
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simple sensitive UFLC-MS/MS method for the simultaneous quantification of artesunate, dihydroartemisinin and quercetin in rat plasma and its application to pharmacokinetic studies
Nethravathi Puttappa†
 *a,
Karthik Yamjalab,
Narenderan S. T.b,
Suresh Kumar Raman†*a,
Gowthamarajan Kuppusamya,
Basuvan Babub and
P. Ram Kumarc
*a,
Karthik Yamjalab,
Narenderan S. T.b,
Suresh Kumar Raman†*a,
Gowthamarajan Kuppusamya,
Basuvan Babub and
P. Ram Kumarc
aDepartment of Pharmaceutics, JSS College of Pharmacy, JSS Academy of Higher Education & Research, Ooty, Nilgiris, Tamil Nadu, India. E-mail: nethra0809@gmail.com; sureshcoonoor@yahoo.com
bDepartment of Pharmaceutical Analysis, JSS College of Pharmacy, JSS Academy of Higher Education & Research, Ooty, Nilgiris, Tamil Nadu, India
cNetmeds Marketplace Limited, Chennai, Tamil Nadu, India
First published on 17th December 2019
Abstract
An ultrafast liquid chromatography-tandem mass spectrometry (UFLC-MS/MS) method was developed for the simultaneous estimation of artesunate (ART), dihydroartemisinin (DHA, an active metabolite of ART) and quercetin (QRT) in rat plasma. The separation was achieved using a Zorbax C18 column (3 μm, 50 mm × 4.6 mm) as a stationary phase with a mobile phase of 0.1% formic acid (10% by volume) and methanol (90% by volume) at a flow rate of 0.4 mL min−1 and an injection volume of 10 μL. Artemisinin (ATM) was used as the internal standard (IS). Mass detection was performed by electrospray ionization (ESI)-tandem mass spectrometry via multiple reaction monitoring (MRM) in positive mode except for QRT, where negative ionization was used. The extraction recoveries of ART, DHA, and QRT from plasma were found to be 91.05–99.62%, 95.12–98.56% and 89.35–98.90%, respectively. The developed method was validated and successfully applied to the quantitative analysis of ART, DHA and QRT in plasma samples after the oral administration of ART and ART–QRT pure drugs to rats at the dose of 5 mg kg−1 each. The results reveal that the developed method can be further used for the quantification of the proposed combination drugs in nanoformulations.
1. Introduction
Malaria, a tropical disease caused by the plasmodium parasite, is affecting about 219 million people globally. Due to the insistent lack of an effective vaccine, the battle against malaria depends mostly on chemotherapy and chemoprophylaxis. However, the effectiveness of the current chemotherapy has been seriously reduced due to the resistance shown by the parasite to the drugs. Thus, the WHO recommended the use of artemisinin-based combination therapies (ACTs) as a current first-line treatment to prevent the emergence of antimalarial drug resistance.1,2 However later, ACTs tended to show poor treatment compliance, which contributed to the emergence of drug resistance.3 Thus, there is a need for improvement in the therapeutic potential of the existing antimalarial drugs by using them in combination and developing a suitable drug delivery system.Artesunate is a hemisuccinate derivative of artemisinin, which is recommended for the treatment of severe malaria in many countries by WHO and it is effective against the chloroquine-resistant strains of P. falciparum that can cause malaria. However, the major drawbacks of artesunate include poor water solubility, low bioavailability and a short half-life, which make it difficult to deliver it effectively to the intracellular space, leading to its poor efficacy and thereby significantly restricting its clinical applications.4,5 Since ancient times, phytomedicines have played a vital role in the treatment of numerous diseases including malaria. These plant-derived products along with synthetic antimalarial drugs have previously been demonstrated to show potent combination therapies to fight malaria as they can benefit from the synergistic effects of artemisinin derivatives. Among such plant-based antimalarial compounds, flavonoids (polyphenols) have been found to be active against malarial parasites and they are abundantly present in medicinal and dietary food plants.6 In regard to the above-mentioned information, we developed the concept of a dual-drug-loaded self-nanoemulsifying drug delivery system to treat malaria infection.7,8
