
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable preparation of sunlight active α-Fe2O3 nanoparticles using iron containing ionic liquids for photocatalytic applications†
Komala,
Harmandeep Kaura,
Money Kaintha,
Sher Singh Meena b and
Tejwant Singh Kang*a
b and
Tejwant Singh Kang*a
aDepartment of Chemistry, University Grants Commission (UGC) Centre for Advanced Studies-II, Guru Nanak Dev University, Amritsar-143005, Punjab, India. E-mail: tejwant.chem@gndu.ac.in
bSolid State Physics Division, Bhabha Atomic Research Centre, Mumbai 400 085, India
First published on 18th December 2019
Abstract
Inspired by the nano-segregation of ionic liquids (ILs) into bi-continuous structures constituting of arrays of ionic and non-ionic components, herein, a new and sustainable strategy for preparation of mesh-like nano-sheet α-Fe2O3 nanoparticles and their photo-catalytic activity under sunlight, is presented. For the purpose, metal (iron) containing ionic liquids (MILs), 1-alkyl-3-methylimidazolium tetrachloroferrates, [Cnmim][FeCl4], (n = 4, 8 and 16), which not only act as precursors and solvents but also as structure directing agents have been used. Thus prepared NPs show MIL dependent structural, photophysical and magnetic properties. The catalytic efficiency of NPs has been tested for the photo-degradation of organic dyes (Rhodamine B) in aqueous solution under sunlight. The NPs are found to exhibit comparable catalytic efficiency under sunlight as compared to that observed under high intensity visible lamplight, without showing a decline in their catalytic efficiency even after 4 catalytic cycles. It is anticipated that the present work will provide a new platform for preparation of sunlight active nanomaterials for photo-catalytic applications with control over the structural and physical properties via varying the molecular structure of MILs.
1. Introduction
Iron oxide nanoparticles (NPs) have attracted great attention from the scientific community due to their potential applications in bio-sensors,1 bio-electronics,2 drug delivery,3 memory devices,4 gas sensors,5 magnetic resonance imaging,3 lithium-ion batteries6 etc. There are different crystalline forms of iron oxides (FeO, Fe3O4 and Fe2O3), and of these Fe2O3 exists in different polymorphs such as, α-Fe2O3 (hematite), β-Fe2O3, ε-Fe2O3 and γ-Fe2O3 (maghemite).7 Among these polymorphic forms, α-Fe2O3 is the most frequently occurring polymorph of iron(III) oxide. Its promising properties such as low cost, high thermal stability, resistance towards corrosion, tuneable magnetic and optical properties, non-toxic/benign nature towards environment as well as living tissues makes α-Fe2O3 worthy to investigate.2,4–7 Till date, α-Fe2O3 NPs with diverse morphologies such as nanoflowers,8 nanotubes,9 nanodiscs,10 polyhedrals,10 nanoplates5 and needles,11 which show morphology dependent properties, have been reported.For the preparation of α-Fe2O3 NPs, different synthetic routes such as hydrothermal/solvothermal, gas–solid growth route, microwave method and thermal oxidation at high temperature etc., have been employed in past.8–11 The utilized methods have a disadvantage of being multi-step and non-economical where some of these methods are cumbersome too. A low yield of the product and additional cost of solvent removal from final product adds to the disadvantages. Not only this, the washing of the product using organic solvents (methanol, ethanol and acetone etc.) to remove the impurities renders the process as a non-environment friendly. Further, one of the most investigated application of α-Fe2O3 NPs is their use as catalyst for photo-degradation of organic dyes in aqueous medium under visible light, where, in general high intensity (300 W or 1000 W)12–23 Xe lamps have been used as light source. The use of sunlight as a light source unambiguously would add to the sustainability of catalytic process at large scale. Therefore new sustainable methods for the preparation of sun-light active α-Fe2O3 NPs must be devised for expanding the base of catalytic applications of these NPs.
In past two decades, ionic liquids (ILs), being greener and designer solvents, has attracted great interest of researchers all over the globe for the synthesis of nanomaterials as one of their application.24 The asymmetric nature of ions with delocalised electrostatic charges25 and abundance of non-covalent interactions (i.e. van der Waals interactions, π–π stacking, hydrogen bonding and electrostatic interactions)26,27 along with the tuneable nature of their properties by choice of their constituent ions28 make ILs as impressive solvents and templating agents for the synthesis of nanomaterials. In past, ILs21,29–31 have been employed as templates for synthesis of α-Fe2O3 nano-structured materials, whereas there exists only two reports wherein metal (iron) based ILs22,32 (MILs) have been used as precursors for preparation of α-Fe2O3 nanomaterials. However, in both the cases, either solvothermal22 or hydrothermal approaches32 have been utilized and the effort to make the process as a sustainable one is still lacking. Moreover, the nano-segregation of MILs into polar and non-polar domains have not been exploited for preparation of α-Fe2O3 nano-structured materials, which is expected to play an important role as the precursor ion, [FeCl4]−, would remain in close vicinity of imidazolium head groups much away from alkyl chains. The oxidation of [FeCl4]− present in the ionic network of bi-continuous structures of MIL would give rise to α-Fe2O3 specifically affected by ionic network of MIL. This approach would be different than that reported in literature where MILs have been exploited as precursor as well as template for the preparation of α-Fe2O3 using hydrothermal and solvothermal methods. In such cases, the structural network of MIL around the growing NPs would be different than that present in neat MILs, which would certainly affect the shape and size of prepared NPs.
