
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of squafosacin F: stereodivergent approach to mono-tetrahydrofuran acetogenins†
Koichiro Ota *,
Sumika Kohno,
Tomoko Yamashita,
Atsuko Miura,
Kazuo Kamaike and
Hiroaki Miyaoka*
*,
Sumika Kohno,
Tomoko Yamashita,
Atsuko Miura,
Kazuo Kamaike and
Hiroaki Miyaoka*
School of Pharmacy, Tokyo University of Pharmacy and Life Sciences, 1432-1 Horinouchi, Hachioji, Tokyo 192-0392, Japan. E-mail: miyaokah@toyaku.ac.jp; Fax: +81-42-676-3073; Tel: +81-42-676-3080
First published on 5th December 2019
Abstract
Annonaceous acetogenins have a wide range of potential biological activities. The development of simple and diversity-oriented approaches to their synthesis is therefore important. We have achieved the first total synthesis of squafosacin F and assigned its absolute configuration. The key steps were an acid-mediated tandem intramolecular double cyclization to build the hydroxy-flanked mono-tetrahydrofuran core and decoration with the desired functionalities of the target natural product via highly stereoselective reactions.
Introduction
In the past 35 years a vast number of annonaceous acetogenins have been isolated from the leaves, seeds, roots, and bark of tropical plants of the Annonaceae family.1 Acetogenins consist of four substructures, namely an α,β-unsaturated-γ-lactone, one to three tetrahydrofuran (THF) units with flanking hydroxy groups, a long alkyl tail, and an alkyl spacer linking the γ-lactone and tetrahydrofuran moieties. Annonaceous acetogenins are well-known compounds with potent biological activities, e.g., antiviral, antineoplastic, antimalarial, immunosuppressive, and anticoagulant effects.2 Many of the biological activities of acetogenins are attributed to their inhibitory effects on mitochondrial NADH-ubiquinone oxidoreductase (complex I).3 Detailed structure–activity relationship studies have shown that the γ-lactone moiety and the hydroxy-flanked tetrahydrofuran moiety alone do not inhibit mitochondrial complex I activity, but inhibitory activity occurs when these components are linked by an alkyl spacer. The presence of a polar functional group such as a hydroxy group on the alkyl spacer is not necessary. A chain length of C13 is important for achieving the most potent inhibitory activity.4cis-Solamin is a particularly active mono-tetrahydrofuran acetogenin.5 It was isolated from a root extract of Annona muricata L. and its structure was determined by Cavé et al. in 1998. It has a C13 spacer and therefore shows strong mitochondrial complex I inhibitory activity. Because of its unique structure and biological activity, many studies of syntheses of this compound and its analogs have been performed.6 In 2018, we reported its formal synthesis via tandem cyclization of a diepoxy ester to achieve stereoselective formation of the hydroxy-flanked mono-tetrahydrofuran core.7
In 2008, Wu et al. isolated squafosacin F from Annona squamosa L. and determined its structure. This compound is a mono-tetrahydrofuran acetogenin with a C13 spacer (Fig. 1).8
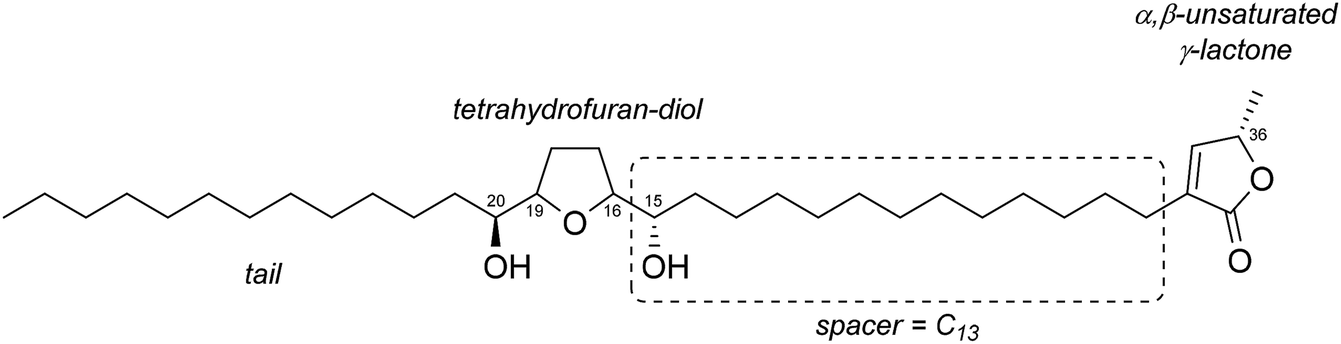 | ||
| Fig. 1 Structure of mono-THF acetogenin squafosacin F. | ||
Squafosacin F is a diastereomer of cis-solamin but its stereochemistry is still uncertain. The signals at δH 3.85 (3H)/δC 83.2, 82.1, and 71.8, and δH 3.40 (1H)/δC 74.3 in the 1H and 13C NMR spectra of this compound indicate that squafosacin F has a tetrahydrofuran moiety flanked by two hydroxy groups in threo and erythro configurations.9 The relative configuration in the important tetrahydrofuran region could not be determined because of the complex signals at C-17 and C-18, and it was not possible to determine whether the configuration was threo/cis/erythro or threo/trans/erythro. However, the absolute configurations of the oxymethine group were identified as 15S and 20S by using the modified Mosher's method.10 This showed that the tetrahydrofuran was in a cis configuration. On the basis of this analysis, the absolute configuration of squafosacin F was identified as either 15S/16S/19R/20S (1) or 15S/16R/19S/20S (2) (Fig. 2). Although squafosacin F is expected to have potent biological activity, similar to that of cis-solamin, the synthesis of squafosacin F has not yet been reported. We undertook the synthesis of squafosacin F to prove that our developed tandem cyclization, which was used in the formal synthesis of cis-solamin,7 provides a comprehensive method for mono-tetrahydrofuran acetogenin synthesis. Here, report the first total synthesis of squafosacin F and determination of its absolute configuration.