To obtain the pharmacokinetic profile of the proposed hypotheses, it is essential to develop a sensitive bioanalytical method suitable for the simultaneous determination of ART–QRT in biological samples. The quantification of ART and DHA is difficult due to the absence of chromophore groups in their structures (Fig. 1) and the required lengthy derivatization technique.9,10 From a literature survey, it was found that various LC-MS/MS methods have been reported for the determination of ART individually or along with its metabolite dihydroartemisinin (DHA) from biological samples.11–14 Similarly, the LC-MS/MS methods have been reported for the characterization or quantification of QRT in biological samples or in a variety of plant extracts.15,16 However, these methods are not suitable for the simultaneous quantitation of ART, DHA, QRT and IS in the biological matrix. The reported methods suggest their individual advantages of a short runtime and improved method sensitivity and sample treatment. Moreover, it is essential to have one method that is economical and has significant advantages over a group of methods for the estimation of selected drugs. Hence, an ultrafast high-performance liquid chromatography-tandem mass spectrometry (UFLC-MS/MS) method was developed for the simultaneous analysis of ART, DHA and QRT in rat plasma.17,18
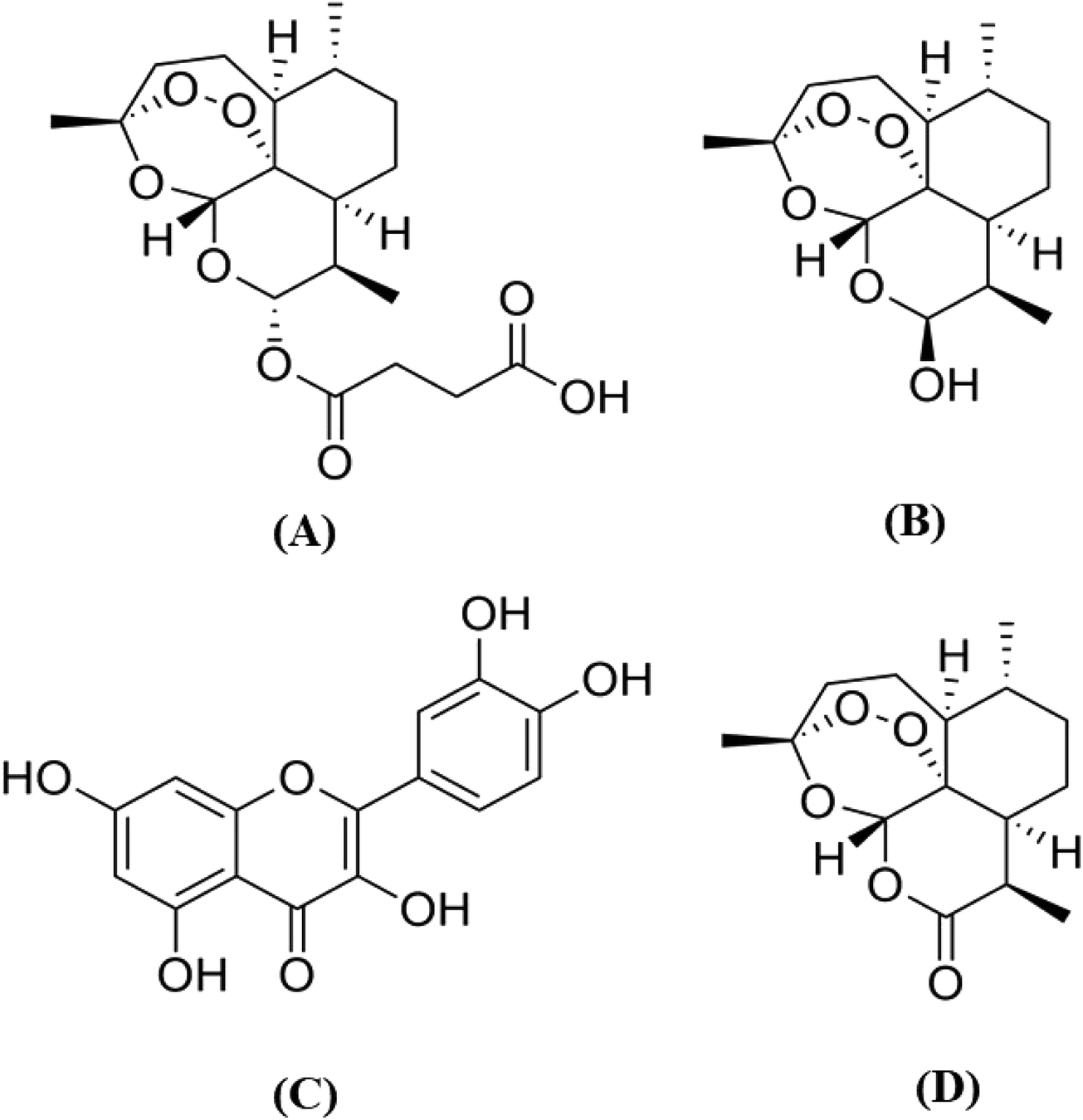 | ||
| Fig. 1 Chemical structures of (A) artesunate, (B) dihydroartemisinin, (C) quercetin and (D) artemisinin (IS). | ||
In this paper, we reported the development of a simple and rapid method for the quantification of ART, dihydroartemisinin (DHA) and the active metabolites of ART and QRT simultaneously from rat plasma with artemisinin (ATM) as an internal standard (IS). The solid-phase extraction procedure was employed followed by triple quadrupole mass spectrometry (UFLC-MS/MS) analysis, which is accurate and sensitive with a wide detection range and low detection limit.19 The established analytical method can be successfully applied for analysing the pharmacokinetics of ART–QRT in rat plasma following oral administration and then, it can be used for the routine analysis of ART–QRT in a lipid-based formulation.
2. Experimental
2.1 Chemicals and reagents
Artesunate (a synthetic derivative of artemisinin; 99.35%), dihydroartemisinin (the active metabolite of artesunate, 99.86%) and artemisinin (internal standard, 98.92%) were obtained from IPCA Laboratories Ltd., (Mumbai, India). Quercetin (98.75%) was purchased from Sigma-Aldrich (St. Louis USA). LC-MS grade methanol and formic acid were purchased from SD Fine Chemicals (Mumbai, India). Milli-Q RO system (Millipore, India) was employed to obtain ultrapure water.2.2 Preparation of stock solutions
Precise amounts of ART, QRT, DHA and IS were dissolved in methanol to achieve a concentration of 1 μg mL−1 for the stock solution. Subsequently, a series of standard working solutions were prepared by diluting the stock solution with methanol and ultrapure millipore water (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) to get the concentrations of 1–1000 ng mL−1. A 100 ng mL−1 solution of IS was prepared in methanol and ultrapure millipore water (50:50).
:50) to get the concentrations of 1–1000 ng mL−1. A 100 ng mL−1 solution of IS was prepared in methanol and ultrapure millipore water (50:50).