Therefore, to achieve our target of sustainable preparation of α-Fe2O3 NPs, we relied on metal (iron) containing imidazolium based ionic liquids (MILs), [Cnmim][FeCl4], with varying length of alkyl chain (n = 4, 8 and 16) via a simple grinding approach followed by calcination. The sustainability of the process lies in the fact that contrary to previous reports where ILs have been used as templates21,29–31 or template as well as precursor of metal ion,22,32 the employed strategy utilizes MILs as templates, precursor of metal ions as well as solvent for the preparation of α-Fe2O3 NPs. This limits the use of organic solvent, a separate precursor of metal ions and reduces the number of steps adding sustainability to the process. The synthesized α-Fe2O3 NPs have been characterized by X-ray diffraction (XRD), Raman spectroscopy, transmission electron microscopy (TEM), UV-visible and photoluminescence spectroscopy (PL). The synthesized NPs have shown MIL dependent structural, photo-physical and magnetic properties, which have not been reported till date, to best of our knowledge. Importantly, as catalyst, α-Fe2O3 NPs have shown efficient photo-degradation of RhB under sunlight as compared to that reported under high intensity visible light (Xe lamp 300 W), catalyzed by α-Fe2O3 NPs. This along with our previous report on preparation of Ag@AgBr JNPs33 for sustainable photo-degradation of organic dyes in water under sunlight, would open up a new avenue of research in the field of MILs for sustainable preparation of a variety of nanomaterials for different applications.
2. Experimental
2.1. Materials
Ferric chloride hexahydrate (FeCl3·6H2O, 98%), 1-methylimidazole (>99%), 1-chlorobutane (>99%), 1-chlorooctane (>99%), 1-chlorohexadecane (>99%) and Rhodamine B (RhB) (>95%) were purchased from Sigma-Aldrich. Dichloromethane (AR grade), diethyl ether (AR grade), hexane (AR grade), and ethyl acetate (AR grade) were purchased from SD Fine-Chem. Ltd., India. Sodium hydroxide (NaOH) (>99%) and hydrogen peroxide (30 wt%) were purchased from Spectrochem, India. The detailed synthetic procedure and characterization data for iron containing ionic liquids, [Cnmim][FeCl4], with varying length of alkyl chain (n = 4, 8 and 16) are provided in Annexure S1 and Fig. S1–S5 (ESI).†2.2. Preparation of iron oxide (α-Fe2O3) NPs
For the preparation of α-Fe2O3 NPs, 2 mmol of [Cnmim][FeCl4] (n = 4, 8 and 16) and 8 mmol of NaOH were grounded for 20 min in a pestle-mortar at room temperature. Thus obtained thick viscous paste was washed several times with excess of distilled water to remove the residual MIL, if any. The obtained powder, after air drying for 24 h, was calcined at 600 °C for 4 h to increase the crystallinity of the NPs. The prepared NPs were coded as α-Fe-4, α-Fe-8 and α-Fe-16 depending up on the alkyl chain length (n = 4, 8 and 16) of the MILs used for their preparation.2.3. Characterization of α-Fe2O3 NPs
Powder X-ray diffraction pattern of the prepared NPs was recorded at room temperature by using a Cu-target (λ = 1.54 Å) on Rigaku Xpert Pro X-ray diffractometer at a voltage of 40 kV and at a current of 30 mA. The scan was performed in the 2θ range 20–80° with a step size of 0.02°. Raman spectra were obtained using Renishaw Raman spectrometer equipped with 488 nm Ar-ion laser in the range of 100–1500 cm−1 by placing the prepared NPs on a glass slide. The size, morphology and lattice structure of prepared NPs was investigated by JEM-2100 Transmission Electron Microscope (TEM) at a working voltage of 200 kV. For the TEM measurement, the NPs were dispersed in ethanol followed by ultrasonication in a bath sonicator for 15 minutes. A drop of dispersion was placed on the carbon coated grid (300 mesh) and the samples were dried at room temperature for 24 hours before the measurements. Optical absorption spectra were recorded on UV-Vis spectrophotometer (UV-1800 SHIMADZU) in the wavelength range 200–800 nm using a quartz cuvette of path length 1 cm. Photoluminescence spectra were recorded on a PerkinElmer spectrophotometer using the excitation wavelength of 300 nm employing an excitation and emission slit width of 2.5, each. The measurements were made using a quartz cuvette of path length 1 cm in the wavelength range of 300–550 nm. For both UV-Vis and photoluminescence measurements, spectra were recorded by dispersing equal amount of NPs prepared from different precursors in distilled water as solvent. Mössbauer spectra were recorded at room temperature with a conventional spectrometer operated in constant acceleration mode in transmission geometry with Co57 source in Rh matrix of 45 mCi. The recorded Mössbauer spectra were fitted using the WinNormos site fit program. The calibration of the velocity scale was done by using an enriched α-57 Fe metal foil. The isomer shift values are relative to Fe metal foil (δ = 0.0 mm s−1). The investigation on magnetic properties were made using Microsense EV-90 vibrating sample magnetometer (VSM) in the applied magnetic field of −15 to +15 kOe at room temperature.2.4. Photocatalytic activity of α-Fe2O3 NPs
10 mg of prepared α-Fe2O3 NPs was added to 30 ml of 1 × 10−5 M Rhodamine B (RhB) solution in water in the presence of 0.5 ml of H2O2 (final concentration 0.5 vol%). Before irradiation under sunlight (normal irradiance value of 5.90 kW h per m2 per day), the samples were stirred in dark for 30 min to reach adsorption–desorption equilibrium. The solution was then irradiated under sunlight and 1 ml of sample was taken out of the cell after certain interval of time followed by measurement of UV-Vis spectra after centrifugation for 5 min at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. Every care has been taken to keep sample away from sunlight/visible light during the process.