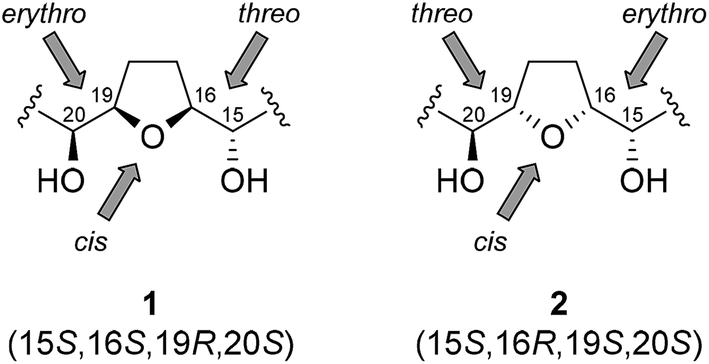 | ||
| Fig. 2 Possible structures of erythro/cis/threo tetrahydrofuran-diol. | ||
Results and discussion
Our retrosynthetic analysis of the candidate compound 1 is shown in Scheme 1. We envisioned that 1 could be obtained from terminal alkene A via a ruthenium-catalyzed Alder–ene reaction11 with ethyl (S)-4-hydroxypent-2-ynoate (3). We anticipated that the synthesis of threo/cis/erythro tetrahydrofurandiol A could achieved by elongation of the appropriate alkyl tail and introduction of the spacer unit via a stereoinversion at the C-15 position to give erythro/cis/erythro tetrahydrofuran-γ-lactone B. We expected that tetrahydrofuran-γ-lactone B could be obtained by using our developed acid-mediated tandem reaction for opening of the epoxide group in diepoxy ester C, which in turn could be synthesized by stepwise asymmetric epoxidation from the known allylic alcohol 4.12 | ||
| Scheme 1 Retrosynthetic analysis of 1. | ||
The synthesis of 1 started with the stereoselective synthesis of diepoxy ester 7 (Scheme 2). The symmetric E,E-diallylic alcohol 4 (ref. 12) was transformed to a monoacetate, which was then converted to epoxy alcohol 5 via a Sharpless asymmetric epoxidation under standard conditions.13 The optical purity of 5 was determined to be >95% ee from the 1H NMR spectrum in the presence of Chirabite-AR.14 Protection of the primary hydroxy group in epoxy alcohol 5 with a methoxymethyl group and subsequent deacetylation gave E-allylic alcohol 6, which was converted to an epoxy alcohol in 86% yield via sharpless asymmetric epoxidation. We obtained the unsaturated ester with excellent selectivity (>95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, E/Z) by oxidation and then a Wittig reaction. Unexpected problems arose during reduction of the resulting unsaturated ester. Initially, we attempted to hydrogenate the double bond by using Pd/C, but the main reaction was epoxide ring opening and almost no diepoxy ester 7 was obtained (<5% yield). This problem was overcome by using a rhodium catalyst.15 The order of addition of the reagents was important. Blowing hydrogen into a suspension of the unsaturated ester and Rh/Al2O3 in THF gave diepoxy ester 7 in moderate yield (51%) but was accompanied by formation of an epoxide-opened byproduct. Slow addition of the unsaturated ester to a suspension of pre-hydrogen-adsorbed Rh/Al2O3 in THF provided ester 7 in 80% yield over two steps from the epoxy alcohol. This suggests that hydrogen adsorption on the rhodium catalyst was not required for epoxide opening and that hydrogenation was faster than epoxide opening.
:5, E/Z) by oxidation and then a Wittig reaction. Unexpected problems arose during reduction of the resulting unsaturated ester. Initially, we attempted to hydrogenate the double bond by using Pd/C, but the main reaction was epoxide ring opening and almost no diepoxy ester 7 was obtained (<5% yield). This problem was overcome by using a rhodium catalyst.15 The order of addition of the reagents was important. Blowing hydrogen into a suspension of the unsaturated ester and Rh/Al2O3 in THF gave diepoxy ester 7 in moderate yield (51%) but was accompanied by formation of an epoxide-opened byproduct. Slow addition of the unsaturated ester to a suspension of pre-hydrogen-adsorbed Rh/Al2O3 in THF provided ester 7 in 80% yield over two steps from the epoxy alcohol. This suggests that hydrogen adsorption on the rhodium catalyst was not required for epoxide opening and that hydrogenation was faster than epoxide opening.
 | ||
Scheme 2 Synthesis of diepoxyester 7. Reagents and conditions: (a) Ac2O, pyridine, r.t., 52%; (b) TBHP, D-(−)-DIPT, Ti(OiPr)4, 4 Å molecular sieves, CH2Cl2, –20 °C, 98% (>95% ee); (c) MOMCl, DIPEA, CH2Cl2, reflux; (d) K2CO3, MeOH, r.t., 88% (2 steps); (e) TBHP, D-(−)-DIPT, Ti(OiPr)4, 4 Å molecular sieves, CH2Cl2, –20 °C, 86%; (f) Dess–Martin periodinane, NaHCO3, CH2Cl2, r.t. then Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2Et, r.t.; (g) H2, Rh/Al2O3, THF, r.t., 80% (2 steps). CHCO2Et, r.t.; (g) H2, Rh/Al2O3, THF, r.t., 80% (2 steps). | ||
The ester was efficiently synthesized by catalytic hydrogenation using a rhodium catalyst with pre-stored hydrogen. NMR analysis of the diepoxy ester gave some unexpected results. In a previous study of cis-solamin synthesis,7 tandem cyclization caused by a small amount of an acid contaminant proceeded to form tetrahydrofuran-γ-lactone when NMR spectroscopic analysis of the corresponding diepoxy ester was performed in deuterated chloroform as the solvent. This observation triggered development of the acid-mediated tandem cyclization. However, in this case, diepoxy ester 7 was stable, despite the deuterated solvent, and no tetrahydrofuran-γ-lactone 8 was formed. This indicates that the acid sensitivity of the reaction using an E,E-diepoxide differed from that of the previously reported reaction using a Z,Z-diepoxide. Although a possible decrease in the reactivity was a concern, the tandem cyclization proceeded smoothly when E,E-diepoxy ester 7 was treated with 10-camphorsulfonic acid (0.2 equiv.) and water (1.0 equiv.) at room temperature in dichloromethane for 1.5 h; tetrahydrofuran-γ-lactone 8 was obtained with controlled stereochemistry at the C-15, C-16, C-19, and C-20 positions (91% yield; Scheme 3). The stereochemistry of 8 was confirmed by X-ray crystallographic analysis of the dinitrobenzoate 9,16 which was obtained by condensation of 3,5-dinitrobenzoic acid, removal of the methoxymethyl protecting group, and concomitant 1,2-acyl migration (Fig. 3).
 | ||
| Scheme 3 Synthesis of tetrahydrofuran-γ-lactone 8. Reagents and conditions: (a) 10-camphorsulfonic acid, H2O, CH2Cl2, r.t., 91%; (b) 3,5-dinitrobenzoic acid, WSC, DMAP, CH2Cl2, r.t.; (c) BF3·OEt2, Me2S, CH2Cl2, –30 °C, 81% (2 steps). | ||
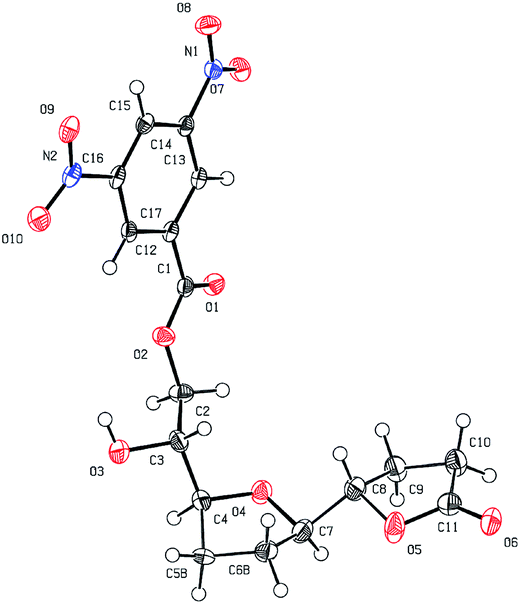 | ||
| Fig. 3 ORTEP drawing of dinitrobenzoate 9. | ||
This transformation can be explained by the mechanism shown in Scheme 4. First, acidic hydrolysis of the ester generates a carboxyl group in situ. The protonated epoxide oxygen acts as a leaving group, which leads to a sequential intramolecular SN2-like cyclization of the carbonyl oxygen to form erythro/cis/erythro tetrahydrofuran-γ-lactone 8.