2.3 Preparation of calibration curves and quality control samples
The calibration curves were constructed over the concentration range of 1–1000 ng mL−1 for ART, QRT and DHA along with the internal standard (ATM 100 ng mL−1) using blank rat plasma. Four quality control samples of high (500 ng mL−1), medium (100 ng mL−1), low (5 ng mL−1) and lower limits (1 ng mL−1) were prepared by spiking the standard working solutions of ART, QRT and DHA into Wistar rat plasma (blank) using ATM (100 ng mL−1) as an internal standard. The solutions were labelled and stored at −70 ± 2 °C until analysis.202.4 Extraction procedure of ART, DHA, QRT and ATM (IS) from rat plasma
A solid-phase extraction (SPE) method was employed for the extraction of samples from rat plasma samples. SAM PREP SPE C18 (3 mL, 100 mg) cartridges were procured from Ranbaxy Fine Chemicals Ltd., India; they were pre-treated with methanol (1 mL), followed by ultrapure water (1 mL). Drug-free rat plasma (0.5 mL) was spiked with 0.5 mL of the drug mixture solution along with IS (200 ng mL−1) and vortexed (1 min) before transferring to the extraction column. Furthermore, the cartridge was washed with 0.9 mL of ultrapure water, and the sample was eluted with 100 μL of methanol. The eluent obtained (10 μL) was then injected into the UFLC-MS/MS system.212.5 Chromatographic conditions
The analysis of ART, QRT, DHA and ATM was carried out by using UFLC-MS/MS (Shimadzu 8030, Tokyo) equipped with an LC-20AD pump, CTO-20AC column oven, CBM-20 controller, SPD-M20 PDA detector, electrospray ionization (ESI) interface, and SIL-20AC autosampler.The separation of analytes was achieved on a Zorbax C18 (50 mm × 4.6 mm; 5 μm) stationary phase at room temperature. The separation was carried out using 0.1% formic acid and methanol in the ratio of 10:90 v/v as the mobile phase with a 0.4 mL min−1 flow rate using the internal standard (IS) as ATM (100 ng mL−1). The analysis was carried out using 10 μL as an injection volume.
UFLC-MS/MS was operated in dual mode (positive and negative ions) with the ESI interface. The quantification of analytes was performed using multiple reaction monitoring (MRM) mode. In the positive and negative modes, the mass spectrometer parameters were as follows: block temperature and desolvation temperature maintained at 350 °C and 250 °C, respectively, with a detector voltage of 1.3 kV and CID gas at 230 kPa. Nitrogen was employed as the carrier gas (15 L min−1) and drying gas (3 L min−1). The MRM transitions of ART, DHA, QRT and ATM were found to be 407.2 → 261.0, 307.1 → 261.0, 301.1 → 151.0 and 283.2 → 265.05, respectively. The collision energies used were −7, −12, 22 and −15 eV for ART, DHA, QRT and ATM, respectively.10,22
2.6 Pharmacokinetic study
The pharmacokinetic studies were carried out using Wistar rats (either sex) weighing 180–250 g. The study protocol was approved by the IAEC (Institutional Animal Ethical Committee-CEUTICS/01/2017-2018). Food and water were provided ad libitum to the animals and before the experiment started, they were made to fast for 12 h. Twelve Wistar rats were assigned randomly into two groups containing six animals each. Group I received an ART aqueous suspension and Group II received an ART–QRT aqueous suspension orally (dose equivalent of 5 mg kg−1 each). Around 0.5 mL of blood aliquots samples were collected via the retro-orbital sinus at different time intervals for a pre-dose sample at 0 h followed by post-dose samples (0.25, 0.5, 1.0, 2.0, 4.0, 6.0, 8.0 and 12 h). The blood samples were collected in a heparinized centrifuge tube followed by centrifugation (4000 rpm for 15 min); isolated rat plasma samples were stored at −70 ± 2 °C until analysis.24,25The pharmacokinetic (PK) assessments were calculated by non-compartmental analysis (NCA) with the help of pK solver excel add-on. The PK parameters such as area under the plasma concentration–time curve from 0 to 12 h (AUC 0–12 h), maximum plasma concentration (Cmax), elimination half-life (T1/2), the time to reach Cmax (Tmax), elimination rate constant (Kel), area under the curve from 0 h extrapolated to infinity (AUC 0–∞), the apparent volume of distribution (Vz/F) and apparent total plasma clearance (CL/F) were determined.26
2.7 Statistical analysis
The data obtained were expressed as the mean ± standard deviation (SD) and subjected to statistical analysis using GraphPad prism software (version 6.01; GraphPad Software, Inc.) by one-way analysis of variance (ANOVA). The differences were considered to be significant at p < 0.05.3. Results and discussion
3.1 Mass spectrometric detection
The optimization of the ionization modes for ART, DHA, QRT and IS was carried out by dissolving the selected drugs in methanol, followed by injection into UFLC-MS/MS. The analytes ART, DHA, QRT and ATM (IS) were ionised by the ESI technique with the mobile phase; ART and DHA yielded [M + Na]+ ions, whereas QRT and IS yielded [M – H]− and [M + H]+ ions, respectively, along with their most stable fragment ions. In accordance to the published articles, it was observed that sodium ions are more stable and provide strong interactions with the oxygen atoms in the molecules.27 Therefore, the mass transitions selected for quantitation were m/z 407.2 → 261.10, 307.1 → 261.0, 305.2 →151.0, and 283.2 → 265.05 for ART, DHA, QRT and IS, respectively (Fig. 2).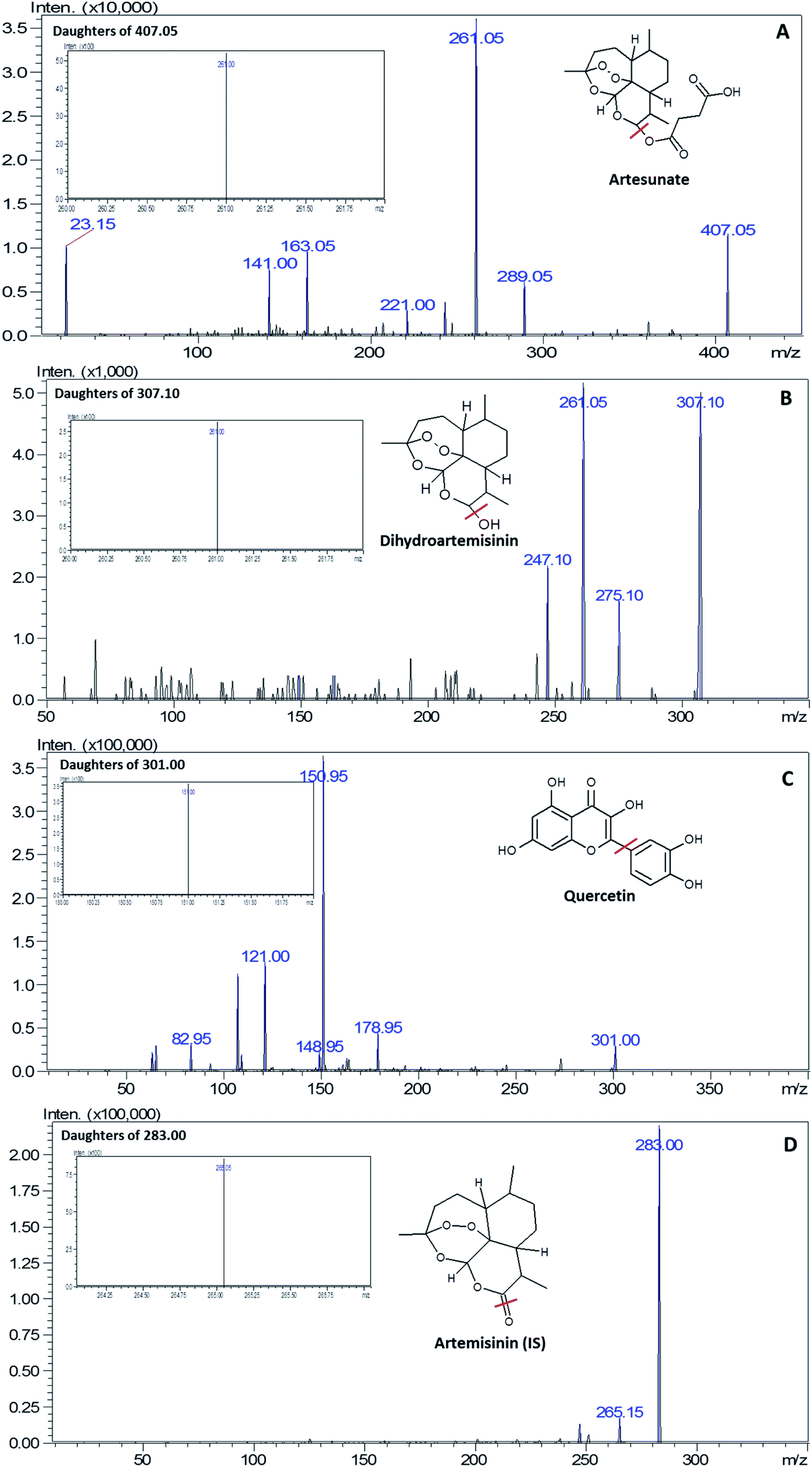 | ||
| Fig. 2 Product ion scan and MRM (image shown inside the box) spectra of (A) artesunate, (B) dihydroartemisinin, (C) quercetin and (D) artemisinin (IS). | ||
3.2 Chromatographic separation
The chromatographic separation and ionization of ART, DHA, QRT and IS were achieved by a Zorbax C18 (50 mm × 4.6 mm; 5 μm) column at a flow rate of 0.4 mL min−1. The mobile phase consisted of methanol and 0.1% formic acid at the ratio of 90:10 v/v. The method sensitivity increased with the increase in formic acid by producing good ionization of all the analytes. Based on the optimized LC and MS conditions, ART, DHA, QRT and IS were separated at the retention times of 1.709, 1.747, 1.533 and 1.739 min, respectively, with the total run time of 4 min. The target analytes were unaffected by the endogenous substances from the rat plasma and were free from interferences, as shown in Fig. 3.
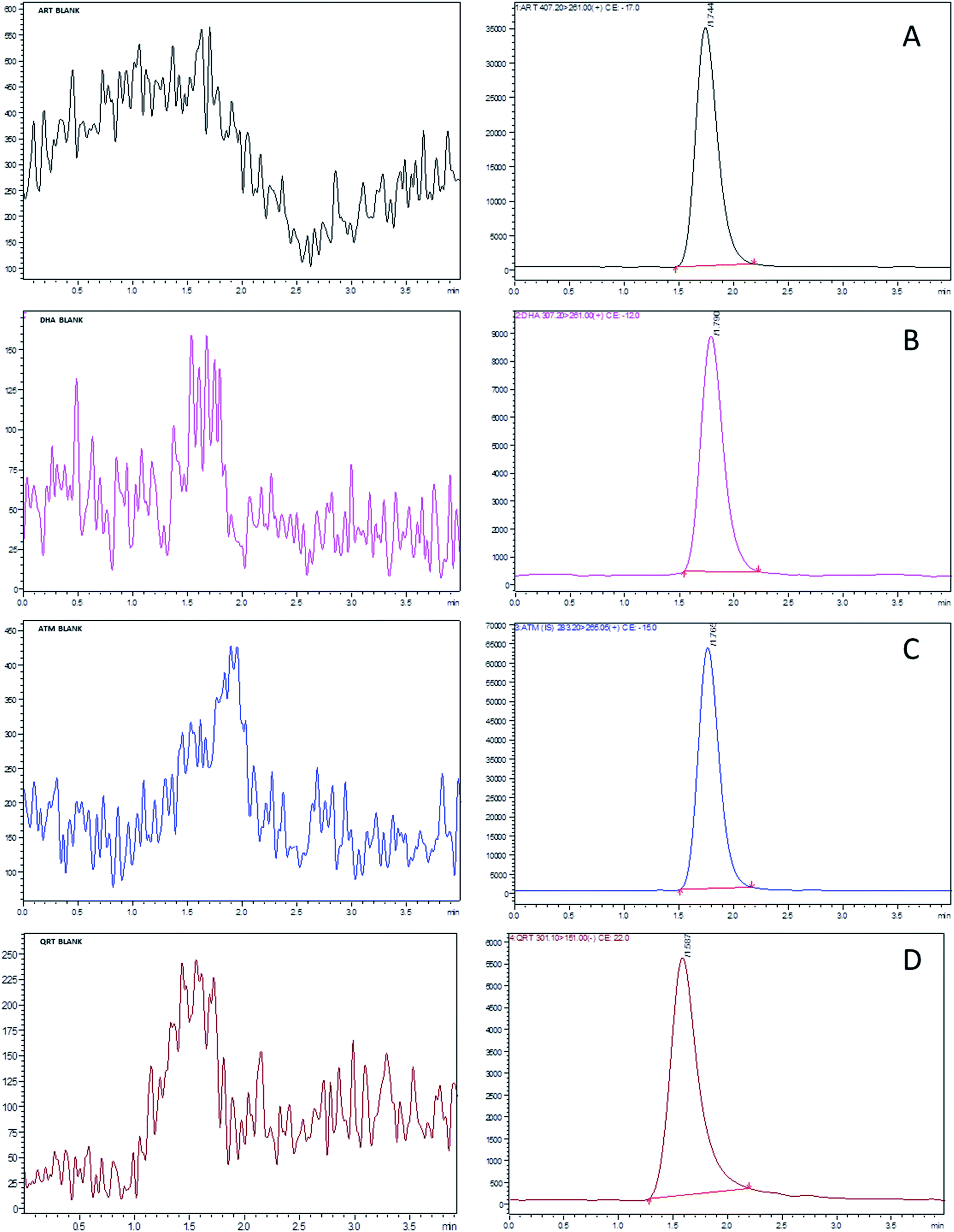 | ||
| Fig. 3 Standard UFLC-MS/MS chromatograms of (A) artesunate, (B) dihydroartemisinin, (C) quercetin and (D) artemisinin (IS) at a concentration of LLOQ (1 ng mL−1) and blank (images on left). | ||
During the process of validation, the protein precipitation technique (PPT) and liquid–liquid extraction (LLE) and solid phase extraction (SPE) methods were applied to determine the limit of detection (LOD) for ART, DHA and QRT. The recovery of the selected drugs from plasma samples was less than 50% in PPT. The sample extraction procedure using SPE cartridges was simple and less laborious when compared to LLE. The LOD values of ART, DHA and QRT were 0.5, 0.7 and 1.0, respectively, by solid-phase extraction. The extraction recoveries of ART, DHA and QRT from plasma were found to be 91.05–99.62%, 95.12–98.56% and 89.35–98.90%, respectively. Hence, further validation of the method was carried out using the SPE method.
3.3 Method validation