000 rpm. Every care has been taken to keep sample away from sunlight/visible light during the process.
3. Results and discussions
3.1. X-ray diffraction
Before going into detailed characterization of NPs, the complete removal of residual MILs from α-Fe2O3 NPs was checked using FT-IR spectra (Fig. S6, ESI†) recorded on Agilent Cary 630 FTIR spectrometer. Fig. 1A shows the X-ray diffraction pattern of prepared NPs. Nine diffraction peaks corresponding to (012), (104), (110), (113), (024), (116), (122), (214) and (300) planes indicate the formation of rhombohedral α-Fe2O3 phase (hematite) (JCPDS card no. 86-0550).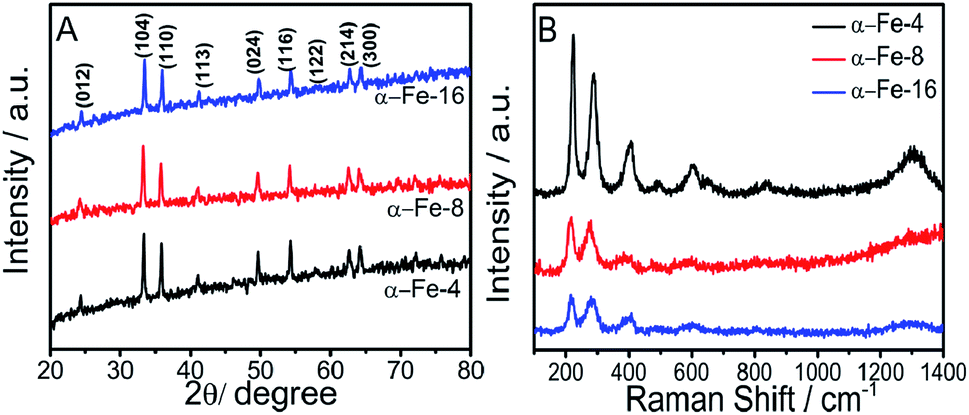 | ||
| Fig. 1 (A) XRD pattern; and (B) Raman spectra of prepared α-Fe2O3 NPs prepared using MILs having different alkyl chain lengths. | ||
The average crystallite size of NPs is calculated using Debye–Scherrer's equation.34 The crystallite size decreases sharply by changing the alkyl chain length of MIL from butyl (α-Fe-4, 40 nm) to octyl (α-Fe-8, 34.4 nm) whereas the change in crystallite size while going from octyl (α-Fe-8, 34.4 nm) to hexadecyl (α-Fe-16, 32.1 nm) alkyl chain is not so significant. An increase in half-line width of diffraction peak (values provided in Table S1, ESI†) while going from α-Fe-4 to α-Fe-16 suggests the decrease in crystallinity. In general, various physicochemical properties of MILs such as viscosity, dielectric constant, refractive index, surface tension and polarity etc.35–37 undergo a change with the extension of their alkyl chain length, which could affect the size and crystallinity of NPs. The inter-planar distance of (104) plane (as a representative) increases on moving from α-Fe-4 to α-Fe-16 (Table S1, ESI†), which suggest the expansion of lattice with decrease in size of crystallites in comparison to bulk α-Fe2O3 (d104 = 2.70 Å).38 In addition to this, energy dispersive X-ray spectroscopic (EDAX) measurements also confirm the formation of α-Fe2O3 as can be seen form Fig. S7 (ESI).† The crystal space group and the phase purity of the synthesized α-Fe2O3 NPs have been examined by the Raman spectroscopy. Raman spectra (Fig. 1B), shows the presence of seven bands corresponding to two A1g and five Eg modes of D63d space group, which is consistent with the literature reports for the Raman spectra of hematite NPs.38 A slight shift of the first three Raman bands towards lower wavenumber is observed while moving from α-Fe-4 to α-Fe-16, which is assignable to quantum confinement effect.38
3.2. Morphological studies
The morphology and average particle size of α-Fe2O3 NPs have been investigated by transmission (TEM) and high-resolution transmission electron (HR-TEM) microscopy (Fig. 2(A–F)). The average particle size (histograms, inset Fig. 2(A–C) and Table S1†) of NPs decreases while moving for α-Fe-4 to α-Fe-16. This is in line with the results obtained from XRD and Raman spectroscopic measurements which indicate that the alkyl chain length of MILs plays an important role in the growth of NPs. The average particle size obtained from TEM measurements is found to be on higher specifically in case of α-Fe-4 NPs, which is assigned to relatively less amphiphilic character of [C4mim][FeCl4] offered by small butyl chain that resulted in aggregation of NPs.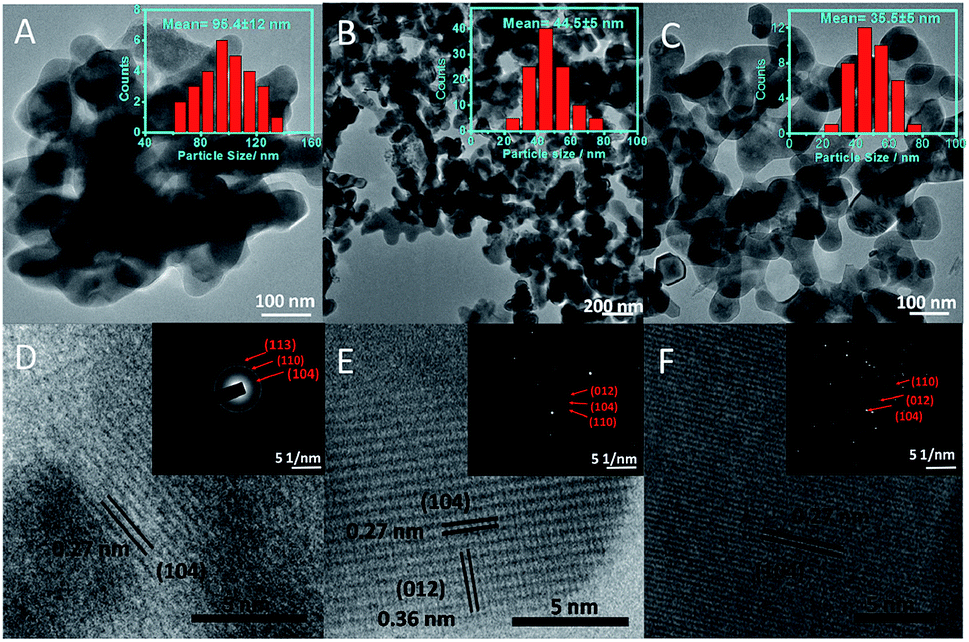 | ||
| Fig. 2 (A–C) Transmission electron microscopic (TEM) images of α-Fe-4, α-Fe-8 and α-Fe 16, respectively; (D–F) HR-TEM images showing the lattice planes of synthesized α-Fe2O3 NPs. Inset of (A–C) show the histograms showing the mean particle diameter along with standard deviation (S.D.); inset of (D–F) shows their respective selected area electron diffraction (SAED) pattern. | ||