 | ||
| Scheme 4 Acid-catalyzed one-step construction of tetrahydrofuran-diol. | ||
Having obtained tetrahydrofuran-γ-lactone 8, we introduced the appropriate alkyl linker, spacer, and γ-lactone to achieve the synthesis of 1. First, we attached an alkyl linker to the tetrahydrofuran core. Half-reduction of 8 followed by a Wittig reaction of the resulting hemiacetal yielded an unsaturated alcohol as an inconsequential mixture (Scheme 5). Triol 10 was obtained by hydrogenation of the double bond, acid deprotection, protection as the acetonide derivative, and then treatment with acetic anhydride and pyridine to provide the corresponding acetate. Mild acidic deprotection of the acetonide and subsequent silylation of the resulting diol furnished the secondary alcohol, which underwent mesylation; subsequent treatment with tetrabutylammonium fluoride at 0 °C for 10 min generated the alkoxide anion species in situ. The reaction mixture was warmed to 50 °C and then stirred at this temperature for 12 h. Subsequent cyclization to a sulfonate moiety proceeded smoothly in a one-pot system to afford epoxide 11 with complete inversion of the configuration at C-15. The conversion of epoxide 11 to 1 was performed by using the method reported by Brown et al.17 Introduction of the alkyl spacer into epoxide 11 was achieved by a copper-catalyzed Grignard reaction to give terminal alkene 12. Deacetylation of 12 and a subsequent ruthenium-catalyzed Alder–ene reaction11 with ethyl (S)-4-hydroxypent-2-ynoate (3) afforded the corresponding γ-lactone. Finally, reduction of the isolated double bond by using p-toluenesulfonylhydrazide and NaOAc furnished 1 with the 15S/16S/19R/20S configuration.
 | ||
| Scheme 5 Synthesis of 1. Reagents and conditions: (a) DIBAH, CH2Cl2, –78 °C; (b) nonyltriphenylphosphonium bromide, BuLi, THF, 0 °C to r.t.; (c) H2, Pd/C, MeOH, r.t.; (d) HCl, MeOH, 0 °C, 82% (4 steps); (e) 2,2-dimethoxypropane, pTsOH·H2O, r.t.; (f) Ac2O, pyridine, r.t.; (g) 80% AcOH, 50 °C; (h) TBSCl, Et3N, DMAP, CH2Cl2, r.t.; (i) MsCl, Et3N, CH2Cl2, r.t.; (j) TBAF, THF, 50 °C, 87% (6 steps); (k) CH2CH(CH2)9MgBr, CuI, THF, −60 °C, 79%; (l) DIBAH, CH2Cl2, –78 °C; (m) ethyl (S)-4-hydroxypent-2-ynoate (3), CpRu(MeCN)3PF6, DMF, r.t.; (n) TsNHNH2, NaOAc, THF/H2O, 60 °C, 65% (3 steps). | ||
The 1H NMR spectra of natural squafosacin F8 and synthetic compound 1 are similar. However, careful comparison of the 13C NMR spectra revealed minor discrepancies at three positions, namely C-14: δC 33.2 vs. 34.2; C-18: δC 25.6 vs. 24.3; and C-20: δC 71.5 vs. 72.3 (Table 1).
| Position | Natural squafosacin F δa [ppm] | Synthetic 1 δb [ppm] | Synthetic 2 δb [ppm] |
|---|---|---|---|
| a 100 MHz.b 125 MHz.c Assignments may be interchanged. | |||
| 1 | 173.9 | 173.9 | 173.9 |
| 2 | 134.3 | 134.4 | 134.2 |
| 3 | 25.3 | 25.2 | 25.2 |
| 4 | 27.4 | 27.4 | 27.3 |
| 5–13 | 25.6–29.7 | 25.7–29.7 | 26.0–29.7 |
| 14 | 33.2c | 34.2c | 33.1c |
| 15 | 74.4 | 74.5 | 74.3 |
| 16 | 83.2 | 82.7 | 83.3 |
| 17 | 28.6 | 28.4 | 28.6 |
| 18 | 25.6 | 24.3 | 25.5 |
| 19 | 82.1 | 82.2 | 82.2 |
| 20 | 71.5 | 72.3 | 71.4 |
| 21 | 32.5c | 33.1c | 32.5c |
| 22–29 | 25.6–29.7 | 25.7–29.7 | 26.0–29.7 |
| 30 | 31.9 | 31.9 | 31.9 |
| 31 | 22.7 | 22.7 | 22.6 |
| 32 | 14.1 | 14.1 | 14.1 |
| 33 | 148.9 | 148.8 | 148.8 |
| 34 | 77.4 | 77.4 | 77.4 |
| 35 | 19.2 | 19.2 | 19.2 |
The differences between the 13C NMR spectroscopic shifts were slight, therefore we synthesized another candidate compound, i.e., 2, from triol 13, which was an intermediate in our previous synthesis of cis-solamin,7 by using a similar reaction sequence to that shown in Scheme 6. Synthesis of 2 with the 15S/16R/19S/20S configuration was successful. Comparison of the 13C NMR spectra showed that the spectrum of 2 was a better match than that of 1 for the spectrum of natural squafosacin F. The spectral and physical properties of 2, namely the 1H and 13C NMR spectra, IR spectrum, HRMS results, and optical rotation, perfectly matched those of natural squafosacin F. The first total synthesis of squafosacin F (2) had therefore been accomplished and its absolute configuration was identified as 15S,16R,19S,20S,36S.
 | ||
| Scheme 6 Synthesis of 2. Reagents and conditions: (a) 2,2-dimethoxypropane, pTsOH·H2O, r.t.; (b) Ac2O, pyridine, r.t.; (c) 80% AcOH, 50 °C; (d) TBSCl, Et3N, DMAP, CH2Cl2, r.t.; (e) MsCl, Et3N, CH2Cl2, r.t.; (f) TBAF, THF, 50 °C, 75% (6 steps); (g) CH2CH(CH2)9MgBr, CuI, THF, −60 °C, 77%; (h) ethyl (S)-4-hydroxypent-2-ynoate (3), CpRu(MeCN)3PF6, DMF, r.t.; (i) TsNHNH2, NaOAc, THF/H2O, 60 °C, 89% (2 steps). | ||
Conclusions
In conclusion, a diversity-oriented synthetic approach enabled the first total synthesis of squafosacin F (2) and the absolute configuration was established with the help of a 13C NMR spectroscopic comparison of both possible configurations at C-16 and C-19. The salient features of the developed synthesis are tandem cyclization of a diepoxy ester with a Brønsted acid and 1 equiv. of water, introduction of a long alkyl linker and spacer, and α,β-unsaturated-γ-lactone formation by a ruthenium-catalyzed Alder–ene reaction. In addition, our developed acid-mediated tandem cyclization can be used to control the stereochemistry of the tetrahydrofuran moiety flanked by two hydroxy groups by selecting an appropriate configuration of the diepoxy ester. This synthetic method can be used to construct natural mono-tetrahydrofuran acetogenins with various configurations. We hope that this strategy will open up a new approach to the preparation of natural mono-tetrahydrofuran acetogenins and of various analogs with similar structures.Experimental section