The chromatographic peaks obtained were symmetric and showed no interferences from endogenous substances, thus indicating the specificity of the method. The accuracy of the method was determined by the percentage recovery of the analytes in the samples. The comparison of the mean peak area response obtained from the extracted rat plasma samples with the peak area response of the standard (spiked standard solutions) was used to calculate recovery. Four QC levels were used to determine the precision and accuracy of the experiments for the rat plasma samples and the results obtained were within the acceptable range, indicating that the developed method is precise and accurate for the analysis of analytes in the samples (n = 6). The validation results are presented in Table 1.| Biological matrix | Analyte | QCs (ng mL−1) | Mean concentration found (ng mL−1) ± SD | Recovery (%) | Absolute matrix effect | Intraday | Inter-day | ||
|---|---|---|---|---|---|---|---|---|---|
| Accuracy (% nominal) | Precision (% RSD) | Accuracy (% nominal) | Precision (% RSD) | ||||||
| Rat plasma | ART | 1 | 0.91 ± 0.07 | 91.05 | 0.91 | 89.30 | 7.69 | 89.00 | 8.10 |
| 5 | 4.61 ± 0.11 | 92.20 | 0.95 | 93.54 | 2.38 | 91.77 | 5.08 | ||
| 100 | 95.87 ± 3.54 | 95.87 | 0.96 | 96.07 | 3.69 | 93.14 | 4.35 | ||
| 500 | 498.10 ± 6.03 | 99.62 | 1.13 | 97.92 | 1.21 | 98.76 | 1.62 | ||
| DHA | 1 | 0.95 ± 0.09 | 95.12 | 0.96 | 93.10 | 9.47 | 92.00 | 10.89 | |
| 5 | 4.77 ± 0.14 | 95.40 | 0.91 | 94.08 | 2.93 | 93.98 | 6.88 | ||
| 100 | 98.70 ± 3.08 | 98.70 | 0.99 | 96.30 | 3.12 | 97.05 | 4.01 | ||
| 500 | 492.80 ± 7.83 | 98.56 | 1.09 | 99.06 | 1.59 | 97.67 | 5.82 | ||
| QRT | 1 | 0.89 ± 0.03 | 89.35 | 0.90 | 87.10 | 3.37 | 87.80 | 5.07 | |
| 5 | 4.55 ± 0.09 | 91.00 | 0.96 | 90.77 | 1.97 | 90.01 | 3.95 | ||
| 100 | 98.90 ± 9.01 | 94.29 | 0.95 | 97.08 | 1.11 | 98.01 | 2.11 | ||
| 500 | 494.50 ± 5.12 | 98.90 | 1.15 | 98.10 | 1.03 | 97.30 | 2.04 | ||
The analytes were spiked into the rat plasma and the samples were extracted using the SPE method to determine the calibration curves. The linearity for ART, DHA and QRT was carried over the concentration range between 1 and 1000 ng mL−1. The linearity of the analytes was calculated by plotting a graph response factor vs. the concentration of the standard solution. The regression analysis results showed that the correlation coefficient was >0.999 for the analytes in the plasma. The LLOQ (lowest limit of quantitation) value for ART, DHA and QRT in the samples was 1 ng mL−1, with acceptable accuracy and precision (Table 2). Four different concentration levels (n = 6) of QCs were used to conduct the stability tests of QCs at different storage conditions, such as −70 ± 2 °C (freeze–thaw, 3 cycles), 25 °C (short term for 6 h), −70 ± 2 °C (long term for 30 days) and 25 °C (stock solution stability for 6 h). For freeze–thaw stability, the rat plasma samples containing the analytes were frozen at −70 °C for 24 h and thawed. After completion, the rat plasma samples with analytes were re-frozen for at least 24 h under similar conditions, and the process was repeated at least three times; after the completion of the third cycle, the samples were injected into the UFLC-MS/MS system for analysis and compared with freshly prepared QCs in the rat plasma. A similar procedure was followed for the stock solution and the short-term stability study except that the rat plasma samples were stored at 25 °C for 6 h. Long-term stability was evaluated by analysing the stored rat plasma samples. The plasma samples with the analytes were considered stable if the nominal values were within the standard limits, i.e., ±20%. The obtained results indicated that all the analytes (ART, DHA, QRT and ART) were stable under all the test conditions (Table 3).