Despite of alteration in various physicochemical properties of MILs with variation in alkyl chain length, the electronic as well as steric properties of MILs are also expected to play an important role in controlling the size as well as shape of NPs. Further, α-Fe2O3 NPs are found to be interconnected with each other in the form of nano-sheets where the void spaces in the interconnected network increase while moving from α-Fe-4 and α-Fe-16. No doubt, the prepared α-Fe2O3 NPs show excellent magnetic and photo-catalytic properties (discussed later), however, it is expected that the mechanism of formation of α-Fe2O3 NPs would be different as compared to previously employed methods, which utilizes some solvent and other conditions for directional growth of NPs.21,22,29–32 It has been established that most of the ILs having sufficiently long alkyl chain (equal to or >6 carbon atoms) segregates in polar and non-polar domains,39–42 and with the increase in length of alkyl chain, the extent of such segregation increases (Scheme 1).43 It is natural to assume that the precursor ions, [FeCl4]−, would stay in close proximity of imidazolium head group39 (Scheme 1) and therefore the formation and growth of [Fe(OH)4]− in the presence of NaOH would happen in polar domains of MILs by virtue of adsorption of cationic part of MILs on the surface of growing [Fe(OH)4]− NPs by electrostatic interactions29,44 as shown in Scheme 1, supplemented by hydrogen bonding interactions between the H-atom at C-2 position of imidazolium ring of MIL and oxygen atom of O–Fe. Therefore, the size of the segregated nano-domains which increases with increase in alkyl chain length of ILs, would eventually control the shape and size of [Fe(OH)4]− NPs, which upon calcinations transforms to α-Fe2O3 NPs. The higher extent of segregation of [C16mim][FeCl4] into polar and non-polar domains results in relatively larger voids in interconnected network of α-Fe-16 NPs as compared to other investigated NPs.
 | ||
| Scheme 1 Pictorial representation showing the role of nano-segregated polar and non-polar domains of MILs in formation of interconnected network of α-Fe2O3 NPs. | ||
In addition to this, the properties of MILs (surface tension and viscosity) also changes with the variation of alkyl chain length from butyl to hexadecyl.36,37 The increase in viscosity of MIL with alkyl chain length would prevent the NPs from agglomerating, while the decrease of surface tension with extension of alkyl chain length would result in the reduction of energy barrier to nucleation, thereby the rate of nucleation increases as compared to growth rate of NPs resulting in formation of relatively small NPs. The results obtained here are similar to that observed in case of α-Fe2O3 NPs prepared in binary solvent mixtures of an IL, [C2mim][C2OSO3] and ethylene glycol,34 where the formation of solute–solvent nano-domains were found to effect the growth of prepared NPs. HR-TEM (Fig. 2(D and E)) reveals the presence of lattice planes having an inter-planar distance of 0.27 nm, which corresponds to the (104) plane of the rhombohedral α-Fe2O3 phase.22 The selected area diffraction pattern (SAED) of prepared α-Fe2O3 NPs (inset, Fig. 2), shows the presence of (104), (110) and (012) planes and confirms the existence of rhombohedral form of α-Fe2O3 NPs. The prepared NPs are found to be polycrystalline in nature, where the crystallinity increases while going from α-Fe-4 to α-Fe-16 as judged from increased diffraction intensity.