General experimental procedures
Melting points (mp) were measured using the Yanaco melting point apparatus MP-S3 and were uncorrected. Optical rotations were measured with a JASCO P-1030 polarimeter. IR spectra were recorded with a JASCO FT-IR/620 spectrometer. Single crystal X-ray diffraction was recorded using a MacScience Co., Ltd DIP 2020 Image Plate. 1H and 13C NMR spectra were recorded on a Bruker Biospin AVANCE III HD 400 (400 MHz for 1H, 100 MHz for 13C) and a Bruker Biospin AVANCE III HD 500 (500 MHz for 1H, 125 MHz for 13C). The reported chemical shifts (δ) in parts per million (ppm) were relative to the internal CHCl3 (7.26 ppm for 1H and 77.0 ppm for 13C); the coupling constant (J) values were measured in hertz. The coupling patterns are denoted as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and br (broad). HR-ESI-MS spectra were obtained using a Micromass LCT spectrometer with a time-of-flight (TOF) analyzer. Elemental analysis data were obtained using an Elementar Vario EL. Precoated silica gel plates with a fluorescent indicator (Merck 60 F254) were used for analytical and preparative thin-layer chromatography (TLC). Flash column chromatography was performed using Kanto Chemical silica gel 60N (spherical, natural) 40–50 μm. All reagents (Aldrich, Kanto, TCI, and Wako) and solvents were of commercial quality and were used as received.:1 to 0:1) to give monoacetate 16 (668 mg, 52% yield) as a colorless oil, diacetate (445 mg, 28% yield) as a colorless oil, and 4 (100 mg, 10% recovered yield): Rf 0.25 (hexane/EtOAc = 1:1); IR (neat) νmax 3417, 3022, 2926, 2850, 1740, 1672, 1236, 1024 cm−1; 1H NMR (CDCl3, 400 MHz) δ 5.76 (1H, m), 5.68–5.65 (2H, m), 5.59 (1H, m), 4.51 (2H, dd, J = 0.6, 6.4 Hz), 4.11–4.07 (2H, m), 2.17–2.14 (4H, m), 2.06 (3H, s), 1.54 (1H, brs); 13C NMR (CDCl3, 100 MHz) δ 170.9 (C), 135.4 (CH), 131.9 (CH), 129.7 (CH), 124.4 (CH), 65.1 (CH2), 63.6 (CH2), 31.7 (CH2), 31.4 (CH2), 21.0 (CH3); MS (ESI-TOF) m/z 207 [M + Na]+ (100); HRMS (ESI-TOF) m/z [M + Na]+ calcd for C10H16O3Na 207.0997, found 207.0994; anal. calcd for C10H16O3: C, 65.19; H, 8.75. Found: C, 65.07; H, 8.80.:2 to 0:1) to give epoxyalcohol 5 (2.97 g, 98% yield, >95% ee) as a colorless oil: Rf 0.30 (hexane/EtOAc = 1:2); [α]25D +19.9 (c 1.68, CHCl3); IR (neat) νmax 3444, 2933, 1739, 1672, 1239 cm−1; 1H NMR (CDCl3, 400 MHz) δ 5.78 (1H, m), 5.63 (1H, m), 4.52 (2H, d, J = 6.2 Hz), 3.89 (1H, ddd, J = 2.6, 5.2, 12.4 Hz), 3.64 (1H, m), 2.97 (1H, dt, J = 2.3, 5.8 Hz), 2.92 (1H, dt, J = 4.0, 2.6 Hz), 2.30–2.15 (2H, m), 2.06 (3H, s), 1.72–1.65 (2H, m), 1.61 (1H, brs); 13C NMR (CDCl3, 100 MHz) δ 170.8 (C), 134.6 (CH), 124.8 (CH), 64.9 (CH2), 61.6 (CH2), 58.5 (CH), 55.3 (CH), 30.8 (CH2), 28.6 (CH2), 20.9 (CH3); MS (ESI-TOF) m/z 223 [M + Na]+ (100); HRMS (ESI-TOF) m/z [M + Na]+ calcd for C10H16O4Na 223.0946, found 223.0939; anal. calcd for C10H16O4: C, 59.98; H, 8.05. Found: C, 59.83; H, 8.14.To a stirred solution of the crude MOM ether in methanol (46.3 mL) was added K2CO3 (9.61 g, 69.5 mmol) at room temperature. After stirring the mixture for 30 min, the mixture was diluted with Et2O, and then was passed through a pad of silica gel and then concentrated in vacuo. The residue was purified with flash column chromatography on silica gel (hexane/EtOAc = 1:2) to give allylic alcohol 6 (2.65 g, 88% yield for 2 steps) as a colorless oil: Rf 0.60 (hexane/EtOAc = 1:2); [α]25D +16.5 (c 0.83, CHCl3); IR (neat) νmax 3433, 2991, 2933, 2863 cm−1; 1H N/MR (CDCl3, 400 MHz) δ 5.72–5.69 (2H, m), 4.66 (1H, d, J = 7.5 Hz), 4.64 (1H, d, J = 7.5 Hz), 4.12–4.08 (2H, m), 3.70 (1H, dd, J = 3.8, 11.6 Hz), 3.57 (1H, dd, J = 5.4, 11.6 Hz), 3.38 (3H, s), 2.94 (1H, ddd, J = 2.3, 3.8, 5.8 Hz), 2.86 (1H, dt, J = 2.1, 5.8 Hz), 2.30–2.18 (2H, m), 1.71–1.67 (2H, m), 1.52 (1H, brs); 13C NMR (CDCl3, 100 MHz) δ 131.4 (CH), 130.0 (CH), 96.5 (CH2), 67.8 (CH2), 63.4 (CH2), 56.7 (CH), 55.9 (CH), 55.3 (CH3), 31.1 (CH2), 28.6 (CH2); MS (ESI-TOF) m/z 225 [M + Na]+ (100); HRMS (ESI-TOF) m/z [M + Na]+ calcd for C10H18O4Na 225.1103, found 225.1104; anal. calcd for C10H18O4: C, 59.39; H, 8.97. Found: C, 59.21; H, 8.97.
After a suspension of 5% Rh/Al2O3 (308 mg) in THF (268 mL) was stirred under hydrogen atmosphere at room temperature for 30 min, a solution of above crude α,β-unsaturated ester in THF (40.0 mL) was added to the stirred suspension. After stirring for 2 h, argon was blown into the reaction mixture in order to remove hydrogen and then diluted with Et2O, filtered through silica gel pad, and concentrated in vacuo. The residue was purified with flash column chromatography on silica gel (hexane/EtOAc = 2:1) to give bisepoxyester 7 (1.15 g, 51% yield for 2 steps) as a colorless oil: Rf 0.30 (hexane/EtOAc = 1:1); [α]25D +39.5 (c 0.28, CHCl3); IR (neat) νmax 2981, 2935, 1733, 1640 cm−1; 1H NMR (CDCl3, 400 MHz) δ 4.66 (1H, d, J = 7.6 Hz), 4.64 (1H, d, J = 7.6 Hz), 4.14 (2H, q, J = 7.2 Hz), 3.73 (1H, dd, J = 3.5, 11.7 Hz), 3.56 (1H, dd, J = 5.5, 11.7 Hz), 3.37 (3H, s), 2.96 (1H, ddd, J = 2.3, 3.5, 5.6 Hz), 2.90 (1H, m), 2.80–2.74 (2H, m), 2.47–2.41 (2H, m), 1.96 (1H, m), 1.82–1.60 (5H, m), 1.26 (3H, t, J = 7.2 Hz); 13C NMR (CDCl3, 100 MHz) δ 172.8 (C), 96.6 (CH2), 67.7 (CH2), 60.5 (CH2), 58.0 (CH), 57.5 (CH), 56.7 (CH), 55.5 (CH), 55.3 (CH3), 30.4 (CH2), 28.2 (CH2), 28.0 (CH2), 27.1 (CH2), 14.2 (CH3); MS (ESI-TOF) m/z 311 [M + Na]+ (100); HRMS (ESI-TOF) m/z [M + Na]+ calcd for C14H24O6Na 311.1471, found 311.1459; anal. calcd for C14H24O6: C, 58.32; H, 8.39. Found: C, 58.16; H, 8.29.