| Analyte | Internal standard (100 ng mL−1) | Equation for plasma sample | Linear range (ng mL−1) | Correlation coefficient (R2) | LOD (ng mL−1) | LLOQ (ng mL−1) |
|---|---|---|---|---|---|---|
| ART | ATM | y = 0.0183x + 0.148 | 1–1000 | 0.9923 | 0.5 | 1.0 |
| DHA | y = 0.0042x + 0.0702 | 0.9948 | 0.7 | 1.0 | ||
| QRT | y = 0.0016x − 0.0104 | 0.9992 | 1.0 | 1.0 |
| Stability test | QCs (ng mL−1) | ART | DHA | QRT |
|---|---|---|---|---|
| Mean ± SD (ng mL−1); accuracy (% nominal); precision (% RSD) | Mean ± SD (ng mL−1); accuracy (% nominal); precision (% RSD) | Mean ± SD (ng mL−1); accuracy (% nominal); precision (% RSD) | ||
| Freeze–thaw (3 cycles at −70 ± 2 °C) | 1 | 0.93 ± 0.15; 93.00; 16.12 | 0.94 ± 0.11; 94.00; 11.70 | 0.87 ± 0.13; 87.00; 14.94 |
| 5 | 4.72 ± 0.12; 94.40; 2.52 | 4.80 ± 0.21; 96.00; 4.37 | 4.53 ± 0.33; 90.60; 7.28 | |
| 100 | 95.30 ± 3.66; 95.30; 3.84 | 97.85 ± 6.07; 97.85; 6.20 | 98.00 ± 8.12; 98.00; 8.28 | |
| 500 | 494.70 ± 8.67; 98.94; 1.75 | 493.20 ± 5.40; 98.64; 1.09 | 492.00 ± 7.17; 98.40; 1.45 | |
| Short term (25 °C for 6 h) | 1 | 0.96 ± 0.14; 96.00; 14.58 | 0.97 ± 0.08; 97.00; 8.24 | 0.97 ± 0.09; 97.50; 9.23 |
| 5 | 4.81 ± 0.11; 96.20; 2.28 | 4.85 ± 0.15; 97.00; 3.09 | 4.80 ± 0.19; 96.00; 3.95 | |
| 100 | 97.75 ± 4.10; 97.75; 4.19 | 98.00 ± 5.5; 98.00; 5.61 | 98.75 ± 6.9; 98.75; 6.98 | |
| 500 | 496.00 ± 9.22; 99.20; 1.85 | 496.90 ± 10.8; 99.38; 2.17 | 497.10 ± 11.30; 99.42; 2.27 | |
| Stock solution (25 °C for 6 h) | 1 | 0.98 ± 0.01; 98.00; 1.02 | 0.98 ± 0.05; 98.70; 5.06 | 0.97 ± 0.02; 97.90; 2.04 |
| 5 | 4.82 ± 0.09; 96.40; 1.86 | 4.88 ± 0.15; 97.60; 3.07 | 4.90 ± 0.11; 98.00; 2.24 | |
| 100 | 97.87 ± 3.17; 97.87; 3.23 | 98.03 ± 4.21; 98.03; 4.29 | 98.25 ± 3.82; 98.25; 3.88 | |
| 500 | 496.20 ± 8.02; 99.24; 1.61 | 495.90 ± 9.89; 99.18; 1.99 | 496.30 ± 7.11; 99.26; 1.43 |
3.4 Pharmacokinetic study
The method developed was validated and applied successfully for the quantitative analysis of ART, DHA and QRT in plasma samples after the oral administration of the ART aqueous suspension and the combination of the ART–QRT aqueous suspension to rats at the dose of 5 mg kg−1 each. The mean plasma concentration–time profiles of ART, DHA, and QRT after a single oral administration of free ART (5 mg kg−1) or a combination of free ART and QRT (1:1 w/w; 5 mg kg−1 ART, 5 mg kg−1 QRT) are shown in Fig. 4 and Table 4. After the oral administration of the ART solution, the Cmax values of ART and DHA were found to be 29.6 ± 6.2 ng mL−1 and 62.72 ± 13.20 ng mL−1, respectively. However, when ART was given in combination with QRT, the Cmax values of ART and DHA were 52.2 ± 12.2 ng mL−1 and 112.13 ± 22.60 ng mL−1, respectively. Hence the results demonstrated the enhanced bioavailability after the oral administration of ART when combined with QRT. The reason can be that quercetin has been proven to modulate the P-gp efflux pumps and/or CYP450 enzyme function in the intestine region, which can help improve the absorption of the drug.28 Hence ART–QRT can become an alternative combination therapy in the treatment of malaria.