3.3. Optical studies
The prepared α-Fe2O3 NPs show two dominant absorption bands, one in the UV region at 220–275 nm and another in the visible region i.e. 555–570 nm (Fig. 3). The band observed in UV region is due to the ligand to metal charge transitions (LMCT) from O2− (2p) to Fe3+ (3d),44 while the band in visible region is assigned to 6A1 + 6A1 to 4T1 + 4T1 transition, called as ‘double excitation process’.45–48 The transition observed in visible region is the reason for the colour of the NPs.30 As can be seen from Fig. 3A, a hypsochromic shift of 25 nm (from 586 nm to 559 nm) and enhancement in absorbance is observed while going from α-Fe-4 to α-Fe-16 NPs. | ||
| Fig. 3 (A) UV-Vis spectra of synthesized α-Fe2O3 NPs; and (B) photoluminescence spectra of synthesized α-Fe2O3 NPs, [λexc = 300 nm]. | ||
This can be assigned to decrease in particle size. Further, the band gap energy of NPs has been calculated from UV-Visible spectra (Fig. S8, ESI†), using Tauc's equation22
| (ahν)2 = b(hν − Eg)n | (1) |
3.4. Magnetic studies
Fig. 4A and S9 (ESI)† shows the room temperature Mössbauer spectra of synthesized α-Fe2O3 NPs. The values of Mössbauer spectral parameters calculated from fitting of spectra are tabulated in Table S2, ESI.† A decrease in value of hyperfine field (Hhf) on moving from α-Fe-4 to α-Fe-16, is observed, which can be attributed to decrease in size of NPs. Further a decrease in value of quadrupole splitting (Δ) with the extension of alkyl chain length of MILs indicates increase in distortions from cubic structure due to surface effects on smaller particles.54 The values of isomer shift (δ) are consistent with literature report of hematite and indicates that Fe3+ is in high spin state.55 Along with a sextet, the presence of small central doublet indicates the superparamagnetic nature of synthesised NPs.56–58 Besides this, magnetic measurements of synthesized α-Fe2O3 NPs were also carried out using Vibrating Sample Magnetometer (VSM) at room temperature and the obtained magnetic moment (M) vs. applied field (H) hysteresis loops obtained are provided in Fig. 4B, which is indicative of weak ferromagnetic nature of NPs. Usually, hematite is antiferromagnetic in nature below Morin temperature (Tm = 263 K) and exhibit weak ferromagnetic behaviour in temperature range between 263 and 960 K (i.e. Néel temperature).59 | ||
| Fig. 4 (A) Mössbauer spectra of α-Fe-16; (B) M–H hysteresis loop of α-Fe2O3 NPs synthesized using MIL of different alkyl chain lengths; (C) variation of Hhf and MS values with the alkyl chain length of MIL; inset of B shows the values of MS, Mr and HC for respective α-Fe2O3 NPs. | ||
This transition is due to slight canting of spins from trigonal [111] axis towards basal (111) plane.60 This slight canting of spins results from the anisotropic exchange interaction, also named as Dzialoshinski–Moriya interaction.60,61 The values of saturation magnetisation (MS), remanant magnetisation (Mr) and coercivity (HC) are provided in inset of Fig. 4B.
The values of MS and Mr decrease with decrease in size of NPs, which is attributable to increase in surface area and surface disorders. This reduces the inter-particle interactions resulting in decrease in MS and Mr.62 The HC increases with decrease in particle size, which is in accordance with the literature reports as well.63 The negligible values of Mr and HC could be due to presence of superparamagnetic character as also confirmed from Mössbauer analysis.64 It is observed that apart from controlling the size, structural and optical properties of α-Fe2O3 NPs, the alkyl chain length of MILs also controls the magnetic properties of α-Fe2O3 NPs.
3.5. Photocatalytic activity of synthesized α-Fe2O3 NPs
The synthesized NPs have been tested for the photocatalytic degradation of RhB dye under sunlight in the presence of H2O2. The Fig. 5A shows the time dependent absorbance spectra of aqueous solution of RhB dye in the presence of α-Fe-16 as catalyst (as a representative) under sunlight in the presence of H2O2, whereas Fig. 5B shows the photocatalytic efficiency of different α-Fe2O3 NPs. It is observed that about 98% of RhB dye degrades in the presence of α-Fe-16 as catalyst in 105 min, while 92% and 86% of dye degrades in the presence of α-Fe-8 and α-Fe-4 NPs, respectively in the same time frame. Moreover, only 5% degradation of RhB dye in the absence of catalyst and H2O2 and 8% using MIL ([C16min][FeCl4] as representative) and H2O2 under sunlight (Fig. 5B) is observed, which confirms the role of α-Fe2O3 NPs. On the other hand, the degradation rate is found to be increased upto 11% in the presence of H2O2 only. This could be due to the highly oxidative hydroxyl radicals formed by the photo decomposition of H2O2.65 Therefore, it is established that the degradation efficiencies of α-Fe2O3 NPs have a direct relation with the size and extent of agglomeration of synthesized NPs, which could be related to increase in number of surface sites of synthesized NPs. | ||
| Fig. 5 (A) Absorption spectra of the aqueous solution of RhB in the presence of α-Fe-16 as photocatalyst exposed to sunlight for different times; (B) photocatalytic performances of different synthesized α-Fe2O3 NPs and from literature report;22 (C) comparison of reaction rates along with rate constants for different synthesized α-Fe2O3 NPs and from literature report;22 (D) recycling efficiency of α-Fe-16 as photocatalyst for 4 repeated catalytic cycles; (E) 1 × 10−5 M solution of RhB dye in water; (F) dispersed α-Fe2O3 NPs in RhB dye solution; (G) removal of α-Fe2O3 NPs after degradation of RhB dye using magnet; and (H) literature reports for degradation of RhB dye by α-Fe2O3 NPs using different lamps. Ref. 12–23 are provided in reference section. The data point under circle represents this work. | ||
To have further insight into the phenomenon, the rate constants for degradation of RhB have also been calculated using first order rate equation:
|
lnCo/Ct = kt
| (2) |
Co/Ct vs. t gives a straight line and slope of line gives rate constant (k). The values of rate constants (inset Fig. 5C) are found to increase on moving from α-Fe-4 to α-Fe-16. This is assignable to smaller size and larger surface to volume ratio of α-Fe-16 NPs, which results in the presence of relatively more number of active sites required for high catalytic efficiency. A decrease in agglomeration and increase in size of voids in interconnected network of α-Fe-16 NPs as compared to other investigated NPs offers relatively large available surface for the dye to get adsorbed and catalyzed. A relative decrease in UV-Vis absorption of RhB in the presence of different catalysts after stirring in dark for 30 min follows the same trend (Fig. S10, ESI†) in which their catalytic activity varies indicating that the surface area of synthesised NPs governs the catalytic efficiency. Therefore, the recycling ability of α-Fe-16 as photocatalyst, as a representative, is also investigated and α-Fe-16 is found to exhibit almost no significant change in catalytic efficiency for at least 4 repeated photocatalytic cycles (Fig. 5D). Owing to magnetically separable nature of α-Fe2O3 NPs when dispersed in solvent (Fig. 5E), it is very easy to separate and reuse α-Fe2O3 NPs for further catalytic cycles. The catalytic efficiency of α-Fe-16 NPs is also compared with the literature reports. Fig. 5H shows the comparison of rate constants obtained for photo-degradation of RhB using α-Fe2O3 NPs prepared by different methods. It is observed that α-Fe-16 NPs exhibited comparative or even higher catalytic efficiency towards photo-degradation of RhB as compared to most of the exploited α-Fe2O3 NPs in the presence of high intensity visible light despite their non-specific shape. Large surface area owing to near spherical shape supplemented by the presence of voids may be assigned as a reason for high catalytic efficiency. It is observed that the obtained rate constant for degradation of RhB is higher or comparable in comparison to some of the literature reports, where degradation of Rh B dye occurred with a comparable rate constants with few exceptions under visible light (under 300 W Xe lamp, λ > 400 nm), (Fig. 5H)12–23 while in our case, degradation rate constant is found to be 0.030/min under sunlight. Obviously, the synthetic method used for preparation of NPs, which affects their size, morphology and crystallinity, plays an important role in governing their photocatalytic efficiency. It is important to mention that the photo-degradation of RhB, as a representative, using α-Fe2O3 NPs can be scaled up for large scale industrial applications, where the use of sunlight would not only be cost-effective but also renders the process as a sustainable one. In this way, the present work along with the earlier reports on sustainable methods of preparation and catalytic applications of NPs33,66 would pave the way for preparation and applications of new nanomaterials using MILs in an environment friendly manner.
4. Conclusions
A new approach for the preparation of magnetic and sunlight active α-Fe2O3 NPs for photo-degradation of organic dyes (RhB as a representative) is established employing metal (iron) containing ionic liquids (MILs). The morphological, photophysical, magnetic as well catalytic properties of NPs are found to be dependent upon the length of the alkyl chain of MIL used, which acts as a precursor, solvent as well as capping agent for the preparation of NPs. The prepared NPs are found to be catalytically active under sun-light for photo-degradation of RhB in aqueous solutions. Interestingly, when compared, it is observed that the catalytic efficiency of NPs under sunlight is higher or comparable to that observed while using α-Fe2O3 NPs with specific shapes and sizes and under high intensity visible light, which adds to the utility of prepared NPs. Magnetically separable nature of NPs renders the recycling process during different catalytic cycles very easy without the loss of NPs which resulted in negligible loss of catalytic efficiency over repeated number of catalytic cycles. The present report along with the earlier reported work on preparation of nanomaterials using ILs via hand grinding,34,66,67 represents a simple, greener and economical method for the synthesis of α-Fe2O3 NPs which show MIL dependent characteristics physicochemical properties of NPs and provides alternate way to manipulate the properties at nano level.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Komal is thankful to UGC, Govt. of India for award of SRF. H. K. is thankful to CSIR, Govt. of India for award of JRF. Authors are thankful to UGC, India, for their UGC-CAS program awarded to the Department of Chemistry, Guru Nanak Dev University, Amritsar. The infrastructure facility utilized for carrying out this work under the UPE grant is highly acknowledged. We are thankful to Mr Hirdesh, Dept. of Physics, GNDU, Amritsar for performing Raman measurements.Notes and references
- C. S. K. S. Patel, M. Z. Anwar, A. Kumar, S. V. Otari, R. T. Pagolu, S. Y. Kim, I. W. Kim and J. K. Lee, Biochem. Eng. J., 2018, 132, 1–8 CrossRef.
- U. Kostiv, V. Patsula, M. Šlouf, I. M. Pongrac, S. Škokić, M. D. Radmilović, I. Pavičić, I. V. Vrček, S. Gajović and D. Horák, RSC Adv., 2017, 7, 8786–8797 RSC.
- M. Kluenker, M. N. Tahir, R. Dören, M. Deuker, P. Komforth, S. Plana-Ruiz, B. Barton, S. I. Shylin, V. Ksenofontov, M. Panthöfer, N. Wiesmann, J. Herzberger, A. Möller, H. Frey, J. Brieger, U. Kolb and W. Treme, Chem. Mater., 2018, 30, 4277–4288 CrossRef CAS.
- T. Ninjbadgar, S. Yamamoto and T. Fukuda, Solid State Sci., 2004, 6, 879–885 CrossRef CAS.
- J. Ma, J. Teo, L. Mei, Z. Zhong, Q. Li, T. Wang, X. Duan, J. Lian and W. Zheng, J. Mater. Chem., 2012, 22, 11694–11700 RSC.
- J. Zhang, T. Huang, Z. Liu and A. Yu, Electrochem. Commun., 2003, 29, 17–20 CrossRef.
- R. Kant, D. Kumar and V. Dutta, RSC Adv., 2015, 5, 52945–52951 RSC.
- C. Y. Cao, J. Qu, W. S. Yan, J. F. Zhu, Z. Y. Wu and W. G. Song, Langmuir, 2012, 28, 4573–4579 CrossRef CAS PubMed.
- C. J. Jia, L. D. Sun, Z. G. Yan, L. P. You, F. Luo, X. D. Han, Y. C. Pang, Z. Zhang and C. H. Yan, Angew. Chem., Int. Ed., 2005, 44, 4328–4333 CrossRef CAS PubMed.
- M. Sorescu, R. A. Brand, D. Mihaila-Tarabasanu and L. Diamandescu, J. Appl. Phys., 1999, 85, 5546–5548 CrossRef CAS.
- H. Yang, X. Mao, Y. Guo, D. Wang, G. Ge, R. Yang, X. Qiu, Y. Yang, C. Wang, Y. Wang and G. Liu, CrystEngComm, 2010, 12, 1842–1849 RSC.
- S. Bharathi, D. Nataraj, D. Mangalaraj, Y. Masuda, K. Senthil and K. Yong, J. Phys. D: Appl. Phys., 2009, 43, 1–9 Search PubMed.
- B. Showa, N. Mukherjee and A. Mondal, RSC Adv., 2016, 6, 75347–75358 RSC.
- R. Satheesh, K. Vignesh, A. Suganthi and M. Rajarajan, J. Environ. Chem. Eng., 2014, 2, 1956–1968 CrossRef CAS.
- M. A. Mahadik, S. S. Shinde, K. Y. Rajpure and C. H. Bhosale, Mater. Res. Bull., 2013, 48, 4058–4065 CrossRef CAS.
- Q. Tian, W. Wu, L. Sun, S. Yang, M. Lei, J. Zhou, Y. Liu, X. Xiao, F. Ren, C. Jiang and V. A. L. Roy, ACS Appl. Mater. Interfaces, 2014, 6, 13088–13097 CrossRef CAS PubMed.
- X. Zhou, Q. Xu, W. Lei, T. Zhang, X. Qi, G. Liu, K. Deng and J. Yu, Small, 2014, 10, 674–679 CrossRef CAS PubMed.
- H. Yin, Y. Zhao, Q. Hua, J. Zhang, Y. Zhang, X. Xu, Y. Long, J. Tang and F. Wang, Front. Chem., 2019, 7, 58 CrossRef CAS PubMed.
- S. Li, G. Qin, X. Meng, Y. Ren and L. Zuo, J. Mater. Sci., 2013, 48, 5744–5749 CrossRef CAS.
- A. A. Gobouri, Res. Chem. Intermed., 2016, 42, 5099–5113 CrossRef CAS.
- B. Xu, B. Huang, H. Cheng, Z. Wang, X. Qin, X. Zhang and Y. Dai, Chem. Commun., 2012, 48, 6529–6531 RSC.
- L. Xu, J. Xia, K. Wang, L. Wang, H. Li, H. Xu, L. Huang and M. He, Dalton Trans., 2013, 42, 6468–6477 RSC.
- X. Zhou, J. Lan, G. Liu, K. Deng, Y. Yang, G. Nie, J. Yu and L. Zhi, Angew. Chem., Int. Ed., 2012, 51, 178–182 CrossRef CAS PubMed.
- P. Wasserscheid and T. Welton, Ionic Liquid in Synthesis, Wiley, New York, 2003 Search PubMed.
- A. A. H. Padua, M. F. C. Gomes and J. N. A. C. Lopes, Acc. Chem. Res., 2007, 40, 1087–1096 CrossRef CAS PubMed.
- T. Ueki and M. Watanabe, Macromolecules, 2008, 41, 3739–3749 CrossRef CAS.
- J. C. Schleicher and A. M. Scurto, Green Chem., 2009, 11, 694–703 RSC.
- M. Zhen, J. Yu and S. Dai, Adv. Mater., 2010, 22, 261–285 CrossRef PubMed.
- S. Xie, H. Jia, F. Lu, N. Sun, J. Yu, S. Liu and L. Zheng, CrystEngComm, 2015, 17, 1210–1218 RSC.
- J. Lian, X. Duan, J. Ma, P. Peng, T. Kim and W. Zheng, ACS Nano, 2009, 3, 3749–3761 CrossRef CAS PubMed.
- C. M. Lee, H. J. Jeong, S. T. Lim, M. H. Sohn and D. W. Kim, ACS Appl. Mater. Interfaces, 2010, 2, 756–759 CrossRef CAS PubMed.
- L. Xu, J. Xia, L. Wang, J. Qian, H. Li, K. Sun and M. He, Chem.–Eur. J., 2014, 20, 2244–2253 CrossRef CAS PubMed.
- G. Singh, Komal, G. Singh, M. Kaur and T. S. Kang, J. Mater. Chem. A, 2019, 7, 5185–5189 RSC.
- P. Shikha, B. S. Randhawa and T. S. Kang, RSC Adv., 2015, 5, 51158–51168 RSC.
- T. Singh and A. Kumar, J. Phys. Chem. B, 2008, 112, 12968–12972 CrossRef CAS PubMed.
- R. Alcalde, G. García, M. Atilhan and S. Aparicio, Ind. Eng. Chem. Res., 2015, 54, 10918–10924 CrossRef CAS.
- G. Law and P. R. Watson, Langmuir, 2001, 17, 6138–6141 CrossRef CAS.
- K. V. Manukyan, Y. S. Chen, S. Li, P. Rouvimov, X. Li, S. Dong, X. Liu, J. K. Furdyna, A. Orlov, G. H. Bernstein, W. Porod, S. Roslyakov and A. S. Mukasyan, J. Phys. Chem. C, 2014, 118, 16264–16271 CrossRef CAS.
- R. E. D. Sesto, T. M. McCleskey, A. K. Burrell, G. A. Baker, J. D. Thompson, B. L. Scott, J. S. Wilkes and P. Williams, Chem. Commun., 2008, 447–449 RSC.
- L. S. Ott, M. L. Cline, M. Deetlefs, K. R. Seddon and R. G. Finke, J. Am. Chem. Soc., 2005, 127, 5758–5759 CrossRef CAS PubMed.
- D. Kogelnig, A. Stojanovic, F. v. d. Kammer, P. Terzieff, M. Galanski, F. Jirsa, R. Krachler, T. Hofmann and B. K. Keppler, Inorg. Chem. Commun., 2010, 13, 1485–1488 CrossRef CAS.
- C. P. Cabry, L. D'Andrea, K. Shimizu, I. Grillo, P. Li, S. Rogers, D. W. Bruce, J. N. C. Lopes and J. M. Slattery, Faraday Discuss., 2018, 206, 265 RSC.
- S. Li, J. L. Bañuelos, J. Guo, L. Anovitz, G. Rother, W. R. Shaw, P. C. Hillesheim, G. Dai, G. A. Baker and P. T. Cummings, J. Phys. Chem. Lett., 2012, 3, 125–130 CrossRef CAS.
- A. K. Burrell, R. E. D. Sesto, S. N. Baker, T. M. McCleskeya and G. A. Baker, Green Chem., 2007, 9, 449–454 RSC.
- Y. P. He, Y. M. Miao, C. R. Li, S. Q. Wang, L. Cao, S. S. Xie, G. Z. Yang, B. S. Zou and C. Burda, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 1–9 Search PubMed.
- T. Hashimoto, T. Yamada and T. Yoko, J. Appl. Phys., 2002, 80, 3184–3190 CrossRef.
- D. M. Sherman, Int. J. Geogr. Inf. Sci., 1985, 70, 1262–1269 CAS.
- N. Tsuda, K. Nasu, A. Fujimori and K. Siratori, Electronic Conduction in Oxide, Springer, Berlin, 2006 Search PubMed.
- A. S. H. Tolbert and A. P. Alivisatos, Science, 1994, 265, 373–376 CrossRef PubMed.
- T. Takagahara and K. Takeda, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 15578–15581 CrossRef CAS PubMed.
- B. Zou, W. Huang, M. Han, S. F. Y. Li, X. Wu, Y. Zhang, J. Zhang, P. Wu and R. Wang, J. Phys. Chem. Solids, 1997, 58, 1315–1320 CrossRef CAS.
- B. Zou, G. Tang, G. Zhang, W. Chen, G. Zhang, L. Xiao, Y. Zhang, T. Li and H. Fei, Acta Phys. Sin., 1993, 42, 1127 CAS.
- F. C. Zhang and T. M. Rice, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 3759 CrossRef CAS PubMed.
- S. S. Shinde, S. S. Meena, S. M. Yusuf and K. Y. Rajpure, J. Phys. Chem. C, 2011, 115, 3731–3736 CrossRef CAS.
- A. Paesano, S. C. Zanatta, S. N. De Medeiros, L. F. Cótica and J. B. M. Da Cunha, Hyperfine Interact., 2005, 161, 211–220 CrossRef CAS.
- W. Kundig and H. Bommel, Phys. Rev., 1966, 142, 327 CrossRef.
- B. Ganguly, F. E. Huggins, K. R. P. M. Rao and G. P. Huffman, J. Catal., 1993, 142, 552–560 CrossRef CAS.
- C. Frandsen and S. Mørup, J. Magn. Magn. Mater., 2003, 266, 36–48 CrossRef CAS.
- X. M. Liu, S. Y. Fu, H. M. Xiao and C. J. Huang, J. Solid State Chem., 2005, 178, 2798–2803 CrossRef CAS.
- Q. A. Pankhurst, C. E. Johnson and M. F. Thomas, J. Phys. C: Solid State Phys., 2002, 19, 7081–7098 CrossRef.
- I. Dzyaloshinsky, Phys. Status Solidi, 1971, 4, 763–772 Search PubMed.
- T. Moriya, Phys. Rev., 1960, 120, 91–98 CrossRef CAS.
- M. C. Varma, A. M. Kumar, G. S. V. R. K. Choudary and K. H. Rao, Int. J. Nanosci., 2012, 11, 1240003 CrossRef.
- X. He, W. Zhong, C.-T. Au and Y. Du, Nanoscale Res. Lett., 2013, 8, 446 CrossRef PubMed.
- N. Panda, H. Sahoo and S. Mohapatra, J. Hazard. Mater., 2011, 185, 359 CrossRef CAS PubMed.
- Komal, P. Shikha and T. S. Kang, New J. Chem., 2017, 41, 7407–7416 RSC.
- L. Wang, L. Chang, B. Zhao, Z. Yuan, Z. Shao and W. Zheng, Inorg. Chem., 2008, 47, 1443–1452 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Annexure S1, Fig. S1–S10, Tables S1 and S2. See DOI: 10.1039/c9ra09678g |
| This journal is © The Royal Society of Chemistry 2019 |