:10) to give tetrahydrofuran 8 (28.9 mg, 91% yield) as a colorless oil: Rf 0.30 (hexane/EtOAc = 1:4); [α]25D +14.4 (c 0.83, CHCl3); IR (neat) νmax 3434, 2948, 2889, 1767, 1644 cm−1; 1H NMR (CDCl3, 400 MHz) δ 4.67 (1H, d, J = 7.0 Hz), 4.66 (1H, d, J = 7.0 Hz), 4.44 (1H, m), 3.99 (1H, m), 3.90 (1H, m), 3.75–3.67 (2H, m), 3.57 (1H, m), 3.39 (3H, s), 2.69 (1H, brd, J = 4.0 Hz), 2.60–2.45 (2H, m), 2.30 (1H, m), 2.15–1.92 (4H, m), 1.80 (1H, m); 13C NMR (CDCl3, 100 MHz) δ 177.0 (C), 97.2 (CH2), 81.2 (CH), 80.2 (CH), 79.9 (CH), 72.3 (CH), 70.4 (CH2), 55.5 (CH3), 28.1 (CH2), 27.3 (CH2), 26.7 (CH2), 23.9 (CH2); MS (ESI-TOF) m/z 283 [M + Na]+ (100); HRMS (ESI-TOF) m/z [M + Na]+ calcd for C12H20O6Na 283.1158, found 283.1150; anal. calcd for C12H20O6: C, 55.37; H, 7.75. Found: C, 55.23; H, 7.81.To a solution of above crude dinitrobenzoate in CH2Cl2 (2.56 mL) were added Me2S (0.256 mL, 3.50 mmol) and BF3·OEt2 (0.0285 mL, 0.230 mmol) at −30 °C. After stirring for 1.5 h, the reaction mixture was quenched with Et3N (0.0107 mL, 0.0769 mmol) and warmed to room temperature. The mixture was diluted with Et2O, washed with saturated aqueous NaHCO3 solution, H2O, and brine, dried over anhydrous MgSO4 and Na2SO4, and then concentrated in vacuo. The residue was purified with flash column chromatography on silica gel (hexane/EtOAc = 1:2) to give dinitrobenzoate 9 (25.5 mg, 81% yield for two steps) as a white needle: 1H NMR (CDCl3, 400 MHz) δ 9.24 (1H, m), 9.19–9.15 (2H, m), 4.63 (1H, dd, J = 3.0, 11.6 Hz), 4.57–4.43 (2H, m), 4.20–3.92 (3H, m), 2.60–2.53 (2H, m), 2.40–2.22 (2H, m), 2.18–1.92 (4H, m), 1.52 (1H, bs); ESI-MS m/z 433 [M + Na]+ (100); HR-ESI-MS m/z 433.0858 (calcd for C17H18N2O10Na, 433.0859).
To a stirred suspension of nonyltriphenylphosphonium bromide (8.00 g, 17.0 mmol) in THF (41.0 mL) was added BuLi (1.64 M solution in hexane, 9.30 mL, 15.3 mmol) dropwise at 0 °C and the mixture was stirred for 1.5 h at same temperature. A solution of the above crude hemiacetal in THF (20.0 mL) was then added to the mixture at 0 °C and then warmed to room temperature. After stirring for 2 h, the reaction mixture was quenched with saturated aqueous NH4Cl solution, diluted with EtOAc, washed with H2O and brine, dried over anhydrous Na2SO4, and then concentrated in vacuo. The residue was passed through a pad of silica gel (hexane/EtOAc = 1:1) and then concentrated in vacuo to give a crude diol which was used for the next step without further purification.
To a solution of above crude diol in MeOH (117 mL) was added Pd/C (5%, 117 mg), and then was stirred under H2 atmosphere. After stirring for 1 h, the reaction mixture was diluted with Et2O and passed through a pad of silica gel and then concentrated in vacuo. The residue was passed through a pad of silica gel (hexane/EtOAc = 1:2) and then concentrated in vacuo. The residue was passed through a pad of silica gel (toluene/EtOAc = 2:1) and then concentrated in vacuo to give a crude diol which was used for the next step without further purification.
To a solution of above crude diol in CH2Cl2 (22.1 mL) were added Me2S (7.37 mL, 99.6 mmol) and BF3·OEt2 (1.36 mL, 11.0 mmol) at −30 °C. After stirring for 30 min, the reaction mixture was quenched with Et3N (3.08 mL, 22.1 mmol) and warmed to room temperature. The mixture was diluted with Et2O, washed with saturated aqueous NaHCO3 solution, H2O, and brine, dried over anhydrous MgSO4 and Na2SO4, and then concentrated in vacuo. The residue was purified with flash column chromatography on silica gel (hexane/EtOAc = 1:4 to 0:1) to give triol 10 (575 mg, 57% yield for four steps) as a white plate: Rf 0.30 (CHCl3/MeOH = 15:1); mp 76−77 °C; [α]25D +2.7 (c 1.04, CHCl3); IR (KBr) νmax 3393, 3283, 2925, 2850, 1153 cm−1; 1H NMR (CDCl3, 400 MHz) δ 3.99 (1H, dt, J = 4.1, 7.1 Hz), 3.89 (1H, m), 3.87–3.81 (2H, m), 3.71 (1H, dd, J = 3.9, 11.2 Hz), 3.61 (1H, dd, J = 6.4, 11.2 Hz), 2.00–1.87 (3H, m), 1.82 (1H, m), 1.54–1.20 (22H, m), 0.88 (3H, t, J = 6.7 Hz); 13C NMR (CDCl3, 100 MHz) δ 82.4 (CH), 80.2 (CH), 73.4 (CH), 72.2 (CH), 63.9 (CH2), 33.2 (CH2), 31.9 (CH2), 29.7 (CH2) × 2, 29.64 (CH2), 29.63 (CH2), 29.61 (CH2), 29.56 (CH2), 29.3 (CH2), 26.3 (CH2), 26.0 (CH2), 24.0 (CH2), 22.7 (CH2), 14.1 (CH3); MS (ESI-TOF) m/z 353 [M + Na]+ (100); HRMS (ESI-TOF) m/z [M + Na]+ calcd for C19H38O4Na 353.2668, found 353.2672; anal. calcd for C19H38O4: C, 69.05; H, 11.59. Found: C, 69.28; H, 11.54.
Acetic anhydride (3.10 mL, 32.8 mmol) was added to a stirred solution of the above crude acetonide in pyridine (3.10 mL, 38.5 mmol) at room temperature. After stirring for 12 h, the mixture was concentrated in vacuo to give a crude acetate which was used for the next step without further purification.
To the above crude acetate was added 80% AcOH aq. (3.10 mL) at room temperature. The mixture was warmed to 50 °C and after 40 min recooled to room temperature. The mixture was diluted with EtOAc, washed with saturated aqueous NaHCO3 solution H2O and brine, dried over anhydrous MgSO4 and Na2SO4, and then concentrated in vacuo to give a crude diol which was used for the next step without further purification.
To a stirred solution of the above crude diol in CH2Cl2 (0.618 mL) were added Et3N (0.258 mL, 1.85 mmol), TBSCl (140 mg, 0.929 mmol), and DMAP (3.8 mg, 0.0311 mmol) at 0 °C and then warmed to room temperature. After stirring for 3.5 h, the mixture was quenched with saturated aqueous NaHCO3 solution, diluted with Et2O, washed with H2O, and brine, dried over anhydrous MgSO4 and Na2SO4, and then concentrated in vacuo to give a crude TBS ether which was used for the next step without further purification.
To a stirred solution of the above crude TBS ether in CH2Cl2 (3.10 mL) were added Et3N (0.172 mL, 1.23 mmol) and MsCl (0.048 mL, 0.620 mmol) at 0 °C and then warmed to room temperature. After stirring for 45 min, the mixture was quenched with saturated aqueous NaHCO3 solution, diluted with Et2O, washed with H2O, and brine, dried over anhydrous MgSO4 and Na2SO4, and then concentrated in vacuo to give a crude mesylate which was used for the next step without further purification.
To a stirred solution of the above crude mesylate in THF (3.10 mL) were added TBAF (1.00 M solution in THF, 0.773 mL, 0.773 mmol) at room temperature. After stirring for 1 h, the mixture was warmed to 50 °C and stirred for 12 h. The mixture was diluted with Et2O, washed with H2O, and brine, dried over anhydrous MgSO4 and Na2SO4, and then concentrated in vacuo. The residue was purified with flash column chromatography on silica gel (hexane/EtOAc = 6:1) to give epoxide 11 (95.3 mg, 87% yield for six steps) as a white waxy solid: Rf 0.60 (hexane/EtOAc = 1:1); mp 31–32 °C; [α]25D −10.5 (c 0.19, CHCl3); IR (KBr) νmax 2925, 2854, 1741, 1466, 1370, 1239 cm−1; 1H NMR (CDCl3, 400 MHz) δ 4.98 (1H, ddd, J = 4.4, 4.7, 8.8 Hz), 3.93 (1H, m), 3.83 (1H, m), 2.94 (1H, ddd, J = 2.6, 4.3, 7.1 Hz), 2.73 (1H, dd, J = 4.3, 5.2 Hz), 2.65 (1H, dd, J = 2.6, 5.2 Hz), 2.07 (3H, s), 2.05–1.76 (4H, m), 1.37–1.15 (22H, m), 0.88 (3H, t, J = 7.0 Hz); 13C NMR (CDCl3, 100 MHz) δ 170.7 (C), 80.8 (CH), 78.7 (CH), 74.4 (CH), 53.8 (CH), 44.0 (CH2), 31.9 (CH2), 30.9 (CH2), 29.6 (CH2), 29.6 (CH2) × 2, 29.6 (CH2), 29.5 (CH2) × 2, 29.3 (CH2), 28.1 (CH2), 26.8 (CH2), 25.3 (CH2), 22.7 (CH2), 21.2 (CH3), 14.1 (CH3); MS (ESI-TOF) m/z 377 [M + Na]+ (100); HRMS (ESI-TOF) m/z [M + Na]+ calcd for C21H38O4Na 377.2668, found 377.2664; anal. calcd for C21H38O4: C, 71.14; H, 10.80. Found: C, 70.97; H, 10.95.
:1, 1.40 mL) was added to the reaction mixture. The mixture was diluted with Et2O, washed with saturated aqueous NH4Cl solution, H2O, and brine, dried over anhydrous MgSO4 and Na2SO4, and then concentrated in vacuo. The residue was purified with flash column chromatography on silica gel (hexane/EtOAc = 7:1) to give alcohol 12 (56.8 mg, 79% yield) as a colorless oil: Rf 0.35 (hexane/EtOAc = 7:1); [α]25D −11.4 (c 0.75, CHCl3); IR (neat) νmax 3545, 2925, 2854, 1742, 1640, 1466, 1370, 1238 cm−1; 1H NMR (CDCl3, 400 MHz) δ 5.81 (1H, ddt, J = 10.1, 16.9, 6.8 Hz), 3.98 (1H, dt, J = 4.9, 5.7 Hz), 3.73 (1H, q, J = 6.2 Hz), 3.32 (1H, dt, J = 5.7, 6.3 Hz), 2.07 (3H, s), 2.06–2.00 (2H, m), 1.94–1.75 (3H, m), 1.68 (1H, m), 1.59–1.17 (44H, m), 0.87 (3H, t, J = 7.0 Hz); 13C NMR (CDCl3, 100 MHz) δ 170.9 (C), 139.3 (CH), 114.1 (CH2), 82.8 (CH), 80.5 (CH), 74.5 (CH), 74.1 (CH), 33.8 (CH2), 31.9 (CH2), 31.1 (CH2), 29.7 (CH2), 29.65 (CH2), 29.62 (CH2) × 2, 29.57 (CH2) × 4, 29.54 (CH2), 29.47 (CH2) × 2, 29.46 (CH2), 29.3 (CH2), 29.1 (CH2), 28.9 (CH2), 27.6 (CH2), 26.6 (CH2), 25.7 (CH2), 25.4 (CH2), 22.7 (CH2), 21.2 (CH3), 14.1 (CH3); MS (ESI-TOF) m/z 531 [M + Na]+ (100); HRMS (ESI-TOF) m/z [M + Na]+ calcd for C32H60O4Na 531.4389, found 531.4391; anal. calcd for C32H64O4: C, 75.54; H, 11.89. Found: C, 75.37; H, 11.84.[CpRu(CH3CN)3]+PF6− (2.1 mg, 0.00484 mmol) was added to a stirred solution of the above crude diol and (S)-4-hydroxypent-2-ynoate 3 (10.1 mg, 0.0711 mmol) in DMF (0.948 mL). The solution was allowed to stir at room temperature for 1 h before the reaction mixture was diluted with Et2O and passed through a plug of silica gel. The mixture was washed with 1.0 M HCl aq., H2O, and brine, dried over anhydrous MgSO4 and Na2SO4, and then concentrated in vacuo to give a crude butenolide.
To a solution of the above crude butanolide and TsNHNH2 (618 mg, 3.32 mmol) in DME (4.80 mL) was added a solution of NaOAc (311 mg, 3.79 mmol) in H2O (4.80 mL). The mixture was heated at 80 °C for 12 h, and then cooled to room temperature. The mixture was diluted with Et2O, washed with H2O and brine, dried over anhydrous MgSO4 and Na2SO4, and then concentrated in vacuo. The residue was purified with flash column chromatography on silica gel (hexane/EtOAc = 1:1) to give butenolide 1 (17.4 mg, 65% yield for three steps) as a white waxy solid: Rf 0.40 (hexane/EtOAc = 1:1); mp 89−90 °C; [α]25D +13.2 (c 0.19, CHCl3); IR (KBr) νmax 3450, 2920, 2849, 1734, 1468 cm−1; 1H NMR (CDCl3, 500 MHz) δ 6.98 (1H, dt, J = 1.5, 1.6 Hz), 4.99 (1H, ddq, J = 6.8, 13.6, 1.7 Hz), 3.90 (1H, dt, J = 3.2, 7.3 Hz), 3.83 (1H, m), 3.81 (1H, m), 3.44 (1H, m), 2.27 (1H, dt, J = 8.1, 1.7 Hz), 2.25 (1H, dt, J = 8.1, 1.7 Hz), 1.98–1.90 (2H, m), 1.88–1.66 (4H, m), 1.58–1.19 (44H, m), 1.40 (3H, d, J = 6.9 Hz), 0.88 (3H, t, J = 6.9 Hz); 13C NMR (CDCl3, 125 MHz) δ 173.9 (C), 148.8 (CH), 134.4 (C), 82.7 (CH), 82.2 (CH), 77.4 (CH), 74.5 (CH), 72.3 (CH), 34.2 (CH2), 33.1 (CH2), 31.9 (CH2), 29.69 (CH2) × 2, 29.66 (CH2) × 3, 29.64 (CH2), 29.60 (CH2), 29.57 (CH2) × 2, 29.56 (CH2) × 2, 29.5 (CH2), 29.34 (CH2), 29.28 (CH2), 29.2 (CH2), 28.4 (CH2), 27.4 (CH2), 25.9 (CH2), 28.7 (CH2), 25.2 (CH2), 24.3 (CH2), 22.7 (CH2), 19.2 (CH3), 14.1 (CH3); MS (ESI-TOF) m/z 587 [M + Na]+ (100); HRMS (ESI-TOF) m/z [M + Na]+ calcd for C35H64O5Na 587.4651, found 587.4655; anal. calcd for C35H64O5: C, 74.42; H, 11.42. Found: C, 74.32; H, 11.55.
Acetic anhydride (0.369 mL, 3.90 mmol) was added to a stirred solution of the above crude acetonide in pyridine (0.369 mL, 4.58 mmol) at room temperature. After stirring for 12 h, the mixture was concentrated in vacuo to give a crude acetate which was used for the next step without further purification.
To the above crude acetate was added 80% AcOH aq. (0.369 mL) at room temperature. The mixture was warmed to 50 °C and after 40 min recooled to room temperature. The mixture was diluted with EtOAc, washed with saturated aqueous NaHCO3 solution H2O and brine, dried over anhydrous MgSO4 and Na2SO4, and then concentrated in vacuo to give a crude diol which was used for the next step without further purification.
To a stirred solution of the above crude diol in CH2Cl2 (0.0740 mL) were added Et3N (0.0310 mL, 0.222 mmol), TBSCl (16.7 mg, 0.111 mmol), and DMAP (0.5 mg, 0.00409 mmol) at 0 °C and then warmed to room temperature. After stirring for 3.5 h, the mixture was quenched with saturated aqueous NaHCO3 solution, diluted with Et2O, washed with H2O, and brine, dried over anhydrous MgSO4 and Na2SO4, and then concentrated in vacuo to give a crude TBS ether which was used for the next step without further purification.
To a stirred solution of the above crude TBS ether in CH2Cl2 (0.369 mL) were added Et3N (0.0206 mL, 0.148 mmol) and MsCl (0.00571 mL, 0.0738 mmol) at 0 °C and then warmed to room temperature. After stirring for 1 h, the mixture was quenched with saturated aqueous NaHCO3 solution, diluted with Et2O, washed with H2O, and brine, dried over anhydrous MgSO4 and Na2SO4, and then concentrated in vacuo to give a crude mesylate which was used for the next step without further purification.
To a stirred solution of the above crude mesylate in THF (0.369 mL) were added TBAF (1.00 M solution in THF, 0.0923 mL, 0.0923 mmol) at room temperature. After stirring for 1 h, the mixture was warmed to 50 °C and stirred for 12 h. The mixture was diluted with Et2O, washed with H2O, and brine, dried over anhydrous MgSO4 and Na2SO4, and then concentrated in vacuo. The residue was purified with flash column chromatography on silica gel (hexane/EtOAc = 2:1) to give epoxide 14 (9.8 mg, 75% yield for six steps) as a colorless oil: Rf 0.55 (hexane/EtOAc = 1:1); [α]25D −13.4 (c 0.95, CHCl3); IR (neat) νmax 2925, 2854, 1739, 1467, 1372, 1240, 1075, 1026 cm−1; 1H NMR (CDCl3, 400 MHz) δ 4.87 (1H, dt, J = 7.7, 5.3 Hz), 4.06 (1H, dt, J = 5.3, 6.6 Hz), 3.91 (1H, dt, J = 4.8, 6.6 Hz), 2.99 (1H, ddd, J = 2.7, 4.0, 4.8 Hz), 2.79 (1H, dd, J = 4.0, 4.9 Hz), 2.58 (1H, dd, J = 2.7, 4.9 Hz), 2.09 (3H, s), 2.06–1.96 (2H, m), 1.76 (1H, m), 1.65 (1H, m), 1.35–1.19 (22H, m), 0.88 (3H, t, J = 6.7 Hz); 13C NMR (CDCl3, 100 MHz) δ 170.9 (C), 80.1 (CH), 78.9 (CH), 75.1 (CH), 53.1 (CH), 45.3 (CH2), 31.9 (CH2), 31.0 (CH2), 29.6 (CH2), 29.6 (CH2) × 2, 29.5 (CH2), 29.4 (CH2) × 2, 29.3 (CH2), 27.8 (CH2), 27.6 (CH2), 25.4 (CH2), 22.6 (CH2), 21.1 (CH3), 14.1 (CH3); MS (ESI-TOF) m/z 377 [M + Na]+ (100); HRMS (ESI-TOF) m/z [M + Na]+ calcd for C21H38O4Na 377.2668, found 377.2671; anal. calcd for C21H38O4: C, 71.14; H, 10.80. Found: C, 71.08; H, 10.72.
:1, 36.6 mL) was added to the reaction mixture. The mixture was diluted with Et2O, washed with saturated aqueous NH4Cl solution, H2O, and brine, dried over anhydrous MgSO4 and Na2SO4, and then concentrated in vacuo. The residue was purified with flash column chromatography on silica gel (hexane/EtOAc = 7:1) to give alcohol 15 (658 mg, 77% yield) as a white waxy solid: Rf 0.20 (hexane/EtOAc = 4:1); mp 75−76 °C; [α]25D −9.3 (c 0.77, CHCl3); IR (KBr) νmax 3434, 2918, 2850, 1642, 1467 cm−1; 1H NMR (CDCl3, 400 MHz) δ 5.81 (1H, ddt, J = 10.2, 17.1, 6.6 Hz), 4.99 (1H, ddt, J = 2.2, 17.1, 1.6 Hz), 4.92 (1H, ddt, J = 2.2, 10.2, 1.2 Hz), 3.87 (1H, ddd, J = 3.4, 6.0, 9.2 Hz), 3.85–3.77 (2H, m), 3.39 (1H, dt, J = 5.4, 6.6 Hz), 2.16–1.79 (8H, m), 1.63 (1H, m), 1.57–1.19 (39H, m), 0.88 (3H, t, J = 6.7 Hz); 13C NMR (CDCl3, 100 MHz) δ 139.2 (CH), 114.1 (CH2), 83.2 (CH), 82.1 (CH), 74.3 (CH), 71.5 (CH), 33.8 (CH2), 33.2 (CH2), 32.5 (CH2), 31.9 (CH2), 29.71 (CH2), 29.66 (CH2), 29.64 (CH2), 29.63 (CH2), 29.61 (CH2), 29.57 (CH2), 29.56 (CH2) × 2, 29.53 (CH2), 29.47 (CH2), 29.3 (CH2), 29.1 (CH2), 28.9 (CH2), 28.6 (CH2), 28.0 (CH2), 26.0 (CH2), 25.6 (CH2), 25.2 (CH2), 22.7 (CH2), 14.0 (CH3); MS (ESI-TOF) m/z 489 [M + Na]+ (100); HRMS (ESI-TOF) m/z [M + Na]+ calcd for C30H58O3Na 489.4284, found 489.4284; anal. calcd for C30H58O3: C, 77.19; H, 12.52. Found: C, 76.91; H, 12.22.To a solution of the above crude butanolide and TsNHNH2 (3.83 g, 20.6 mmol) in DME (25.7 mL) was added a solution of NaOAc (2.11 g, 25.7 mmol) in H2O (25.7 mL). The mixture was heated at 80 °C for 3 h, and then cooled to room temperature. The mixture was diluted with Et2O, washed with H2O and brine, dried over anhydrous MgSO4 and Na2SO4, and then concentrated in vacuo. The residue was purified with flash column chromatography on silica gel (hexane/EtOAc = 1:1) to give butenolide 2 (260 mg, 89% yield for two steps) as a white waxy solid: Rf 0.40 (hexane/EtOAc = 1:1); mp 86−87 °C; [α]25D +4.5 (c 0.39, CHCl3); IR (KBr) νmax 3440, 2920, 2851, 1740, 1469 cm−1; 1H NMR (CDCl3, 500 MHz) δ 6.98 (1H, dt, J = 1.6, 1.5 Hz), 4.99 (1H, ddq, J = 6.8, 13.6, 1.6 Hz), 3.87 (1H, ddd, J = 3.4, 6.0, 9.3 Hz), 3.82 (1H, dt, J = 8.2, 6.7 Hz), 3.80 (1H, ddd, J = 3.6, 6.8, 9.3 Hz), 3.39 (1H, dt, J = 6.5, 6.7 Hz), 2.27 (1H, dt, J = 8.0, 1.6 Hz), 2.25 (1H, dt, J = 8.0, 1.6 Hz), 2.02–1.81 (2H, m), 1.92 (2H, brs), 1.68–1.56 (2H, m), 1.54–1.18 (44H, m), 1.40 (3H, d, J = 6.8 Hz), 0.88 (3H, t, J = 6.9 Hz); 13C NMR (CDCl3, 125 MHz) δ 173.9 (C), 148.8 (CH), 134.3 (C), 83.2 (CH), 82.1 (CH), 77.4 (CH), 74.3 (CH), 71.6 (CH), 33.2 (CH2), 32.6 (CH2), 31.9 (CH2), 29.70 (CH2), 29.66 (CH2) × 2, 29.64 (CH2), 29.62 (CH2), 29.60 (CH2), 29.57 (CH2) × 3, 29.6 (CH2), 29.53 (CH2), 29.49 (CH2), 29.34 (CH2), 29.28 (CH2), 29.2 (CH2), 28.6 (CH2), 27.4 (CH2), 26.0 (CH2), 25.6 (CH2), 25.3 (CH2), 25.2 (CH2), 22.7 (CH2), 19.2 (CH3), 14.1 (CH3); MS (ESI-TOF) m/z 587 [M + Na]+ (100); HRMS (ESI-TOF) m/z [M + Na]+ calcd for C35H64O5Na 587.4651, found 587.4659; anal. calcd for C35H64O5: C, 74.42; H, 11.42. Found: C, 74.17; H, 11.31.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) S. D. Jolad, J. J. Hoffman, K. H. Schram, J. R. Cole, M. S. Tempesta, G. R. Kriek and R. B. Bates, J. Org. Chem., 1982, 47, 3151 CrossRef; (b) F. Q. Alali, X.-X. Liu and J. L. McLaughlin, J. Nat. Prod., 1999, 62, 504 CrossRef CAS PubMed; (c) N. Li, Z. Shi, Y. Tang, J. Chen and X. Li, Beilstein J. Org. Chem., 2008, 4, 1 Search PubMed.
- (a) J. K. Rupprecht, Y.-H. Hui and J. L. McLaughlin, J. Nat. Prod., 1990, 53, 237 CrossRef CAS PubMed; (b) X. Fang, M. J. Rieser, Z. Gu, G. Zhao and J. L. McLaughlin, Phytochem. Anal., 1993, 4, 27 CrossRef CAS; (c) S. Hoppen, U. Emde, T. Friedrich, L. Grubert and U. Koert, Angew. Chem., Int. Ed., 2000, 39, 2099 CrossRef CAS; (d) N. Ichimaru, M. Murai, M. Abe, T. Hamada, Y. Yamada, S. Makino, T. Nishioka, H. Makabe, A. Makino, T. Kobayashi and H. Miyoshi, Biochemisty, 2005, 44, 816 CrossRef CAS PubMed.
- (a) T. Friedrich, P. van Heek, H. Leif, T. Ohnishi, E. Forche, B. Kunze, R. Jansen, W. Trowitzsch-Kienast, G. Höfle, H. Reichenbach and H. Weiss, Eur. J. Biochem., 1994, 219, 691 CrossRef CAS PubMed; (b) M. Degli Esposti, Biochim. Biophys. Acta, 1998, 1364, 222 CrossRef CAS PubMed; (c) H. Miyoshi, Biochim. Biophys. Acta, 1998, 1364, 236 CrossRef CAS PubMed.
- M. Abe, M. Murai, N. Ichimaru, A. Kenmochi, T. Yoshida, A. Kubo, Y. Kimura, A. Moroda, H. Makabe, T. Nishioka and H. Miyoshi, Biochemistry, 2005, 44, 14898 CrossRef CAS PubMed.
- C. Gleye, P. Duret, A. Laurens, R. Hocquemiller and A. Cavé, J. Nat. Prod., 1998, 61, 576 CrossRef CAS PubMed.
- (a) H. Makabe, Y. Hattori, A. Tanaka and T. Oritani, Org. Lett., 2002, 4, 2613 CrossRef; (b) T. J. Donohoe and S. Butterworth, Angew. Chem., Int. Ed., 2005, 44, 4766 CrossRef CAS PubMed; (c) H. Göksel and C. B. W. Stark, Org. Lett., 2006, 8, 3433 CrossRef PubMed; (d) H. Konno, Y. Okuno, H. Makabe, K. Nosaka, A. Onishi, Y. Abe, A. Sugimoto and K. Akaji, Tetrahedron Lett., 2008, 49, 782 CrossRef CAS.
- K. Ota, T. Yamashita, S. Kohno, A. Miura, K. Kamaike and H. Miyaoka, Org. Biomol. Chem., 2018, 16, 3018 RSC.
- C.-C. Liaw, Y.-L. Yang, M. Chen, F.-R. Chang, S.-L. Chen, S.-H. Wu and Y.-C. Wu, J. Nat. Prod., 2008, 71, 764 CrossRef CAS PubMed.
- (a) L. Born, F. Lieb, J. P. Lorentzen, H. Moeschler, M. Nonfon, R. Soellner and D. Wendisch, Planta Med., 1990, 56, 312 CrossRef CAS PubMed; (b) Y. Fujimoto, C. Murasaki, H. Shimada, S. Nishioka, K. Kakinuma, S. Singh, M. Singh, Y. K. Gupta and M. Sahai, Chem. Pharm. Bull., 1994, 42, 1175 CrossRef CAS.
- B. N. Su, E. J. Park, Z. H. Mbwambo, B. D. Santarsiero, A. D. Mesecar, H. H. S. Fong, J. M. Pezzuto and A. D. Kinghorn, J. Nat. Prod., 2002, 65, 1278 CrossRef CAS PubMed.
- B. M. Trost, G. D. Probst and A. Schoop, J. Am. Chem. Soc., 1998, 120, 9228 CrossRef CAS.
- V. M. T. Carneiro, C. M. Avila, M. J. Balunas, W. H. Gerwick and R. A. Pilli, J. Org. Chem., 2014, 79, 630 CrossRef CAS PubMed.
- T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974 CrossRef CAS.
- T. Ema, D. Tanida and T. Sakai, Org. Lett., 2006, 8, 3773 CrossRef CAS PubMed.
- D. R. Williams, M. G. Fromhold and J. D. Earley, Org. Lett., 2001, 3, 2721 CrossRef CAS PubMed.
- K. Ota, CCDC 1950967: CSD Communication, 2019, DOI:10.5517/ccdc.csd.cc23h4f6.
- (a) A. R. L. Cecil, Y. Hu, M. J. Vicent, R. Duncan and R. C. D. Brown, J. Org. Chem., 2004, 69, 3368 CrossRef CAS PubMed; (b) S. B. A. Ghani, J. M. Chapman, B. Figadëre, J. M. Herniman, G. J. Langley, S. Niemann and R. C. D. Brown, J. Org. Chem., 2009, 74, 6924 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1950967. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra09762g |
| This journal is © The Royal Society of Chemistry 2019 |