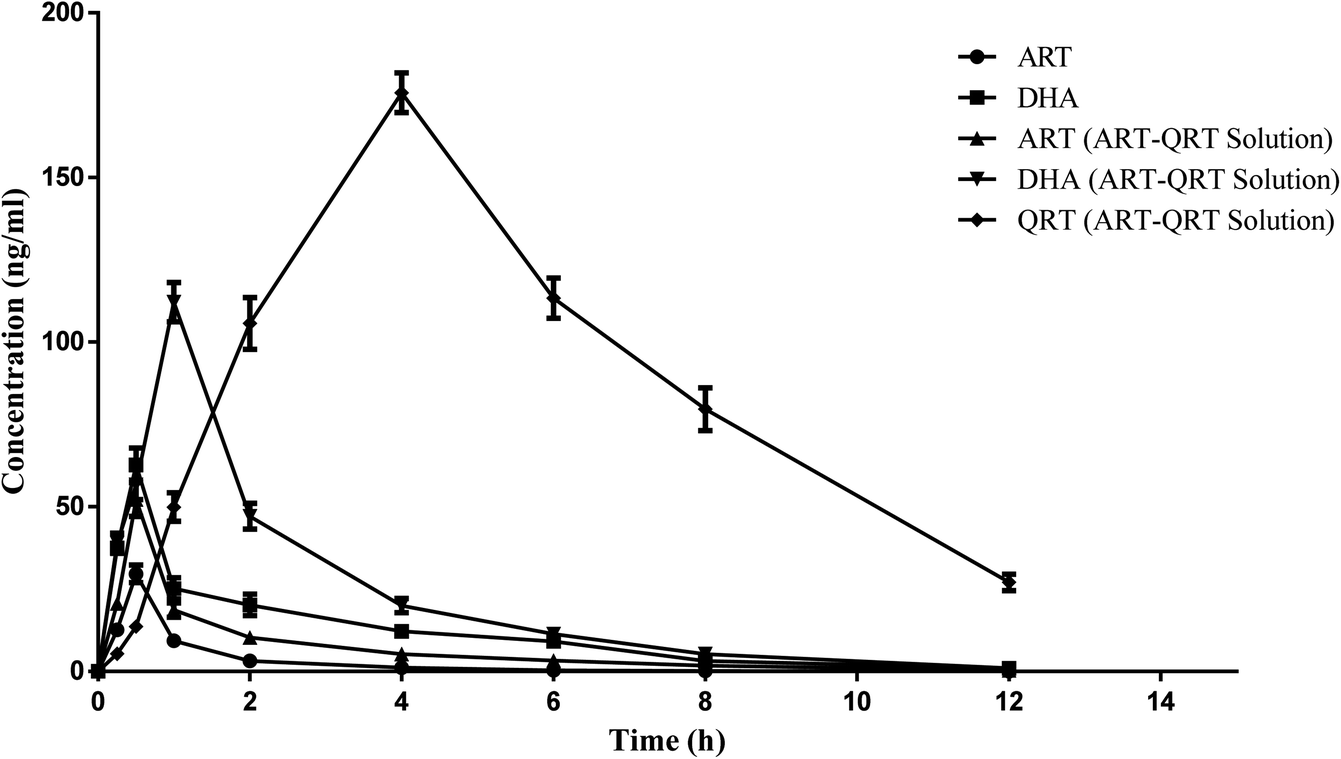 | ||
| Fig. 4 Mean plasma concentration–time profiles after the oral administration of artesunate solution, artesunate and artesunate–quercetin solution. | ||
| Parameters | ART solution | ART–QRT solution | |||
|---|---|---|---|---|---|
| ART | DHA | ART | DHA | QRT | |
| a The data represent mean ± SD, n = 3.b Significant difference at p < 0.05. | |||||
| Cmax (ng mL−1) | 29.60 ± 6.2 | 62.72 ± 13.20 | 52.20 ± 12.20b | 112.13 ± 22.60b | 175.73 ± 26.60 |
| T1/2 (h) | 0.85 ± 0.18 | 2.28 ± 0.41 | 1.75 ± 0.38b | 1.69 ± 0.24 | 2.97 ± 0.35 |
| Tmax (h) | 0.50 ± 0.12 | 0.50 ± 0.09 | 0.50 ± 0.13 | 1.00 ± 0.22b | 4.00 ± 0.90 |
| AUC (0–12 h) (ng h mL−1) | 29.02 ± 5.32 | 135.88 ± 26.32 | 77.16 ± 15.21b | 266.14 ± 54.19b | 1073.53 ± 151.65 |
| AUC (0–∞) (ng h mL−1) | 29.02 ± 5.02 | 138.91 ± 27.01 | 77.96 ± 14.85b | 268.53 ± 55.14b | 1189.44 ± 163.75 |
| Kel (h−1) | 0.81 ± 0.15 | 0.30 ± 0.08 | 0.39 ± 0.07 | 0.41 ± 0.09 | 0.23 ± 0.04 |
| MRT (h) | 1.29 ± 0.25 | 3.20 ± 0.78 | 2.56 ± 0.66 | 2.67 ± 0.76 | 6.36 ± 1.46 |
| Vz/F (ng mL−1) | 0.21 ± 0.02 | 0.11 ± 0.02 | 0.16 ± 0.04 | 0.04 ± 0.01 | 0.01 ± 0.005 |
| Cl/F (ng h mL−1) | 0.17 ± 0.04 | 0.03 ± 0.006 | 0.06 ± 0.01 | 0.01 ± 0.004 | 0.004 ± 0.001 |
4. Conclusion
A rapid and sensitive UFLC-MS/MS method was developed and validated for the simultaneous quantification of ART, DHA and QRT in rat plasma using the SPE technique. The method was rapid, sensitive, reliable and reproducible for routine analysis. The developed method could be successfully used to evaluate the pharmacokinetic parameters after the peroral administration of the ART–QRT solution to rats. The pharmacokinetic data of the ART–QRT solution showed better systemic absorption than the pure drug solution. The analysis required a small volume (500 μL) of plasma with a short analysis run time of 4.0 min, which is highly beneficial in a pharmacokinetic study. Therefore, the developed UFLC-MS/MS method can be an excellent technique for the further evaluation of pharmacokinetic properties and the therapeutic potential of the new nanopreparations of antimalarial drugs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author Ms Nethravathi Puttappa thanks Lady Tata Memorial Trust, Mumbai, India for providing Junior and Senior Research Fellowship (2017–2020) to carry out the research study. The authors would like to thank Department of Science and Technology- Fund for improvement of science and technology infrastructure in universities and higher educational institutions (DST-FIST), New Delhi for their infrastructure support to our department.References
- N. Puttappa, R. S. Kumar, G. Kuppusamy and A. Radhakrishnan, Acta Trop., 2019, 195, 103–114 CrossRef CAS PubMed.
- World Health Organization, World Malaria Report, 2018, avalaible at https://www.who.int/malaria/publications/world-malaria-report-2018/en/ Search PubMed.
- M. Imwong, K. Suwannasin, C. Kunasol, K. Sutawong and M. Mayxay, et al., Lancet Infect. Dis., 2017, 17, 491–497 CrossRef PubMed.
- E. E. Chinaeke, S. A. Chime, F. C. Kenechukwu and M. C. C. Goymann, et al., J. Drug Delivery Sci. Technol., 2014, 24, 69–77 CrossRef CAS.
- Q. Li and P. Weina, Pharmaceu, 2010, 3, 2322–2332 CrossRef CAS PubMed.
- D. R. Reddy, A. Khurana, S. Bale, R. Ravirala and V. S. Reddy, et al., SpringerPlus, 2016, 5, 1618 CrossRef PubMed.
- N. Puttappa, R. S. Kumar and K. Yamjala, Med. Hypotheses, 2017, 109, 176–180 CrossRef CAS PubMed.
- N. Puttappa, R. S. Kumar, B. Babu, K. Yamjala and S. T. Narenderan, J. Chromatogr. Sep. Tech., 2019, 10, 56 Search PubMed.
- B. Li, J. Zhang, X. Z. Zhou, J. Y. Li and Y. J. Yang, et al., J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 997, 146–153 CrossRef CAS PubMed.
- U. Duthaler, J. Keiser and J. Huwyler, J. Mass Spectrom., 2011, 46(2), 172–181 CrossRef CAS PubMed.
- S. L. Choon, N. K. Nair, S. M. Mansor, P. L. Olliaro and V. Navaratnam, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 857, 308–314 CrossRef PubMed.
- S. L. Choon, N. K. Nair, S. M. Mansor, P. L. Olliaro and V. Navaratnam, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 877, 558–562 Search PubMed.
- G. Yuanchao, L. Qigui, M. Victor and W. Peter, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 867, 213–218 CrossRef PubMed.
- B. Sofia, E. Therese, B. Antje, V. H. Cornelia and A. Michael, et al., Bioanalysis, 2014, 6(18), 2357–2369 CrossRef PubMed.
- C. D. Venkatapura, G. K. Burdipad and L. V. Gollapalle, Chin. J. Integr. Med., 2011, 9, 1022–1030 CrossRef PubMed.
- C. Yeqing, Y. Hongli, W. Hao, P. Yaozong and W. Kuilong, et al., Molecules, 2015, 20, 18352–18366 CrossRef PubMed.
- W. Hanpithakpong, B. Kamanikom, A. M. Dondorp, P. Singhasivanon and N. J. White, et al., J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 876(1), 61–68 CrossRef CAS PubMed.
- S. Sabarinath, M. Rajanikanth, K. P. Madhusudanan and R. C. Gupta, J. Mass Spectrom., 2003, 38, 732–742 CrossRef CAS PubMed.
- M. C. K. Geditz, G. Heinkele, A. hmed, P. G. Kremsner and R. Kerb, et al., Anal. Bioanal. Chem., 2014, 406(17), 4299–4308 CrossRef CAS PubMed.
- I. P. Teja, D. Siriyanonda, R. Siripokasupkul, R. Apinan and N. Chanarat, et al., Molcules, 2010, 15, 8747–8768 CrossRef.
- A. Dubala, J. S. Nagarajan, C. S. Vimal and R. George, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2013, 921, 49–55 CrossRef PubMed.
- V. D. Kumar, P. R. P. Verma, S. K. Singh and S. Viswanathan, Biomed. Chromatogr., 2015, 29, 1731–1736 CrossRef PubMed.
- US Food and Drug Administration, Center for Drug Evaluation and Research, 2001, accessed 23 May 2019 available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatatoryinformation/Guidances/UCM070107pdf Search PubMed.
- T. H. Tran, Y. Guo, D. Song, R. S. Bruno and X. Lu, J. Pharm. Sci., 2014, 103, 840–852 CrossRef CAS PubMed.
- P. Dwivedi, R. Khatik, K. Khandelwal, I. Taneja and K. S. Raju, et al., Int. J. Pharm., 2014, 466, 321–327 CrossRef CAS PubMed.
- N. S. Ambhore, K. Yamjala, S. Mohire, K. R. Raju and S. Mulukutla, et al., J. Pharm. Biomed. Anal., 2016, 120, 57–64 CrossRef CAS.
- S. Grimalt, O. J. Pozo, J. M. Marin, J. V. Sancho and F. Hernandez, et al., Mass Spectrom., 2005, 16, 1619 CrossRef CAS PubMed.
- J. S. Choi, Y. J. Piao and K. W. Kang, Arch. Pharmacal Res., 2011, 34, 607–611 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |