to the corresponding anion XX, followed by protonation to give the uncyclized Giese adduct 84. Alternatively, radical cyclisation onto the benzene ring gives radical XXI and the same sequential process of reduction and protonation would then afford the cyclised product 82 or 83. Interestingly, even though the negative charge in XXII can in theory be delocalised around the 6-membered ring, protonation of XXII does not seem to occur at the alternative sites to give more stable conjugated diene isomeric products, presumably for the same kinetic control reason as in the Birch reduction.74 The relative abundance of each observed product was dependent upon the electron-donating abilities of benzene ring in 81, steric hindrance of olefin 2, and the electrophilicity of radical XIX. The rate of radical cyclisation is increased by having a more electron-rich benzene ring and a less sterically demanding Michael acceptor. Using Michael acceptors such as styrene and acrylonitrile increases the electrophilicity of radical XIX, allowing for more efficient reductive BET to prematurely terminate radical cyclisation and favourably giving 84. In addition, acid substrates that do not bear an N-Boc group failed to undergo cyclisation, indicating the necessity of the N-Boc group to promote cyclisation. The stereochemistry of 82 and 83 was rationalised by using the two 6-membered transition states (Scheme 29C), where the N-Boc protected amino group occupies the equatorial position. Dipeptides containing C-terminal tyrosine were also subjected to the reaction conditions, forming ring-constrained peptides. The yields in these cases were diminished, presumably due to low solubility of the resulting peptides in the medium.
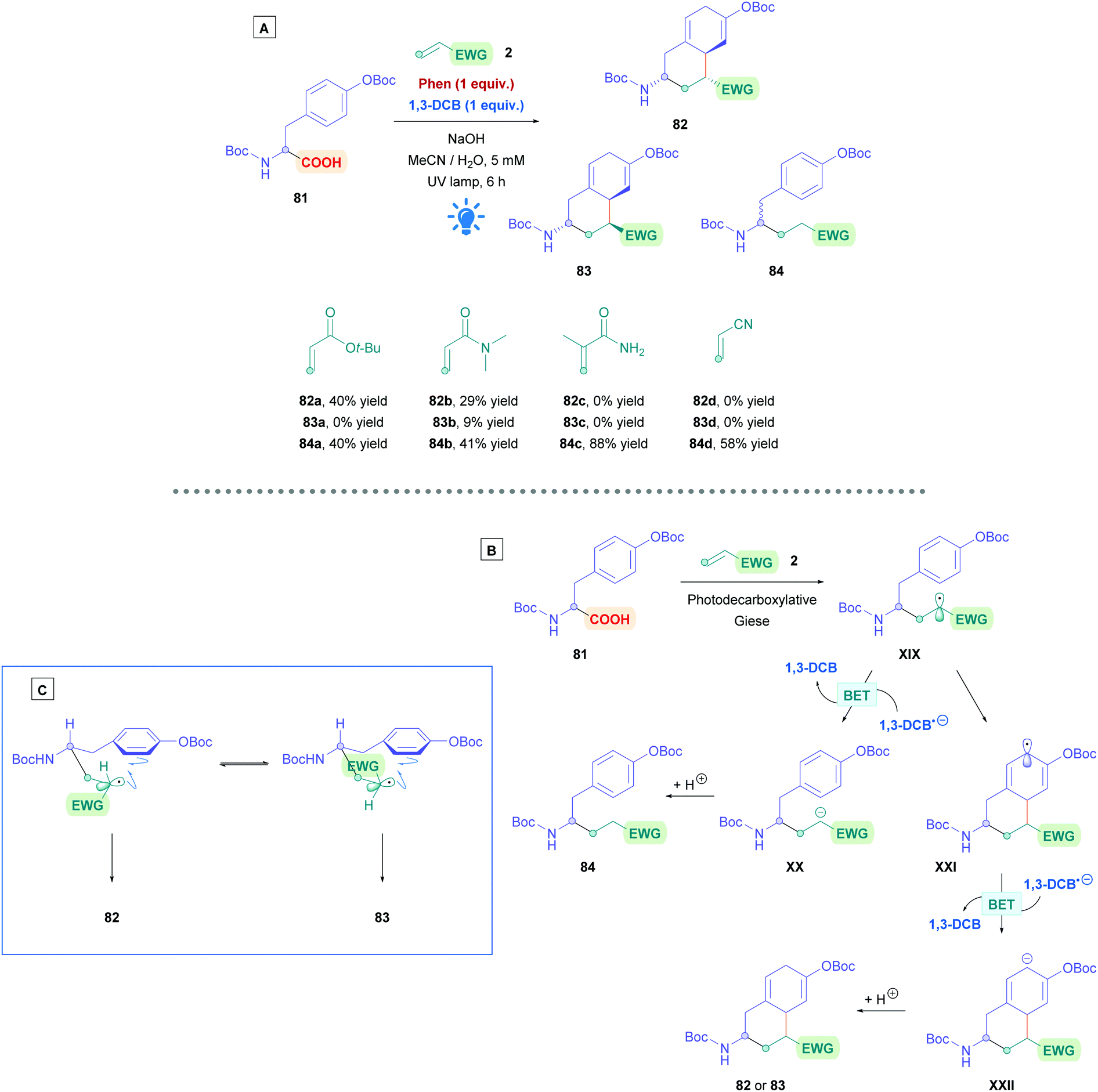 | ||
| Scheme 29 (A) Ar/DCA-catalysed direct DGR of O-Boc protected tyrosine. (B) Proposed mechanism for the Ar/DCA-catalysed direct DGR of O-Boc protected tyrosine. (C) Proposed rationale for sterochemistry. | ||
Radical polymerisation using this coupled photochemical system has also been explored (Scheme 28B). Yoshimi employed complex carboxylic acids to function as radical initiators which were incorporated as the polymer chain ends.72g Such acids included sugars, steroids, and peptides, thus adding unique complexity to polymer chains under mild conditions (e.g., 67a). Excellent conversions were observed throughout for the different acid substrates when using Phen and 1,4-DCB as catalysis partners. Sequential radical polymerisation of two or more different Michael acceptor monomers could also be attained. Good participation was observed for more sterically hindered Michael acceptors such as ethyl acrylate and t-butyl acrylate, with greater conversion for increasing sterics. A drawback of this protocol was the lack control of the molecular weight distributions, resulting in high polydispersities (Mw/Mn) being observed throughout (2.16–3.87), where Mw is the weight-average molecular weight, and Mn is the number-average molecular weight. The Mn value was shown to be dependent on the concentration of Phen and 1,4-DCB and types of electron acceptors. Using 1,4-DCN instead of 1,4-DCB resulted in more efficient BET which led to increased efficiency for chain termination.
Yoshimi also developed a strategic protocol for the O-alkylation of serine and threonine from serinyl and threoninyl acetic acids (70) (Scheme 28C).72i The synthesis of 70 involved a two-step procedure, firstly involving alkylation with α-bromo t-butyl acetate, followed by deprotection of the t-butyl group to expose the free carboxylic acid. No epimerisation of the stereogenic centre was observed during the synthesis. Dual-catalytic system involving biphenyl (BP) and 1,4-dicyanonaphthalene (1,4-DCN) were found to provide the highest yields of 71 (44–66%) compared to other catalytic systems. The use of NaOH was avoided to prevent base-promoted epimerisation and hydrolysis. The Michael acceptors used were predominantly acrylate- and acrylamide-protected amino acids and carbohydrates (e.g., 71a and b), giving their respective Giese adducts with moderate yields (up to 66%) without racemisation.
Furthermore, this photocatalytic system has been shown to induce intramolecular decarboxylative Giese cyclisation, generating macrocyclic lactones, lactams, and ketones ranging from 16- to 21-membered rings with excellent yields throughout (76–84%) (Scheme 28D left).72a,b The 16-membered lactone 72 was first transformed into a linear carboxylic acid possessing either a tethered acrylate amide (73), acrylate ester (75), or α,β-unsaturated ketone (77). Under high dilution conditions, photoinduced Giese cyclisation with Phen and 1,4-DCB, 73 and 75 gave their corresponding ring-expanded, 18-membered lactam (74) and lactone (76), respectively, with excellent yields (Conditions A). Lower loadings of the catalysis partners required prolonged reaction times, thus 20 equiv. of Phen and 1,4-DCB were used to complete cyclisation within 6 h. Photoinduced Giese cyclisation of 77 gave the corresponding macrocyclic ketone (78) of the same ring size (16-membered) as the original lactone 72. In this case, the slower radical cyclisation onto the less electron-deficient olefin required a combination of BP and 1,4-DCN as catalysis partners, since the radical anion derived from BP is longer lived than for Phen, allowing for more efficient BET of the α-carbonyl radical intermediate (Conditions B). To highlight the applicability of these methodologies, the same transformations that were applied to 72 could also be applied to 76 to generated 20-membered lactams and lactones, as well as 18-membered ketones.
Later developments introduced the synthesis for larger macrocyclic lactones (Scheme 28D right).72j Light-promoted decarboxylative Giese cyclisation of (Z)-alkene 79 afforded 25- (80a), 27- (80b), and 29-membered (80c) (Z)-selective lactones, and efficient hydrogenation afforded their corresponding saturated lactones. Precursor 79 could be easily prepared using a combination of Wittig chemistry and a more efficient installation of acryloyl groups, compared to their previous report.72b
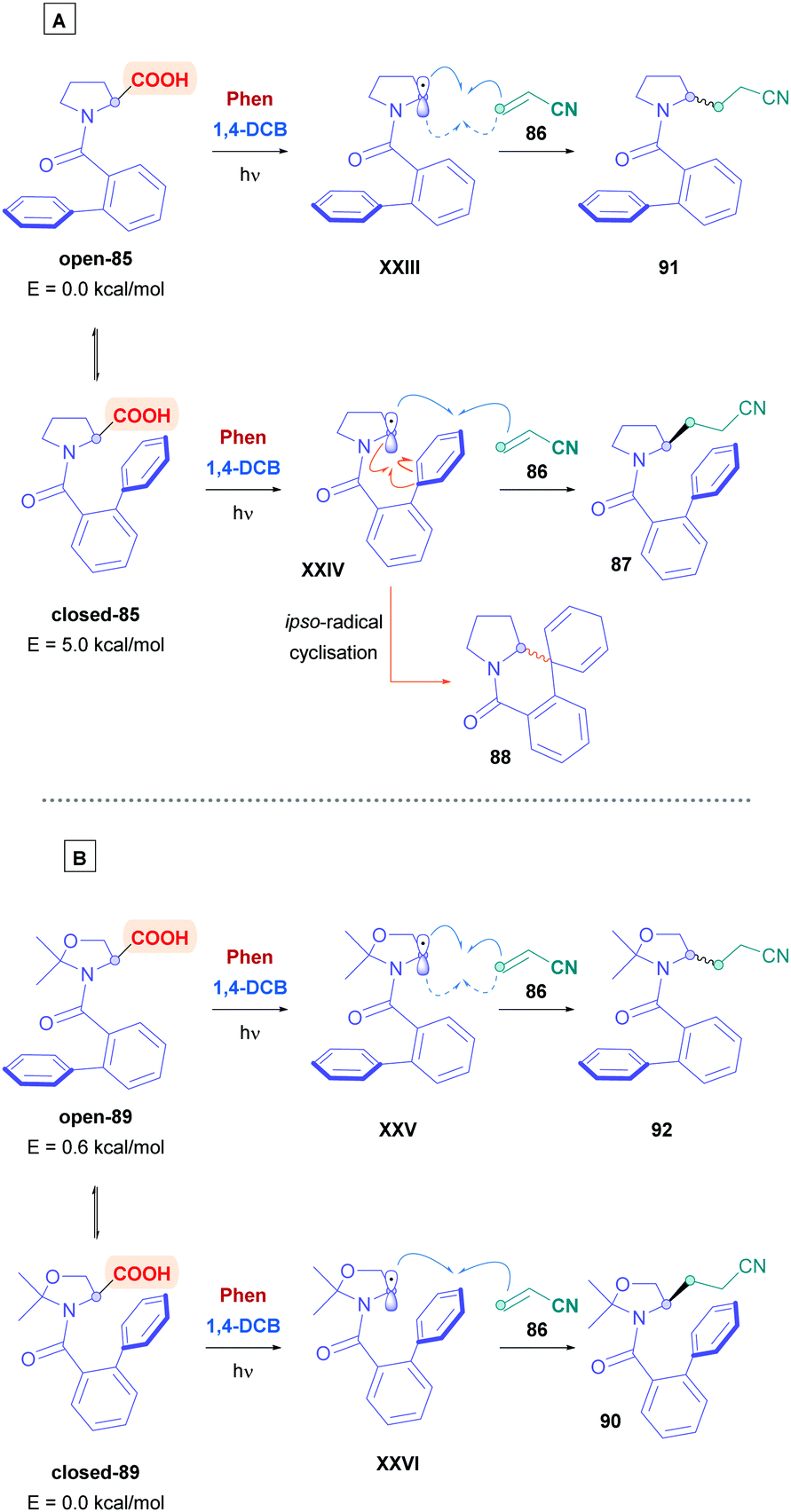
Amino acids are frequently used for radical decarboxylation processes. However, generation of the α-amino radicals scrambles the stereochemistry, often giving products as racemates. The process can be rendered stereoselective through the use of chiral ligands, an example of which was demonstrated by the MacMillan group using synergistic Ir photoredox and chiral Ni catalysis.75 Another tactic to control the stereochemistry of the process involves memory of chirality (MOC),76 which can make use of the inherent chirality of the substrate rather than of another chiral source. Yoshimi used this strategy to provide a novel, stereospecific intermolecular Giese addition of N-protected proline derivatives to acrylonitrile (Scheme 30).77 Choosing N-(2-phenyl)benzoyl-L-proline (85) as the acid substrate, with a catalytic system comprising of Phen and 1,4-DCB, reaction with acrylonitrile (86) gave the corresponding Giese adduct 87 with excellent yields (85%) but little of the stereochemistry was retained (48% ee). In addition, formation of corresponding spirodihydroisoquinolinone 88 was observed as a side-product, resulting from ipso-radical cyclisation.78 Stereospecificity, however, was dramatically improved when introducing ways to tightly control the biphenyl amide conformation. Using cyclic substrates 89 containing a gem-methyl substituent, which is derived from N-(2-phenyl)benzoyl-L-threonine methyl ester, the authors achieved >79% enantiomeric excess of Giese product 90. Employing higher concentrations of acrylamide diminished the amount of 90, whereas bulky Michael acceptors, e.g., methyl acrylates, t-butyl acrylates and N-t-butyl acrylamide, tended to increase formation of their corresponding derivatives of 90.
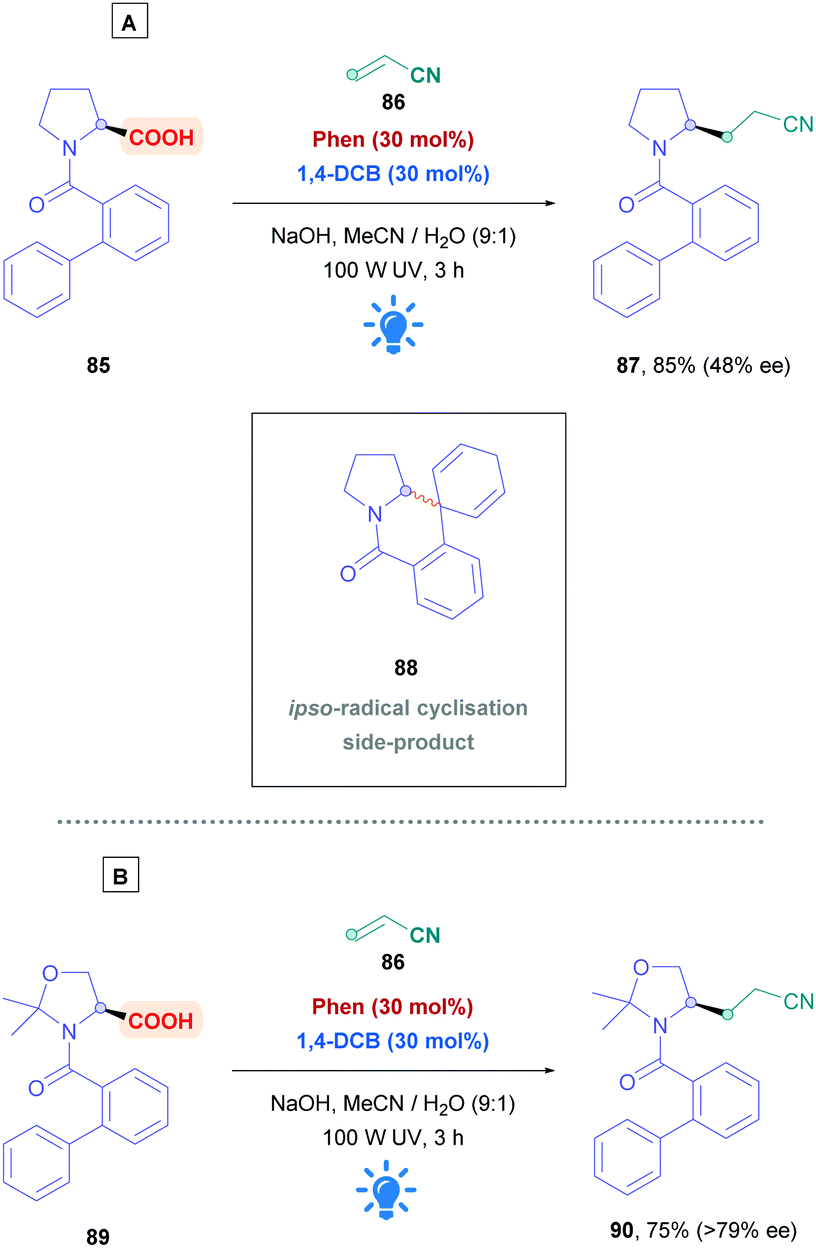 | ||
| Scheme 30 Diastereoselective Ar/DCA-catalysed direct DGR using memory of chirality (MOC) on acid substrates (A) 85 and (B) 89. | ||
The stereoselectivity of the reaction is dictated by the axial chirality of the acid substrate about the CO (carbonyl)-C (phenyl) bond in the ground state (Scheme 31A). Acid substrate 85 can adopt two conformers of open-85 and closed-85, with the closed state facilitating enantioselectivity by preventing bottom-side insertion of the olefin due to steric hindrance. Molecular orbital calculations (using HF/3-21G basis set) revealed that open-85 is favoured over closed-85 with a stabilisation energy of 5.0 kcal mol−1. Introducing a dimethyl unit, such as 89 (Scheme 31B), inverted the energy difference towards closed-89 (−0.6 kcal mol−1), thus favouring enantioselectivity.
 | ||
| Scheme 31 Proposed mechanism to rationalised stereochemistry for acid substrates (A) 85 and (B) 89. | ||
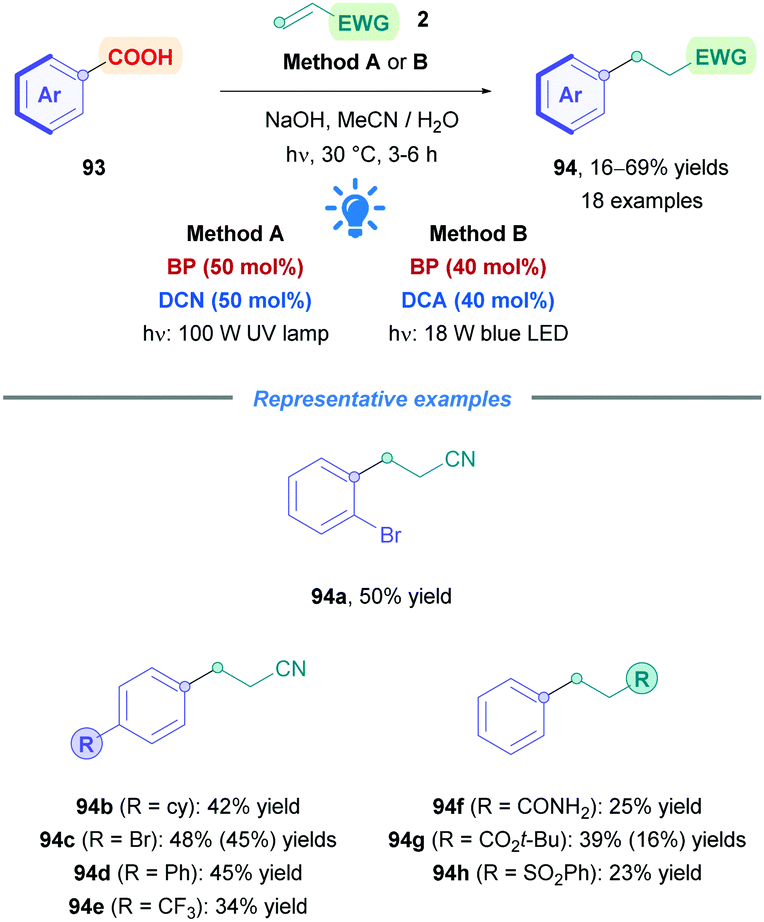
Very recently, Yoshimi successfully initiated direct photoinduced decarboxylation of benzoic acids (93), resulting in the formation of aryl radicals and their subsequent conjugate addition onto Michael acceptors (Scheme 32).72l This was achieved using BP and DCN (Method A), with slight heating to promote CO2 extrusion. Alternatively, BP and DCA could be used together under visible blue light irradiation (Method B). However, lower yields were observed using Method B due to the lower solubility of DCA in aqueous MeCN solution. Benzoic acids possessing alkyl groups and halogens proceeded well with low to moderate yields (16–69%) (94a–h). Benzoic acids with appropriately protected phenolic hydroxy groups were also successful substrates, and electron-rich aryls, such as those bearing methoxy groups, did not give their corresponding adducts 94. Decreased yields were observed for aryl with para-substituted EWGs (94e) due to the weaker nucleophilicity of the aryl radicals. Acrylonitrile was the acceptor of choice for this process, yet other acceptors included t-butyl acrylate, acrylamide (93f), and phenyl vinyl sulphone (93h). Higher concentrations of Michael acceptor (100 mM, 5 equiv.) were necessary to overcome competitive phenyl radical H-abstraction from MeCN. However, the high acceptor concentration also introduced the increased propensity for polymerisation, thus high concentrations of photocatalysts were required to counteract this phenomenon.
 | ||
| Scheme 32 Ar/DCA-catalysed direct DGR of aryl carboxylic acids. Yields in parentheses correspond to the reactions by Method B. | ||
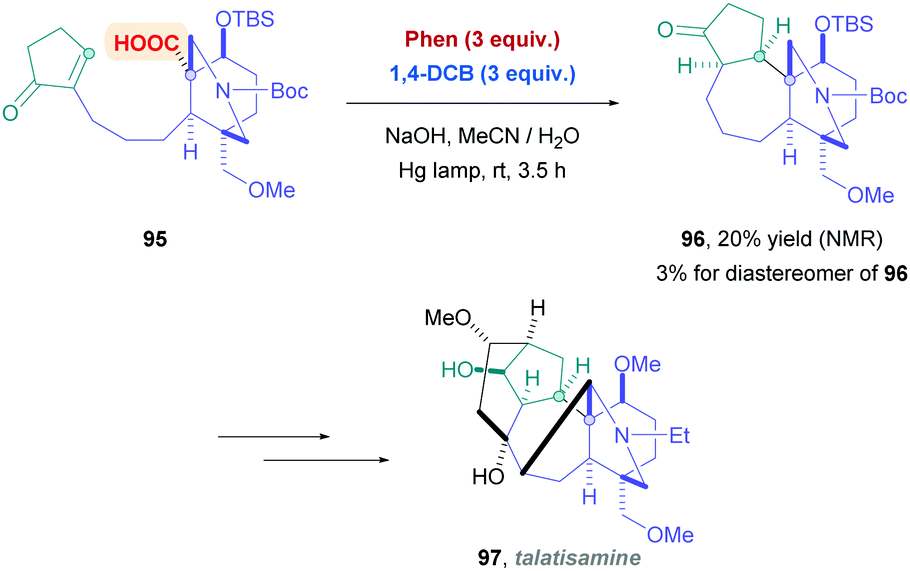
A recent application of this reagent system developed by Yoshimi was demonstrated by Inoue and co-workers for the synthesis of talatisamine precursor compound 96 (Scheme 33).79 The group was seeking to find a more efficient means to construct the complex ABCE-ring scaffold of 96 through an intramolecular decarboxylative Giese-type process. Intermediate 96 has the potential to be used toward the synthesis of C19-diterpenoid alkaloid talatisamine (97) and other derivatives. The more common iridium or acridinium photocatalysts gave complex mixtures, but a combination of Phen and 1,4-DCB induced the stereospecific, decarboxylative 7-endo radical cyclisation product 96, albeit with low yields (20%).
 | ||
| Scheme 33 Ar/DCA-catalysed direct DGR for the total synthesis of talatisamine. | ||
An unfortunate drawback for these dual catalytic systems covered in this section is the need for UV light irradiation, which ultimately diminishes the substrate scope. However, many organophotoredox catalysts that can be activated by visible light have been developed, and this will be the subject for the next section.
 | ||
| Fig. 1 Structure of 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzlPN). | ||
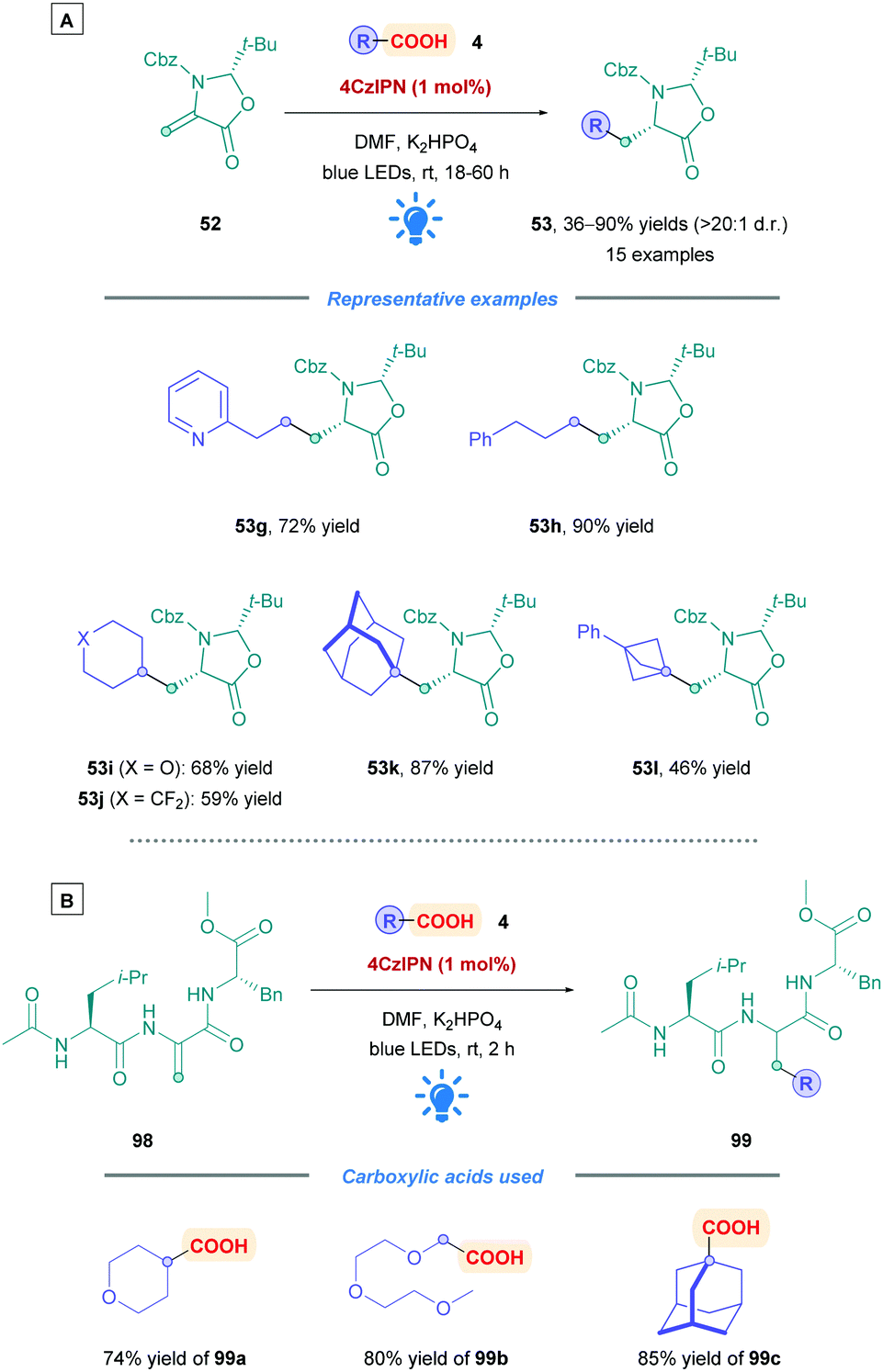
Alongside the previously described work of Gómez-Suárez,49 Schubert and Zhang also inserted radicals onto dehydroalanine derivative (Dha) derivative 52, but using 4CzIPN as the PC rather than the Ir-based [Ir-4][PF6] used in Gómez-Suárez's work (Scheme 34A).80 Although the investigation into the carboxylic acid scope was not as extensive, a good range of functional groups were well tolerated, including the presence of a pyridine such as in 53g. Cyclic and acyclic primary, secondary and tertiary carboxylic acids (53h–l) all proceeded to give their corresponding Giese adducts with moderate to excellent yields (46–90%) with excellent diastereoselectivity (>20:1 d.r.). The acid substrates tested were those containing functional groups of pharmaceutical interest, including polyethylene glycol (PEG) derivatives, and fluorinated substrates. Long irradiation times were required (up to 60 h) to ensure complete consumption of Dha derivative 52. The researchers also applied the reaction conditions to the functionalisation of peptides containing a Dha residue (98) (Scheme 34B). An example each for primary, secondary, and tertiary carboxylic acids (99a–c) afforded the desired products with excellent yields (74–85%). The influence of the functionalisation process on the peptide physicochemical properties was illustrated by the incorporation of a PEG2 unit (using 99b) to enhance solubility, or the addition of the adamantyl group (using 99c) to increase lipophilicity. Interestingly, 2 h of irradiation was sufficient to access the functionalise peptides 99.
 | ||
| Scheme 34 4CzlPN-catalysed direct DGR onto Dha derivatives for (A) individual amino acids and (B) peptides. | ||
 | ||
| Fig. 2 Structre of 2,3-dicyanopyrazine (2,3-DPZ). | ||
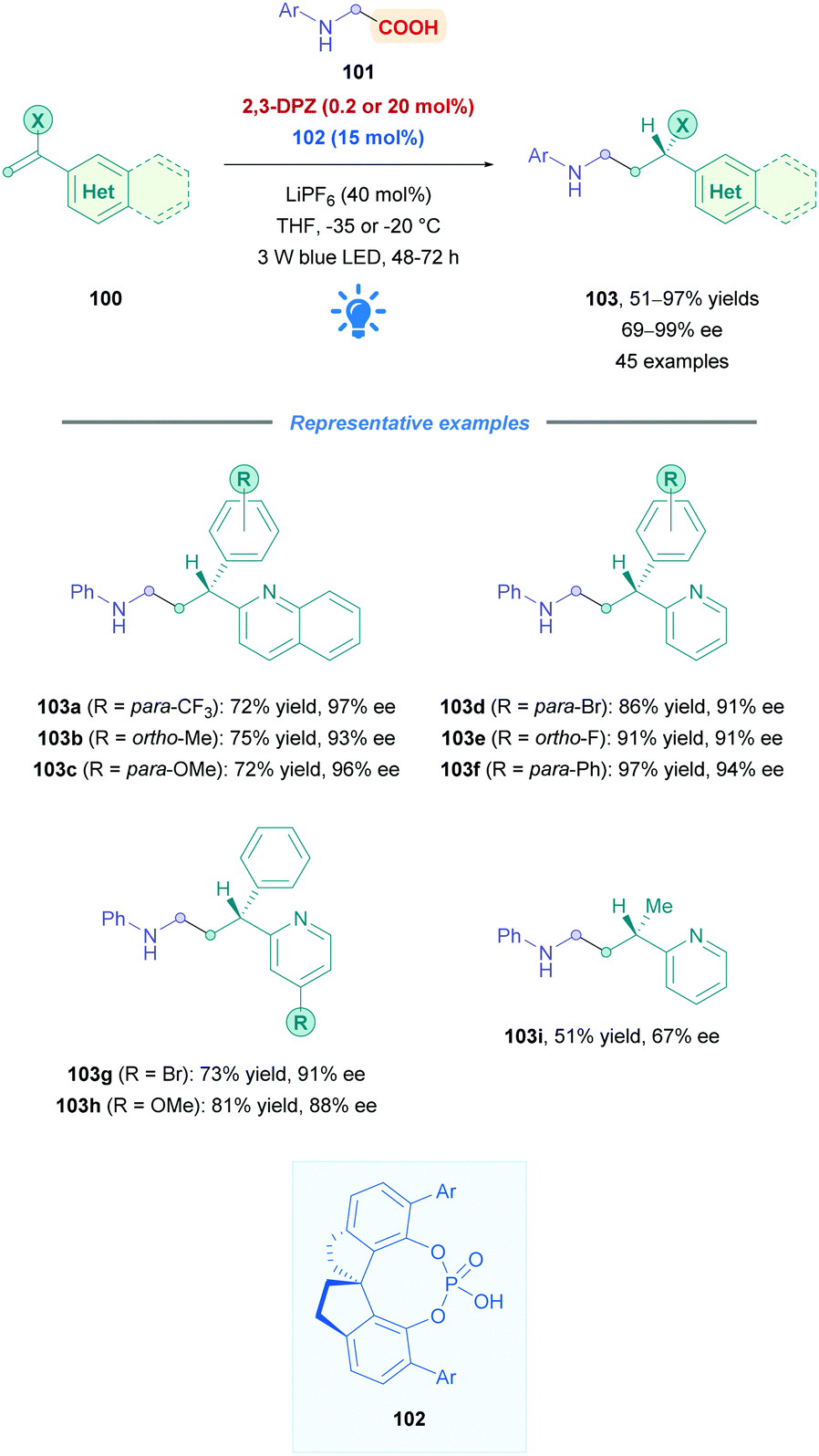
One of the major challenges regarding all types of Giese additions is achieving stereocontrol of the resulting Giese adduct outside of cases where substrate-controlled stereospecific radical addition is possible (such as cases presented in Scheme 30). To achieve this, Jiang and co-workers employed 2,3-DPZ as the oxidative PC to initiate addition of radicals derived from N-aryl glycine derivatives 101 onto 2-vinyl-pyridine/quinoline derivatives 100 (Scheme 35).82 In the presence of catalytic amounts of chiral 1,1′-spirobiindane-7,7′-diol (SPINOL)-based phosphoric acid 102. Good to excellent yields (51–97%) of 3-(2-pyridine/quinoline)-3-substituted amines 103 were obtained with high enantioselectivities. A variety of EWGs and EDGs (103a–f) on the N-aryl and aryl-vinyl groups were generally well tolerated. A noticeable exception was observed for the N-aryl bearing the strongly electron donating methoxy group, where the reaction temperature was raised to 0 °C to compensate for the seemingly slower reaction rates. Functional group variation on the pyridine moiety was also well tolerated (103g and h), regardless of the substitution pattern. The low reaction temperatures indicate the highly facile nature of this protocol and are presumably also important for high enantioselectivities, though it does introduce some limitations with regards to practicality.
 | ||
| Scheme 35 Enantionselective DPZ-catalysed direct DGR of N-aryl-α-amino acids onto vinyl pyridine/quinoline derivatives. | ||
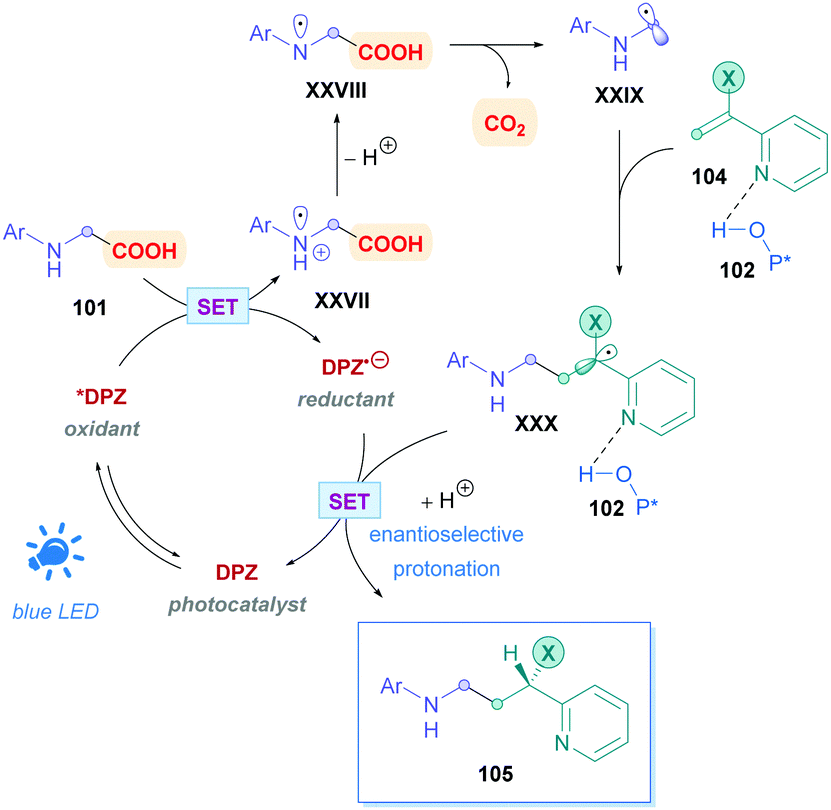
The proposed mechanism (Scheme 36) for this process is unique in that the photoinduced *DPZ (Ered1/2 = +0.91 V vs. SCE)83 is insufficient to oxidise carboxylates via a SET process. Instead, *DPZ oxidises the N-aryl amine to an aminium radical cation XXVII, which was further verified by Stern–Volmer analysis. A cascade reaction then proceeds involving a deprotonation, HAT, and decarboxylation to generate α-amino radical XXVIII. Conjugate addition of XXIX then proceeds onto olefin 104, which is activated by 102via H-bonding, giving rise to intermediate radical XXX. The additive LiPF6 increases the acidity of 102 and thus improves the overall yield and enantioselectivity. Upon reductive SET of the complex containing XXX and 102 to the corresponding pro-chiral anion, the all-important enantioselective protonation furnishes the desired chiral Giese adduct 105. While the work constitutes excellent progress for enantioselective direct DGRs, it should be noted that because the decarboxylation is initiated via the oxidation of N-aryl amine and not the carboxyl anion, using 2,3-DPZ as a photocatalyst severely limits the substrate scope. Indeed, the N-aryl group is essential and N-Boc protected amino acids do not undergo decarboxylation in the presence of 2,3-DPZ.
 | ||
| Scheme 36 Proposed mechanism for the enantionselective DPZ-catalysed direct DGR of N-aryl-α-amino acids onto vinyl pyridine/quinoline derivatives. | ||
Flavin catalysis has recently seen application in direct DGRs. The Macmillan group utilised flavin catalysis in their pioneering work on the site- and chemoselective bioconjugation of Michael acceptors onto C-termini of native peptides (Scheme 37).85 The group exploited the discrepancy in oxidation potentials between internal and C-terminal carboxylates, allowing for exclusive C-terminal functionalisation. It was recognised that carboxylate-containing residues such as internal Glu and Asp would be resistant to oxidative decarboxylation due to the formation of an unstabilised carbon-centred radical (Ered1/2 = +1.25 V vs. SCE),86 yet formation of a stabilised α-amino radical would allow preferential C-terminal decarboxylation (Ered1/2 = ∼+0.95 V vs. SCE).87
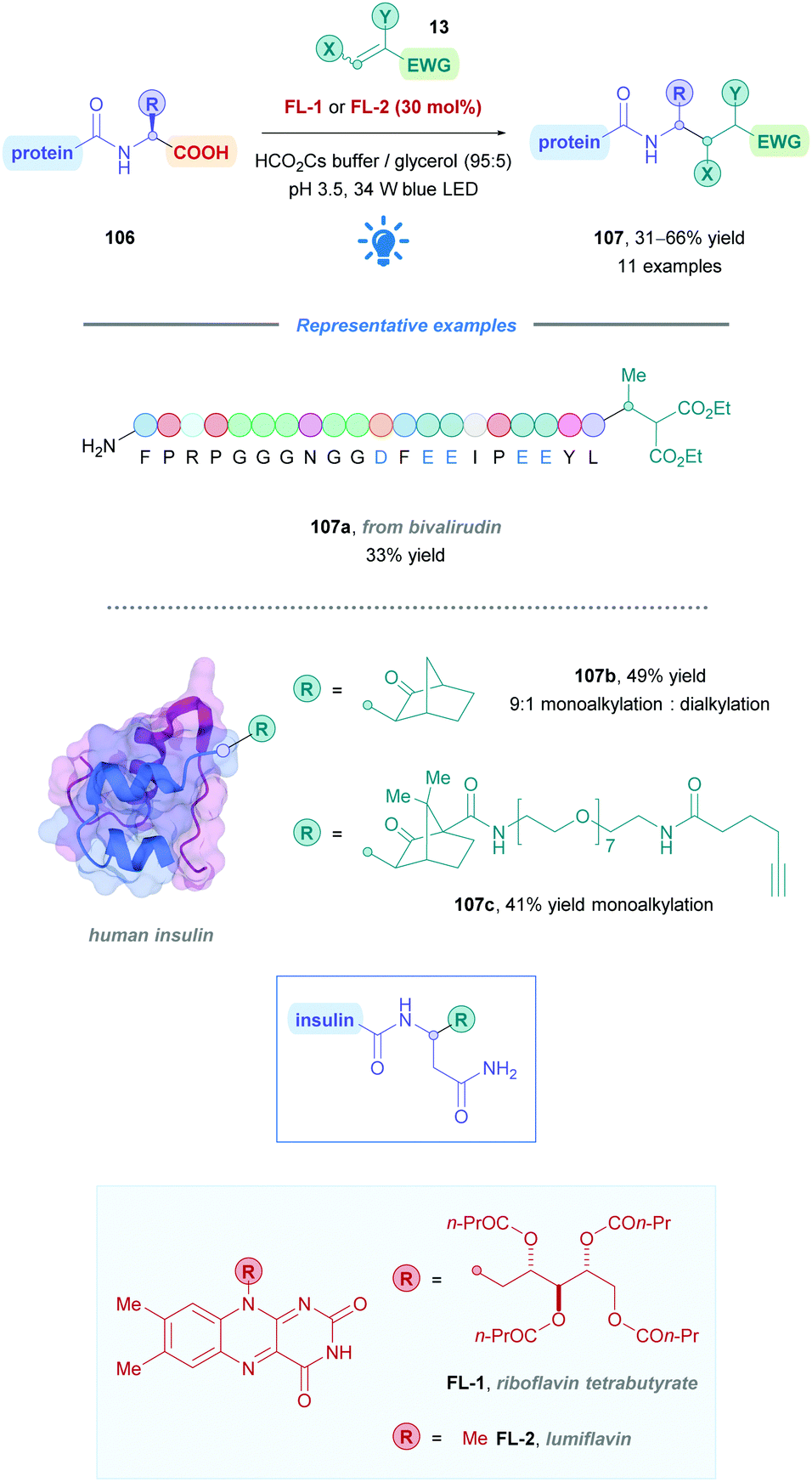
Using biological substrates required aqueous buffer, high dilution, and mild temperatures, thus a water-compatible PC was necessary to achieve the desired functionalisation. Attempts to use more commonly used PCs such as Ir- and Ru-based catalysts, and organic dyes proved to be unfruitful. The group thus turned their attention to flavin catalysis, as they are known to facilitate acetate decarboxylation via a two-electron pathway in aqueous media.88 A survey of flavin catalysts and reaction conditions gave rise to the methodology presented in Scheme 37, which further unveiled that riboflavin tetrabutyrate (FL-1) and lumiflavin (FL-2) were the most effective PCs.
Functional group tolerance was assessed by employing peptides possessing amino acid residues commonly found on protein surfaces. Moderate to good yields (31–66%) were obtained for a variety of native peptides of increasing complexity (such as 107a), demonstrating overall broad functional group tolerance (yields of >25% is sufficient to render the protocol a productive bioconjugation method). Importantly, exclusive C-terminal functionalisation was observed in all cases, including Glu- and Asp-bearing peptides where no side-chain decarboxylation was detected. The authors found that a low pH of 3.5 was critical to ensure protonation of Lys and His residues, as these were hypothesised to engage in deleterious oxidation reactions at higher pH.
Tyr residues were also subject to unwanted side-oxidation when PC FL-1 was employed. This could be somewhat improved by using the less oxidising lumiflavin (FL-2) as an alternative catalyst (Scheme 37).
The functionalisation of human insulin through this protocol is a particularly neat illustration for the potential of this bioconjugation method. Insulin is composed of two parent peptide chains (each with a potentially reactive C-terminus) that are linked by two redox-sensitive disulfide bridges, as well as containing four Tyr residues. A 49% yield of a predominantly (9:1) A-chain mono-alkylated insulin was nevertheless obtained (107b), with no detectable impact on disulphide bridges or other amino acid residues. An alkyne-substituted tag was also installed by this method (107c), allowing for further elaboration of the conjugated protein (Scheme 37).
Although no other examples of direct DGRs using flavin catalysis is known, this study underscores the potential for flavin catalysis and warrants further investigation.
 | ||
| Fig. 3 Structure of thioxanthone (TX). | ||
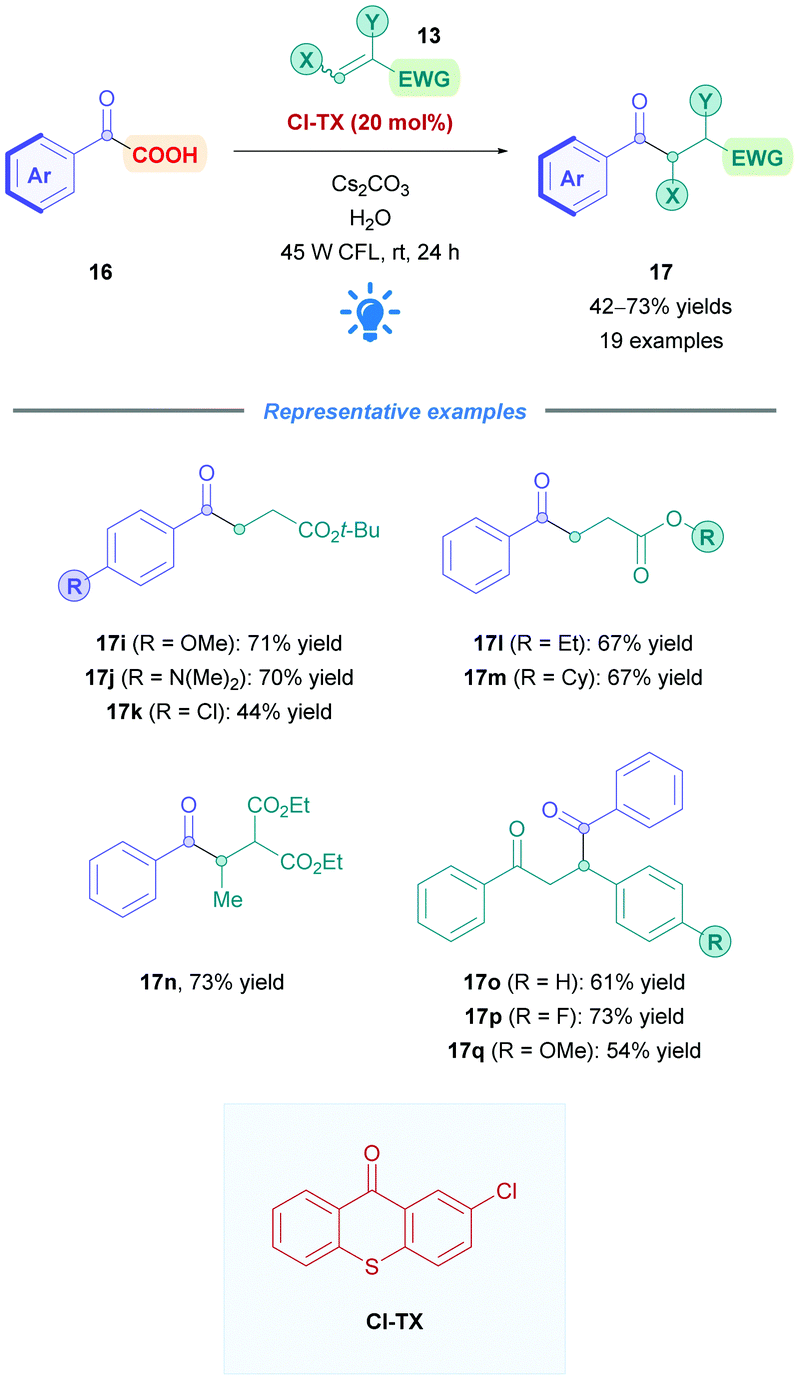
Li and co-workers recently employed the TX derivative 2-chloro-thioxanthane-9-one (Cl-TX) as a photocatalyst for the direct DGR of aryl α-keto acids 16 onto mainly acrylate derivatives 13 (Scheme 38), achieving moderate to excellent yields (42–73%).93 Acid substrates (16) bearing electron-donating (17i–j), and -withdrawing (17k) groups were well tolerated, and acrylates of different ester moieties and substitution patterns (e.g., 17l–n) reacted smoothly. α,β-Unsaturated ketones also proceeded well (17o–q). During optimisation studies, it was found that TX gave lower yields, possible due to its lower excited-state redox potential (Ered1/2(TX*/TX˙−) = +1.34 V vs. SCE in DMF)94 than Cl-TX (Ered1/2(Cl-TX*/Cl-TX˙−) = +1.468 vs. SCE in DMF).93 The mechanism of the reaction is predicted to proceed via the pathway analogous to that depicted in Scheme 5.
 | ||
| Scheme 38 Thioxanthone-catalysed direct DGR of α-keto acids. | ||
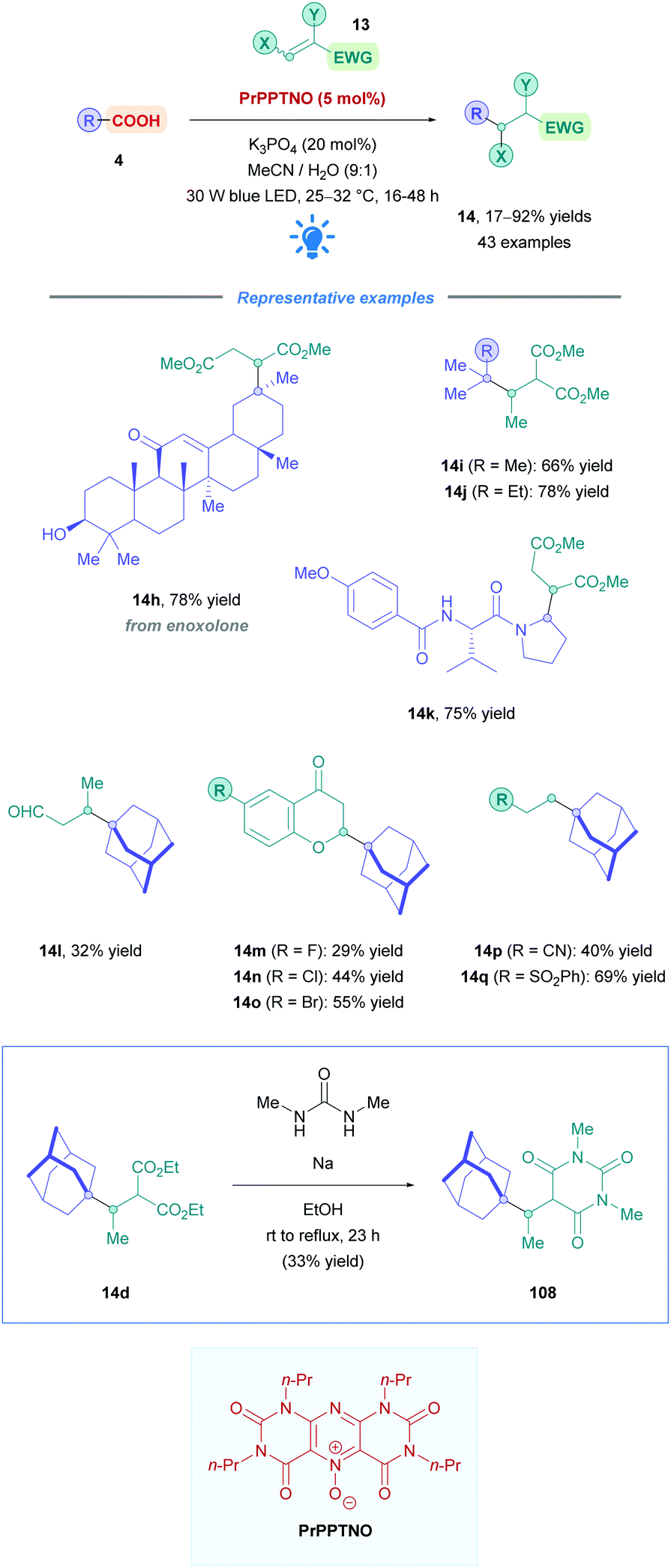 | ||
| Scheme 39 PrPPTNO-catalysed direct DGR. | ||
The proposed reaction mechanism follows the same pathway as that of the general mechanism shown in Scheme 5. However, it should be highlighted that PrPPTNO serves as a pre-catalyst, whereby a deoxygenation step generates the active PC PrPPT (Scheme 40). Indeed, initial catalytic screening revealed that PrPPT was just as effective as its N-oxide analogue PrPPTNO.
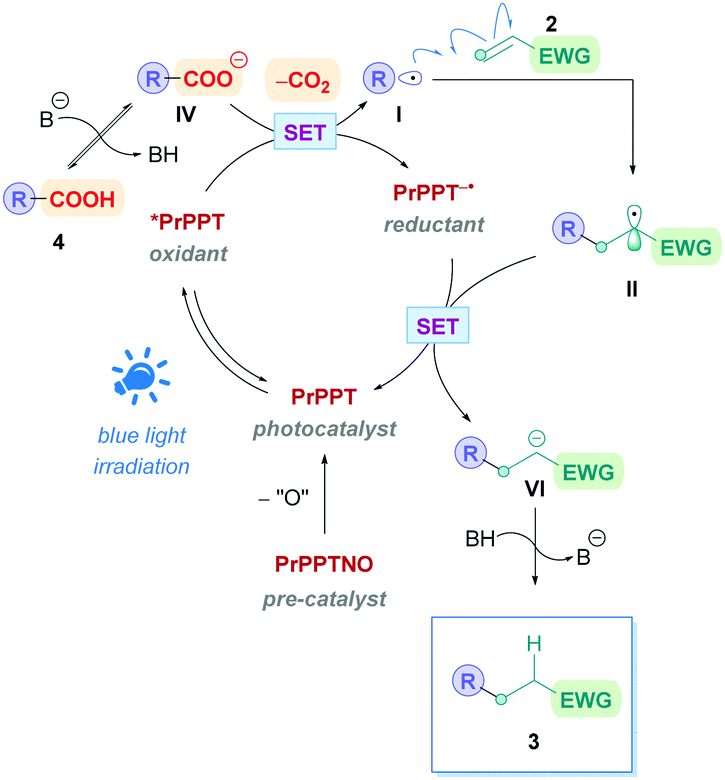 | ||
| Scheme 40 Proposed mechanism of PrPPTNO-catalysed direct DGR. | ||
The first use of an acridinium PC for direct DGRs was reported by Koike, Akita and co-workers,100 where they employed the acridinium catalyst 9-mesitylene-10-methylacridinium perchlorate ([Acr-Mes]ClO4, Fukuzumi catalyst, Acr-1). However, only four carboxylic acid substrates (4) were examined using the same Michael acceptor, diminished yields were obtained for secondary acids, and an oxygen-free environment was necessary. Inspired by this work, Gonzalez-Gomez and Ramirez sought to improve the reaction conditions and further expand the scope (Scheme 41).101 Minor modifications to the reaction conditions, including increased blue light irradiation and no protective atmosphere, gave improved yields within shorter reaction times. A change of solvent from MeOH to an MeCN/H2O mixture, together with an increase in reaction concentration, enabled secondary acids to undergo DGRs with moderate to excellent yields (e.g., 14v, 78%). These improved conditions were then applied to tertiary acids (14l and 14q–s), α-amino, α-oxy and aryl α-keto acids (14t–u) with good overall yields (54–78%). Interestingly, the group was able for the first time to report the DGR of aliphatic α-keto acids with moderate yields (14u, 54%).
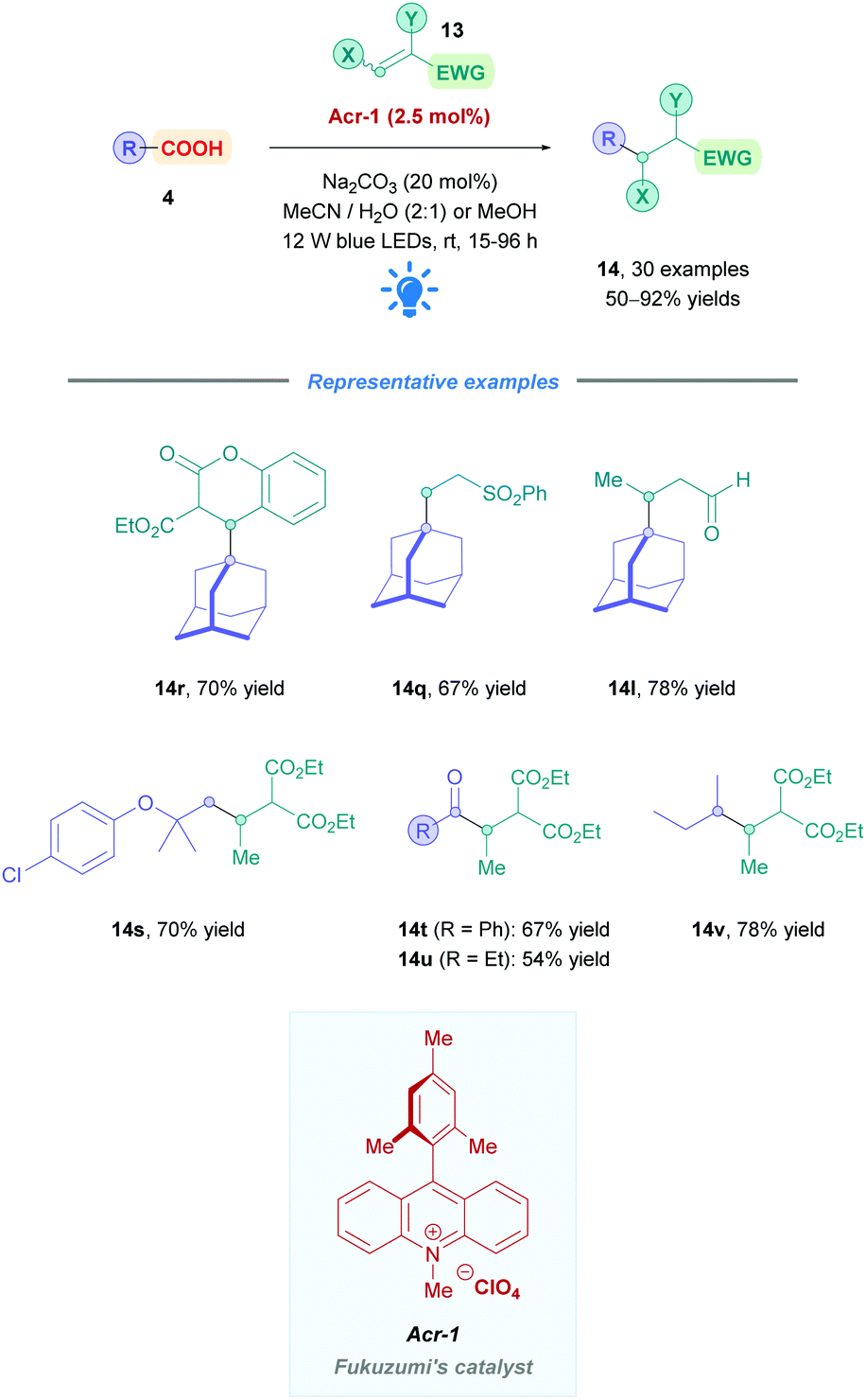 | ||
| Scheme 41 Acr-catalysed direct DGR. | ||
Acids that were unsuccessful included α-hydroxy acids, electron-poor phenoxyacetic acids, simple primary aliphatic acids and phenyl acetic acids. With regards to the Michael acceptors, more sterically demanding Michael acceptor substrates proved futile, and acceptors substituted at the α-position by a single ester group also failed to react, possibly because the corresponding α-acyl radical was unable to re-oxidise the [Acr-Mes]˙ species to recycle the catalyst. In addition, β-phenyl-substituted acceptors were unsuccessful likely due to the competitive formation of the more stable benzylic radical, which is also unable to recycle the catalyst.
Wang and co-workers also employed Fukuzumi's catalyst to prepare enantioenriched α-deuterated α-amino acids.102 Preparation of isotopically enriched biological compounds are highly valuable within the life sciences, as they provide a powerful tool for the elucidation of enzymatic mechanisms,103 biosynthetic pathways,95a,104 and structural information within peptides and proteins.102,105 Incorporation of deuterium can also enhance metabolic stability for peptide-based therapeutics.
Similar to the works of Gómez-Suárez (Scheme 19),49 Schubert (Scheme 34),80 and their respective co-workers, the Wang group also employed Dha derivative 52 to synthesise a diverse range of deuterated non-canonical amino acids (110) (Scheme 42).102 Using PC Acr-1, moderate to excellent yields (46–92%) were observed for primary, secondary, and tertiary acids with broad functional group tolerance. They also included sugar-derived carboxylic acids (110c), as well as late-stage functionalisations of carboxylic acid-containing pharmaceuticals (110d). Across the board, over 90% D-incorporation and diastereomeric ratios >20:1 were obtained. Some substrates required the use of the less oxidising carbazolyldicyanobenzene PC 4CzlPN instead of Acr-1. It is interesting to note that during their optimisation using a glycosyl radical precursor, the group did not achieve product formation using 4CzIPN. However, Schubert80 reported the highest yields using 4CzIPN during their optimisation using the same acceptor, but with a primary carboxylic acid radical precursor. These results illustrate the necessity to tailor the choice of the PC to the system considered, as well as the cross-applicability of PCs that enable the expansion of a given reaction's scope. Following Giese addition, the resulting adducts could be transformed to the corresponding α-deuterated α-amino acids by reacting with concentrated HCl, as showcased through the acid hydrolysis of 110e to α-deuterated Leu 111.
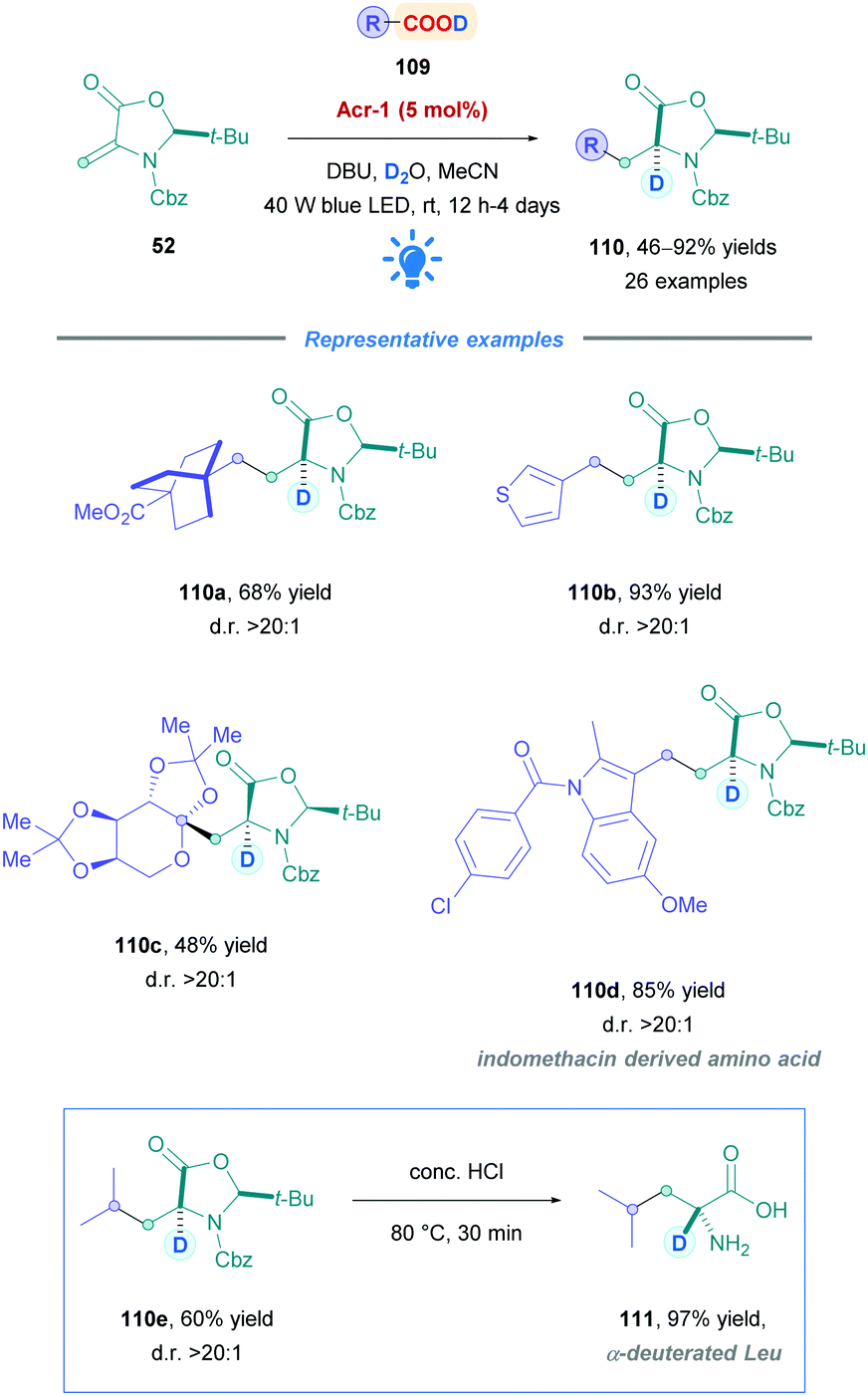 | ||
| Scheme 42 Acr-Catalysed direct DGR onto Dha. | ||
Another methodology to prepare unnatural amino acids was reported by Shah and co-workers at Merck, whereby they performed DGRs onto dehydroalanine derivative 112 to generate β-alkyl α-amino acids (Scheme 43).106 A high-throughput experimentation screen of PCs unveiled that catalyst Acr-2 was the most effective. The reaction conditions were chosen to be compatible with parallel library synthesis and purification, allowing for a rapid assessment of the substrate scope. Dehydroalanine derivatives 112 were bis-Boc protected, as mono-Boc substrates were found to be not sufficiently reactive. Two reaction set-ups focusing either on library synthesis throughput (Method A, parallel two steps synthesis) or on enhanced process scalability (Method B, single steps with intermediate purification) were developed using different photoreactors. Low to excellent yields of 113 (4–90%) were observed for a wide range of acids bearing functionalities such as nitriles, alkynes, aryl halides or heteroaromatics (113a). α-Difluoro acids gave diminished yields, possibly due to the less nucleophilic nature of the corresponding radicals, however, the difluoro moiety is tolerated in other positions (113b). A variety of saturated 6-membered cyclic systems were also compatible (113c), giving excellent yields of Giese adducts; however, some limitations were observed for 4- and 5-membered saturated systems. The group further demonstrated the applicability of their methodology by the synthesis of alkyne-functionalised protected amino acid 113d of potential interest for the synthesis of linkable peptides.
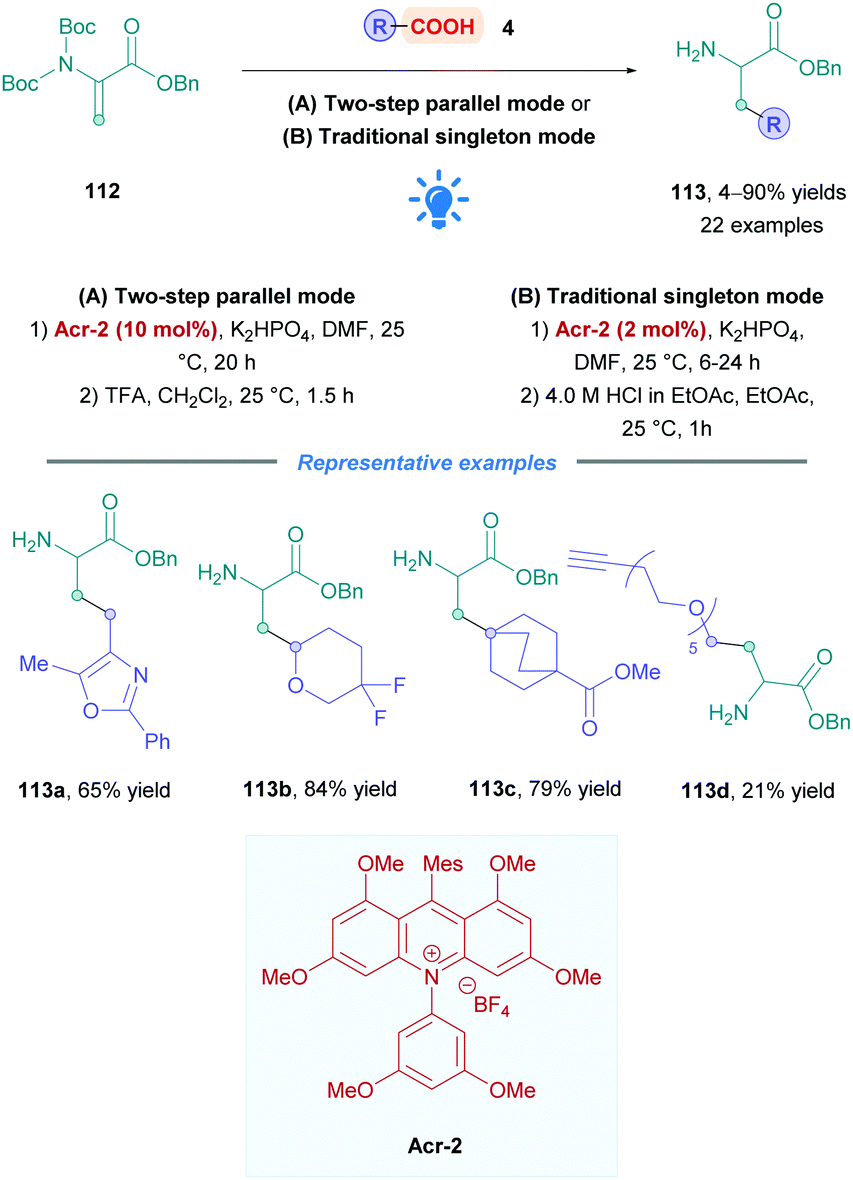 | ||
| Scheme 43 Acr-Catalysed direct DGR onto dihydroalanines. | ||
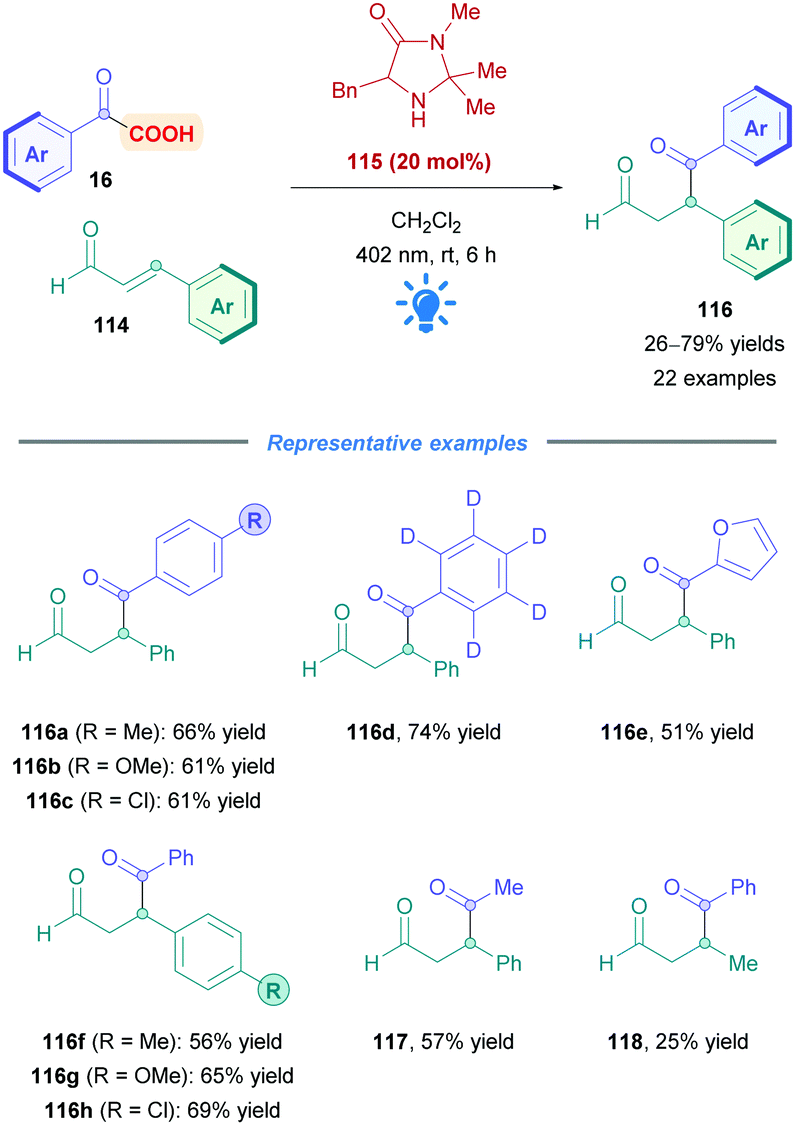 | ||
| Scheme 44 Iminium-catalysed direct DGR of α-keto acids onto α,β-unsaturated aldehydes. | ||
The proposed mechanism is illustrated in Scheme 45. Initial reaction of the amine catalyst 115 with aldehyde 114 generates the iminium cation, which subsequently forms a close ion pair intermediate XXXI with the carboxylate of 16. Photoexcitation of XXXI gives rise to the photoactive EDA-complex XXXII, which undergoes SET followed by intersystem crossing (XXXIII) which induces decarboxylation and radical–radical recombination (XXXIV) to give enamine XXXV. Using cinnamaldehyde derivatives as radical acceptors ensures the presence of an extended π-system, allowing favourable π,π-interactions in the EDA complex. Further evidence for the necessity of the extended π-system can be found when using crotonaldehyde as the radical acceptor, which only gave 25% of the desired product 118. Final hydrolysis of XXXV completes the catalytic cycle and furnishes the desired Giese adduct 116.
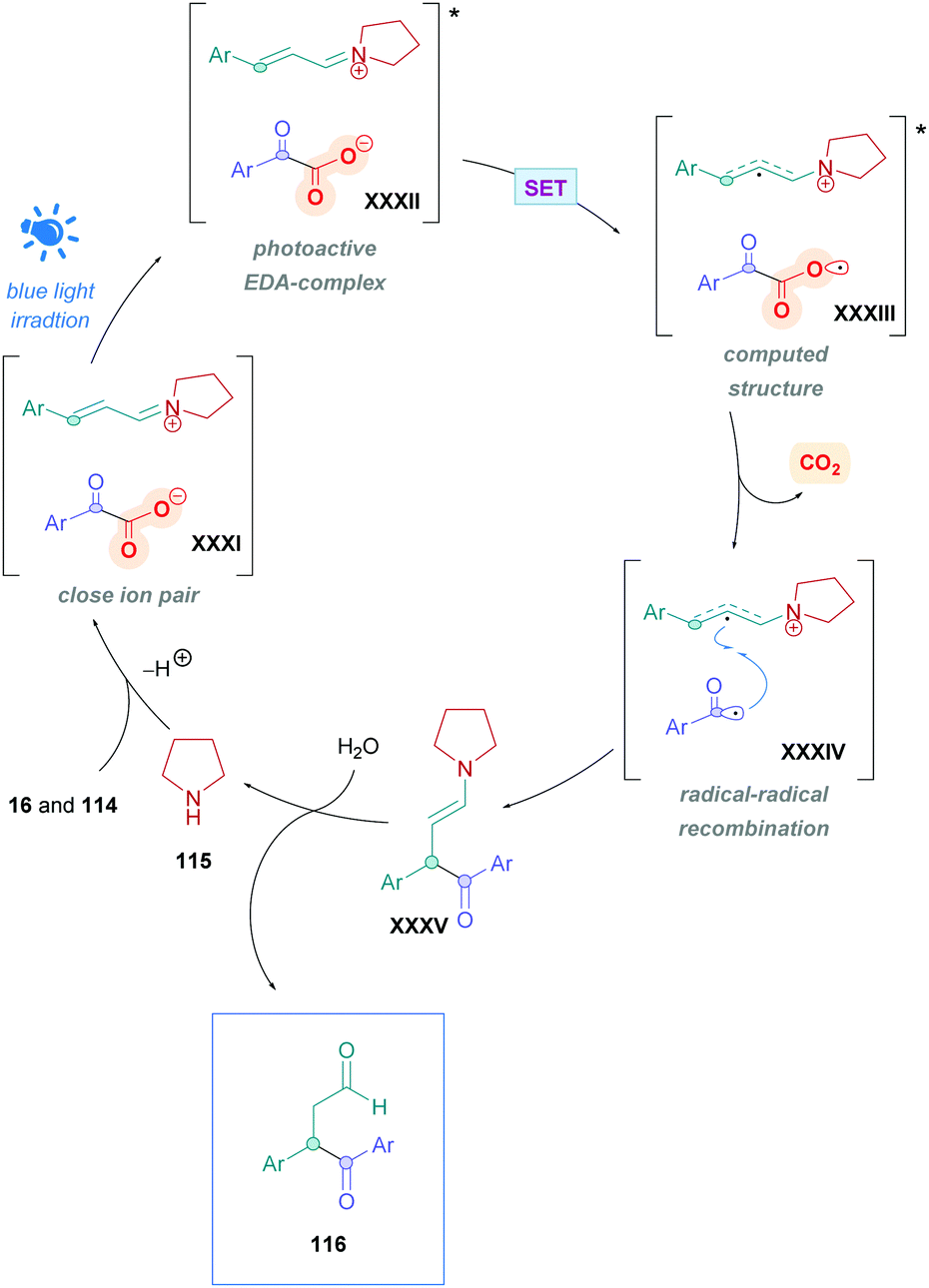 | ||
| Scheme 45 Proposed mechanism for iminium-catalysed direct DGR of α-keto acids onto α,β-unsaturated aldehydes. The structure of amine catalyst 115 has been simplified for clarity. | ||
2.3 Synergistic dual photocatalysis
Due to the often highly oxidising conditions required to achieve decarboxylation, sensitive functional groups are susceptible under these reaction conditions and thus frequently require protection. This issue can be bypassed using milder reductive decarboxylation methodologies through the implementation of redox-active esters, avoiding the need for acid deprotonation or energetically demanding homolysis of O–H via a HAT process. As stated previously, however, these esters require preparation and isolation in most cases, leading to additional steps.
To address these issues, Larionov and co-workers recently devised a mild dual-catalytic system to facilitate the direct DGR of carboxylic acids without the need for pre-activation by deprotonation.110 Employing their recently described new class of 9-arylacridine photocatalysts,111Acr-3 or Acr-4 were coupled with an efficient Cu(I)/Cu(0) catalytic cycle to access an impressive array of Giese adducts (Scheme 46A). Moderate to excellent yields were obtained for a variety of primary (120b, 81%), secondary (121, 76%) and tertiary (120a, c–f, and 122, 52–83%) carboxylic acids with good functional group tolerance for halogens and (un)saturated heterocycles. Moreover, they showcased an extensive array of Michael acceptors that can undergo efficient Giese participation with moderate to excellent yields (51–91%). Less electron-poor olefins, such as acrolein derivatives (120a–f) and (a)cyclic α,β-unsaturated ketones (120c and d) were successfully converted, using piperazine as additive for the copper catalyst. However, N,N,N′,N′-tetramethyl-1,3-propylenediamine (TMPDA) was the optimal additive when investigating the scope of more electron-deficient substrates, which includes acrylonitrile, phenyl vinyl sulfones (121), vinyl phosphonates (122) and vinyl phosphine oxides. The group also showcased many examples of their protocol applied to naturally occurring and medicinally relevant compounds (120a-b, e-f) which possess multiple sites for potential oxidation, often significantly reducing the number of steps compared to previously reported syntheses.
 | ||
| Scheme 46 Synergistic Acr/Cu-catalysed Direct DGR of carboxylic acids with proposed mechanisms. (A) Proposed mechanism via Giese addition of I. (B) Proposed mechanism via Cu(0) interception. | ||
Rigorous mechanistic studies unveiled the plausible presence of two competing pathways (Scheme 46B and C). Both pathways begin with hydrogen-bonded coordination of the acridine Acr and carboxylic acid 4 to form the activated complex 123. Irradiation of visible light then induces decarboxylation from the singlet excited state of 123via a proton-coupled electron-transfer (PCET) process to generate the alkyl radical I and acridinyl radical Acr-H˙.112 It should be noted that decarboxylation does not proceed via an acridinium carboxylate salt.112Acr-H˙ then participates in reductive SET with the Cu(I) catalyst 125 to produce Cu(0) species 126 and acridinium Acr-H+. Meanwhile, radical I can follow one of two distinct routes. As depicted in Scheme 46B, I can participate in conjugate addition with Michael acceptor 124 to generate the radical intermediate XXXVI, which subsequently cross-terminates with 126 to afford Cu(I) enolate 127. The product 128 is then furnished through protonation by acridinium Acr-H+, which in turn regenerates the acridine catalyst Acr and Cu(I) catalyst 125 simultaneously, thus closing both catalytic cycles. Alternatively, radical I is first intercepted by Cu(0) species 126 to generate organocopper intermediate 129 (Scheme 46C). Installation of I onto Michael acceptor 124 can proceed to copper enolate 127′ either via direct Michael addition (130a) or via Cu(III) intermediate 130b. The choice of amine additive was also paramount to the success of the reaction. Tertiary aliphatic amines were shown to perform better than secondary and primary diamines, and aromatic diamines. Evidence obtained during mechanistic studies suggests that the amine additive acts as a ligand, rather than a base, to stabilise the Cu(0) species and prevent aggregation.113
 | ||
| Scheme 47 Ruthenium/amine co-catalysed direct DGR of α-keto acids onto α,β-unsaturated aldehydes. | ||
The proposed mechanism is illustrated in Scheme 48. Initial photoexcitation of Ru(II) to *Ru(II), followed by its reductive quenching by acid 16 leads to the acyl radical XXXVIIvia decarboxylation. Meanwhile, chiral amine catalyst 131 reacts with aldehyde 114 to generate iminium ion XXXVIIIin situ. Subsequent enantioselective conjugate addition of radical XXXVII onto XXXVIII gives radical XXXIX, which is then reduced by Ru(I) to close the catalytic cycle and to give enamine XL. Finally, hydrolysis of XL furnishes the final Giese adduct 116.
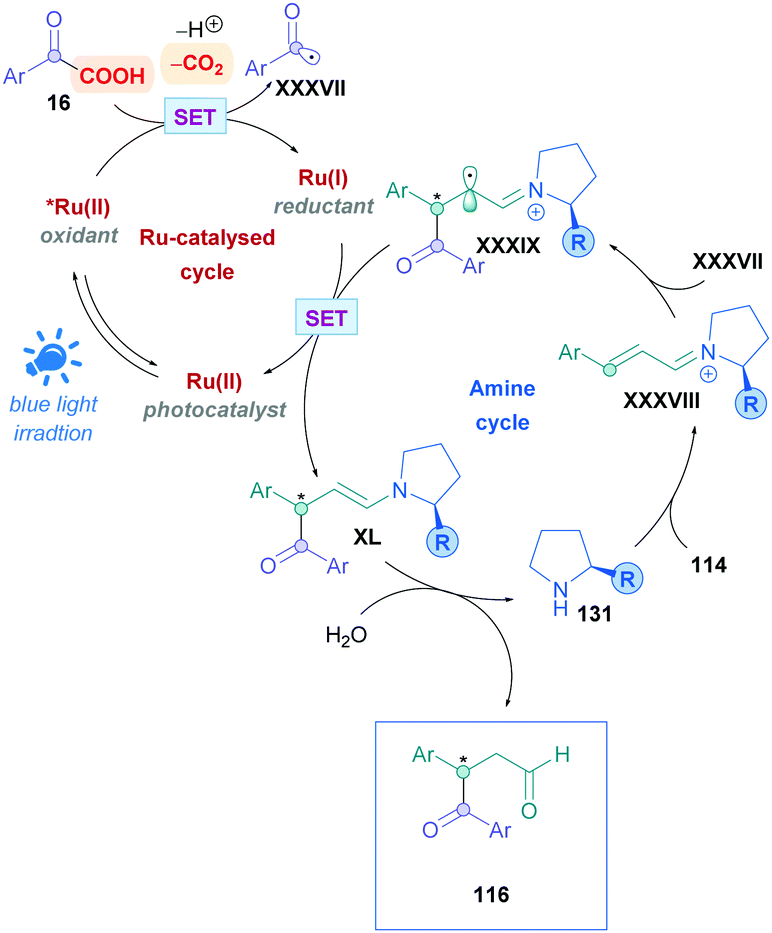 | ||
| Scheme 48 Proposed mechanism of Ruthenium/amine co-catalysed direct DGR of α-keto acids onto α,β-unsaturated aldehydes. The structure of amine catalyst 131 has been simplified for clarity. | ||
2.4 Heterogeneous photocatalysis
Homogenous catalysis, as opposed to heterogeneous catalysis, is often characterised by its better tunability, higher activity and selectivity. Homogeneous catalysis has been dominating in the field of photoredox catalysis, and despite the advantages stated above, difficulties pertaining to catalyst separation, recycling, thermal stability, and cost have emerged as potential limitations for the wider application of some homogeneous PCs.117 To overcome these limitations, there has been considerable research focussed towards the development of heterogenous catalysts, which can easily be separated and recycled.An appealing approach to harness the advantages of homogeneous and heterogenous catalysis could consist of anchoring soluble catalysts onto insoluble supports. This approach, however, often suffers from reduction in catalytic activity and selectivity due to limited number of accessible reaction sites, leading to the heterogenised systems being unable to achieve the performance of their soluble counterparts.117b Alternative heterogeneous systems capable of acting as PCs have instead been used, such as semiconductors, conjugates microporous polymers, covalent organic frameworks, and metal organic frameworks.117b A semiconductor heterogenous photocatalysts (Het-PCs) operate using the same electron and energy transfer pathways that govern homogenous photocatalytic reactions. When a Het-PC particle absorbs a photon of sufficient energy, an electron is excited from the valence band (VB) to the conducting band (CB), simultaneously establishing an oxidising and reducing species (the energy difference between the CB and VB is known as the band gap) (Scheme 49). The electrons in the CB can participate in reductive SET with electron-acceptor substrates, and the generated holes (h+) in the VB can oxidise electron donors.
 | ||
| Scheme 49 General mechanism of action for semiconductor heterogenous photocatalysis. | ||
:1.118
The first example of TiO2 being used for direct DGRs was reported by Albini and co-workers in 1998.119 They demonstrated one example by coupling 4-methoxyphenylacetic acid to maleic acid, achieving 65% yield of the Giese adduct after 60 h of UV irradiation. By 2012, Walton and co-workers had conducted a more extensive study concerning application of TiO2 P25 for direct DGRs.120 Under dry and anaerobic conditions in MeCN solvent, they showed that α-oxy acids could act as suitable radical precursors for addition onto maleic anhydride, maleimides, and acrylamide. Annulation was also observed when using α-phenoxy acids.
Later, the same group expanded upon their preliminary work to investigate: (1) the necessary structural features of the acids that undergo efficient Giese addition; (2) the Michael acceptor scope, paying attention towards functional group tolerance; (3) alternative catalyst forms and supports; and (4) the mechanism of the reaction (Scheme 50A).121 Alkylations of N-phenylmaleimide using simple carboxylic acids that generated primary radicals gave low yields, and moderate improvements were made with acids that gave secondary and tertiary radicals. However, a significant amount of maleimide reduction to the succinimide was observed. By contrast, acids that generated primary and secondary radicals stabilised by an α-heteroatom (α-alkoxy or α-amino) gave improved yields (14w-aa, 29–75%), yet radicals stabilised by α-aryl moieties gave depleted yields accompanied by homocoupling (e.g., 14ab, 22%). In addition, succinimide formation was observed for all cases. The catalyst was removed by filtration, emphasising the procedural simplicity.
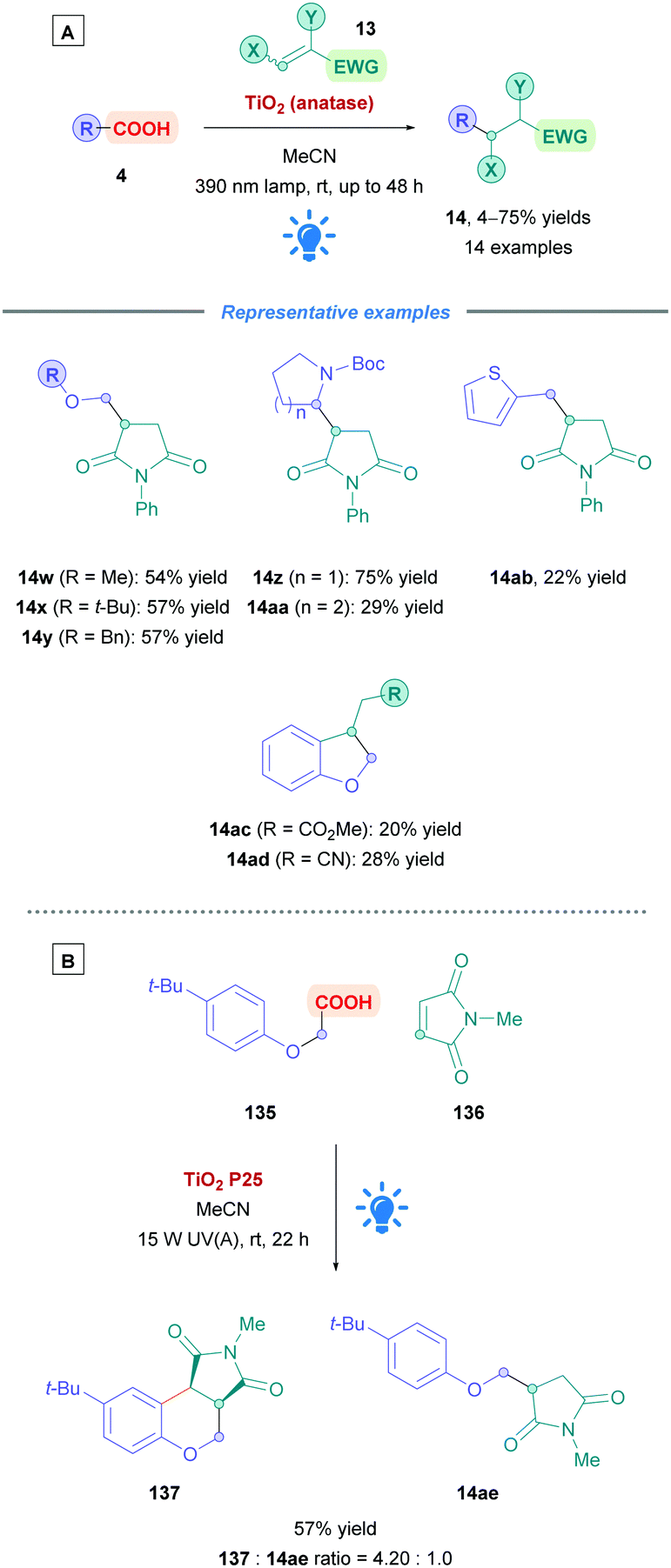 | ||
| Scheme 50 TiO2-Catalysed direct DGR of carboxylic acids onto maleimides and other Michael acceptors with (A) stabalised radical precursors and (B) O-aryl-α-oxy acids. | ||
The group also tested the efficiencies of different forms of the TiO2 catalyst using the reaction of phenoxyacetic acid with acrylamide. Standard TiO2 P25 gave a yield of 47%, but employing TiO2 PC500, which has a 6 times greater surface area compared to P25, led to a decrease in yield to 39%.122 Even lower yields (9%) were observed when using TiO2 Photospheres (consisting of hollow Pyrex beads with an external coating of rutile titania) due to difficulties in achieving uniform dispersion of the Photospheres throughout the reaction mixture. In contrast, the best yield (73%) was observed when the reaction was performed in Schlenk tubes coated with a fine internal layer of TiO2 by a sol–gel process.123 However, the authors noted that the coat could only be used three times due to the poor adherence of the catalyst on the tube surface – they therefore reasoned the conventional P25 catalyst to be the best compromise.
They next investigated the reaction of aryloxyacetic acids (e.g., 135) with N-substituted maleimides (e.g., 136) to give N-substituted-3,4-dihydrochromeno-[3,4]pyrrole-1,3-dione derivatives (e.g., 137) together with the expected Giese adduct (e.g., 14ae) (Scheme 50A). Generally, moderate to excellent yields were observed for substrates bearing electron-withdrawing or -donating substituents on the aryl ring. Selectivity for the chromenopyrrole product 137 can be increased by using combination of a denser dispersion of P25 and longer reaction times. Conversely, the standard Giese adduct 14ae is favoured when using the sol–gel TiO2-coated tubes, and in the presence of EWGs on the aryl moiety. The cis isomer of 137 was obtained in every case, as determined by 1H NMR.124
Finally, the group demonstrated examples of intramolecular Giese reactions to give the adduct such as 14ac and 14ad (Scheme 50A), where low yields (20–28%) were obtained for substrates with tethered olefins bearing ester substituents. However, moderate yields (44–58%) were achieved when applying the sol–gel tube reactions.
With regards to mechanistic studies (Scheme 51A), it is proposed that initial decarboxylation of carboxylate XLI is induced by hole capture to give the desired radical XLII. Conjugate addition of XLII onto the N-substituted maleimide 138 gives rise to the imidoalkyl radical XLIII. From here, SET with the CB of the TiO2 particle is proposed to generate the corresponding enolate XLIV (Pathway A). Subsequent protonation125 would afford the final Giese adduct 139. When using aryloxyacetic acids, Pathway A becomes competitive with Pathway B, where a ring closure (6-endo-trig) of the electrophilic radical XLIII onto the aryl ring occurs. Increasing the electron density on the aryl ring increases its reactivity with the electrophilic radical XLIII, thus favouring annulation.126 Aromatisation of the resulting cyclohexadienyl-type radical XLV by SET gives the final cyclised product 140. The aromatisation process could take place through hole capture with TiO2 to give oxonium XLVI, followed by proton loss. Alternatively, SET to maleimide might take place, as suggested by the work of Hoffman.121,127 A series of protonation and electron-transfer steps of the resulting maleimide radical anions would then give the corresponding succinimides, thus explaining the significant yields of succinimides observed for these reactions. However, a high succinimide:139 ratio was generally observed, indicating that the maleimide acceptor was also being directly reduced by TiO2 under these conditions.
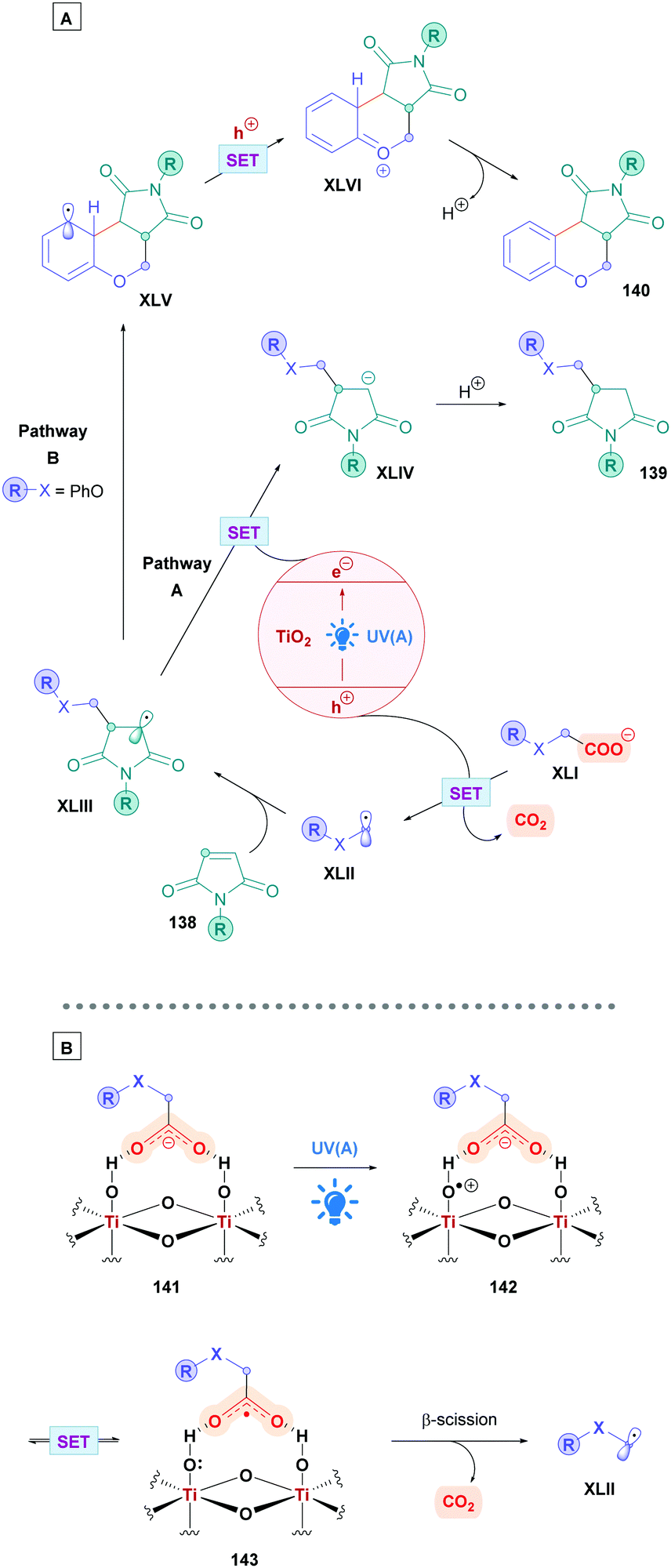 | ||
| Scheme 51 (A) Proposed mechanism for TiO2-catalysed direct DGR of carboxylic acids with maleimides. (B) Proposed mechanism for the SET oxidation between the carboxylic acid and the TiO2 surface. | ||
The decarboxylation process is believed to take place through specified interactions between the acid and TiO2 surface (Scheme 51B). Initial dissociation of the acid onto the TiO2 binds the carboxylate to the surface (141).128 UV(A) irradiation then generates the electron–hole pair (142), which can migrate through the bulk of the semiconductor to the surface. A unique feature of the TiO2 surface is that it is extensively hydroxylated,129 and it is believed that the VB-holes are trapped by these moieties.121,130 Thus, electron transfer between the ‘trapped’ holes and the carboxylate π-system gives the surface-bound carboxyl radical 143. The authors then proposed a competition between decarboxylation via β-scission of the surface-bound radical to give the free radical species 143 and BET to 142. The preferred step is dependent upon the stability of the free radical. For acids that generate more stabilised radicals, decarboxylation is favoured, and those less stabilised favour BET to the carboxylate. This can explain the higher yields observed for the more stabilised radicals, however other factors might be at play to decide reaction outcomes. For instance, the group were unsuccessful when trying to convert vinylacetic, ethynylacetic, and cyanoacetic acids, whilst these species should form stabilised radicals. Interestingly, when using vinylacetic acid, the corresponding allyl radical, was observed by EPR experiments.
As mentioned above, the Walton group encountered difficulties when attempting to generate the less stable aliphatic radicals, and low yields of the corresponding Giese adducts were obtained. Recently, however, Nocera and Zhu were successful in overcoming these limitations, reporting the photocatalytic hydroalkylation of electron-deficient olefins (Scheme 52).131 Most noteworthy was their successful incorporation of primary alkyl radicals including  , achieving 22–87% for a whole range of Michael acceptors. The production of
, achieving 22–87% for a whole range of Michael acceptors. The production of  is known from the photooxidative degradation of acetic acid by TiO2,132 where it is a useful intermediate in aerobic oxidation and pollutant degradation conversion processes.131,133 The authors used purely anatase titania nanoparticles instead of P25 (mixture of anatase and rutile forms). Decarboxylative Giese methylation showed broad scope for α-aryl acrylates (e.g., 14ag), with excellent functional group tolerance for electron-withdrawing and -donating groups on the aryl moiety. Aryl-substituted enones, acrylonitriles, and acrylamides also proceeded well. In addition, 2- and 4-vinylpyridines gave moderate yields (e.g., 14af). The aryl substituent was not necessary however, with Michael acceptors such as fumarate, phenyl sulfone and cyclohexanone affording their methylated products. Trisubstituted olefins were also successful (e.g., 14ah), although lower efficiency was observed for tetrasubstituted olefins.
is known from the photooxidative degradation of acetic acid by TiO2,132 where it is a useful intermediate in aerobic oxidation and pollutant degradation conversion processes.131,133 The authors used purely anatase titania nanoparticles instead of P25 (mixture of anatase and rutile forms). Decarboxylative Giese methylation showed broad scope for α-aryl acrylates (e.g., 14ag), with excellent functional group tolerance for electron-withdrawing and -donating groups on the aryl moiety. Aryl-substituted enones, acrylonitriles, and acrylamides also proceeded well. In addition, 2- and 4-vinylpyridines gave moderate yields (e.g., 14af). The aryl substituent was not necessary however, with Michael acceptors such as fumarate, phenyl sulfone and cyclohexanone affording their methylated products. Trisubstituted olefins were also successful (e.g., 14ah), although lower efficiency was observed for tetrasubstituted olefins.
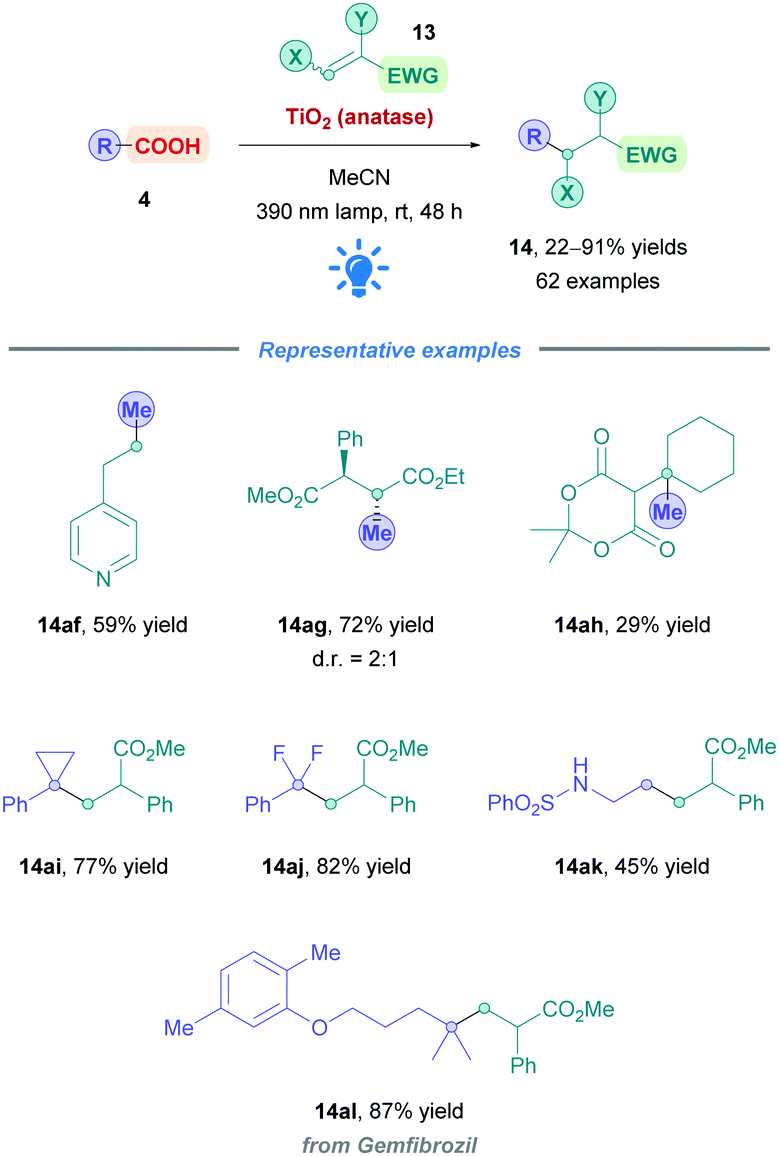 | ||
| Scheme 52 TiO2-Catalysed direct DGR of carboxylic acids for methylation and alkylation. | ||
A broad range of carboxylic acids were explored, including primary, secondary, tertiary, benzylic, and homobenzylic acids which all gave synthetically useful yields (41–91%). Furthermore, α-heteroatom carboxylic acids, e.g., alkyl ether, aromatic, ether, ester, thioester, and amide, gave excellent yields overall without over-oxidation. Cyclic acids, including cyclopropane (14ai) and cyclobutene, were also well tolerated, in addition to primary acids bearing other functional groups in the α-position, e.g., gem-difluoro (14aj), sulfonamide (14ak), ester, carboxylic acid, and ketone. Interestingly, no intramolecular cyclisation was observed for acids bearing alkenes and alkynes, showcasing the facile intermolecular addition of the primary radicals. α-Heterocyclic substrates and phenyl glyoxylic acid also gave their corresponding adducts. Finally, the group demonstrated the utility of their protocol through the alkylation of drug molecule Gemfibrozil (14al).
The mechanism (Scheme 53) is proposed to proceed through adsorption of the acid 4 onto the anatase titanium dioxide surface, with the two carboxyl oxygens coordinating to neighbouring titanium(IV) centres of 144, generating complex 145.134 UV(A) irradiation leads to generation of an electron–hole pair within the anatase, allowing for oxidative cleavage of the C–C bond and inducing decarboxylation to give the alkyl radical I. The kinetics of the bond dissociation is facilitated through inner-sphere hole transfer from the VB to the carboxylate.135 Evidence for the reduction of the TiO2 catalyst (146) was attained using UV-vis diffusive absorbance spectroscopy of the TiO2 catalyst, showing broad absorbance from 500 to 1100 nm (consistent with a blue coloration observed during the reaction) and reduced absorbance at the band edge (<400 nm).136 Radical I then undergoes conjugate addition to the olefin 2, generating the carbon-centred radical II. Reductive SET from the CB of 146 with II produces anion VI, followed by proton transfer from the acetic acid (confirmed by deuterium labelling experiments) to close the catalytic cycle and furnish the desired Giese adduct 3. The formation of VI to 3 could alternatively proceed through a concerted proton-coupled electron transfer. The proximity of the photogenerated hole in VB and an electron in CB prevents dimerization of the I and redox side reactions.
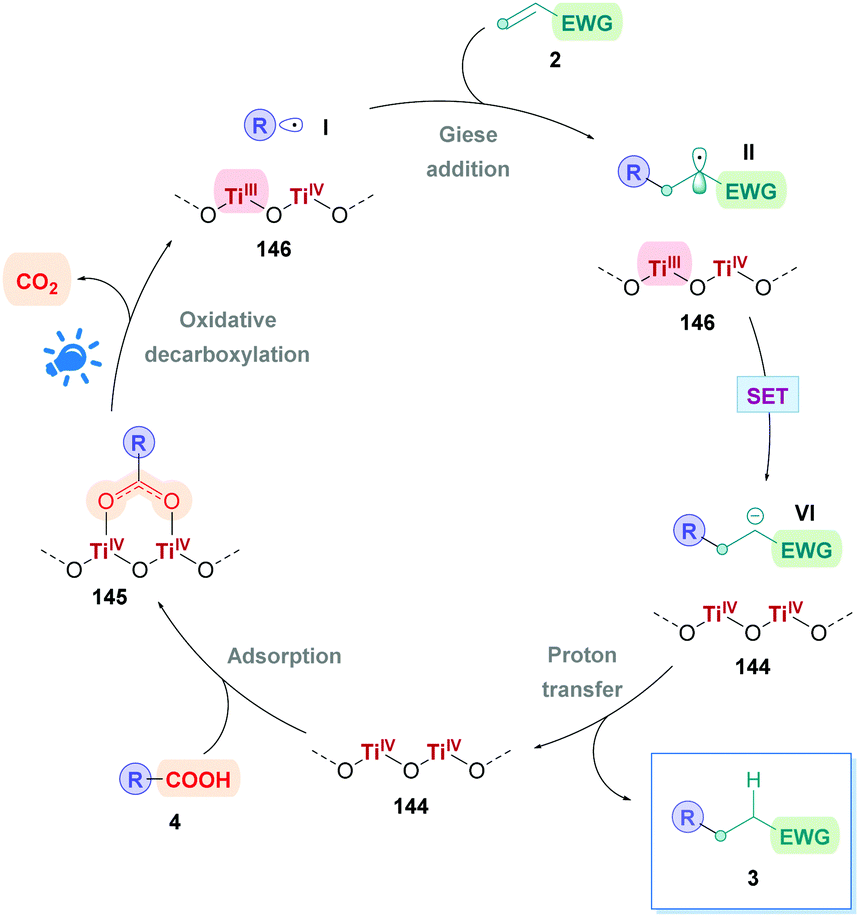 | ||
| Scheme 53 Proposed mechanism for the inner-spehere, TiO2 direct DGR of alkyl carboxylic acids. | ||
Another key feature of this reaction is the inner-sphere direct electron transfer mediated oxidation of the carboxylic acid, without the need for an ancillary base, which is often required for a majority of homogenous photocatalytic systems involving outer-sphere oxidation of carboxylates. The absence of base also permits the application of this transformation to carboxylic acids bearing alkyl halide moieties, with excellent yields achieved and without loss of halide functionality.
The drawback of using TiO2 catalysts is the requirement of higher energy UV light in order to achieve photocatalytic activity, which ultimately limits the substrate scope. Extension into the visible-light region can be achieved by doping TiO2 with metal cations (Cd, Ce, Mn, Bi etc.) and non-metallic anions.137 However, in addition to these additives often being expensive and toxic, the thermal stability of these catalysts is reduced and create undesired electron traps.137b Furthermore, while heterogenous systems benefit from higher stability and easier separation, homogenous systems are more attractive due to better fine-tuning capacities, enhanced activity and selectivity, as well as convenient solution-phase reaction monitoring and mechanistic studies.138 Recently, McIntosh and co-workers synthesised bimetallic Ti–O–Ti bridged amine bis(phenolate) complexes that displayed homogeneous photocatalytic activity for the generation of 1O2 when irradiated with blue light (420 nm).138 Further investigation into the tunability and utility of TiO2-derived catalysts would be beneficial for direct DGRs, as well as other organic transformations.
Since the submission of this manuscript, Inoue and co-workers published a photocatalytic direct DGR involving Pt-doped TiO2 to couple densely oxygenated structures to Michael acceptors.139
3. Silver catalysis (Kochi conditions)
In the 1970s, Kochi pioneered oxidative decarboxylation using readily available persulfate salts.140 The presence of strong acids and catalytic Ag accelerates the rate-limiting decomposition of the persulfate ion to generate a highly oxidising persulfate radical (Ered1/2 = +2.5 to +3.1 V vs. the normal hydrogen electrode (NHE)).141 Minisci and co-workers famously utilised Kochi's conditions to enable decarboxylative radical addition onto heteroaromatics.142It is only in recent years that researchers have begun to use Ag(I)-catalysed Kochi conditions to facilitate decarboxylative radical addition onto electron-deficient olefins to generate cyclic143 and fluorinated144 products. The only known example to date of Kochi-type conditions being applied to direct DGR transformations was demonstrated by the Baran group during their total synthesis of (−)-maximiscin (150, Scheme 54).145 The group desired to perform a key C–C bond formation step through decarboxylation of lactone intermediate 147 to synthesise 149 using phenyl vinyl sulfone (148) as the chosen Michael acceptor. Initially, they attempted to use a reductive manifold, employing their Ni-catalysed decarboxylative Giese protocol15e which was shown to be unsuccessful due to competing HAT processes. Further extensive screening of a variety of redox strategies, including photocatalysis and C–H activation, surprisingly led them to Kochi-type conditions as a simple solution that could both form the desired C–C bond and aldehyde moiety of 149 in one-pot, with Fe2(SO4)2 serving as a unique co-catalyst. Mechanistically, it is believed that initial saponification of lactone 147 gives rise to the carboxylate XLVII, rendering it compatible for Ag-catalysed decarboxylation followed by addition of resulting radical XLVIII onto 148. Radical intermediate XLIX then undergoes a 1,5-HAT process to generate α-oxy radical L. The Fe2(SO4)2 additive is believed to then facilitate Fe-catalysed oxidation of L, furnishing the desired product 149.146 Moreover, the reaction was surprisingly clean with no further purification required before the next step, and suffered little detriment in yields when applied on a gram scale (91%) with excellent diastereoselectivity (>20:1 d.r.).
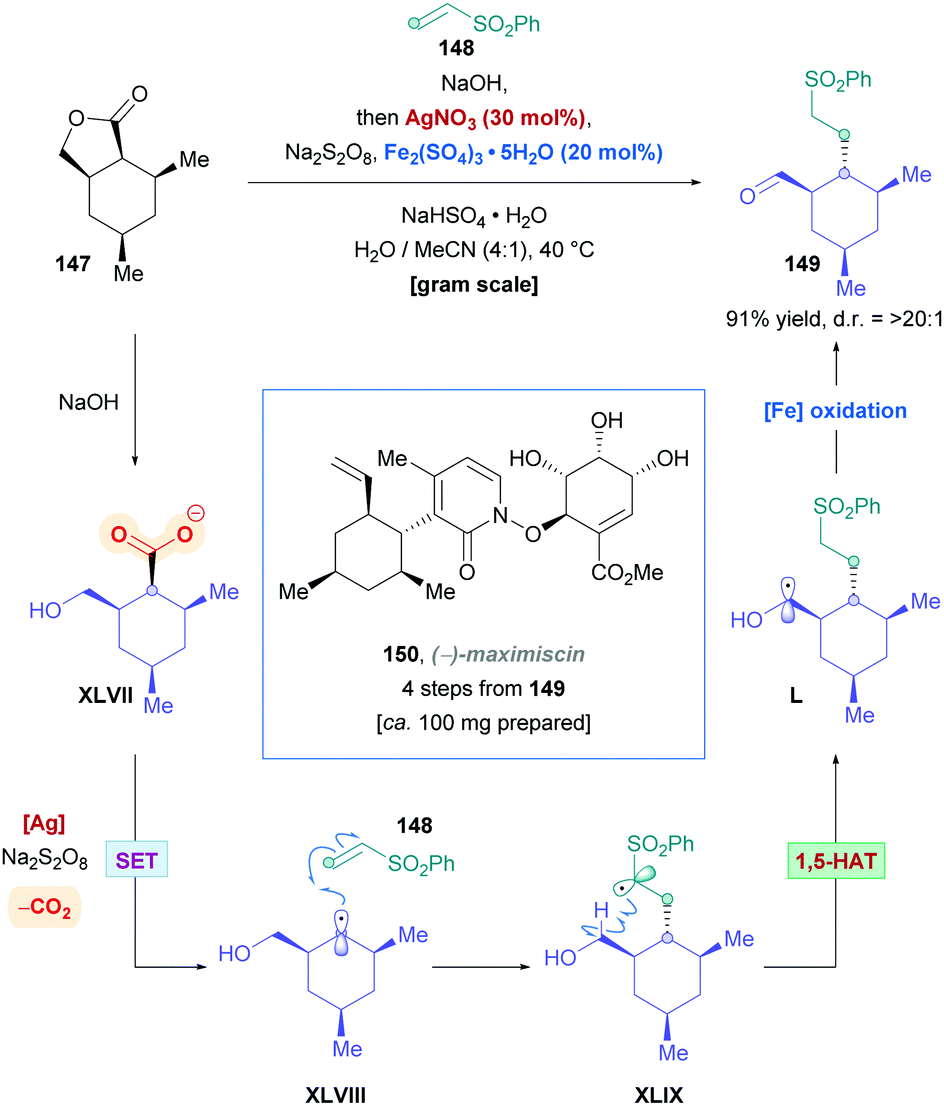 | ||
| Scheme 54 Direct DGR using oxidative silver catalysis (Kochi conditions) for the total synthesis of (−)-maximiscin. | ||
It should be noted that while Baran's example shown in Scheme 54 is, to our knowledge, the only example of a true direct DGR using Kochi conditions, there are a few examples of interrupted Giese reactions, in which the radical conjugate addition intermediate is trapped by an arene,143a–g alkyne,143h or nitrile143i to form a new cyclic system (e.g., Scheme 55). These examples typically use either Ag-mediated Kochi conditions, or related metal-free conditions. In addition, Ag-catalysed decarboxylative allylations have also been reported.147
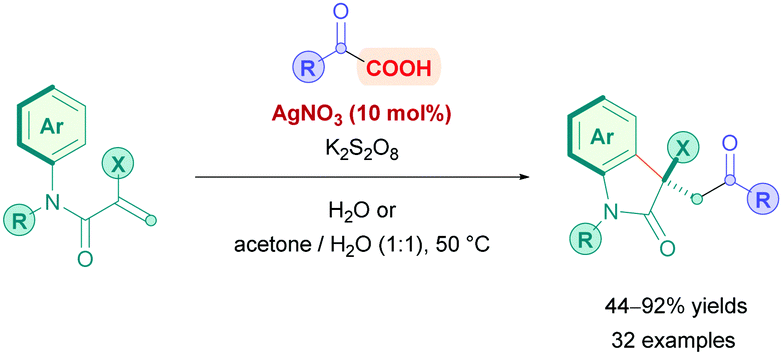 | ||
| Scheme 55 Kochi conditions used for an interrupted Giese where the conjugate addition intermediate is trapped by an arene for cyclisation. | ||
4. Organo-electrochemistry
Recently, organo-electrochemistry has been re-established as an intense area of research.148 Electrochemistry provides organic chemists access to highly selective reactions under relatively mild and tunable conditions.148a In addition, the use of an electric current negates the need for stoichiometric oxidants and reductants.148a As far as we are aware, however, electrochemical oxidative decarboxylation149 has yet to be used for direct DGRs, though generating carbon-centred radicals using decarboxylative Kolbe10 electrolysis for trifluoromethylations150 and tandem annulations151 are known. Wang and co-workers have recently reported using electrochemistry for the indirect DGR of NHPI esters, which proceeds through a reductive decarboxylation mechanism.152The closest reaction to a direct DGR was reported by Markó in 2008 (Scheme 56),151b building on work by Schäfer and co-workers.151a A Kolbe decarboxylation of 141 or 142 followed by a Giese-type radical conjugate addition forms a radical intermediate. However, instead of HAT to form the Giese adduct, here the radical intermediate is trapped by an alkyl radical formed from another acid RCO2H (4). A variety of 5- and 6-membered rings (143 or 144) were formed in this way (Scheme 56).
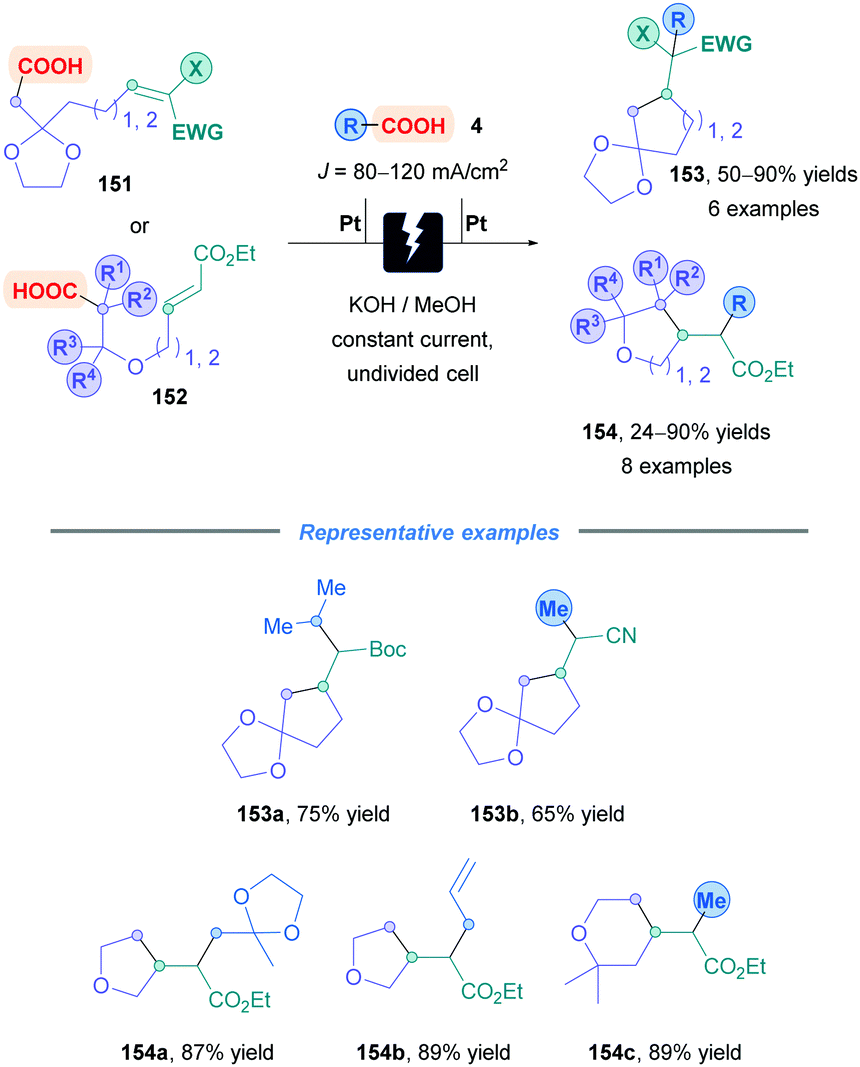 | ||
| Scheme 56 Marko's organo-electrochemistry method for Kolbe decarboxylation followed by radical cyclisation. | ||
5. Conclusions
In this review, we have summarised the latest developments on Giese reactions that proceed via direct decarboxylation of carboxylic acids. Different methodologies to facilitate decarboxylation including photocatalysis involving transition metal- and organo-based systems, heterogenous photocatalysis, dual catalysis as well as silver catalysis were discussed. The mild conditions associated with these methodologies grant organic chemists access to a diverse range of novel C–C coupled products which are often not otherwise attainable by conventional nucleophilic protocols. While most of the progress made in the past decade has been related to photocatalysis, Baran's synthesis of (−)-maximiscin145 demonstrates that non-photocatalytic methods can offer additional and complementary protocols. As to future development of the field, we envisage that research towards developing direct DGRs using organo-electrochemistry would further advance the field.Additional improvements could also be made to direct DGRs, such as further development of enantioselective DGRs, improved chemoselective protocols for enhanced functional group tolerance (i.e. for oxidation-prone and base-labile substrates), and the use of more sustainable reaction conditions. Indeed, base-free Giese methods using cheap and available titanium oxide photocatalysts currently have the drawback of requiring UV irradiation rather than visible light irradiation, and enantioselective protocols are currently still rather limited in substrate scope.
The direct DGR has so far been exploited in a plethora of applications, emphasising their utility in organic synthesis. Applications include chemoselective bioconjugation of peptides,85 synthesis of unnatural amino acids,49,80,102,106 polymerisation,72g macrocyclisations38,72a,72b,72j and natural product/drug molecule synthesis,26,43,50,79,145 though there is no doubt that further applications will be forthcoming as the field progresses.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Engineering and Physical Sciences Research Council and GSK for financial support [industrial CASE PhD studentship to DMK; grant code: EP/V519522/1].Notes and references
- For recent reviews on radical chemistry, see: (a) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed; (b) J. M. Smith, S. J. Harwood and P. S. Baran, Acc. Chem. Res., 2018, 51, 1807–1817 CrossRef CAS PubMed; (c) S. Ni, N. M. Padial, C. Kingston, J. C. Vantourout, D. C. Schmitt, J. T. Edwards, M. M. Kruszyk, R. R. Merchant, P. K. Mykhailiuk, B. B. Sanchez, S. Yang, M. A. Perry, G. M. Gallego, J. J. Mousseau, M. R. Collins, R. J. Cherney, P. S. Lebed, J. S. Chen, T. Qin and P. S. Baran, J. Am. Chem. Soc., 2019, 141, 6726–6739 CrossRef CAS PubMed.
- For selected reviews on two-electron conjugate additions, see: (a) B. E. Rossiter and N. M. Swingle, Chem. Rev., 1992, 92, 771–806 CrossRef CAS; (b) J. Leonard, E. Díez-Barra and S. Merino, Eur. J. Org. Chem., 1998, 2051–2061 CrossRef CAS; (c) M. P. Sibi and S. Manyem, Tetrahedron, 2000, 56, 8033–8061 CrossRef CAS; (d) J. L. Vicario, D. Badía and L. Carrillo, Synthesis, 2007, 2065–2092 CrossRef CAS; (e) A. Alexakis, J. E. Bäckvall, N. Krause, O. PàMies and M. DiéGuez, Chem. Rev., 2008, 108, 2796–2823 CrossRef CAS PubMed; (f) S. R. Harutyunyan, T. Den Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824–2852 CrossRef CAS PubMed; (g) A. G. Csákÿ, G. D. L. Herrán and M. C. Murcia, Chem. Soc. Rev., 2010, 39, 4080–4102 RSC; (h) K. Zheng, X. Liu and X. Feng, Chem. Rev., 2018, 118, 7586–7656 CrossRef CAS PubMed; (i) D. Pichon, J. Morvan, C. Crévisy and M. Mauduit, Beilstein J. Org. Chem., 2020, 16, 212–232 CrossRef CAS PubMed.
- (a) B. Giese and S. Lachhein, Angew. Chem., Int. Ed. Engl., 1981, 20, 967 CrossRef; (b) B. Giese, Angew. Chem., Int. Ed. Engl., 1983, 22, 753–764 CrossRef; (c) B. Giese and J. Dupuis, Angew. Chem., Int. Ed. Engl., 1983, 22, 622–623 CrossRef; (d) B. Giese, J. A. González-Gómez and T. Witzel, Angew. Chem., Int. Ed. Engl., 1984, 23, 69–70 CrossRef.
- For selected examples showcasing decarboxylative Giese-type reactions to form carbon-heteroatom bonds, see: (a) S. B. Lang, K. C. Cartwright, R. S. Welter, T. M. Locascio and J. A. Tunge, Eur. J. Org. Chem., 2016, 3331–3334 CrossRef CAS PubMed; (b) J. Davies, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2017, 56, 13361–13365 CrossRef CAS PubMed; (c) H. Jiang and A. Studer, Angew. Chem., Int. Ed., 2017, 56, 12273–12276 CrossRef CAS PubMed; (d) V. R. Yatham, P. Bellotti and B. König, Chem. Commun., 2019, 55, 3489–3492 RSC; (e) N. X. Xu, B. X. Li, C. Wang and M. Uchiyama, Angew. Chem., Int. Ed., 2020, 59, 10639–10644 CrossRef CAS PubMed.
- B. Giese, Angew. Chem., Int. Ed. Engl., 1983, 95, 771–782 CrossRef CAS.
- (a) K. U. Ingold and J. K. Kochi, Free Radicals, Wiley, New York, 1973 Search PubMed; (b) H. Knoll, Z. Chem., 1982, 22, 245–252 CrossRef CAS.
- A. L. Gant Kanegusuku and J. L. Roizen, Angew. Chem., Int. Ed., 2021, 60, 21116–21149 CrossRef CAS PubMed.
- For selected examples on interrupted direct DGRs, see: (a) X.-F. Xia, S.-L. Zhu, C. Chen, H. Wang and Y.-M. Liang, J. Org. Chem., 2016, 81, 1277–1284 CrossRef CAS PubMed; (b) Q.-F. Bai, C. Jin, J.-Y. He and G. Feng, Org. Lett., 2018, 20, 2172–2175 CrossRef CAS PubMed; (c) Y. Shi, H. Xiao, X.-H. Xu and Y. Huang, Org. Biomol. Chem., 2018, 16, 8472–8476 RSC; (d) C. Shu, R. S. Mega, B. J. Andreassen, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 15430–15434 CrossRef CAS PubMed; (e) Y. Yu, W. Yuan, H. Huang, Z. Cai, P. Liu and P. Sun, J. Org. Chem., 2018, 83, 1654–1660 CrossRef CAS PubMed; (f) G. Chen, C. Li, J. Peng, Z. Yuan, P. Liu and X. Liu, Org. Biomol. Chem., 2019, 17, 8527–8532 RSC; (g) H. Yang, G. Wei and Z. Jiang, ACS Catal., 2019, 9, 9599–9605 CrossRef CAS; (h) R. S. Mega, V. K. Duong, A. Noble and V. K. Aggarwal, Angew. Chem., 2020, 132, 4405–4409 CrossRef; (i) Y. Su, R. Zhang, W. Xue, X. Liu, Y. Zhao, K.-H. Wang, D. Huang, C. Huo and Y. Hu, Org. Biomol. Chem., 2020, 18, 1940–1948 RSC; (j) H. Zhao, N. Ni, X. Li, D. Cheng and X. Xu, Polyhedron, 2021, 206, 115337 CrossRef CAS; (k) Y. Y. Cheng, J. X. Yu, T. Lei, H. Y. Hou, B. Chen, C. H. Tung and L. Z. Wu, Angew. Chem., Int. Ed., 2021, 60, 26822–26828 CrossRef CAS PubMed; (l) L.-L. Liao, G.-M. Cao, Y.-X. Jiang, X.-H. Jin, X.-L. Hu, J. J. Chruma, G.-Q. Sun, Y.-Y. Gui and D.-G. Yu, J. Am. Chem. Soc., 2021, 143, 2812–2821 CrossRef CAS PubMed.
- (a) J. Schwarz and B. König, Green Chem., 2016, 18, 4743–4749 RSC; (b) J. Schwarz and B. König, Green Chem., 2018, 20, 323–361 RSC.
- (a) H. Kolbe, Liebigs Ann. Chem., 1848, 64, 339–341 CrossRef; (b) H. Kolbe, Liebigs Ann. Chem., 1849, 69, 257–294 CrossRef.
- H. Hunsdiecker and C. Hunsdiecker, Chem. Ber., 1942, 75, 291–297 CrossRef.
- (a) D. H. R. Barton, D. Crich and W. B. Motherwell, J. Chem. Soc., Chem. Commun., 1983, 939–941 RSC; (b) D. H. R. Barton, Y. Hervé, P. Potier and J. Thierry, J. Chem. Soc., Chem. Commun., 1984, 1298–1299 RSC; (c) D. H. R. Barton, D. Bridon, I. Fernandaz-Picot and S. Z. Zard, Tetrahedron, 1987, 43, 2733–2740 CrossRef CAS.
- (a) D. H. R. Barton, D. Crich and G. Kretzschmar, Tetrahedron Lett., 1984, 25, 1055–1058 CrossRef CAS; (b) D. H. R. Barton, C. Ching-Yuh and J. C. Jaszberenyi, Tetrahedron Lett., 1992, 33, 5013–5016 CrossRef CAS.
- (a) K. Okada, K. Okamoto, N. Morita, K. Okubo and M. Oda, J. Am. Chem. Soc., 1991, 113, 9401–9402 CrossRef CAS; (b) S. Murarka, Adv. Synth. Catal., 2018, 360, 1735–1753 CrossRef CAS; (c) S. K. Parida, T. Mandal, S. Das, S. K. Hota, S. De Sarkar and S. Murarka, ACS Catal., 2021, 11, 1640–1683 CrossRef CAS.
- For selected examples involving NHPI esters, see: (a) M. J. Schnermann and L. E. Overman, Angew. Chem., Int. Ed., 2012, 51, 9576–9580 CrossRef CAS PubMed; (b) G. L. Lackner, K. W. Quasdorf and L. E. Overman, J. Am. Chem. Soc., 2013, 135, 15342–15345 CrossRef CAS PubMed; (c) C. R. Jamison and L. E. Overman, Acc. Chem. Res., 2016, 49, 1578–1586 CrossRef CAS PubMed; (d) Y. Slutskyy and L. E. Overman, Org. Lett., 2016, 18, 2564–2567 CrossRef CAS PubMed; (e) T. Qin, L. R. Malins, J. T. Edwards, R. R. Merchant, A. J. E. Novak, J. Z. Zhong, R. B. Mills, M. Yan, C. Yuan, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 260–265 CrossRef CAS PubMed; (f) L. Z. Gao, G. Q. Wang, J. Cao, D. D. Yuan, C. Xu, X. W. Guo and S. H. Li, Chem. Commun., 2018, 54, 11534–11537 RSC; (g) C. Zheng, G. Z. Wang and R. Shang, Adv. Synth. Catal., 2019, 361, 4500–4505 CrossRef CAS.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, 1st edn, CRC Press, Boca Raton, FL, 2007 Search PubMed.
- J. D. Griffin, M. A. Zeller and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 11340–11348 CrossRef CAS PubMed.
- (a) W. Zhang, Tetrahedron, 2001, 57, 7237–7262 CrossRef CAS; (b) G. S. C. Srikanth and S. L. Castle, Tetrahedron, 2005, 61, 10377–10441 CrossRef CAS.
- (a) P. Perlmutter, Conjugate Addition Reactions in Organic Synthesis, 1st edn, Pergamon, Oxford, 1992 Search PubMed; (b) P. Renaud and M. Gerster, Angew. Chem., Int. Ed., 1998, 37, 2562–2579 CrossRef CAS; (c) M. P. Sibi and N. A. Porter, Acc. Chem. Res., 1999, 32, 163–171 CrossRef CAS; (d) M. P. Sibi, S. Manyem and J. Zimmerman, Chem. Rev., 2003, 103, 3263–3296 CrossRef CAS PubMed.
- For selected reviews on photoredox catalysis, see: (a) J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS PubMed; (b) J.-P. Goddard, C. Ollivier and L. Fensterbank, Acc. Chem. Res., 2016, 49, 1924–1936 CrossRef CAS PubMed; (c) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed; (d) M. H. Shaw, J. Twilton and D. W. C. Macmillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (e) J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563–2575 CrossRef CAS PubMed; (f) S. Roslin and L. R. Odell, Eur. J. Org. Chem., 2017, 1993–2007 CrossRef CAS; (g) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed; (h) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed; (i) C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed.
- L. Capaldo, L. Buzzetti, D. Merli, M. Fagnoni and D. Ravelli, J. Org. Chem., 2016, 81, 7102–7109 CrossRef CAS PubMed.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (b) T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562–576 RSC; (c) K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 CrossRef CAS PubMed.
- Y. Miyake, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2013, 49, 7854–7856 RSC.
- G. D'Aprano, E. Proynov, M. Lebœuf, M. Leclerc and D. R. Salahub, J. Am. Chem. Soc., 1996, 118, 9736–9742 CrossRef.
- (a) D. A. Smith and S. Vijayakumar, Tetrahedron Lett., 1991, 32, 3617–3620 CrossRef CAS; (b) X. Li, Y.-D. Wu and D. Yang, Acc. Chem. Res., 2008, 41, 1428–1438 CrossRef CAS PubMed; (c) X.-J. Liao, W. Guo and S.-H. Xu, Acta Crystallogr., Sect. E, 2011, 67, o1732 CrossRef CAS PubMed; (d) R. K. Castellano, Y. Li, E. A. Homan, A. J. Lampkins, I. V. Marín and K. A. Abboud, Eur. J. Org. Chem., 2012, 4483–4492 CrossRef CAS.
- L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef CAS PubMed.
- G.-Z. Wang, R. Shang, W.-M. Cheng and Y. Fu, Org. Lett., 2015, 17, 4830–4833 CrossRef CAS PubMed.
- C. Walling and E. R. Briggs, J. Am. Chem. Soc., 1946, 68, 1141–1145 CrossRef CAS PubMed.
- (a) L. J. Gooßen, B. Zimmermann and T. Knauber, Angew. Chem., Int. Ed., 2008, 47, 7103–7106 CrossRef PubMed; (b) R. Shang and L. Liu, Sci. China: Chem., 2011, 54, 1670–1687 CrossRef CAS; (c) T. Patra and D. Maiti, Chem. – Eur. J., 2017, 23, 7382–7401 CrossRef CAS PubMed.
- J.-Q. Chen, R. Chang, Y.-L. Wei, J.-N. Mo, Z.-Y. Wang and P.-F. Xu, J. Org. Chem., 2018, 83, 253–259 CrossRef CAS PubMed.
- C. C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. Macmillan and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 11270–11273 CrossRef CAS PubMed.
- S. Y. Abbas, P. Zhao and L. E. Overman, Org. Lett., 2018, 20, 868–871 CrossRef CAS PubMed.
- Y. Slutskyy, C. R. Jamison, G. L. Lackner, D. S. Müller, A. P. Dieskau, N. L. Untiedt and L. E. Overman, J. Org. Chem., 2016, 81, 7029–7035 CrossRef CAS PubMed.
- A. Millet, Q. Lefebvre and M. Rueping, Chem. – Eur. J., 2016, 22, 13464–13468 CrossRef CAS PubMed.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- F. Monti, A. Baschieri, I. Gualandi, J. J. Serrano-Pérez, J. M. Junquera-Hernández, D. Tonelli, A. Mazzanti, S. Muzzioli, S. Stagni, C. Roldan-Carmona, A. Pertegás, H. J. Bolink, E. Ortí, L. Sambri and N. Armaroli, Inorg. Chem., 2014, 53, 7709–7721 CrossRef CAS PubMed.
- A. Gualandi, E. Matteucci, F. Monti, A. Baschieri, N. Armaroli, L. Sambri and P. G. Cozzi, Chem. Sci., 2017, 8, 1613–1620 RSC.
- S. J. McCarver, J. X. Qiao, J. Carpenter, R. M. Borzilleri, M. A. Poss, M. D. Eastgate, M. M. Miller and D. W. C. Macmillan, Angew. Chem., Int. Ed., 2017, 56, 728–732 CrossRef CAS PubMed.
- P. Fernandez-Rodriguez, F. Legros, T. Maier, A. Weber, M. Méndez, V. Derdau, G. Hessler, M. Kurz, A. Villar-Garea and S. Ruf, Eur. J. Org. Chem., 2021, 782–787 CrossRef CAS.
- S. Zhang, Z. M. Tan, H. N. Zhang, J. L. Liu, W. T. Xu and K. Xu, Chem. Commun., 2017, 53, 11642–11645 RSC.
- C. Cai, S. Yu, B. Cao and X. Zhang, Chem. – Eur. J., 2012, 18, 9992–9998 CrossRef CAS PubMed.
- L. Gingipalli, J. Boerth, D. Emmons, T. Grebe, H. Hatoum-Mokdad, B. Peng, L. Sha, S. Tentarelli, H. Wang, Y. Wu, X. Zheng, S. Edmondson and A. Gopalsamy, Org. Lett., 2020, 22, 3418–3422 CrossRef CAS PubMed.
- S. Inuki, K. Sato, T. Fukuyama, I. Ryu and Y. Fujimoto, J. Org. Chem., 2017, 82, 1248–1253 CrossRef CAS PubMed.
- T. Guo, L. Zhang, Y. Fang, X. Jin, Y. Li, R. Li, X. Li, W. Cen, X. Liu and Z. Tian, Adv. Synth. Catal., 2018, 360, 1352–1357 CrossRef CAS.
- (a) K. Schlüter, R. D. Walter, B. Bergmann and T. Kurz, Eur. J. Med. Chem., 2006, 41, 1385–1397 CrossRef PubMed; (b) C. T. Behrendt, A. Kunfermann, V. Illarionova, A. Matheeussen, T. Gräwert, M. Groll, F. Rohdich, A. Bacher, W. Eisenreich, M. Fischer, L. Maes and T. Kurz, ChemMedChem, 2010, 5, 1673–1676 CrossRef CAS PubMed.
- A. Noble, R. S. Mega, D. Pflästerer, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 2155–2159 CrossRef CAS PubMed.
- (a) D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CrossRef CAS PubMed; (b) A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382–1414 CrossRef CAS PubMed.
- M. Zhang, P. He and Y. Li, Chem. Res. Chinese Univ., 2021, 37, 1044–1054 CrossRef CAS.
- K. Merkens, F. J. Aguilar Troyano, J. Djossou and A. Gómez-Suárez, Adv. Synth. Catal., 2020, 362, 2354–2359 CrossRef CAS.
- J. C. Deforest, R. A. Samame, G. Suryn, A. Burtea and S. D. Rychnovsky, J. Org. Chem., 2018, 83, 8914–8925 CrossRef CAS PubMed.
- C. R. J. Stephenson, T. P. Yoon and D. W. C. MacMillan, Visible Light Photocatalysis in Organic Chemistry, 1st edn, Wiley-VCH, Weinheim, 2018 Search PubMed.
- C. B. Larsen and O. S. Wenger, Chem. – Eur. J., 2018, 24, 2039–2058 CrossRef CAS PubMed.
- K. S. Kjær, N. Kaul, O. Prakash, P. Chábera, N. W. Rosemann, A. Honarfar, O. Gordivska, L. A. Fredin, K.-E. Bergquist, L. Häggström, T. Ericsson, L. Lindh, A. Yartsev, S. Styring, P. Huang, J. Uhlig, J. Bendix, D. Strand, V. Sundström, P. Persson, R. Lomoth and K. Wärnmark, Science, 2019, 363, 249–253 CrossRef PubMed.
- (a) C. A. Parker and E. J. Bowen, Proc. R. Soc. Lond. A, 1953, 220, 104–116 CrossRef; (b) C. G. Hatchard and C. A. Parker, Proc. R. Soc. Lond. A, 1956, 235, 518–536 CrossRef CAS.
- (a) C. Weller, S. Horn and H. Herrmann, J. Photochem. Photobiol., A, 2013, 268, 24–36 CrossRef CAS; (b) D. M. Mangiante, R. D. Schaller, P. Zarzycki, J. F. Banfield and B. Gilbert, ACS Earth Space Chem., 2017, 1, 270–276 CrossRef CAS; (c) G. C. O’Neil, L. Miaja-Avila, Y. I. Joe, B. K. Alpert, M. Balasubramanian, D. M. Sagar, W. Doriese, J. W. Fowler, W. K. Fullagar, N. Chen, G. C. Hilton, R. Jimenez, B. Ravel, C. D. Reintsema, D. R. Schmidt, K. L. Silverman, D. S. Swetz, J. Uhlig and J. N. Ullom, J. Phys. Chem. Lett., 2017, 8, 1099–1104 CrossRef PubMed; (d) J. E. Vernia, M. R. Warmin, J. A. Krause, D. L. Tierney and M. J. Baldwin, Inorg. Chem., 2017, 56, 13029–13034 CrossRef CAS PubMed; (e) S. Straub, P. Brünker, J. Lindner and P. Vöhringer, Angew. Chem., Int. Ed., 2018, 57, 5000–5005 CrossRef CAS PubMed.
- Y. Abderrazak, A. Bhattacharyya and O. Reiser, Angew. Chem., Int. Ed., 2021, 60, 21100–21115 CrossRef CAS PubMed.
- G. Feng, X. Wang and J. Jin, Eur. J. Org. Chem., 2019, 6728–6732 CrossRef CAS.
- For selected reviews on TBADT, see: (a) C. Tanielian, Coord. Chem. Rev., 1998, 178–180, 1165–1181 CrossRef CAS; (b) M. D. Tzirakis, I. N. Lykakis and M. Orfanopoulos, Chem. Soc. Rev., 2009, 38, 2609 RSC; (c) D. Ravelli, S. Protti and M. Fagnoni, Acc. Chem. Res., 2016, 49, 2232–2242 CrossRef CAS PubMed.
- For selected reviews on POMs, see: (a) T. Yamase and T. Usami, J. Chem. Soc., Dalton Trans., 1988, 183–190 RSC; (b) S. Protti, D. Ravelli, M. Fagnoni and A. Albini, Chem. Commun., 2009, 7351–7353 RSC; (c) T. Basile, L. Capaldo, D. Ravelli and P. Quadrelli, Eur. J. Org. Chem., 2020, 1443–1447 CrossRef CAS.
- (a) M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34–48 CrossRef; (b) J. J. Borrás-Almenar, E. Coronado and A. Mueller, Polyoxometalate Molecular Science, Kluwer Academic Publisher, Dordrecht, The Netherlands, 2003 CrossRef; (c) D.-L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105–121 RSC; (d) N. Dupré, P. Rémy, K. Micoine, C. Boglio, S. Thorimbert, E. Lacôte, B. Hasenknopf and M. Malacria, Chem. – Eur. J., 2010, 16, 7256–7264 CrossRef PubMed; (e) S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- D. C. Duncan, T. L. Netzel and C. L. Hill, Inorg. Chem., 1995, 34, 4640–4646 CrossRef CAS.
- D. Ravelli, M. Fagnoni, T. Fukuyama, T. Nishikawa and I. Ryu, ACS Catal., 2018, 8, 701–713 CrossRef CAS.
- (a) I. Texier, J. A. Delaire and C. Giannotti, Phys. Chem. Chem. Phys., 2000, 2, 1205–1212 RSC; (b) S. Montanaro, D. Ravelli, D. Merli, M. Fagnoni and A. Albini, Org. Lett., 2012, 14, 4218–4221 CrossRef CAS PubMed.
- V. D. Waele, O. Poizat, M. Fagnoni, A. Bagno and D. Ravelli, ACS Catal., 2016, 6, 7174–7182 CrossRef CAS.
- D. W. Manley and J. C. Walton, Org. Lett., 2014, 16, 5394–5397 CrossRef CAS PubMed.
- (a) T. Yamase, N. Takabayashi and M. Kaji, J. Chem. Soc., Dalton Trans., 1984, 793–799 RSC; (b) R. F. Renneke, M. Pasquali and C. L. Hill, J. Am. Chem. Soc., 1990, 112, 6585–6594 CrossRef CAS.
- M. A. Fox, Photoinduced Electron Transfer: Part B. Experimental Techniques and Medium Effects, Elsevier, Amsterdam and New York, 1988 Search PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- D. Ravelli, A. Albini and M. Fagnoni, Chem. – Eur. J., 2011, 17, 572–579 CrossRef CAS PubMed.
- G. Guirado, C. N. Fleming, T. G. Lingenfelter, M. L. Williams, H. Zuilhof and J. P. Dinnocenzo, J. Am. Chem. Soc., 2004, 126, 14086–14094 CrossRef CAS PubMed.
- For selected reviews on organophotocatalysis, see: (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (b) E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed; (c) A. Vega-Peñaloza, J. Mateos, X. Companyó, M. Escudero-Casao and L. Dell'Amico, Angew. Chem., Int. Ed., 2021, 60, 1082–1097 CrossRef PubMed.
- (a) Y. Yoshimi, M. Masuda, T. Mizunashi, K. Nishikawa, K. Maeda, N. Koshida, T. Itou, T. Morita and M. Hatanaka, Org. Lett., 2009, 11, 4652–4655 CrossRef CAS PubMed; (b) K. Nishikawa, Y. Yoshimi, K. Maeda, T. Morita, I. Takahashi, T. Itou, S. Inagaki and M. Hatanaka, J. Org. Chem., 2013, 78, 582–589 CrossRef CAS PubMed; (c) Y. Yoshimi, S. Hayashi, K. Nishikawa, Y. Okita, K. Maeda, T. Morita and T. Itou, Res. Chem. Intermed., 2013, 39, 397–402 CrossRef CAS; (d) Y. Yoshimi, S. Washida, Y. Okita, K. Nishikawa, K. Maeda, S. Hayashi and T. Morita, Tetrahedron Lett., 2013, 54, 4324–4326 CrossRef CAS; (e) K. Maeda, H. Saito, K. Osaka, K. Nishikawa, M. Sugie, T. Morita, I. Takahashi and Y. Yoshimi, Tetrahedron, 2015, 71, 1117–1123 CrossRef CAS; (f) K. Osaka, M. Sugie, M. Yamawaki, T. Morita and Y. Yoshimi, J, Photochem. Photobiol., A, 2016, 317, 50–55 CrossRef CAS; (g) M. Yamawaki, A. Ukai, Y. Kamiya, S. Sugihara, M. Sakai and Y. Yoshimi, ACS Macro Lett., 2017, 6, 381–385 CrossRef CAS; (h) Y. Yoshimi, J. Photochem. Photobiol., A, 2017, 342, 116–130 CrossRef CAS; (i) T. Yamamoto, T. Iwasaki, T. Morita and Y. Yoshimi, J. Org. Chem., 2018, 83, 3702–3709 CrossRef CAS PubMed; (j) T. Iwasaki, Y. Tajimi, K. Kameda, C. Kingwell, W. Wcislo, K. Osaka, M. Yamawaki, T. Morita and Y. Yoshimi, J. Org. Chem., 2019, 84, 8019–8026 CrossRef CAS PubMed; (k) K. Osaka, A. Usami, T. Iwasaki, M. Yamawaki, T. Morita and Y. Yoshimi, J. Org. Chem., 2019, 84, 9480–9488 CrossRef CAS PubMed; (l) S. Kubosaki, H. Takeuchi, Y. Iwata, Y. Tanaka, K. Osaka, M. Yamawaki, T. Morita and Y. Yoshimi, J. Org. Chem., 2020, 85, 5362–5369 CrossRef CAS PubMed.
- R. M. Borg, D. R. Arnold and T. S. Cameron, Can. J. Chem., 1984, 62, 1785–1802 CrossRef CAS.
- (a) H. E. Zimmerman and P. A. Wang, J. Am. Chem. Soc., 1990, 112, 1280–1281 CrossRef CAS; (b) H. E. Zimmerman and P. A. Wang, J. Am. Chem. Soc., 1993, 115, 2205–2216 CrossRef CAS.
- Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. Macmillan, J. Am. Chem. Soc., 2016, 138, 1832–1835 CrossRef CAS PubMed.
- For selected examples using the MOC strategy, see: (a) T. Kawabata, K. Yahiro and K. Fuji, J. Am. Chem. Soc., 1991, 113, 9694–9696 CrossRef CAS; (b) K. Fuji and T. Kawabata, Chem. – Eur. J., 1998, 4, 373–376 CrossRef CAS; (c) H. Zhao, D. C. Hsu and P. R. Carlier, Synthesis, 2005, 1–16 Search PubMed.
- Y. Ozaki, T. Yamada, T. Mizuno, K. Osaka, M. Yamawaki, H. Maeda, T. Morita and Y. Yoshimi, Tetrahedron, 2019, 75, 130493 CrossRef CAS.
- T. Yamada, Y. Ozaki, M. Yamawaki, Y. Sugiura, K. Nishino, T. Morita and Y. Yoshimi, Tetrahedron Lett., 2017, 58, 835–838 CrossRef CAS.
- K. Minagawa, D. Kamakura, K. Hagiwara and M. Inoue, Tetrahedron, 2020, 76, 131385 CrossRef CAS.
- O. Zhang and J. W. Schubert, J. Org. Chem., 2020, 85, 6225–6232 CrossRef CAS PubMed.
- F. Bureš, RSC Adv., 2014, 4, 58826–58851 RSC.
- Y. Yin, Y. Dai, H. Jia, J. Li, L. Bu, B. Qiao, X. Zhao and Z. Jiang, J. Am. Chem. Soc., 2018, 140, 6083–6087 CrossRef CAS PubMed.
- L. Lin, X. Bai, X. Ye, X. Zhao, C.-H. Tan and Z. Jiang, Angew. Chem., Int. Ed., 2017, 56, 13842–13846 CrossRef CAS PubMed.
- V. Srivastava, P. K. Singh, A. Srivastava and P. P. Singh, RSC Adv., 2021, 11, 14251–14259 RSC.
- S. Bloom, C. Liu, D. K. Kölmel, J. X. Qiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. Macmillan, Nat. Chem., 2018, 10, 205–211 CrossRef CAS PubMed.
- Z. Zuo and D. W. C. Macmillan, J. Am. Chem. Soc., 2014, 136, 5257–5260 CrossRef CAS PubMed.
- M. Galicia and F. J. González, J. Electrochem. Soc., 2002, 149, D46–D50 CrossRef CAS.
- M. Novak, A. Miller, T. C. Bruice and G. Tollin, J. Am. Chem. Soc., 1980, 102, 1465–1467 CrossRef CAS.
- V. I. Timofeev, R. N. Chuprov-Netochin, V. R. Samigina, V. V. Bezuglov, K. A. Miroshnikov and I. P. Kuranova, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2010, 66, 259–263 CrossRef CAS PubMed.
- I. Carmichael, W. P. Helman and G. L. Hug, J. Phys. Chem. Ref. Data, 1987, 16, 239–260 CrossRef CAS.
- N. F. Nikitas, P. L. Gkizis and C. G. Kokotos, Org. Biomol. Chem., 2021, 19, 5237–5253 RSC.
- (a) K. Iijima, I. Oonishi, S. Fujisawa and S. Shibata, Bull. Chem. Soc. Jpn., 1987, 60, 3887–3890 CrossRef CAS; (b) L. T. Okano, T. C. Barros, D. T. H. Chou, A. J. Bennet and C. Bohne, J. Phys. Chem. B, 2001, 105, 2122–2128 CrossRef CAS; (c) Ò. Rubio-Pons, L. Serrano-Andrés, D. Burget and P. Jacques, J. Photochem. Photobiol., A, 2006, 179, 298–304 CrossRef.
- D.-L. Zhu, Q. Wu, D. J. Young, H. Wang, Z.-G. Ren and H.-X. Li, Org. Lett., 2020, 22, 6832–6837 CrossRef CAS PubMed.
- D.-L. Zhu, R. Xu, Q. Wu, H.-Y. Li, J.-P. Lang and H.-X. Li, J. Org. Chem., 2020, 85, 9201–9212 CrossRef CAS PubMed.
- (a) Y. Maki, I. Oyabu, S. Ohara, M. Sako, Y. Kitade and K. Hirota, Chem. Pharm. Bull., 1989, 37, 3239–3242 CrossRef CAS; (b) R. Hauptmann, A. Petrosyan, F. Fennel, M. A. Argüello Cordero, A. E. Surkus and J. Pospech, Chem. – Eur. J., 2019, 25, 4325–4329 CrossRef CAS PubMed.
- F. El-Hage, C. Schöll and J. Pospech, J. Org. Chem., 2020, 85, 13853–13867 CrossRef CAS PubMed.
- S. P. Pitre, C. D. McTiernan and J. C. Scaiano, ACS Omega, 2016, 1, 66–76 CrossRef CAS PubMed.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- S. Fukuzumi, K. Ohkubo, T. Suenobu, K. Kato, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 2001, 123, 8459–8467 CrossRef CAS PubMed.
- T. Chinzei, K. Miyazawa, Y. Yasu, T. Koike and M. Akita, RSC Adv., 2015, 5, 21297–21300 RSC.
- N. P. Ramirez and J. C. Gonzalez-Gomez, Eur. J. Org. Chem., 2017, 2154–2163 CrossRef CAS.
- P. Ji, Y. Zhang, Y. Dong, H. Huang, Y. Wei and W. Wang, Org. Lett., 2020, 22, 1557–1562 CrossRef CAS PubMed.
- J. E. Rose, P. D. Leeson and D. Gani, J. Chem. Soc., Chem. Commun., 1992, 1784–1786 RSC.
- (a) T. Furuta, H. Takahashi and Y. Kasuya, J. Am. Chem. Soc., 1990, 112, 3633–3636 CrossRef CAS; (b) J. E. Baldwin, R. M. Adlington, D. G. Marquess, A. R. Pitt, M. J. Porter and A. T. Russell, Tetrahedron, 1996, 52, 2515–2536 CrossRef CAS.
- L.-Y. Lian and D. A. Middleton, Prog. Nucl. Magn. Reson. Spectrosc., 2001, 39, 171–190 CrossRef CAS.
- A. A. Shah, M. J. Kelly and J. J. Perkins, Org. Lett., 2020, 22, 2196–2200 CrossRef CAS PubMed.
- Y.-Q. Zou, F. M. Hörmann and T. Bach, Chem. Soc. Rev., 2018, 47, 278–290 RSC.
- T. Morack, C. Mück-Lichtenfeld and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 1208–1212 CrossRef CAS PubMed.
- (a) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (b) J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429–1439 CrossRef CAS PubMed; (c) C. B. Kelly, N. R. Patel, D. N. Primer, M. Jouffroy, J. C. Tellis and G. A. Molander, Nat. Protoc., 2017, 12, 472–492 CrossRef CAS PubMed; (d) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. Macmillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS; (e) E. B. McLean and A.-L. Lee, Tetrahedron, 2018, 74, 4881–4902 CrossRef CAS; (f) W.-J. Zhou, Y.-H. Zhang, Y.-Y. Gui, L. Sun and D.-G. Yu, Synthesis, 2018, 3359–3378 CrossRef CAS.
- H. T. Dang, G. C. Haug, V. T. Nguyen, N. T. H. Vuong, V. D. Nguyen, H. D. Arman and O. V. Larionov, ACS Catal., 2020, 10, 11448–11457 CrossRef CAS.
- V. T. Nguyen, V. D. Nguyen, G. C. Haug, N. T. H. Vuong, H. T. Dang, H. D. Arman and O. V. Larionov, Angew. Chem., Int. Ed., 2020, 59, 7921–7927 CrossRef CAS PubMed.
- V. T. Nguyen, V. D. Nguyen, G. C. Haug, H. T. Dang, S. Jin, Z. Li, C. Flores-Hansen, B. S. Benavides, H. D. Arman and O. V. Larionov, ACS Catal., 2019, 9, 9485–9498 CrossRef CAS.
- N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS.
- W. Yao, E. A. Bazan-Bergamin and M. Y. Ngai, ChemCatChem, 2021, e202101292 Search PubMed.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- J.-J. Zhao, H.-H. Zhang, X. Shen and S. Yu, Org. Lett., 2019, 21, 913–916 CrossRef CAS PubMed.
- (a) D. J. Cole-Hamilton, Science, 2003, 299, 1702–1706 CrossRef CAS PubMed; (b) S. Gisbertz and B. Pieber, ChemPhotoChem, 2020, 4, 456–475 CrossRef CAS; (c) Y. Okada, Chem. Rec., 2021, 21, 1–17 CrossRef PubMed.
- T. Ohno, K. Sarukawa, K. Tokieda and M. Matsumura, J. Catal., 2001, 203, 82–86 CrossRef CAS.
- L. Cermenati, M. Mella and A. Albini, Tetrahedron, 1998, 54, 2575–2582 CrossRef CAS.
- D. W. Manley, R. T. McBurney, P. Miller, R. F. Howe, S. Rhydderch and J. C. Walton, J. Am. Chem. Soc., 2012, 134, 13580–13583 CrossRef CAS PubMed.
- D. W. Manley, R. T. McBurney, P. Miller, J. C. Walton, A. Mills and C. O’Rourke, J. Org. Chem., 2014, 79, 1386–1398 CrossRef CAS PubMed.
- M. Noorjahan, M. Pratap Reddy, V. Durga Kumari, B. Lavédrine, P. Boule and M. Subrahmanyam, J. Photochem. Photobiol., A, 2003, 156, 179–187 CrossRef CAS.
- A. Mills, N. Elliott, G. Hill, D. Fallis, J. R. Durrant and R. L. Willis, Photochem. Photobiol. Sci., 2003, 2, 591–596 CrossRef CAS.
- R. B. Roy and G. A. Swan, J. Chem. Soc. C, 1969, 1886–1891 RSC.
- B. Kraeutler and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 5985–5992 CrossRef CAS.
- (a) E.-A. I. Heiba, R. M. Dessau and W. J. Koehl, J. Am. Chem. Soc., 1968, 90, 1082–1084 CrossRef CAS; (b) E.-A. I. Heiba, R. M. Dessau and W. J. Koehl, J. Am. Chem. Soc., 1969, 91, 138–145 CrossRef CAS; (c) M. E. Kurz, V. Baru and P. N. Nguyen, J. Org. Chem., 1984, 49, 1603–1607 CrossRef CAS; (d) A. Citterio, R. Sebastiano, A. Maronati, R. Santi and F. Bergamini, J. Chem. Soc., Chem. Commun., 1994, 1517–1518 RSC.
- (a) S. Marinković and N. Hoffmann, Int. J. Photoenergy, 2003, 5, 175–182 CrossRef; (b) S. Marinković and N. Hoffmann, Eur. J. Org. Chem., 2004, 3102–3107 CrossRef.
- M. A. Henderson, Surf. Sci. Rep., 2011, 66, 185–297 CrossRef CAS.
- (a) C. Deiana, E. Fois, S. Coluccia and G. Martra, J. Phys. Chem. C, 2010, 114, 21531–21538 CrossRef CAS; (b) C. Salazar and M. A. Nanny, J. Catal., 2010, 269, 404–410 CrossRef CAS.
- S. H. Szczepankiewicz, A. J. Colussi and M. R. Hoffmann, J. Phys. Chem. B, 2000, 104, 9842–9850 CrossRef CAS.
- Q. Zhu and D. G. Nocera, J. Am. Chem. Soc., 2020, 142, 17913–17918 CrossRef CAS PubMed.
- E. L. Quah, J. N. Wilson and H. Idriss, Langmuir, 2010, 26, 6411–6417 CrossRef CAS.
- (a) D. S. Muggli, S. A. Keyser and J. L. Falconer, Catal. Lett., 1998, 55, 129–132 CrossRef CAS; (b) Q. Chen, J. M. Song, F. Pan, F. L. Xia and J. Y. Yuan, Environ. Technol., 2009, 30, 1103–1109 CrossRef CAS PubMed.
- D. C. Grinter, M. Nicotra and G. Thornton, J. Phys. Chem. C, 2012, 116, 11643–11651 CrossRef CAS.
- (a) J. M. Saveant, J. Am. Chem. Soc., 1992, 114, 10595–10602 CrossRef CAS; (b) A. Houmam, Chem. Rev., 2008, 108, 2180–2237 CrossRef CAS PubMed.
- J. N. Schrauben, R. Hayoun, C. N. Valdez, M. Braten, L. Fridley and J. M. Mayer, Science, 2012, 336, 1298–1301 CrossRef CAS PubMed.
- (a) R. Chauhan, A. Kumar and R. P. Chaudhary, Spectrochim. Acta, Part A, 2012, 98, 256–264 CrossRef CAS PubMed; (b) R. Daghrir, P. Drogui and D. Robert, Ind. Eng. Chem. Res., 2013, 52, 3581–3599 CrossRef CAS; (c) M. Pawar, S. Topcu Sendoğdular and P. Gouma, J. Nanomater., 2018, 1–13 CAS.
- K. Behm, E. Fazekas, M. J. Paterson, F. Vilela and R. D. McIntosh, Chem. – Eur. J., 2020, 26, 9486–9494 CrossRef CAS PubMed.
- D. Kuwana, Y. Komori, M. Nagatomo and M. Inoue, J. Org. Chem., 2022, 87, 730–736 CrossRef CAS PubMed.
- (a) J. M. Anderson and J. K. Kochi, J. Am. Chem. Soc., 1970, 92, 1651–1659 CrossRef CAS; (b) J. M. Anderson and J. K. Kochi, J. Org. Chem., 1970, 35, 986–989 CrossRef CAS.
- P. Neta, R. E. Huie and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 1027–1284 CrossRef CAS.
- (a) F. Fontana, F. Minisci, M. C. Nogueira Barbosa and E. Vismara, J. Org. Chem., 1991, 56, 2866–2869 CrossRef CAS; (b) F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Tetrahedron, 1971, 27, 3575–3579 CrossRef CAS.
- (a) H. Wang, L.-N. Guo and X.-H. Duan, Adv. Synth. Catal., 2013, 355, 2222–2226 CrossRef CAS; (b) W.-P. Mai, G.-C. Sun, J.-T. Wang, G. Song, P. Mao, L.-R. Yang, J.-W. Yuan, Y.-M. Xiao and L.-B. Qu, J. Org. Chem., 2014, 79, 8094–8102 CrossRef CAS PubMed; (c) H. Yang, L.-N. Guo and X.-H. Duan, RSC Adv., 2014, 4, 52986–52990 RSC; (d) K. Sun, S.-J. Li, X.-L. Chen, Y. Liu, X.-Q. Huang, D.-H. Wei, L.-B. Qu, Y.-F. Zhao and B. Yu, Chem. Commun., 2019, 55, 2861–2864 RSC; (e) G. Feng, C. Jin, J.-Y. He and Q.-F. Bai, Synlett, 2020, 1517–1522 CrossRef; (f) Z. Zhang, C. Jia, X. Kong, M. Hussain, Z. Liu, W. Liang, L. Jiang, H. Jiang and J. Ma, ACS Sustainable Chem. Eng., 2020, 8, 16463–16468 CrossRef CAS; (g) Q. Liu, L. Wang, J. Liu, S. Ruan and P. Li, Org. Biomol. Chem., 2021, 19, 3489–3496 RSC; (h) J. Li, W.-J. Hao, P. Zhou, Y.-L. Zhu, S.-L. Wang, S.-J. Tu and B. Jiang, RSC Adv., 2017, 7, 9693–9703 RSC; (i) S.-S. Wang, H. Fu, Y. Shen, M. Sun and Y.-M. Li, J. Org. Chem., 2016, 81, 2920–2929 CrossRef CAS PubMed; (j) K. Yan, D. Yang, W. Wei, F. Wang, Y. Shuai, Q. Li and H. Wang, J. Org. Chem., 2015, 80, 1550–1556 CrossRef CAS PubMed.
- C.-G. Li, Q. Xie, X.-L. Xu, F. Wang, B. Huang, Y.-F. Liang and H.-J. Xu, Org. Lett., 2019, 21, 8496–8500 CrossRef CAS PubMed.
- K. S. McClymont, F.-Y. Wang, A. Minakar and P. S. Baran, J. Am. Chem. Soc., 2020, 142, 8608–8613 CrossRef CAS PubMed.
- C. Walling, Acc. Chem. Res., 1975, 8, 125–131 CrossRef CAS.
- (a) L. Cui, H. Chen, C. Liu and C. Li, Org. Lett., 2016, 18, 2188–2191 CrossRef CAS PubMed; (b) X. Li, R. Zhang, X. Zhang, P. Zhu and T. Yao, Chem. – Asian J., 2020, 15, 1175–1179 CrossRef CAS PubMed.
- For recent reviews on organo-electrochemistry, see: (a) E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308 CrossRef CAS PubMed; (b) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (c) S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef; (d) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS; (e) D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC; (f) C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed; (g) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS; (h) N. Chen, Z. Ye and F. Zhang, Org. Biomol. Chem., 2021, 19, 5501–5520 RSC.
- K. D. Moeller, Tetrahedron, 2000, 56, 9527–9554 CrossRef CAS.
- (a) R. N. Renaud and P. J. Champagne, Can. J. Chem., 1975, 53, 529–534 CrossRef CAS; (b) K. Uneyama and H. Nanbu, J. Org. Chem., 1988, 53, 4598–4599 CrossRef CAS; (c) V. N. Andreev, V. A. Grinberg, A. G. Dedov, A. S. Loktev, N. A. Mayorova, I. I. Moiseev and A. A. Stepanov, Russ. J. Electrochem., 2013, 49, 996–1000 CrossRef CAS; (d) K. Arai, K. Watts and T. Wirth, ChemistryOpen, 2014, 3, 23–28 CrossRef CAS.
- (a) A. Matzeit, H. J. Schäfer and C. Amatore, Synthesis, 1995, 1432–1444 CrossRef CAS; (b) F. Lebreux, F. Buzzo and I. E. Markó, Synlett, 2008, 2815–2820 CAS.
- X. Chen, X. Luo, X. Peng, J. Guo, J. Zai and P. Wang, Chem. – Eur. J., 2020, 26, 3226–3230 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |