
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, cytotoxicity and anti-inflammatory activity of rhamnose-containing ursolic and betulinic acid saponins†
Balla Syllaa,
Serge Lavoieb,
Jean Legaulta,
Charles Gauthier *ac and
André Pichette*a
*ac and
André Pichette*a
aCentre de Recherche sur La Boréalie (CREB), Chaire de Recherche sur Les Agents Anticancéreux D'origine Naturelle, Laboratoire LASEVE, Département des Sciences Fondamentales, Université du Québec à Chicoutimi, 555, Boul. de L’Université, Chicoutimi, Québec, Canada G7H 2B1. E-mail: andre.pichette@uqac.ca
bInstitut des Sciences de la Forêt Tempérée, Université du Québec en Outaouais, 58, Rue Principale, Ripon, Québec, Canada J0V 1V0
cCentre Armand-Frappier Santé Biotechnologie, Institut National de La Recherche Scientifique (INRS), 531, Boul. des Prairies, Laval, Québec, Canada H7V 1B7. E-mail: charles.gauthier@iaf.inrs.ca
First published on 2nd December 2019
Abstract
Betulinic acid and ursolic acid are ubiquitous, naturally-occurring triterpenoids exhibiting various pharmacological activities including cytotoxic and anti-inflammatory activities. However, these triterpenoids display unfavorable pharmacokinetic properties as well as low aqueous solubility. It has been shown that the presence of α-L-rhamnose moieties positively modulates the anticancer activity of secondary metabolites. Herein we report the synthesis and in vitro evaluation of cytotoxic and anti-inflammatory activities of a series of rhamnose-containing ursolic and betulinic acid saponins. Relying on Schmidt's normal and inverse procedures, monorhamnosides, (1→4)-linked dirhamnosides as well as branched trirhamnosides and tetrarhamnosides were synthesized in high yields with full control of stereoselectivity. A betulinic acid saponin bearing a 3-O-α-L-rhamnopyranosyl-(1→4)-α-L-rhamnopyranosyl residue was found to be a potent cytotoxic agent against human colorectal adenocarcinoma cells without damaging the healthy cells (selectivity ratio > 20) whereas rhamnose-containing ursolic acid saponins potently inhibited NO overproduction induced by LPS-stimulated macrophages. Our results reveal that rhamnose-containing ursolic and betulinic acid saponins represent promising therapeutic agents.
Introduction
Betulinic acid (BA, 1) is a natural product-derived compound, and member of the lupane-type triterpenoid family. BA possesses multiple pharmacological activities including cytotoxic, anticancer, anti-inflammatory, antimalarial, and anti-HIV activities.1 Because of its low in vivo toxicity, ubiquitous natural occurrence in the plant kingdom and broad spectrum of pharmacological activities, BA and its derivatives have been thoroughly investigated in the past few years.2–4 Ursolic acid (UA, 2) is another naturally occurring member of the triterpenoid family. UA is found in various plants, fruits, and medicinal herbs.5 UA has been shown to possess a vast spectra of pharmacological activities including anti-metastatic, anti-angiogenic, antioxidant, anti-inflammatory, and antimicrobial activities.6Although BA and UA are promising natural products from a medicinal point of view, their biopharmaceutical development has been hampered because of their low bioavailability and aqueous solubility.2,5 In order to modulate the pharmacokinetic properties and increase their absorption by the organism, polar substituents have been added on the triterpenoid core by taking advantage of the presence of C-3 hydroxyl and C-28 carboxylic acid groups. For instance, C-28 amino acid,7 C-3 phthalate,8 and C-3 carbamate9 as well as ionic derivatives10 of BA have been synthesized and showed improved aqueous solubility and cytotoxic activities compared to BA. Recently, Baran and co-workers have developed a late-stage diversification approach for improving the pharmacokinetic properties of BA via C–H oxidation through a combination of chemical and enzymatic reactions.11
In parallel to these pioneering studies, we1,12–18 and others19–23 have been interested in coupling diverse hydrosoluble sugar moieties at the C-3 and/or C-28 positions of BA and other members of the lupane-type triterpenoid family such as betulin and lupeol. Among other things, we have shown that synthetic BA saponins bearing L-rhamnopyranose (Rha) residues at the C-3 position were potent cytotoxic agents15 devoid of hemolytic activity12 as compared to oleanane-type saponins. BA 3-O-α-L-rhamnopyranoside (3) exhibited higher cytotoxic activities against lung carcinoma (A549) and colorectal adenocarcinoma (DLD-1) than BA itself while being less active against human normal skin fibroblasts (WS1).15 Furthermore, the anticancer activity of a bidesmosidic betulin saponin bearing Rha residues14 was demonstrated in vivo against LLC1 tumor-bearing mice.24 This compound was shown to induce apoptosis in cancer cells via disturbance of mitochondrial electron transfer chain, reduced reactive oxygen species, and decreased membrane potential.24
Although not fully understood, there seems to be a correlation between the presence of Rha residues and the anticancer properties of Rha-containing secondary metabolites. The intraperitoneal administration of Rha was shown to suppress cancer growth in mice.25 Rhamnospicamycin, a Rha-containing analogue of the natural product spicamycin, was shown to be a potent cytotoxic agent against human myeloma cell lines with an IC50 of 120 nM.26 Rha-containing bufadienolides and cardenolides such as gamabufotalin rhamnoside and ouabain, respectively, are potent anticancer agents.27 O'Doherty and co-workers reported the synthesis of Rha-containing digitoxin derivatives that showed excellent selectivity and activity against a panel of 60 human cancer cell lines.28 Structure–activity relationship study highlighted that the α-configuration of the glycosidic linkage as well as the presence of the L-enantiomer was a prerequisite to the anticancer activity of digitoxin glycosides.29 Interestingly, Lou and co-workers have hypothesized that the presence of a Rha-binding lectins on human cells could play an important role in the anticancer activity of Rha-containing solasodine saponins.30
Within this framework, we report here the synthesis of a series of rhamnose-containing (mono, di, tri, and tetra) BA and UA saponins (compounds 3–10, Fig. 1). By designing these compounds, our objective was to significantly improve the aqueous solubility of BA and UA while preserving (or enhancing) their cytotoxic and anti-inflammatory activities through the multiple presentation of rhamnose units on the triterpenoid scaffold. The synthetic saponins were prepared from corresponding naturally occurring BA and UA, which are commercially available at low prices. The saponins were synthesized following a regioselective approach aiming at minimizing the number of steps and protecting groups throughout the synthetic sequence. The Rha-containing saponins were evaluated for their cytotoxic and anti-inflammatory activities and some interesting selectivities were observed.
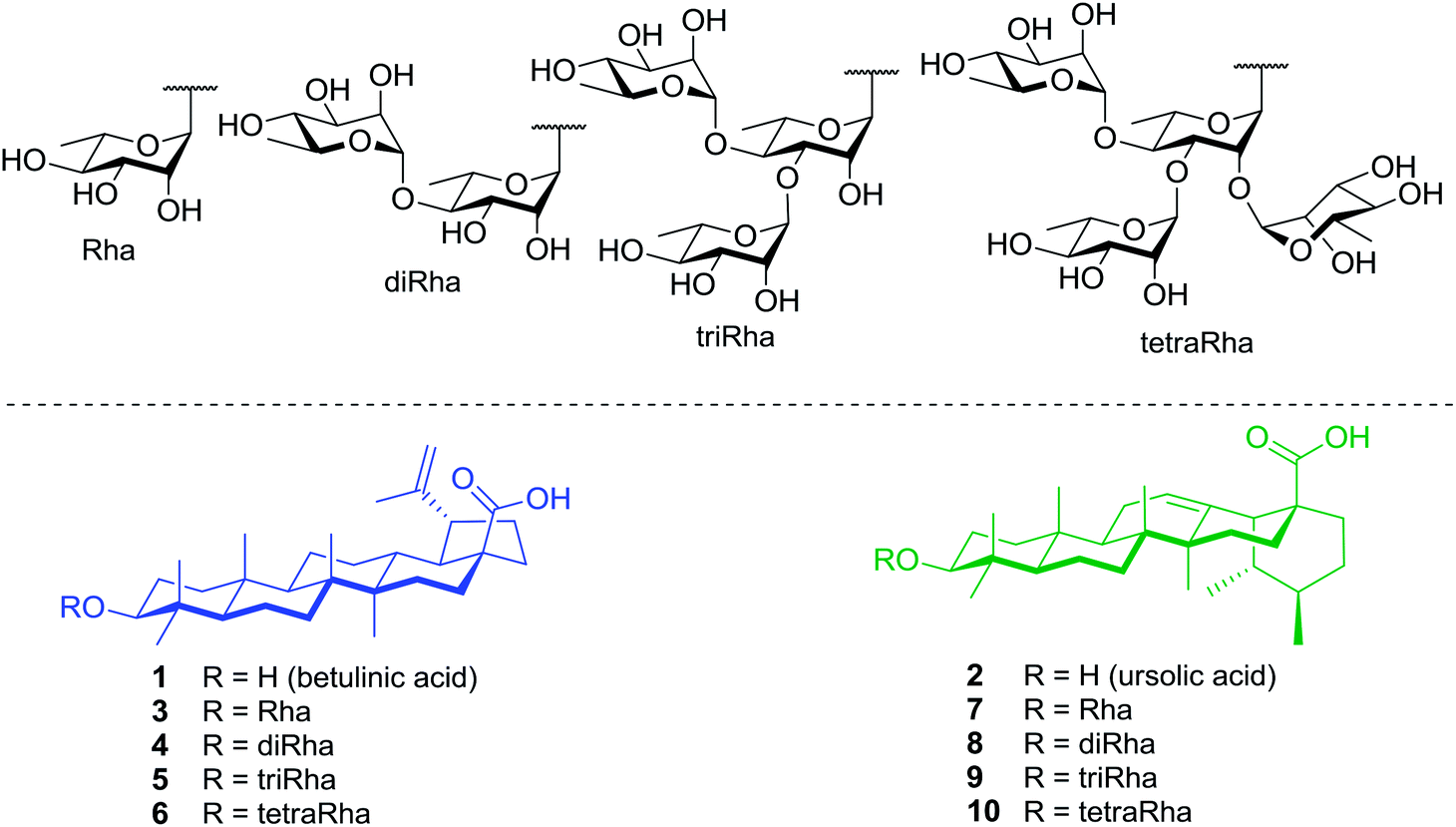 | ||
| Fig. 1 Structures of target rhamnose-containing UA and BA saponins. | ||
Results and discussion
Synthesis of BA saponins 3–6
Our work started with the synthesis of mono- and dirhamnoside-containing BA saponins (Scheme 1). Benzyl betulinate (11)31 was reacted with Schmidt 2,3,4-tri-O-benzoyl-α-L-rhamnopyranosyl trichloroacetimidate donor (12)14 under the promotion of trimethylsilyl trifluoromethanesulfonate (TMSOTf) in anhydrous DCM providing rhamnoside 13 in 76% yield. Relying on the neighbouring participation of the C-2 benzoyl group, full control of stereoselectivity (α-L-rhamnopyranoside) was obtained, as expected. Then, benzoyl groups were removed through Zemplén conditions to give triol 14. Debenzylation of the latter in the presence of catalytic amounts of 10% Pd/C in refluxing EtOAc led to the previously reported BA 3-O-α-L-rhamnopyranoside 3 with an improved overall yield (62% over three steps from derivative 11) without reducing the terminal alkene.15,32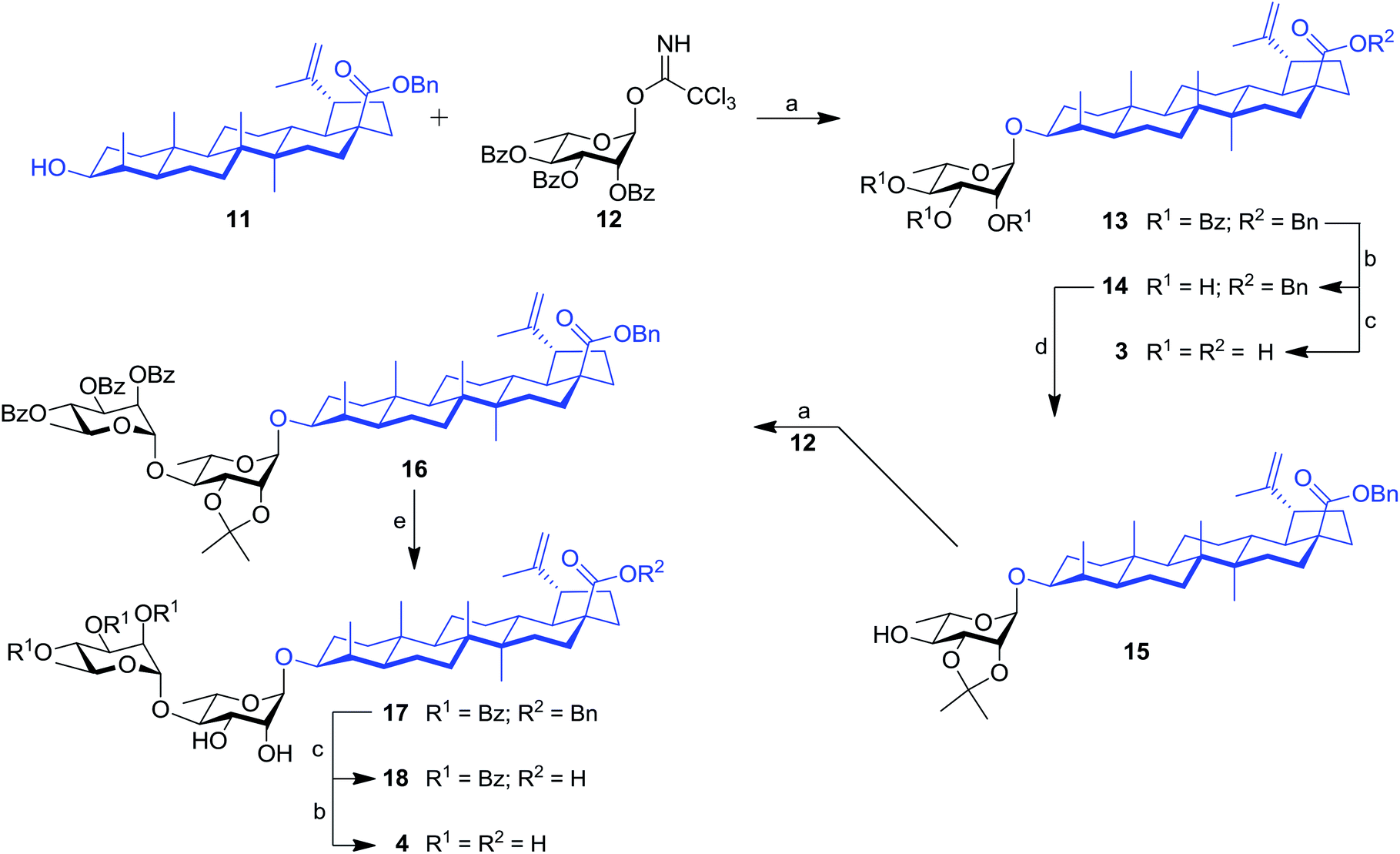 | ||
Scheme 1 Synthesis of mono- and dirhamnoside-containing BA saponins (3 and 4). Reagents and conditions: (a) 12 (1.2–1.5 equiv.), TMSOTf (0.1 equiv.), 4 Å MS, DCM, 0 °C, 30 min, 76% (for 13), 72% (for 16); (b) NaOMe, MeOH/DCM 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, rt, 16 h, 97% (for 14), 85% (for 4); (c) 10% Pd/C, H2, EtOAc, 80 °C, 4 h, 86% (for 18), 82% (for 3, two steps from 13); (d) 2,2-DMP (3 equiv.), PTSA (0.1 equiv.), acetone, rt, 2 h, 62%; (e) 80% aq. HOAc, DCM, rt, 16 h, 63%. :1, rt, 16 h, 97% (for 14), 85% (for 4); (c) 10% Pd/C, H2, EtOAc, 80 °C, 4 h, 86% (for 18), 82% (for 3, two steps from 13); (d) 2,2-DMP (3 equiv.), PTSA (0.1 equiv.), acetone, rt, 2 h, 62%; (e) 80% aq. HOAc, DCM, rt, 16 h, 63%. | ||
Compound 14 was converted into the corresponding 2,3-di-O-isopropylidene derivative 15 following treatment with 2,2-dimethoxypropane (2,2-DMP) under the catalytic action of PTSA. The C-4′ position of derivative 15 was then coupled with trichloroacetimidate (TCA) donor 12 to provide dirhamnoside 16 (72% yield) in exclusive α-form. Cleavage of the isopropylidene group using 80% aqueous HOAc at 80 °C gave key intermediate 17 in 63% yield. Target BA dirhamnoside saponin 4 was obtained following hydrogenolysis and debenzoylation with 73% yield over two steps.
The synthesis of trirhamnoside BA saponin 5 was performed by taking advantage of derivative 17. As depicted in Scheme 2, diol 17 was subjected to regioselective glycosylation33,34 with TCA donor 12 affording C-3′ and C-4′ linked trirhamnoside 19 in 63% yield. The regioselectivity of the reaction at the C-3′ position was confirmed via a 2D NMR HMBC experiment. A small amount of trirhamnoside at the C-2′ position was also isolated at this step (data not shown). Cleavage of protecting groups in compound 19 led to target BA trirhamnoside saponin 5 in 68% yield over two steps.
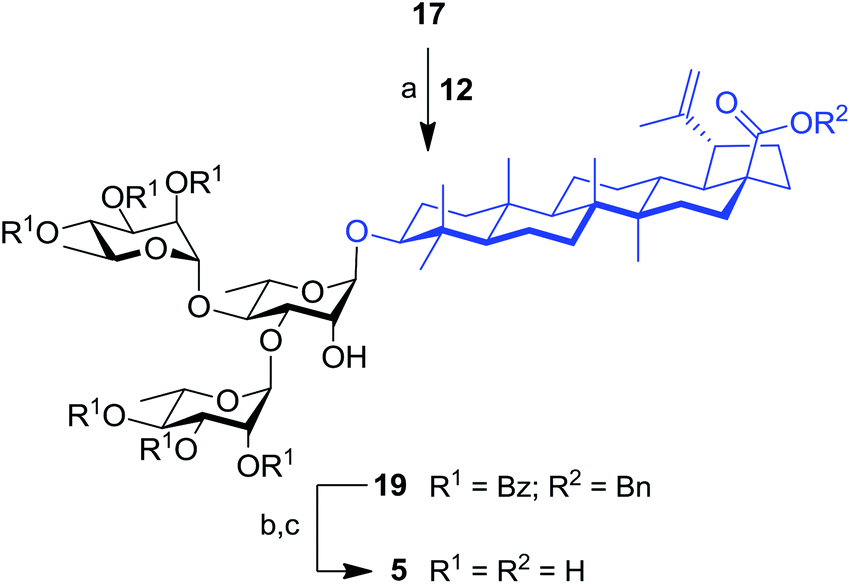 | ||
| Scheme 2 Synthesis of trirhamnoside-containing BA saponin (5). Reagents and conditions: (a) 12 (1.0 equiv.), TMSOTf (0.1 equiv.), 4 Å MS, DCM, 0 °C, 30 min, 63%; (b) 10% Pd/C, H2, EtOAc, 80 °C, 4 h; (c) NaOMe, MeOH/DCM 2:1, rt, 16 h, 68% (two steps). | ||
The synthesis of tetrarhamnoside 6 was our next target. Schmidt's inverse procedure (SIP)35 that is known to minimize donor degradation was preferred for this glycosylation in order to introduce three Rha residues in one step. Indeed, it was previously shown that multiple hydroxyl groups can be simultaneously glycosylated using SIP.17,36 Therefore, as depicted in Scheme 3, triol 14 was subjected to glycosylation with 5.0 equiv. of donor 12 under SIP providing the expected tetrarhamnoside in convenient yield (42%) with full control of stereoselectivity. The latter was deprotected using the aforementioned conditions. Purification using normal phase silica gel followed by solid phase extraction (SPE) furnished target saponin 6 in pure and homogeneous forms.
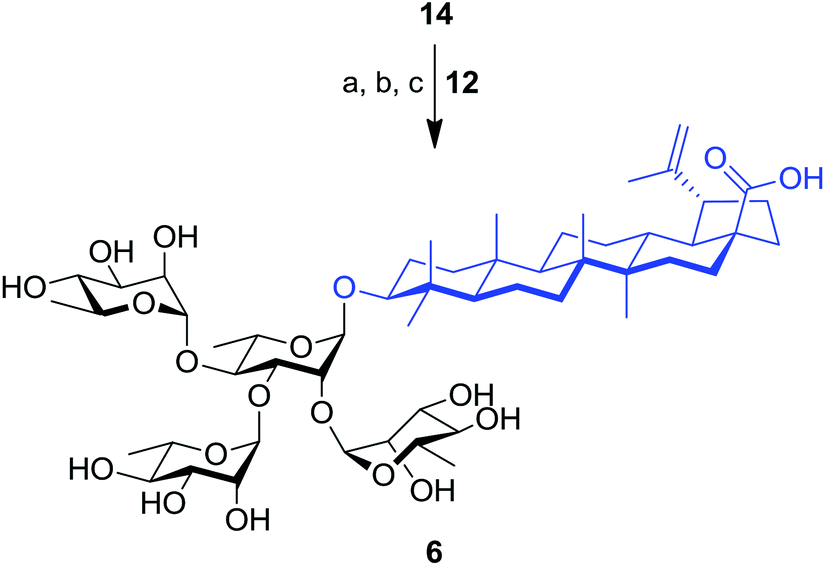 | ||
| Scheme 3 Synthesis of tetrarhamnoside-containing BA (6). Reagents and conditions: (a) inverse procedure, 12 (5.0 equiv.), TMSOTf (0.1 equiv.), 4 Å MS, DCM, −15 to 0 °C, 25 min; (b) 10% Pd/C, H2, EtOAc, 80 °C, 3 h; (c) NaOMe, MeOH/DCM 2:1, rt, 16 h, 37% (three steps). | ||
Synthesis of UA saponins 7–10
These results prompted us to apply a similar synthetic approach to prepare rhamnose-containing UA saponins 7–10. Therefore, as shown in Scheme 4, coupling between benzyl ursolate (20)37 and TCA donor 12 gave compound 21 with 75% yield. Removal of benzoyl groups through Zemplén deacylation gave triol 22. Pd-catalyzed hydrogenolysis afforded UA 3-O-α-L-rhamnopyranoside (7) in 74% yield over two steps. Protection of triol 22 with an isopropylidene group afforded derivative 23, which was rhamnosylated at the C-4′ position providing dirhamnoside 24 in 70% yield. Cleavage of the isopropylidene group generated diol 25 (83% yield), which was subjected to global cleavage of protecting groups giving target dirhamnoside-containing UA saponin 8.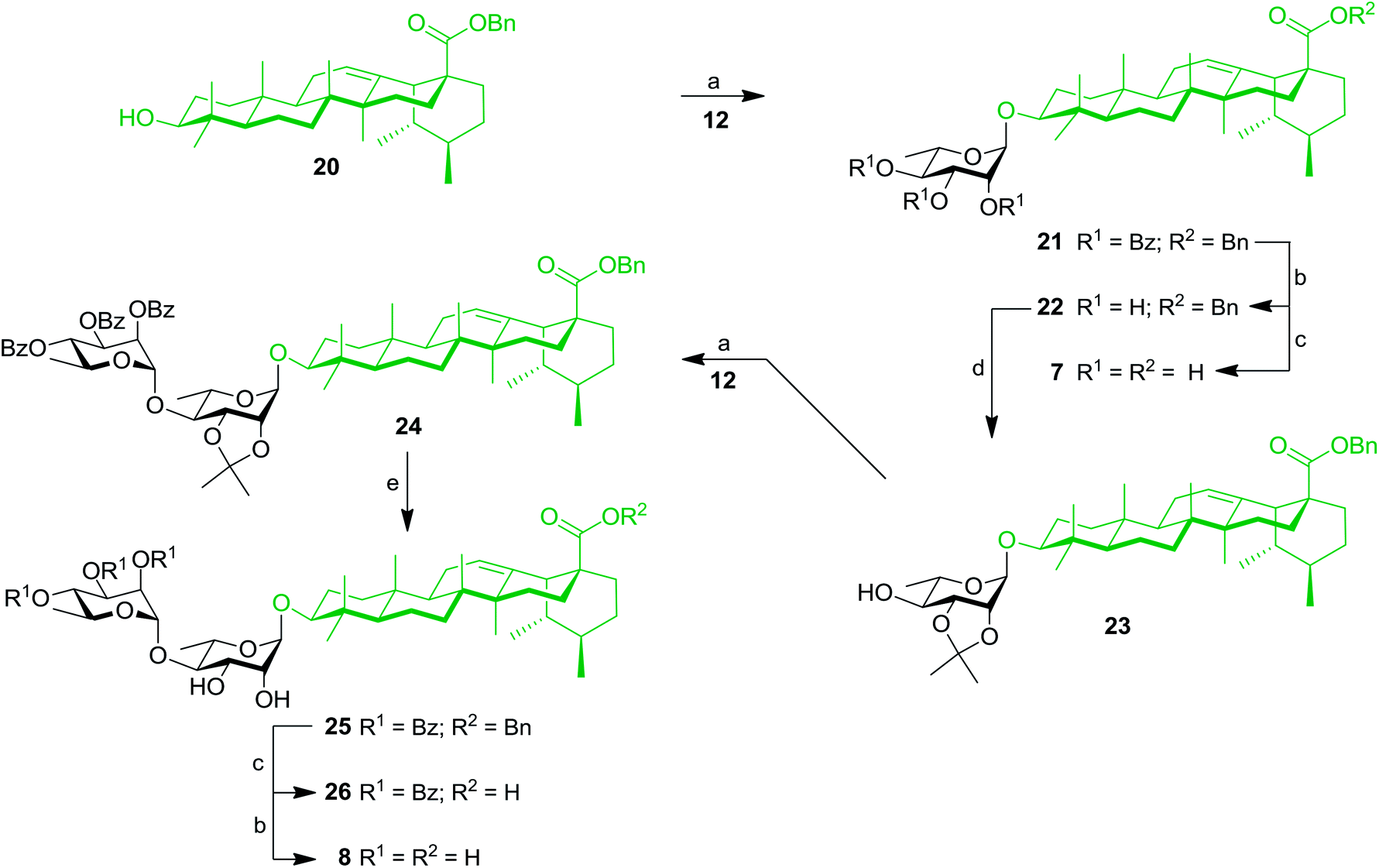 | ||
| Scheme 4 Synthesis of mono- and dirhamnoside-containing UA saponins (7 and 8). Reagents and conditions: (a) 12 (1.2 equiv.), TMSOTf (0.1 equiv.), 4 Å MS, DCM, 0 °C, 30 min, 75% (for 21), 70% (for 24); (b) NaOMe, MeOH/DCM 2:1, rt, 16 h, 98% (for 22), 86% (for 8); (c) 10% Pd/C, H2, EtOAc, 80 °C, 4 h, 89% (for 26), 75% (for 7, two steps from 21); (d) 2,2-DMP (3 equiv.), PTSA (0.1 equiv.), CH3COCH3, rt, 2 h, 74%; (e) 80% aq. HOAc, DCM, rt, 16 h, 83%. | ||
Thereafter, key intermediate 25 was engaged into regioselective glycosylation with TCA donor 12 under TMSOTf promotion (Scheme 5) to give trirhamnoside 27 in 62% yield. The formation of the (1′′′→3′) linkage was proved by a 2D NMR HMBC experiment, which showed strong cross-peaks from H-1′′′ to C-3′. Target trirhamnoside-containing UA saponin 9 was obtained following global deprotection using previously mentioned conditions in 68% yield over two steps.
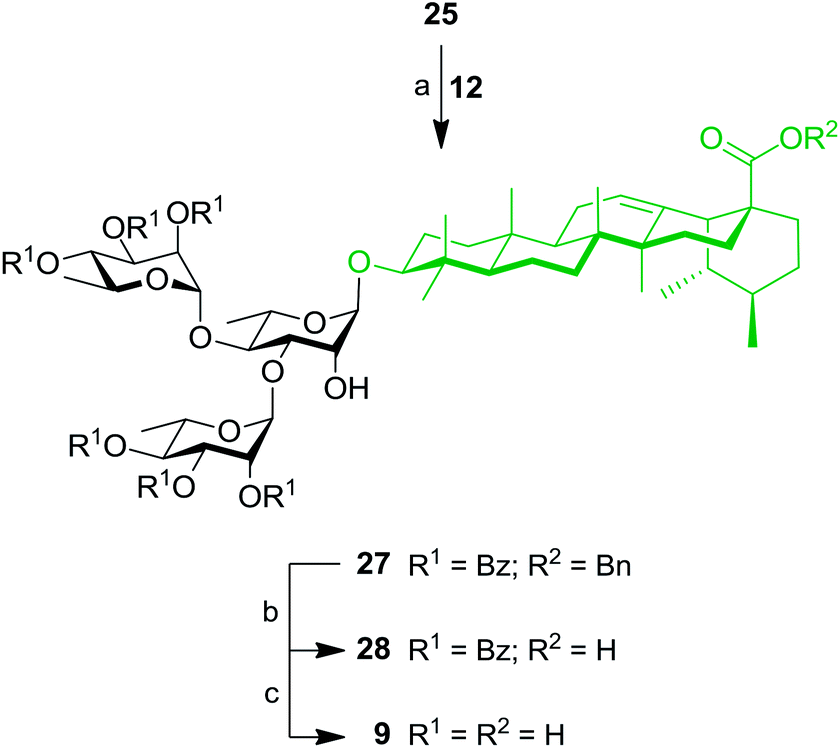 | ||
| Scheme 5 Synthesis of trirhamnoside-containing UA saponin (9). Reagents and conditions: (a) 12 (1.1 equiv.), TMSOTf (0.1 equiv.), 4 Å MS, DCM, 0 °C, 10 min, 62%; (b) 10% Pd/C, H2, EtOAc, 80 °C, 4 h, 81%; (c) NaOMe, MeOH/DCM 2:1, rt, 16 h, 84%. | ||
At this stage, SIP was used for the simultaneous introduction of three Rha residues. As depicted in Scheme 6, triol 22 was reacted with 5.0 equiv. of TCA donor 12 to provide fully protected tetrarhamnoside 29 in a convenient 47% yield. Deprotection of benzyl and benzoyl groups were performed to give target tetrarhamnoside-containing UA saponin 10 in 90% over two steps. All of the synthesized BA and UA saponins as well as parent triterpenoids were evaluated for their cytotoxic and anti-inflammatory activities.
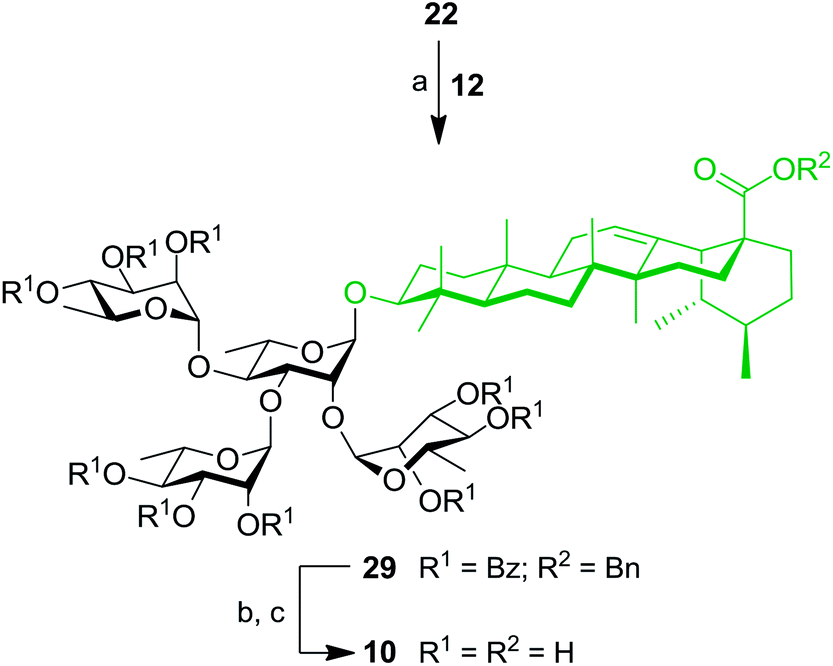 | ||
| Scheme 6 Synthesis of tetrarhamnoside-containing UA saponin (10). Reagents and conditions: (a) inverse procedure, 12 (5.0 equiv.), TMSOTf (0.1 equiv.), 4 Å MS, DCM, −15 to 0 °C, 25 min, 47%; (b) 10% Pd/C, H2, EtOAc, 80 °C, 3 h; (c) NaOMe, MeOH/DCM 2:1, rt, 16 h, 90% (two steps). | ||
Cytotoxic and anti-inflammatory activities of triterpenes and saponins 1–10
The cytotoxicity of compounds 1–10 was evaluated against human colorectal adenocarcinoma (DLD-1) and healthy human skin fibroblasts (WS1), using Hoechst assay as described in the experimental procedure. The results presented in Table 1 are expressed as the concentration inhibiting 50% of cell growth (IC50). Rha-2-Bet, i.e., 28-O-α-L-rhamnopyranosylbetulin 3β-O-α-L-rhamnopyranoside, was used as positive control with IC50 values of 2.08 μM against DLD-1 and 2.5 μM against WS-1.14 BA (1) and UA (2) were found cytotoxic against cancer cells with IC50 respectively of 20 and 13 μM as well as healthy cells with IC50 respectively of 36 and 14 μM. As previously reported, the addition of a single rhamnose moiety to BA (3) at the C-3 position selectively improved cytotoxic activity toward cancer cells with IC50 of 4 μM in comparison with 33 μM for healthy cells.15 Interestingly, saponin 4 containing a dirhamnose grafted to BA at the same position inhibited cancer cell growth, DLD-1 (IC50 = 5 μM), without affecting healthy cells, WS1 (IC50 > 100 μM). In contrast, saponins 5 and 6 containing, respectively, a tri- or a tetra-rhamnose were inactive with IC50 > 100 μM. In the other hand, the addition of a single rhamnose (7) or a dirhamnose (8) at the C-3 position of UA did not improve the selectivity or the cytotoxic activity. As observed with BA, the presence of tri- (9) or tetrarhamnose (10) residues inhibited the cytotoxicity of UA.| Cpd | Cytotoxicity, C50a c (μM) | Anti-inflammatory activity, EC50b c (μM) | |
|---|---|---|---|
| DLD-1 | WS-1 | ||
| a IC50: concentration inhibiting 50% of cell growth. 28-O-α-L-Rhamnopyranosylbetulin 3β-O-α-L-rhamnopyranoside (Rha-2-Bet) was used as positive control.b EC50: efficacy concentration inhibiting 50% of NO overproduction induced by LPS. L-NAME was used as positive control inhibits 54% of NO overproduction at 250 μM.c The results are the mean ± standard deviation of three determination and are representative of three different experiments.d NA: not active; compound was considered inactive when EC50 is ≥50 μM. | |||
| 1 | 20 ± 3 | 36 ± 1 | 14 ± 1 |
| 2 | 13 ± 2 | 14 ± 2 | 9 ± 1 |
| 3 | 4.0 ± 0.5 | 33 ± 6 | NAd |
| 4 | 5 ± 1 | >100 | NAd |
| 5 | >100 | >100 | NAd |
| 6 | >100 | >100 | NAd |
| 7 | 15 ± 1 | 23 ± 1 | 10.5 ± 0.3 |
| 8 | >100 | 70 ± 4 | 16 ± 3 |
| 9 | >100 | >100 | 9.8 ± 0.3 |
| 10 | >100 | >100 | 11 ± 2 |
| Rha-2-Bet | 2.08 ± 0.03 | 2.5 ± 0.3 | 14 ± 1 |
Anti-inflammatory activity of compounds 1–10 was also evaluated using LPS-stimulated RAW 264.7 macrophages, which induces NO overproduction. L-NAME was used as positive control with NO inhibition of 54% at a concentration of 250 μM. Compounds 1, 2, and 7 inhibited NO overproduction induced by LPS with EC50 ranging from 9 to 14 μM but macrophage cytotoxicity appeared at 20 μM. Moreover, compounds 3–6 were found inactive with EC50 > 50 μM. In contrast, all of the UA rhamnosidic derivatives (7–10) inhibited NO overproduction with EC50 ranging from 9.8 to 16 μM. Interestingly, the presence of mono- (7), di- (8), tri- (9) or tetrarhamnoses (10) at the C-3 position of UA decreased gradually the cytotoxicity against healthy cells with IC50 respectively of 23 μM, 70 μM and >100 μM but retained the anti-inflammatory activity.
Conclusions
In summary, we accomplished the straightforward synthesis of a series of rhamnose-containing saponins (3–10) using the bioactive, naturally-occurring BA (1) and UA (2) as aglycones. Monorhamnosides, (1→4)-linked dirhamnosides as well as branched trirhamnosides and tetrarhamnosides were synthesized in high yields relying on both Schmidt's normal and inverse glycosylation procedures. Outstandingly, 3-O-α-L-rhamnopyranosyl-(1→4)-α-L-rhamnopyranosyl-betulinic acid (4) was found to exhibit potent cytotoxic activity against human colorectal adenocarcinoma cells without damaging the healthy cells (selectivity ratio > 20) whereas UA saponins containing more than one sugar unit were inactive against cancer cells. Moreover, we found that rhamnose-containing UA saponins 7–10 potently inhibited NO overproduction induced by LPS-stimulated macrophages. Collectively, our results suggest that the cytotoxic and anti-inflammatory activities of rhamnose-containing saponins are strongly modulated by both the nature of the triterpenes and the number and attachment of sugar units.Experimental section
General methods
Air and water sensitive reactions (glycosylation especially) were performed under nitrogen or Ar atmosphere. Moisture sensitive reagents were introduced in reaction media via a dry syringe. AcroSeal® extra dry solvents over molecular sieves (dichloromethane, ethyl acetate, tetrahydrofuran and methanol) were purchased from Acros Organics. All commercial reagents were used without further purification. Purification of reaction products were carried out using 60–230 mesh silica gel obtained from Silicycle (Canada). Compounds were eluted with reagent grade solvent purchased from Fisher Scientific. Analytical thin-layer chromatography was performed with silica gel 60 F254, 0.25 mm pre-coated TLC plates, supplied by Silicycle (Canada), and visualized using UV light (254 nm) and cerium molybdate (2 g Ce(SO4)4(NH4)4, 5 g MoO4(NH4)2, 200 mL H2O, and 20 mL H2SO4) with charring. All reaction yields are not optimized values. Optical rotations were measured on a Rudolph Research Analytical AUTOPOL IV digital polarimeter. Absorption UV spectra were recorded with an Agilent 8453 diode-array spectrophotometer. The 1D and 2D NMR spectra (1H–1H COSY, HSQC, HMBC and NOESY) were performed using an Avance 400 Bruker spectrometer (400.13 MHz for 1H, 100.61 MHz for 13C spectra) equipped with a 5 mm QNP-probe. All spectra were acquired in CDCl3, CD3OD, DMSO, or in a mixture of CDCl3–CD3OD 1:1. Chemical shifts are reported in ppm (δ) relative to TMS (0 ppm). The modulus of coupling constants (J), extracted from the 1H NMR spectrum, are reported in Hz. HPLC-APCI MS (negative mode) were obtained from an Agilent 1100 series system consisting of a degasser, a quaternary pump, an automatic injector, a temperature-controlled column compartment, a diode array detector and a mass selective detector Agilent G1946 VL model equipped with an APCI source. Analytical separations were performed at a flow rate of 1.0 mL min−1 and a column temperature of 25 °C. Preparative HPLC separation (Agilent 1100) were carried out on a 20.0 × 250 mm C18 column using a multiple wavelength detector and an automatic fraction collector. Chromatographic conditions were the following: gradient elution with H2O:CH3CN (10 → 100%) at flow rate of 20.0 mL min−1. Mass spectral data (HRMS) were obtained at NanoQAM University of Québec at Montréal (UQAM) and department of chemistry at University of Montréal (UdeM), Québec, Canada.
General procedure for normal Schmidt glycosylation
Appropriate triterpene acceptor (1 equiv.) and trichloroacetimidate donor (1.5 equiv.) were premixed with 4 Å molecular sieves in dry DCM (5 mL) under Ar atmosphere. After 30 min, TMSOTf (0.1 equiv.) was added dropwise under Ar while keeping rigorous anhydrous conditions. The reaction was generally stirred during 30 min, after which Et3N (0.2 equiv.) was added to quench the reaction. The suspension was filtered, and the solvents were evaporated under reduced pressure. The resulting residue was purified by flash chromatography on silica gel.General procedure for inverse Schmidt glycosylation
To a stirred suspension of an appropriate triterpene acceptor and 4 Å molecular sieves in dry DCM (5 mL) was added TMSOTf (0.1 equiv.). The mixture was kept at −15 °C under rigorous anhydrous conditions. After 30 min, a solution of trichloroacetimidate donor (5 equiv.) in dry DCM was added to the mixture over 5 min. The mixture was stirred at −15 °C for 25 min, after which the reaction was quenched with Et3N (0.2 equiv.) and filtered. The filtrate was concentrated under reduced pressure to give a residue, which was purified by flash column chromatography.General procedure for removal of benzoyl groups
A freshly prepared solution of NaOMe in MeOH (0.5 M) was added to an appropriate benzoylated compound suspended in dry MeOH or a mixture of MeOH–DCM (2:1 v/v). The mixture was stirred overnight at rt. When TLC indicated the completion, the reaction was neutralized to pH 7 with Dowex G26 (H+ form) resin and filtered. The filtrate was concentrated to dryness and purified by silica gel flash chromatography.
:3, hexanes–EtOAc), [α]20D −15.9 (c 1, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.11 (br d, J = 7.3 Hz, 2H, CH-Bz), 7.99 (br d, J = 7.3 Hz, 2H, CH-Bz), 7.83 (br d, J = 7.3 Hz, 2H, CH-Bz), 7.60 (br t, J = 7.4 Hz, 1H, CH-Bz), 7.51 (br t, J = 7.5 Hz, 1H, CH-Bz), 7.49 (br t, J = 7.5 Hz, 2H, CH-Bz), 7.45–7.30 (m, 7H, CH-Bz, CH-Bn), 7.26 (br t, J = 7.7 Hz, 3H, CH-Bz), 5.82 (dd, J = 10.1, 3.2 Hz, 1H, H-3′), 5.68 (t, J = 10.1 Hz, 1H, H-4′), 5.64 (dd, J = 3.1, 1.5 Hz, 1H, H-2′), 5.16 (d, J = 12.3 Hz, 1H, CH2-Bn), 5.09 (d, J = 12.3 Hz, 1H, CH2-Bn), 5.07 (br s, 1H, H-1′), 4.73 (br s, 1H, H-29a), 4.59 (br s, 1H, H-29b), 4.31 (dq, J = 10.1, 6.1 Hz, 1H, H-5′), 3.19 (t, J = 8.2 Hz, 1H, H-3), 3.03 (td, J = 10.8, 4.5 Hz, 1H, H-19), 2.29 (br d, J = 12.4 Hz, 1H, H-16a), 2.19 (td, J = 12.6, 3.1 Hz, 1H, H-13), 1.68 (s, 3H, H-30), 1.33 (d, J = 6.2 Hz, H-6′), 1.04 (s, 3H, H-23), 0.95 (s, 3H, H-27), 0.92 (s, 3H, H-24), 0.86 (s, 3H, H-25), 0.77 (s, 3H, H-26), 0.71 (br d, J = 8.6 Hz, 1H, H-5). 13C NMR (101 MHz, CDCl3) δ 175.8 (C-28), [165.9, 165.7, 165.6 (CO-Bz)], 150.6 (C-20), 136.5 (C-Bn), [133.4, 133.3, 133.1 (CH-Bz)], [129.9 (2×), 129.8 (2×), 129.7 (2×) (CH-Bz)], [129.6, 129.4, 129.3 (C-Bz)], [128.6 (2×), 128.5 (2×), 128.4 (2×), 128.3 (4×), 128.1 (CH-Bz, CH-Bn)], 109.6 (C-29), 99.7 (C-1′), 90.1(C-3), 72.0 (C-4′), 71.2 (C-2′), 70.2 (C-3′), 66.8 (C-5′), 65.7 (CH2-Bn), 56.6 (C-17), 55.5 (C-5), 50.5 (C-9), 49.4 (C-18), 46.9 (C-19), 42.4 (C-14), 40.7 (C-8), 39.1 (C-4), 38.7 (C-1), 38.2 (C-13), 36.9 (2×, C-22, C-10), 34.3 (C-7), 32.1 (C-16), 30.6 (C-21), 29.6 (C-15), 28.3 (C-23), 25.7 (C-2), 25.5 (C-12), 20.9 (C-11), 19.4 (C-30), 18.3 (C-6), 17.6 (C-6′), 16.4 (C-24), 16.2 (C-25), 15.8 (C-26), 14.7 (C-27). HRMS calcd for C64H77O10 [M + H]+ 1005.5511, found 1005.5507; calcd for C64H76O10Na [M + Na]+ 1022.5777, found 1022.5767.:1, 3 mL) with NaOMe in MeOH (0.5 M, 3 mL) overnight. The reaction was then neutralized to pH 7 with Dowex G26 (H+ form), filtered and purified by flash chromatography (9:1, DCM–MeOH) to give compound 14 (153 mg, 97%) as colorless oil, Rf = 0.5 (CHCl3:MeOH, 7:1), [α]20D −18.9 (c 3, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.39–7.28 (m, 5H, H–Ar), 5.14 (d, J = 12.4 Hz, 1H, CH2-Bn), 5.10 (d, J = 12.4 Hz, 1H, CH2-Bn), 4.77 (s, 1H, H-1′), 4.73 (s, 1H, H-29a), 4.60 (s, 1H, H-29b), 3.90 (s, 1H, H-2′), 3.81–3.68 (m, 2H, H-3′, H-5′), 3.44 (d, J = 9.9 Hz, 1H, H-4′), 3.08–2.97 (m, 2H, H-3, H-19), 2.28 (d, J = 10.4 Hz, 1H, H-16a), 2.18 (t, J = 11.1 Hz, 1H, H-13), 1.68 (s, 3H, H-30), 1.25 (d, J = 6.6 Hz, 3H, H-6′), 0.94 (s, 3H, H-27), 0.86 (s, 3H, H-23), 0.78 (s, 3H, H-25), 0.75 (s, 3H, H-26), 0.71 (s, 3H, H-24), 0.63 (br d, J = 7.3 Hz, 1H, H-5). 13C NMR (101 MHz, CDCl3) δ 175.8 (C-28), 150.5 (C-20), 136.4 (C-Bn), [128.4 (2×), 128.2 (2×), 128.0 (CH-Bn)], 109.6 (C-29), 102.3 (C-1′), 89.3 (C-3), 72.8 (C-4′), 71.8 (C-3′), 71.3 (C-2′), 68.0 (C-5′), 65.7 (CH2-Bn), 56.5 (C-17), 55.4 (C-5), 50.5 (C-9), 49.4 (C-18), 46.9 (C-19), 42.3 (C-14), 40.6 (C-8), 39.0 (C-4), 38.6 (C-1), 38.1 (C-13), 36.9 (C-22), 36.8 (C-10), 34.2 (C-7), 32.1 (C-16), 30.5 (C-21), 29.5 (C-15), 28.1 (C-23), 25.5 (C-12), 25.4 (C-2), 20.8 (C-11), 19.3 (C-30), 18.2 (C-6), 17.4 (C-6′), 16.2 (C-24), 16.1 (C-25), 15.8 (C-26), 14.6 (C-27). HRMS calcd for C43H65O7 [M + H]+ 693.4725, found 693.4698.:1 as solvent. The reaction was neutralized to pH 7 with Dowex G26 (H+ form), filtered and purified by flash chromatography (7:1, DCM–MeOH) to give compound 3 (25 mg, 82%, two steps) as colorless oil, Rf = 0.3 (DCM:MeOH, 7:1). The NMR data are in adequation with those described in the literature.15,32:3, hexanes–EtOAc) to give compound 15 (92 mg, 62%) as colorless oil, Rf = 0.7 (hexanes–EtOAc, 7:3), [α]20D −5.8 (c 2.3, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.42–7.22 (m, 5H, H–Ar), 5.15 (d, J = 12.3 Hz, 1H, CH2-Bn), 5.09 (d, J = 12.3 Hz, 1H, CH2-Bn), 4.97 (s, 1H, H-1′), 4.72 (br s, 1H, H-29a), 4.59 (br s, 1H, H-29b), 4.18–4.09 (m, 2H,, H-2′, H-3′), 3.83 (dq, J = 8.8, 6.3 Hz, 1H, H-5′), 3.42 (dd, J = 8.4, 6.3 Hz, 1H, H-4′), 3.11 (dd, J = 11.4, 4.5 Hz, 1H, H-3), 3.02 (td, J = 10.6, 4.2 Hz, 1H, H-19), 2.27 (br d, J = 12.2 Hz, 1H, H-16a), 2.17 (td, J = 12.7, 3.1 Hz, 1H, H-13), 1.68 (s, 3H, H-30), 1.52 (s, 3H, CH3-iso), 1.37 (s, 3H, CH3-iso), 1.27 (d, J = 6.4 Hz, 3H, H-6′), 0.93 (s, 3H, H-27), 0.91 (s, 3H, H-23), 0.80 (s, 3H, H-25), 0.75 (s, 6H, H-24, H-26), 0.67 (br d, J = 9.6 Hz, 1H, H-5). 13C NMR (101 MHz, CDCl3) δ 175.8 (C-28), 150.6 (C-20), 136.5 (C-Bn), [128.5 (2×), 128.2 (2×), 128.1 (CH-Bn)], 109.6 (C-29), 109.4 (C-iso), 99.8 (C-1′), 89.1 (C-3), 78.1 (C-3′), 75.8 (C-2′), 74.0 (C-4′), 66.5 (C-5′), 65.7 (CH2-Bn), 56.5 (C-17), 55.5 (C-5), 50.5 (C-9), 49.4 (C-18), 46.9 (C-19), 42.4 (C-14), 40.6 (C-8), 39.1 (C-4), 38.7 (C-1), 38.2 (C-13), 36.9 (C-22), 36.9 (C-10), 34.2 (C-7), 32.1 (C-16), 30.6 (C-21), 29.5 (C-15), 28.2 (C-23), 27.9 (CH3-iso), 26.1 (CH3-iso), 25.6 (C-12), 25.5 (C-2), 20.9 (C-11), 19.4 (C-30), 18.2 (C-6), 17.7 (C-6′), 16.3 (C-24), 16.2 (C-25), 15.8 (C-26), 14.7 (C-27). HRMS calcd for C46H69O7 [M + H]+ 733.5038, found 733.5010.:2, hexanes–EtOAc) to give compound 16 (73 mg, 72%) as colorless oil, Rf = 0.6 (hexanes–EtOAc, 8:2), [α]20D +35 (c 2, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.11 (br d, J = 7.7 Hz, 2H, CH-Bz), 7.96 (br d, J = 7.9 Hz, 2H, CH-Bz), 7.80 (br d, J = 8.0 Hz, 2H, CH-Bz), 7.61 (br t, J = 7.6 Hz, 1H, CH-Bz) 7.55–7.22 (m, 13H, CH-Bz, CH-Bn), 5.77 (dd, J = 10.0, 3.3 Hz, 1H, H-4′′), 5.73 (dd, J = 3.2, 1.7 Hz, 1H, H-2′′), 5.67 (t, J = 9.8 Hz, 1H, H-4′′), 5.58 (d, J = 1.4 Hz, 1H, H-1′′), 5.16 (d, J = 12.3 Hz, 1H, CH2-Bn), 5.10 (d, J = 12.3 Hz, 1H, CH2-Bn), 5.03 (s, 1H, H-1′), 4.74 (d, J = 2.4 Hz, 1H, H-29a), 4.61 (s, 1H, H-29b), 4.31 (dd, J = 7.2, 5.4 Hz, 1H, H-3′), 4.19 (dq, J = 9.5, 6.2 Hz, 1H, H-5′′), 4.13 (d, J = 5.5 Hz, 1H, H-2′), 3.97 (dq, J = 10.0, 6.2 Hz, 1H, H-5′), 3.61 (dd, J = 9.9, 7.3 Hz, 1H, H-4′), 3.14 (dd, J = 9.9, 6.1 Hz, 1H, H-3), 3.03 (td, J = 10.7, 4.3 Hz, 1H, H-19), 2.29 (br d, J = 12.3 Hz, 1H, H-16a), 2.19 (td, J = 12.4, 3.3 Hz, 1H, H-13), 1.69 (s, 3H, H-29), 1.55 (s, 3H, CH3-iso), 1.38 (d, J = 5.8 Hz, 3H, H-6′), 1.37 (d, J = 6.0 Hz, 3H, H-6′′), 1.35 (s, 3H, CH3-iso), 0.95 (s, 3H, H-27), 0.93 (s, 3H, H-23), 0.84 (s, 3H, H-25), 0.80 (s, 3H, H-24), 0.77 (s, 3H, H-26), 0.70 (br d, J = 8.5 Hz, 1H, H-5). 13C NMR (101 MHz, CDCl3) δ 175.8 (C-28), [165.8, 165.6, 165.4 (CO-Bz)], 150.6 (C-20), 136.5 (C-Bz), [133.4, 133.3, 133.1, 130.0 (2×), 129.7 (2×), 129.7 (2×) (CH-Bz)], [129.5, 129.3, 129.2 (C-Bz)], 128.53 (2×), 128.49 (2×), 128.41 (2×), 128.3 (4×), 128.1 (CH-Bz, CH-Bn), 109.6 (C-29), 109.5 (C-iso), 99.5 (C-1′), 96.0 (C-1′′), 89.0 (C-3), 78.2 (C-3′), 78.0 (C-4′), 76.3 (C-2′), 71.7 (C-4′′), 70.8 (C-2′′), 70.0 (C-3′′), 67.3 (C-5′′), 65.7 (CH2-Bn), 63.7 (C-5′), 56.6 (C-17), 55.5 (C-5), 50.6 (C-9), 49.5 (C-18), 46.9 (C-19), 42.4 (C-14), 40.7 (C-8), 39.2 (C-4), 38.8 (C-1), 38.2 (C-13), 36.9 (C-22), 36.9 (C-10), 34.3 (C-7), 32.1 (C-16), 30.6 (C-21), 29.6 (C-15), 28.3 (C-23), 28.0 (CH3-iso), 26.5 (CH3-iso), 25.6 (C-12), 25.5 (C-2), 20.9 (C-11), 19.4 (C-30), 18.3 (C-6), 18.1 (C-6′′), 17.7 (C-6′), 16.3 (C-24), 16.2 (C-25), 15.8 (C-26), 14.7 (C-27). HRMS calcd for C73H94O14N [M + NH4]+ 1208.6669, found 1208.6703.:2, hexanes–EtOAc) to give compound 17 (120 mg, 63%) as colorless oil, Rf = 0.5 (hexanes–EtOAc, 8:2), [α]20D +36.4 (c 2, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 7.3 Hz, 2H, CH-Bz), 7.97 (d, J = 7.3 Hz, 2H, CH-Bz), 7.83 (d, J = 7.3 Hz, 2H, CH-Bz), 7.59 (t, J = 7.4 Hz, 1H, CH-Bz), 7.52 (t, J = 7.4 Hz, 1H, CH-Bz), 7.49–7.22 (m, 12H, CH-Bz), 5.81 (dd, J = 10.1, 3.4 Hz, 1H, H-3′′), 5.77 (dd, J = 3.2, 1.8 Hz, 1H, H-2′′), 5.68 (t, J = 9.9 Hz, 1H, H-4′′), 5.48 (br s, 1H, H-1′′), 5.16 (d, J = 12.3 Hz, 1H, CH2-Bn), 5.10 (d, J = 12.3 Hz, 1H, CH2-Bn), 4.85 (s, 1H, H-1′), 4.73 (d, J = 2.4 Hz, 1H, H-29a), 4.60 (br s, 1H, H-29b), 4.29 (dq, J = 9.6, 6.2 Hz, 1H, H-5′′), 4.07 (dd, J = 9.1, 3.3 Hz, 1H, H-3′), 4.01–3.90 (m, 2H, H-2′, H-5′), 3.63 (t, J = 9.3 Hz, 1H, H-4′), 3.08 (dd, J = 10.8, 5.3 Hz, 1H, H-3), 3.03 (dd, J = 10.8, 4.4 Hz, 2H, H-19), 2.29 (br d, J = 12.3 Hz, 1H, H-16a), 2.19 (td, J = 12.8, 3.4 Hz, 1H, H-13), 1.69 (s, 3H, H-30), 1.37 (d, J = 6.8 Hz, 3H, H-6′), 1.36 (d, J = 6.6 Hz, 3H, H-6′′), 0.95 (s, 3H, H-27), 0.89 (s, 3H, H-23), 0.84 (s, 3H, H-25), 0.79 (s, 3H, H-24), 0.76 (s, 3H, H-26), 0.66 (br d, J = 9.1 Hz, 1H, H-5). 13C NMR (101 MHz, CDCl3) δ 175.8 (C-28), [165.9, 165.8, 165.8 (CO-Bz)], 150.6 (C-20), 136.5 (C-Bn), [133.5, 133.3, 133.2 (CH-Bz)], [129.9 (2×), 129.7 (4×) (CH-Bz)], [129.3, 129.3, 129.1 (C-Bz)], 128.6 (2×), 128.5 (2×), 128.4 (2×), 128.31 (2×), 128.25 (2×), 128.1 (CH-Bz, CH-Bn)], 109.6 (C-29), 101.9 (C-1′), 98.9 (C-1′′), 89.6 (C-3), 81.6 (C-4′), 71.9 (C-2′), 71.9 (C-3′), 71.6 (C-4′′), 71.2 (C-2′′), 70.1 (C-3′′), 67.5 (C-5′′), 66.2 (C-5′), 65.7 (CH2-Bn), 56.5 (C-17), 55.5 (C-5), 50.5 (C-9), 49.4 (C-18), 46.9 (C-19), 42.4 (C-14), 40.6 (C-8), 39.1 (C-4), 38.7 (C-1), 38.2 (C-13), 36.9 (C-22), 36.9 (C-10), 34.3 (C-7), 32.1 (C-16), 30.6 (C-21), 29.6 (C-15), 28.2 (C-23), 25.6 (C-12), 25.5 (C-2), 20.9 (C-11), 19.4 (C-30), 18.3 (C-6), 17.9 (C-6′), 17.6 (C-6′′), 16.3 (C-24), 16.1 (C-25), 15.8 (C-26), 14.7 (C-27). HRMS calcd for C70H90O14N [M + NH4]+ 1168.63558, found 1168.63697.:3, hexanes–EtOAc) to give compound 18 (92 mg, 86%) as colorless oil, Rf = 0.2 (hexanes–EtOAc, 7:3), [α]20D +28.3 (c 2.2, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 8.2 Hz, 2H, CH-Bz), 7.97 (d, J = 8.3 Hz, 2H, CH-Bz), 7.82 (d, J = 8.2 Hz, 2H, CH-Bz), 7.62–7.20 (m, 12H, CH-Bz), 5.81 (dd, J = 10.2, 3.2 Hz, 1H, H-3′′), 5.77–5.73 (m, 1H, H-2′′), 5.68 (t, J = 9.9 Hz, 1H, H-4′′), 5.48 (s, 1H, H-1′′), 4.86 (s, 1H, H-1′), 4.75 (br s, 1H, H-29a), 4.62 (br s, 1H, H-29b), 4.29 (dq, J = 9.3, 6.1 Hz, 1H, H-5′′), 4.10–4.05 (m, 1H, H-3′), 4.01–3.92 (m, 2H, H-2′, H-5′), 3.64 (t, J = 9.2 Hz, 1H, H-4′), 3.10 (dd, J = 10.4, 5.2 Hz, 1H, H-3), 3.06–2.96 (m, 1H, H-19), 2.32–2.14 (m, 2H, H-16a, H-13), 1.70 (s, 3H, H-30), 1.38 (d, J = 6.6 Hz, 3H, H-6′), 1.36 (d, J = 6.6 Hz, 3H, H-6′′), 0.98 (s, 3H, H-27), 0.94 (s, 3H, H-26), 0.91 (s, 3H, H-23), 0.87 (s, 3H, H-25), 0.81 (s, 3H, H-24), 0.70 (d, J = 8.5 Hz, 1H, H-5). 13C NMR (101 MHz, CDCl3) δ 181.3 (C-28), [165.9 (2×), 165.8 (CO-Bz)], 150.4 (C-20), [133.5, 133.4, 133.2, 130.0, 129.7 (2×) (CH-Bz)], [129.29, 129.27, 129.1 (C-Bz)], [128.6, 128.4, 128.3 (CH-Bz)], 109.7 (C-29), 101.9 (C-1′), 98.9 (C-1′′), 89.6 (C-3), 81.4 (C-4′), 71.9 (2×, C-3′, C-2′), 71.6 (C-4′′), 71.2 (C-2′′), 70.0 (C-3′′), 67.6 (C-5′′), 66.2 (C-5′), 56.4 (C-17), 55.5 (C-5), 50.5 (C-9), 49.3 (C-18), 46.9 (C-19), 42.4 (C-14), 40.7 (C-8), 39.1 (C-4), 38.7 (C-1), 38.4 (C-13), 37.1 (C-22), 36.9 (C-10), 34.3 (C-7), 32.2 (C-16), 30.6 (C-21), 29.7 (C-15), 28.2 (C-23), 25.6 (C-12), 25.5 (C-2), 20.9 (C-11), 19.4 (C-30), 18.3 (C-6), 17.9 (C-6′), 17.6 (C-6′′), 16.4 (C-24), 16.2 (C-25), 16.0 (C-26), 14.7 (C-27). HRMS calcd for C63H84O14N [M + NH4]+ 1078.5856, found 1078.5901.:1, 3 mL) with NaOMe in MeOH (0.5 M, 3 mL) overnight. The reaction was then neutralized to pH 7 with Dowex G26 (H+ form), filtered and purified by flash chromatography (7:1, DCM–MeOH) to give compound 4 (42 mg, 85%) as colorless oil, Rf = 0.4 (CHCl3:MeOH, 7:1), [α]20D −18.9 (c 3, CHCl3). 1H NMR (400 MHz, DMSO-d6) δ 12.11 (br s, 1H, COOH), 5.06 (s, 1H, H-1′′), 4.95 (s, 1H, OH), 4.76 (br s, 2H, OH), 4.69 (s, 1H, H-29a), 4.65 (br s, 1H, OH), 4.60–4.54 (m, 2H, H-1′, H-29b), 3.69 (s, 1H, H-2′′), 3.60 (s, 1H, H-2′), 3.58–3.51 (m, 2H, H-5′, H-3′), 3.51–3.44 (m, 1H, H-5′′), 3.43–3.30 (m, 2H, H-4′, H-3′′), 3.23–3.14 (m, 1H, H-4′′), 3.04–2.89 (m, 2H, H-3, H-19), 2.23 (br t, J = 10.6 Hz, 1H, H-13), 2.11 (br d, J = 7.9 Hz, 1H, H-16a), 1.64 (s, 3H, H-30), 1.12 (d, J = 6.0 Hz, H-6′), 1.11 (d, J = 6.0 Hz, H-6′′), 0.93 (s, 3H, H-27), 0.86 (s, 6H, H-23, H-26), 0.78 (s, 3H, H-25), 0.70 (s, 3H, H-24). 13C NMR (101 MHz, DMSO-d6) δ 177.2 (C-28), 150.2 (C-20), 109.5 (C-29), 102.5 (C-1′), 101.0 (C-1), 87.6 (C-3), 77.9 (C-4′), 71.8 (C-4′′), 71.5 (C-3′), 71.1 (C-2′), 70.6 (C-3′′), 70.5 (C-2′′), 68.8 (C-5′′), 66.6 (C-5′), 55.3 (C-17), 54.6 (C-5), 49.7 (C-9), 48.4 (C-18), 46.5 (C-19), 41.9 (C-14), 40.1 (C-8), 38.5 (C-4), 38.0 (C-1), 37.4 (C-13), 36.4 (C-10), 36.2 (C-22), 33.7 (C-7), 31.6 (C-16), 30.0 (C-21), 29.1 (C-15), 27.7 (C-23), 25.0 (2×, C-2, C-12), 20.3 (C-11), 18.8 (C-30), 18.0 (C-6′), 17.73 (C-6), 17.68 (C-6′′), 16.1 (C-24), 15.8 (C-25), 15.6 (C-26), 14.3 (C-27). HRMS calcd for C42H72O11N [M + NH4]+ 766.51, found 766.5128.:3, hexanes–EtOAc) to give compound 19 (107 mg, 63%) as colorless oil, Rf = 0.5 (hexanes–EtOAc, 7:3), [α]20D +44.5 (c 1.9, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.02–7.74 (m, 12H, CH-Bz), 7.55–7.06 (m, 17H, CH-Bz, CH-Bn), 6.04 (dd, J = 3.4, 1.7 Hz, 1H, H-2′′′), 5.99 (dd, J = 10.1, 3.2 Hz, 1H, H-3′′′), 5.95 (dd, J = 10.2, 3.1 Hz, 1H, H-3′′), 5.83 (dd, J = 3.5, 1.8 Hz, 1H, H-2′′), 5.75 (t, J = 10.0 Hz, 1H, H-4′′), 5.67 (t, J = 9.9 Hz, 1H, H-4′′′), 5.54 (d, J = 1.9 Hz, 1H, H-1′′), 5.37 (br s, 1H, H-1′′′), 5.16 (d, J = 12.3 Hz, 1H, CH2-Bn), 5.09 (d, J = 12.3 Hz, 1H, CH2-Bn), 4.86 (s, 1H, H-1′), 4.73 (d, J = 2.5 Hz, 1H, H-29a), 4.60 (br s, 1H, H-29b), 4.46 (dq, J = 9.7, 6.2 Hz, 1H, H-5′′′), 4.32 (dq, J = 9.6, 6.3 Hz, 1H, H-5′′), 4.22–4.14 (m, 2H, H-2′, H-3′), 4.03 (dq, J = 9.3, 6.3 Hz, 1H, H-5′), 3.84 (t, J = 9.2 Hz, 1H, H-4′), 3.15–3.07 (m, 1H, H-3), 3.03 (td, J = 10.9, 4.5 Hz, 1H, H-19), 2.29 (br d, J = 12.2 Hz, 1H), 2.18 (td, J = 12.4, 3.2 Hz, 1H), 1.68 (s, 3H, H-30), 1.43 (d, J = 6.2 Hz, 3H, H-6′), 1.36 (d, J = 6.3 Hz, 3H, H-6′′), 1.35 (d, J = 6.2 Hz, 3H, H-6′′′), 0.94 (s, 3H, H-27), 0.91 (s, 3H, H-23), 0.86 (s, 3H, H-25), 0.83 (s, 3H, H-24), 0.76 (s, 3H, H-26), 0.68 (br d, J = 8.2 Hz, 1H, H-5). 13C NMR (101 MHz, CDCl3) δ 175.8 (C-28), [165.9, 165.8, 165.6, 165.5, 165.1, 164.9 (CO-Bz)], 150.6 (C-20), 136.5 (C-Bn), [133.24, 133.23, 133.1, 132.8, 132.7, 132.6, 129.9, 129.82, 129.77, 129.63, 129.59, 129.51, 129.4, 129.3, 128.5, 128.4, 128.35, 128.33, 128.27, 128.21, 128.10, 128.08, 128.0 (CH-Bz, CH-Bn)], 109.6 (C-29), 101.8 (C-1′), 99.7 (C-1′′), 99.5 (C-1′′′), 89.9 (C-3), 81.7 (C-3′), 81.0 (C-4′), 72.0 (C-4′′′), 72.0 (C-4′′), 71.9 (C-2′′), 71.3 (C-2′′′), 71.1 (C-2′), 69.4 (C-3′′′), 69.4 (C-3′′), 67.7 (C-5′′′), 67.6 (C-5′′), 67.1 (C-5′), 65.7 (C-7), 56.6 (C-17), 55.5 (C-5), 50.5 (C-9), 49.5 (C-18), 47.0 (C-19), 42.4 (C-14), 40.7 (C-8), 39.1 (C-4), 38.7 (C-1), 38.2 (C-13), 36.9 (C-22), 36.9 (C-10), 34.3 (C-7), 32.1 (C-16), 30.6 (C-21), 29.7 (C-15), 29.6 (C-15), 28.3 (C-23), 25.7 (C-12), 25.5 (C-2), 20.9 (C-11), 19.4 (C-30), 18.3 (C-6′), 18.3 (C-6), 17.7 (C-6′′), 17.6 (C-6′′′), 16.5 (C-24), 16.2 (C-25), 15.8 (C-26), 14.7 (C-27). HRMS calcd for C97H112O21N [M + NH4]+ 1626.7721, found 1626.7762.:1, 1.5 mL) with NaOMe in MeOH (0.5 M, 1.5 mL) overnight. The reaction was then neutralized to pH 7 with Dowex G26 (H+ form), filtered and purified by flash chromatography (7:1, CHCl3–MeOH to 50:10:1, CHCl3–MeOH–H2O) to give compound 5 (19.5 mg, 84%) as amorphous powder, Rf = 0.3 (26:14:3 CHCl3–MeOH–H2O), [α]20D −6.42 (c, 1.09, 1:1, CHCl3–MeOH). 1H NMR [400 MHz, DMSO-d6 + 1 drop H2O] δ 4.82 (s, 1H, H-1′′), 4.68 (m, 1H, H-29), 4.60 (s, 1H, H-1′′′), 4.58–4.53 (s, 1H, H-1′, H-29b), 3.75–3.55 (m, 6H, H-2′, H-2′′′, H-5′′′, H-3′, H-5′, H-2′), 3.54–3.30 (m, 4H, H-4′, H-3′′′, H-5′′, H-3′′), 3.22–3.14 (m, 2H, H-4′′′, H-4′′), 3.04–2.90 (m, 2H, H-3, H-19), 2.28–2.16 (m, 1H, H-13), 2.14–2.06 (m, 1H, H-16a), 1.63 (s, 3H, H-30), 1.13 (d, J = 6.3 Hz, 3H, H-6′), 1.11 (d, J = 6.3 Hz, 3H, H-6′′), 1.07 (d, J = 6.2 Hz, 3H, H-6′′′), 0.92 (s, 3H, H-27), 0.85 (s, 3H, H-23), 0.85 (s, 3H, H-26), 0.77 (s, 4H, H-25, H-5). 13C NMR [400 MHz, (CD3)2SO + 1 drop H2O] δ 185.5 (C-28), 150.4 (C-20), 109.7 (C-29), 102.9 (C-1′′′), 102.6 (C-1′), 101.8 (C-1′′), 88.0 (C-3), 79.6 (C-3′), 77.4 (C-4′), 71.9 (C-4′′′), 71.7 (C-4′′), 70.7 (C-2′′), 70.7 (C-3′′), 70.5 (C-3′′′), 70.4 (C-2′), 70.4 (C-2′′′), 69.3 (C-5′′), 68.6 (C-5′′′), 67.4 (C-5′), 55.5 (C-17), 54.7 (C-5), 49.9 (C-9), 48.6 (C-18), 46.7 (C-19), 42.1 (C-14), 40.3 (C-8), 38.4 (C-4), 38.0 (C-1), 37.6 (C-13), 36.6 (C-10), 36.5 (C-22), 33.9 (C-7), 31.8 (C-16), 30.2 (C-21), 29.3 (C-15), 27.8 (C-23), 25.2 (C-2), 25.2 (C-12), 20.5 (C-11), 19.0 (C-30), 18.1 (C-6′), 17.9 (C-6), 17.7 (C-6′′), 17.7 (C-6′′′), 16.2 (C-24), 15.9 (C-25), 15.8 (C-26), 14.4 (C-27). HRMS calcd for C48H78O15 [M + Na]+ 917.5232, found 917.5229.:1, 3 mL) to which a freshly prepared solution of NaOMe (0.5 M, 3 mL) was added. After overnight stirring, reaction was neutralized to pH 7 with Dowex G-26 (H+ form) and filtered. The filtrate was concentrated to dryness and purified by normal phase flash chromatography (26:14:3 CHCl3–MeOH–H2O) and by preparative reversed-phase HPLC (Phenomenex Kinetex XB C18 column, gradient elution with H2O:CH3CN 10 → 100% at a flow rate of 20 mL min−1 for 30 min, retention time: 18.944 min) to give 6 (40 mg, 37%, three steps) Rf = 0.19 (26:14:3, CHCl3–MeOH–H2O); [α]20D −37.7 (c 0.1, CHCl3–MeOH 1:1). 1H NMR (400 MHz, CD3OD/CDCl3 1:1) δ 4.97 (s, 1H, H-1′′), 4.93 (s, 1H, H-1′′′′), 4.86 (s, 1H, H-1′′′), 4.82 (s, 1H, H-1′), 4.73 (br s, 1H, H-29), 4.60 (m, 1H, H-29), 3.98 (dd, J = 9.7, 2.8 Hz, 1H, H-3′), 3.93 (br s, 1H, H-2′′′′), 3.90 (br s, 1H, H-2′′′), 3.88 (br s, 1H, H-2′), 3.82 (br s, 1H, H-2′′), 3.80–3.55 (m, 8H, H-5′, H-5′′, H-3′′′′, H-5′′′′, H-5′′′, H-3′′, H-3′′′, H-4′), 3.47–3.37 (m, 3H, H-4′′′, H-4′′, H-4′′′′), 3.10–2.98 (m, 2H, H-3, H-19), 2.32–2.19 (m, 2H, H-13, H-16a), 2.02–1.88 (m, 2H, H-21a, H-22a), 1.69 (s, 3H, H-30), 1.33–1.23 (m, 12H, H-6′′′, H-6′′, H-6′′′′, H-6′), 0.99 (s, 3H, H-27), 0.95 (s, 3H, H-26), 0.92 (s, 3H, H-23), 0.85 (s, 3H, H-25), 0.77 (s, 3H, H-24), 0.72 (br d, J = 9.7 Hz, 1H, H-5). 13C NMR (101 MHz, CD3OD/CDCl3 1:1) δ 179.7 (C-28), 151.1 (C-20), 109.7 (C-29), 103.2 (C-1′′′), 102.7 (C-1′′), 102.6 (C-1′′′′), 101.7 (C-1′), 89.8 (C-3), 80.1 (C-4′), 79.8 (C-3′), 78.8 (C-2′), 73.0 (C-4′′′′), 72.9 (C-4′′′), 72.6 (C-4′′), 71.6 (C-3′′), 71.5 (C-3′′′), 71.4 (C-3′′′′), 71.3 (C-2′′), 71.0 (2×, C-2′′′′, C-2′′′), 69.7 (C-5′′), 69.6 (C-5′′′), 69.3 (C-5′′′′), 68.1 (C-5′), 56.6 (C-17), 55.8 (C-5), 50.9 (C-9), 49.6 (C-18), 47.4 (C-19), 42.8 (C-14), 41.1 (C-8), 39.5 (C-4), 39.0 (C-1), 38.7 (C-13), 37.5 (C-22), 37.3 (C-10), 34.7 (C-7), 32.7 (C-16), 31.0 (C-21), 30.0 (C-15), 28.4 (C-23), 26.0 (C-2), 25.9 (C-12), 21.3 (C-11), 19.5 (C-30), 18.6 (C-6), 18.2 (C-6′), 17.6 (C-6′′′′), 17.6 (C-6′′′), 17.3 (C-6′′), 16.5 (C-24), 16.4 (C-25), 16.2 (C-26), 14.9 (C-27). HRMS calcd for C54H92O19N [M + NH4]+ 1058.62581, found 1058.62553.:2, hexanes–EtOAc) to yield 21 as a white powder (342 mg, 68%): Rf = 0.52 (8:2, hexanes–EtOAc); [α]20D +82.6 (c 1, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 7.2 Hz, 2H, CH-Bz), 7.99 (d, J = 7.2 Hz, 2H, CH-Bz), 7.84 (d, J = 7.2 Hz, 2H, CH-Bz), 7.60 (t, J = 7.4 Hz, 2H, CH-Bz), 7.55–7.22 (m, 13H, CH-Bz, CH-Bn), 5.84 (dd, J = 10.2, 3.3 Hz, 1H, H-3′), 5.69 (t, J = 10.0 Hz, 1H, H-4′), 5.65 (dd, J = 3.2, 1.7 Hz, 1H, H-2′), 5.25 (t, J = 3.1 Hz, 1H, H-12), 5.12 (d, J = 12.5 Hz, 1H, CH2-Bn), 5.09 (br s, 1H, H-1′), 4.99 (d, J = 12.5 Hz, 1H, CH2-Bn), 4.32 (dq, J = 9.7, 6.1 Hz, 1H, H-5′), 3.24 (m, 1H, H-3), 2.27 (d, J = 11.3 Hz, 1H, H-18) 1.34 (d, J = 6.3 Hz, 3H, H-6′), 1.08 (s, 3H, H-27), 1.07 (s, 3H, H-23), 0.97 (s, 3H, H-25), 0.96 (s, 3H, H-24), 0.93 (d, J = 6.1 Hz, 3H, H-30), 0.86 (d, J = 6.4 Hz, 3H, H-29), 0.76 (d, J = 11.2 Hz, 1H, H-5), 0.66 (s, 3H, H-26). 13C NMR (101 MHz, CDCl3) δ 177.3 (C-28), [165.8, 165.7, 165.6 (CO-Bz)], 138.1 (C-13), 136.4 (C-Bn), [133.4, 133.3, 133.1, 129.9, 129.73, 129.66 (CH-Bz)], [129.5, 129.32, 129.25 (C-Bz)], [128.6, 128.4 (2×), 128.3, 128.1, 127.9 (CH-Bz, CH-Bn)], 125.6 (C-12), 99.7 (C-1′), 90.0 (C-3), 72.0 (C-4′), 71.2 (C-2′), 70.2 (C-3′), 66.7 (C-5′), 66.0 (CH2-Bn), 55.4 (C-5), 52.9 (C-18), 48.1 (C-17), 47.5 (C-9), 42.0 (C-14), 39.5 (C-8), 39.1 (C-19), 39.0 (C-4), 38.8 (C-20), 38.6 (C-1), 36.7 (C-10), 36.6 (C-16), 33.0 (C-7), 30.6 (C-21), 28.4 (C-23), 27.9 (C-15), 25.5 (C-2), 24.2 (C-22), 23.5 (C-27), 23.3 (C-11), 21.2 (C-30), 18.3 (C-6), 17.6 (C-6′), 17.01 (C-26), 17.98 (C-29), 16.6 (C-24), 15.5 (C-25). HRMS calcd for C64H76O10Na [M + Na]+ 1027.53307, found 1027.53329.:1, 5 mL) with NaOMe in MeOH (0.5 M, 5 mL) overnight. The reaction was then neutralized to pH 7 with Dowex G26 (H+ form), filtered and purified by flash chromatography (9:1, DCM–MeOH) to give compound 22 (202 mg, 98%) as amorphous solid, Rf = 0.21 (7:1, CHCl3:MeOH), [α]20D +1.3 (c 7, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.36–7.31 (m, 5H, CH-Bn), 5.23 (t, J = 3.1 Hz, 1H, H-12), 5.11 (d, J = 12.5 Hz, 1H, CH2-Bn), 4.97 (d, J = 12.5 Hz, 1H, CH2-Bn), 4.80 (br s, 1H, H-1′), 3.93 (br s, 1H, H-2′), 3.79–3.74 (m, 2H, H-3′, H-5′), 3.47 (m, 1H, H-4′), 3.07 (dd, J = 10.5, 4.4 Hz, 1H, H-3), 2.26 (d, J = 11.1 Hz, 1H, H-18), 1.27 (d, J = 6.0 Hz, 1H, H-6′), 1.06 (s, 3H, H-27), 0.93 (s, 3H, H-30), 0.90 (s, 3H, H-23), 0.89 (s, 3H, H-25) 0.85 (d, J = 6.2 Hz, 3H, H-29), 0.75 (s, 3H, H-24), 0.69 (d, J = 11.1 Hz, 1H, H-5), 0.63 (s, 3H, H-26). 13C NMR (101 MHz, CDCl3) δ 177.3 (C-28), 138.1 (C-13), 136.4 (C-Bn), [128.4 (2×), 128.1 (2×), 127.9 (CH-Bn)], 125.7 (C-12), 102.2 (C-1′), 89.6 (C-3), 73.8 (C-4′), 72.0 (C-3′), 71.3 (C-2′), 67.7 (C-5′), 66.0 (CH2-Bn), 55.4 (C-5), 52.9 (C-18), 48.1 (C-17), 47.5 (C-9), 42.0 (C-14), 39.5 (C-8), 39.1 (C-19), 39.0 (C-4), 38.8 (C-20), 38.6 (C-1), 36.6 (C-22), 36.6 (C-10), 33.0 (C-7), 30.6 (C-21), 28.3 (C-23), 27.9 (C-15), 25.3 (C-2), 24.2 (C-16), 23.6 (C-27), 23.3 (C-11), 21.2 (C-30), 18.2 (C-6), 17.4 (C-6′), 17.04 (C-29), 16.98 (C-26), 16.5 (C-24), 15.5 (C-25). HRMS calcd for C43H64O7Na [M + Na]+ 715.45443, found 715.45649.:1, 2 mL) overnight. The reaction was then neutralized to pH 7 with Dowex G26 (H+ form), filtered and purified by flash chromatography (7:1, DCM–MeOH) to give compound 7 (24 mg, 74%, two steps) as colorless oil, Rf = 0.3 (DCM: MeOH, 7:1), [α]20D +5.7 (c 0.74, CHCl3–MeOH 1:1). 1H NMR (400 MHz, CD3OD/CDCl3 1:1) δ 5.24 (br s, 1H, H-12), 4.77 (br s, 1H, H-1′), 3.89 (dd, J = 3.4, 1.7 Hz, 1H, H-2′), 3.76 (dq, J = 9.5, 6.2 Hz, 1H, H-5′), 3.70 (dd, J = 9.5, 3.3 Hz, 1H, H-3′), 3.39 (t, J = 9.5 Hz, 1H, H-4′), 3.10 (dd, J = 11.2, 4.7 Hz, 1H, H-3), 2.20 (d, J = 11.3 Hz, 1H, H-18), 1.27 (d, J = 6.1 Hz, 3H, H-6′), 1.10 (s, 3H, H-27), 0.96 (m, 3H, H-30), 0.95 (s, 3H, H-23), 0.95 (s, 3H, H-25), 0.88 (d, J = 6.4 Hz, 3H, H-29), 0.83 (s, 3H, H-26), 0.78 (s, 3H, H-24). 13C NMR (101 MHz, CD3OD/CDCl3 1:1) δ 181.1 (C-28), 138.7 (C-13), 125.9 (C-12), 103.2 (C-1′), 89.7 (C-3), 73.4 (C-4′), 71.9 (C-3′), 71.5 (C-2′), 68.7 (C-5′), 55.8 (C-5), 53.3 (C-18), 48.2 (C-17), 48.0 (C-9), 42.5 (C-14), 39.9 (C-8), 39.6 (C-19), 39.4 (C-20), 39.3 (C-4), 39.0 (C-1), 37.3 (C-22), 37.1 (C-10), 33.5 (C-7), 31.1 (C-21), 30.0 (C-), 28.5 (C-23), 28.5 (C-15), 25.8 (C-2), 24.6 (C-16), 23.8 (C-27), 23.7 (C-11), 21.4 (C-30), 18.7 (C-6), 17.5 (C-6′), 17.3 (C-29), 17.2 (C-26), 16.7 (C-24), 15.7 (C-25). HRMS calcd for C36H58O7 [M + H]+ 603.4255, found 603.4273.:3, hexanes–EtOAc) to give compound 23 (392 mg, 74%) as colorless oil, Rf = 0.7 (hexanes–EtOAc, 7:3), [α]20D +6.7 (c 10, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.38–7.28 (m, 5H, CH-Bn), 5.23 (t, J = 3.1 Hz, 1H, H-12), 5.10 (d, J = 12.5 Hz, 1H, CH2-Bn), 4.99 (s, 1H, H-1′), 4.97 (d, J = 12.2 Hz, 1H, CH2-Bn), 3.16–4.10 (m, 2H, H-2′, H-3′), 3.82 (dq, J = 12.6, 6.2 Hz, 1H, H-5′), 3.42 (t, 1H, J = 8.5 Hz, H-4′), 3.15 (dd, J = 10.8, 4.3 Hz, 1H, H-3), 2.26 (d, J = 11.1 Hz, 1H, H-18), 1.99 (dd, J = 12.8, 4.1 Hz, 1H), 1.53 (s, 3H, CH3-iso), 1.37 (s, 3H, CH3-iso), 1.26 (d, J = 6.6 Hz, 3H, H-6′), 1.07 (s, 3H, H-27), 0.94 (s, 6H, H-23, H-30), 0.90 (s, 3H, H-25), 0.85 (d, J = 6.3 Hz, 3H, H-29), 0.77 (s, 3H, H-24), 0.72 (d, 1H, J = 11.3 Hz H-5), 0.63 (s, 3H, H-26). 13C NMR (101 MHz, CDCl3) δ 177.3 (C-28), 138.0 (C-13), 136.3 (C-Bn), [128.4 (2×), 128.1 (2×), 127.9 (CH-Bn)], 125.7 (C-12), 109.3 (C-iso), 99.7 (C-1′), 88.9 (C-3), 78.3 (C-3′), 75.8 (C-2′), 74.1 (C-4′), 66.2 (C-5′), 66.0 (CH2-Bn), 55.3 (C-5), 52.8 (C-18), 48.1 (C-17), 47.5 (C-9), 42.0 (C-14), 39.5 (C-8), 39.1 (C-19), 39.0 (C-4), 38.8 (C-20), 38.6 (C-1), 36.6 (C-10), 36.6 (C-22), 32.9 (C-7), 30.6 (C-21), 28.4 (C-23), 27.9 (CH3-iso), 27.9 (C-15), 26.2 (CH3-iso), 25.4 (C-2), 24.2 (C-16), 23.5 (C-27), 23.2 (C-11), 21.2 (C-30), 18.2 (C-6), 17.5 (C-6′), 17.0 (C-29), 17.0 (C-26), 16.5 (C-24), 15.5 (C-25). HRMS calcd for C46H68O7Na [M + Na]+ 755.48573, found 755.48487.:2, hexanes–EtOAc) to give compound 24 (219 mg, 70%) as colorless oil, Rf = 0.6 (hexanes–EtOAc, 8:2), [α]20D +36.1 (c 2, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 7.2 Hz, 2H, CH-Bz), 7.96 (d, J = 7.3 Hz, 2H, CH-Bz), 7.81 (d, J = 7.3 Hz, 2H, CH-Bz), 7.61 (t, J = 7.5 Hz, 1H, CH-Bz), 7.54–7.46 (m, 3H, CH-Bz), 7.44–7.30 (m, 7H, CH-Bz, CH-Bn), 7.28–7.22 (m, 3H, CH-Bz), 5.77 (dd, J = 10.1, 3.3 Hz, 1H, H-3′′), 5.74 (dd, J = 3.1, 1.8 Hz, 1H, H-2′′), 5.67 (t, J = 9.8 Hz, 1H, H-4′′), 5.59 (d, J = 1.7 Hz, 1H, H-1′′), 5.26 (br s, 1H, H-12), 5.12 (d, J = 12.5 Hz, 1H, CH2-Bn), 5.04 (br s, 1H, H-1′), 4.99 (d, J = 12.5 Hz, 1H, CH2-Bn), 4.31 (dd, J = 7.2, 5.5 Hz, 1H, H-3′), 4.20 (dq, J = 9.6, 6.4 Hz, 1H, H-5′′), 4.14 (d, J = 5.5 Hz, 1H, H-2′), 3.98 (dq, J = 10.0, 6.1 Hz, 1H, H-5′), 3.62 (dd, J = 9.9, 7.3 Hz 1H, H-4′), 3.17 (dd, J = 9.2, 6.9 Hz, 1H, H-3), 2.28 (d, J = 11.3 Hz, 1H, H-18), 2.02 (td, J = 12.7, 3.9 Hz, 1H, H-16), 1.55 (s, 3H, CH3-iso), 1.38 (m, 6H, H-6′, H-6′′), 1.35 (s, 3H, CH3-iso), 1.08 (s, 3H, H-27), 0.96 (s, 3H, H-23), 0.94 (m, 6H, H-30, H-25), 0.86 (d, J = 6.2 Hz, H-29), 0.83 (s, 3H, H-24), 0.75 (br d, J = 11.5 Hz, 1H, H-5), 0.66 (s, 3H, H-26). 13C NMR (101 MHz, CDCl3) δ 177.3 (C-28), [165.7, 165.6, 165.4 (CO-Bz)], 138.1 (C-13), 136.4 (C-Bn), [133.4, 133.3, 133.1, 130.0 (2×), 129.71 (2×), 129.68 (2×) (CH-Bz)], [129.5, 129.3, 129.2 (C-Bz)], [128.5 (2×), 128.4 (4×), 128.25 (2×), 128.16 (2×), 127.9 (CH-Bz, CH-Bn)], 125.7 (C-12), 109.5 (C-iso), 99.5 (C-1′), 96.0 (C-1′′), 89.0 (C-3), 78.2 (C-3′), 77.9 (C-4′), 76.3 (C-2′), 71.7 (C-4′′), 70.8 (C-2′′), 70.0 (C-3′′), 67.3 (C-5′′), 66.0 (C-7), 63.7 (C-5′), 55.4 (C-5), 52.9 (C-18), 48.1 (C-17), 47.6 (C-9), 42.0 (C-14), 39.5 (C-8), 39.09 (C-19), 39.08 (C-4), 38.8 (C-20), 38.7 (C-1), 36.7 (2×, C-22, C-10), 33.0 (C-7), 30.7 (C-21), 28.4 (C-23), 28.0 (CH3-iso), 27.9 (C-15), 26.5 (CH3-iso), 25.5 (C-2), 24.2 (C-16), 23.6 (C-27), 23.3 (C-11), 21.2 (C-30), 18.3 (C-6), 18.1 (C-6′), 17.7 (C-6′′), 17.0 (C-29), 17.0 (C-26), 16.6 (C-24), 15.5 (C-25). HRMS calcd for C73H90O18N [M + NH4]+ 1208.6668, found 1208.6647.:2, hexanes–EtOAc) to give compound 25 (129.1 mg, 83%) as a colorless oil, Rf = 0.5 (hexanes-EtOAc, 8:2), [α]20D +37.7 (c 4.1, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 7.3 Hz, 2H, CH-Bz), 7.97 (d, J = 7.3 Hz, 2H, CH-Bz), 7.82 (d, J = 7.3 Hz, 2H, CH-Bz), 7.58 (t, J = 7.4 Hz, 1H, CH-Bz), 7.51 (t, J = 7.4 Hz, 1H, CH-Bz), 7.48–7.30 (m, 10H, CH-Bz), 7.28–7.22 (m, 2H, CH-Bz), 5.82 (dd, J = 10.1, 3.2 Hz, 1H, H-3′′) 5.79 (m, 1H, H-2′′), 5.68 (t, J = 9.9 Hz, 1H, H-4′′), 5.49 (br s, 1H, H-1′′), 5.25 (br s, 1H, H-12), 5.11 (d, J = 12.5 Hz, 1H, CH2-Bn), 4.99 (d, J = 12.5 Hz, 1H, CH2-Bn), 4.86 (br s, 1H, H-1′), 4.30 (dq, J = 9.6, 6.2 Hz, 1H, H-5′′), 4.07 (dd, J = 9.0, 2.8 Hz, 1H, H-3′), 4.01–3.92 (m, 2H, H-2′, H-5′), 3.64 (m, 1H, H-4′), 3.11 (dd, J = 10.5, 5.4 Hz, 1H, H-3), 2.27 (d, J = 11.1 Hz, 1H, H-18), 1.37 (m, 6H, H-6′, H-6′′), 1.08 (s, 3H, H-27), 0.94 (m, 3H, H-30), 0.93 (s, 3H, H-25), 0.91 (s, 3H, H-23), 0.86 (d, J = 6.4 Hz, 3H, H-29), 0.81 (s, 3H, H-24), 0.71 (br d, J = 11.4 Hz, 1H, H-5), 0.65 (s, 3H, H-26). 13C NMR (101 MHz, CDCl3) δ 177.4 (C-28), [165.9, 165.8, 165.7 (CO-Bz)], 138.1 (C-13), 136.3 (C-Bn), [133.5, 133.3, 133.2, 129.9 (2×), 129.7 (4×) (CH-Bz)], [129.3, 129.2, 129.1 (C-Bz)], [128.5 (2×), 128.4 (4×), 128.3 (2×), 128.1 (2×), 127.9 (CH-Bz, CH-Bn)], 125.7 (C-12), 102.0 (C-1′), 99.0 (C-1′′), 89.5 (C-3), 81.6 (C-4′), 71.9 (C-2′), 71.9 (C-3′), 71.6 (C-4′′), 71.2 (C-2′′), 70.1 (C-3′′), 67.5 (C-5′′), 66.3 (C-5′), 66.0 (CH2-Bn), 55.3 (C-5), 52.9 (C-18), 48.1 (C-17), 47.5 (C-9), 42.0 (C-14), 39.5 (C-8), 39.1 (C-19), 38.9 (C-4), 38.8 (C-20), 38.6 (C-1), 36.6 (C-22), 36.6 (C-10), 33.0 (C-7), 30.6 (C-21), 28.3 (C-23), 27.9 (C-15), 25.4 (C-2), 24.2 (C-16), 23.6 (C-27), 23.3 (C-11), 21.2 (C-30), 18.2 (C-6), 17.9 (C-6′), 17.6 (C-6′′), 17.0 (C-29), 17.0 (C-26), 16.6 (C-24), 15.4 (C-25). HRMS calcd for C70H90O14N [M + NH4]+ 1168.6355, found 1168.6351.:3, hexanes–EtOAc) to give compound 26 (33 mg, 89%) as a colorless oil, Rf = 0.6 (hexanes–EtOAc, 7:3), [α]20D +31.3 (c 3, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 7.7 Hz, 2H, CH-Bz), 7.97 (d, J = 7.8 Hz, 2H, CH-Bz), 7.82 (d, J = 7.8 Hz, 2H, CH-Bz), 7.58 (t, J = 7.4 Hz, 1H, CH-Bz), 7.52 (t, J = 7.4 Hz, 1H, CH-Bz), 7.45 (t, J = 7.6 Hz, 2H, CH-Bz), 7.43–7.36 (m, 3H, CH-Bz), 7.28–7.23 (m, 2H, CH-Bz), 5.81 (dd, J = 10.2, 3.4 Hz, 1H, H-3′′), 5.75 (br s, 1H, H-2′′), 5.68 (t, J = 9.9 Hz, 1H, H-4′′), 5.50 (s, 1H, H-1′′), 5.26 (br s, 1H, H-12), 4.86 (s, 1H, H-1′), 4.29 (dq, J = 9.6, 6.5 Hz, 1H, H-5′′), 4.10 (dd, J = 9.1, 3.3 Hz, 1H, H-3′), 4.01–3.93 (m, 2H, H-2′, H-5′), 3.64 (t, J = 9.2 Hz, 1H, H-4′), 3.13 (dd, J = 9.5, 6.0 Hz, 1H, H-3), 2.19 (br d, J = 11.1 Hz, 1H, H-18), 1.38 (d, J = 6.0 Hz, 3H, H-6′), 1.36 (d, J = 6.5 Hz, 3H, H-6′′) 1.08 (s, 3H, H-27), 0.98 (s, 3H, H-25), 0.95 (s, 3H, H-23), 0.95 (m, 3H, H-30), 0.87 (d, J = 6.3 Hz, 3H, H-29), 0.84 (s, 3H, H-24), 0.79 (s, 3H, H-26), 0.75 (m, 1H, H-5). 13C NMR (101 MHz, CDCl3) δ 182.9 (C-28), [165.9 (2×), 165.8 (CO-Bz)], 137.9 (C-13), [133.5, 133.4, 133.2, 130.0 (2×), 129.7 (4×) (CH-Bz)], [129.3 (2×), 129.1 (C-Bz)], [128.6 (2×), 128.4 (2×), 128.3 (2×) (CH-Bz)], 125.9 (C-12), 101.9 (C-1′), 98.8 (C-1′′), 89.6 (C-3), 81.2 (C-4′), 72.0 (C-2′), 71.9 (C-3′), 71.6 (C-4′′), 71.2 (C-2′′), 70.0 (C-3′′), 67.5 (C-5′′), 66.2 (C-5′), 55.4 (C-5), 52.6 (C-18), 47.9 (C-17), 47.5 (C-9), 41.9 (C-14), 39.5 (C-8), 39.1 (C-19), 39.0 (C-4), 38.8 (C-20), 38.6 (C-1), 36.7 (2×, C-22, C-10), 32.9 (C-7), 30.6 (C-21), 28.4 (C-23), 28.0 (C-15), 25.5 (C-2), 24.1 (C-16), 23.6 (C-27), 23.3 (C-11), 21.2 (C-30), 18.3 (C-6), 18.0 (C-6′), 17.6 (C-6′′), 17.1 (C-26), 17.0 (C-29), 16.6 (C-24), 15.5 (C-25). HRMS calcd for C63H80O14Na [M + Na]+ 1083.54403, found 1083.54598.:1, 3 mL) with NaOMe in MeOH (0.5 M, 3 mL) overnight. The reaction was then neutralized to pH 7 with Dowex G26 (H+ form), filtered and purified by flash chromatography (9:1 DCM–MeOH → 26:14:3 DCM–MeOH–H2O) to give compound 8 (20 mg, 86%) as a colorless oil, Rf = 0.2 (DCM: MeOH, 9:1), [α]20D −40.0 (c 0.5, 1:1, CHCl3–MeOH). 1H NMR (400 MHz, CDCl3/CD3OD 1:1) δ 5.24 (m, 1H, H-12), 5.20 (d, J = 1.3 Hz, 1H, H-1′′), 4.75 (m, 1H, H-1′), 3.98 (dd, J = 3.2, 1.8 Hz, 1H, H-2′′), 3.82 (dd, J = 3.1, 1.7 Hz, 1H, H-2′), 3.79 (dd, J = 9.2, 3.3 Hz, 1H, H-3′), 3.76–3.70 (m, 2H, H-5′, H-5′′), 3.68 (dd, J = 9.5, 3.3 Hz, 1H, H-3′′), 3.52 (t, J = 9.3 Hz, 1H, H-4′), 3.41 (t, J = 9.5 Hz, 1H, H-4′′), 3.11 (dd, J = 10.7, 4.7 Hz, 1H, H-3), 2.20 (br d, J = 11.4 Hz, 1H, H-18), 2.02 (td, J = 13.3, 3.6 Hz, 1H, H-16a), 1.28 (d, J = 6.1 Hz, 6H, H-6′, H-6′′), 1.11 (s, 3H, H-27), 0.96 (m, 9H, H-25, H-30, H-23), 0.88 (d, J = 6.4 Hz, 3H, H-29), 0.84 (s, 3H, H-26), 0.80 (s, 3H, H-24). 13C NMR (101 MHz, CDCl3/CD3OD 1:1) δ 180.3 (C-28), 138.9 (C-13), 126.1 (C-12), 103.3 (C-1′), 102.3 (C-1′′), 89.9 (C-3), 80.4 (C-4′), 73.2 (C-4′′), 72.7 (C-3′), 72.2 (C-2′), 71.7 (C-3′′), 71.5 (C-2′′), 69.6 (C-5′′), 67.5 (C-5′), 56.0 (C-5), 53.5 (C-18), 48.2 (C-9), 48.0 (C-17), 42.6 (C-14), 40.1 (C-8), 39.7 (C-19), 39.6 (C-20), 39.5 (C-4), 39.2 (C-22), 37.5 (C-1), 37.2 (C-10), 33.6 (C-7), 31.2 (C-21), 28.6 (C-15), 28.5 (C-23), 26.0 (C-2), 24.8 (C-16), 23.9 (C-27), 23.8 (C-11), 21.4 (C-30), 18.8 (C-6), 18.2 (C-6′), 17.6 (C-6′′), 17.4 (C-26), 17.3 (C-29), 16.8 (C-24), 15.8 (C-25). HRMS calcd for C42H68O11Na [M + Na]+ 771.4653, found 771.4657.:3, hexanes–EtOAc) to give compound 27 (83 mg, 62%) as colorless oil, Rf = 0.6 (hexanes–EtOAc, 8:2), [α]20D −1.0 (c 4.4, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.02–7.92 (m, 8H, CH-Bz), 7.89 (d, J = 8.0 Hz, 2H, CH-Bz), 7.77 (d, J = 8.0 Hz, 2H, CH-Bz), 7.56–7.23 (m, 19H, CH-Bz), 7.17 (t, J = 7.8 Hz, 2H, CH-Bz), 7.10 (t, J = 7.8 Hz, 2H, CH-Bz), 6.04 (dd, J = 3.5, 1.8 Hz, 1H, H-2′′′), 5.98 (dd, J = 10.1, 3.3 Hz, 1H, H-3′′′), 5.95 (dd, J = 10.2, 3.1 Hz, 1H, H-3′′), 5.83 (dd, J = 3.4, 1.9 Hz, 1H, H-2′′), 5.75 (t, J = 10.0 Hz, 1H, H-4′′), 5.67 (t, J = 9.9 Hz, 1H, H-4′′′), 5.54 (br s, 1H, H-1′′), 5.37 (br s, 1H, H-1′′′), 5.25 (br s, 1H, H-12), 5.10 (d, J = 12.5 Hz, 1H, CH2-Bn), 4.99 (d, J = 12.5 Hz, 1H, CH2-Bn), 4.87 (br s, 1H, H-1′), 4.47 (dq, J = 9.8, 6.3 Hz, 1H, H-5′′′), 4.32 (dq, J = 9.8, 5.9 Hz, 1H, H-5′′), 4.22–4.16 (m, 2H, H-2′, H-3′), 4.04 (dq, J = 9.7, 6.3 Hz, 1H, H-5′), 3.85 (t, J = 9.3 Hz, 1H, H-4′), 3.14 (t, J = 7.9 Hz, 1H, H-3), 2.27 (d, J = 11.1 Hz, 1H, H-18), 1.43 (d, J = 6.1 Hz, 3H, H-6′), 1.36 (d, J = 6.8 Hz, 3H, H-6′′), 1.35 (d, J = 7.1 Hz, 3H, H-6′′′), 1.07 (s, 3H, H-27), 0.96 (s, 3H, H-25), 0.93 (m, 6H, H-23, H-30), 0.85 (m, 6H, H-24, H-29), 0.73 (br d, J = 12.0 Hz, 1H, H-5), 0.65 (s, 3H, H-26). 13C NMR (101 MHz, CDCl3) δ 177.3 (C-28), [165.87, 165.85, 165.6, 165.5, 165.1, 164.9 (CO-Bz)], 138.1 (C-13), 136.4 (C-Bn), [133.25, 133.22, 133.1, 132.84, 132.76, 132.6, 129.9 (4×), 129.81 (2×), 129.77 (4×) (CH-Bz)], [129.71, 129.63, 129.58, 129.51, 129.4, 129.3 (C-Bz)], [128.41 (2×), 128.39 (2×), 128.35 (2×), 128.33 (2×), 128.26, 128.22 (2×), 128.17 (2×), 128.1 (2×), 127.97 (2×), 127.96 (2×) (CH-Bz, CH-Bn], 125.7 (C-12), 101.9 (C-1′), 99.7 (C-1′′), 99.5 (C-1′′′), 89.9 (C-3), 81.7 (C-3′), 81.0 (C-4′), 72.0 (C-4′′), 72.0 (C-4′′′), 72.0 (C-2′′), 71.3 (C-2′′′), 71.1 (C-2′), 69.5 (C-3′′′), 69.5 (C-3′′), 67.7 (C-5′′′), 67.6 (C-5′′), 67.1 (C-5′), 66.0 (C-7), 55.4 (C-5), 52.9 (C-18), 48.1 (C-17), 47.6 (C-9), 42.1 (C-14), 39.6 (C-8), 39.1 (C-19), 39.0 (C-4), 38.8 (C-20), 38.7 (C-1), 36.7 (C-10), 36.7 (C-22), 33.0 (C-7), 30.7 (C-21), 28.4 (C-23), 28.0 (C-15), 25.7 (C-2), 24.3 (C-16), 23.6 (C-27), 23.3 (C-11), 21.2 (C-30), 18.3 (C-6′), 18.3 (C-6), 17.7 (C-6′′), 17.6 (C-6′′′), 17.0 (C-29), 17.0 (C-26), 16.7 (C-24), 15.5 (C-25). HRMS calcd for C97H112O11N [M + NH4]+ 1626.7721, found 1626.7724.:2, hexanes–EtOAc) to give compound 28 (33.2 mg, 81%) as a colorless oil, Rf = 0.3 (hexanes–EtOAc, 8:2), [α]20D +60.6 (c 3.4, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.01–7.92 (m, 8H, CH-Bz), 7.88 (d, J = 7.5 Hz, 2H, CH-Bz), 7.77 (d, J = 7.6 Hz, 2H, CH-Bz), 7.54–7.25 (m, 9H, CH-Bz), 7.16 (t, J = 7.7 Hz, 2H, CH-Bz), 7.10 (t, J = 7.8 Hz, 2H, CH-Bz), 6.04 (br s, 1H, H-2′′′), 6.00 (dd, J = 10.0, 3.0 Hz, 1H, H-3′′′), 5.95 (dd, J = 10.0, 3.0 Hz, 1H, H-3′′), 5.83 (br s, 1H, H-2′′), 5.76 (t, J = 9.9 Hz, 1H, H-4′′), 5.67 (t, J = 10.0 Hz, 1H, H-4′′′), 5.54 (s, 1H, H-1′′), 5.38 (s, 1H, H-1′′′), 5.26 (br s, 1H, H-12), 4.88 (s, 1H, H-1′), 4.47 (dq, J = 10.0, 6.4 Hz, 1H, H-5′′′), 4.32 (dq, J = 9.7, 6.4 Hz, 1H, H-5′′), 4.22–4.15 (m, 2H, H-2′, H-3′), 4.04 (m, H-5′), 3.85 (t, J = 8.7 Hz, 1H, H-4′), 3.15 (t, J = 8.0 Hz, 1H, H-3), 2.19 (br d, J = 10.9 Hz, 1H, H-18), 1.43 (d, J = 6.1 Hz, 3H, H-6′), 1.37 (d, J = 6.1 Hz, 3H, H-6′′), 1.35 (d, J = 6.0 Hz, 3H, H-6′′′), 1.09 (s, 3H, H-27), 0.99 (s, 3H, H-25), 0.96 (m, 6H, H-30, H-23), 0.86 (m, 6H, H-29, H-24), 0.80 (s, 3H, H-26), 0.75 (br d, J = 11.7 Hz, 1H, H-5). 13C NMR (101 MHz, CDCl3) δ 182.5 (C-28), [165.86, 165.85, 165.6, 165.5, 165.1, 164.9 (CO-Bz)], 137.9 (C-13), [133.24, 133.22, 133.1, 132.8, 132.7, 132.6, 129.95 (2×), 129.92 (2×), 129.81 (4×), 129.76 (4×), 129.63, 129.57, 129.51, 129.4, 129.3 (2×), 128.39 (2×), 128.35 (2×), 128.33 (2×), 128.2 (2×), 128.1 (2×), 128.0 (2×), CH-Bz, C-Bz], 125.9 (C-12), 101.9 (C-1′), 99.7 (C-1′′), 99.5 (C-1′′′), 89.9 (C-3), 81.7 (C-3′), 81.0 (C-4′), 72.0 (C-4′′′), 72.0 (C-4′′), 71.9 (C-2′′), 71.3 (C-2′′′), 71.1 (C-2′), 69.4 (C-3′′′), 69.4 (C-3′′), 67.7 (C-5′′′), 67.6 (C-5′′), 67.1 (C-5′), 55.4 (C-5), 52.6 (C-18), 47.9 (C-17), 47.6 (C-9), 42.0 (C-14), 39.5 (C-8), 39.1 (C-19), 39.0 (C-4), 38.8 (C-20), 38.7 (C-1), 36.7 (C-10), 36.6 (C-22), 32.9 (C-7), 30.5 (C-21), 28.4 (C-23), 28.0 (C-15), 25.5 (C-2), 24.2 (C-16), 23.6 (C-27), 23.4 (C-11), 21.2 (C-30), 18.8 (C-6), 18.3 (C-6′), 17.7 (C-6′′), 17.6 (C-6′′′), 17.0 (C-29), 16.9 (C-26), 16.7 (C-24), 15.5 (C-25). HRMS calcd for C90H106O21N [M + NH4]+ 1536.7252, found 1536.7236.:1, 3 mL) with NaOMe in MeOH (0.5 M, 3 mL) overnight. The reaction was then neutralized to pH 7 with Dowex G26 (H+ form), filtered and purified by flash chromatography (9:1, DCM–MeOH → 26:14:3 DCM–MeOH–H2O) to give compound 9 (17 mg, 84%) as a colorless oil, Rf = 0.3 (26:14:3 CHCl3–MeOH–H2O), [α]20D +12.3 (c 1.8, 1:1, CHCl3–MeOH). 1H NMR [400 MHz, CD3OD/CDCl3 1:1] δ 5.23 (br s, H-12), 5.00 (br s, 1H, H-1′′), 4.88 (br s, 1H, H-1′′′), 4.73 (m, 1H, H-1′), 3.96–3.62 (m, 10H, H-2′′′, H-2′, H-3′, H-2′′, H-5′, H-5′′′, H-3′′′, H-5′′, H-4′, H-3′′), 3.46–3.38 (m, 2H, H-4′′, H-4′′′), 3.11 (dd, J = 10.9, 4.7 Hz, 1H, H-3), 1.32–1.26 (m, 9H, H-6′, H-6′′′, H-6′′), 1.11 (s, 3H, H-27), 0.99–0.94 (m, 9H, H-30, H-25, H-23), 0.88 (d, J = 6.4 Hz, 3H, H-29), 0.85 (s, 3H, H-26), 0.81 (s, 3H, H-24). 13C NMR [101 MHz, CD3OD/CDCl3 1:1] δ 179.7 (C-28), 139.2 (C-13), 125.9 (C-12), 103.5 (C-1′′′), 103.3 (C-1′), 102.9 (C-1′′), 90.1 (C-3), 80.9 (C-3′), 80.0 (C-4′), 73.2 (C-4′′′), 73.0 (C-4′′), 71.8 (C-3′′), 71.7 (C-2′′), 71.6 (C-2′′′), 71.6 (C-3′′′), 71.4 (C-2′), 69.9 (C-5′′), 69.6 (C-5′′′), 68.3 (C-5′), 56.0 (C-5), 53.7 (C-18), 48.2 (C-9), 42.7 (C-14), 40.1 (C-8), 39.9 (C-19), 39.7 (C-20), 39.5 (C-4), 39.2 (C-22), 37.3 (C-1), 37.3 (C-10), 33.7 (C-7), 28.7 (C-15), 28.6 (C-23), 26.0 (C-2), 24.9 (C-16), 23.9 (C-27), 23.9 (C-11), 21.5 (C-30), 18.9 (C-6), 18.2 (C-6′), 17.6 (C-6′′′), 17.5 (C-6′′), 17.5 (C-26), 17.5 (C-29), 16.9 (C-24), 15.9 (C-25). HRMS: calcd for C48H82O15N [M + NH4]+ 912.5679, found 912.5678.:3, hexanes-EtOAc); [α]20D +12.3 (c 1, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.07–7.73 (m, 18H, CH-Bz), 7.55–7.06 (m, 32H, CH-Bz, CH-Bn), 6.11–5.94 (m, 5H, H-3′′′, H-2′′′, H-3′′, H-3′′′′, H-2′′), 5.88 (dd, J = 3.4, 1.6 Hz, 1H, H-2′′′′), 5.78–5.67 (m, 3H, H-4′′, H-4′′′′, H-4′′′), 5.60 (br s, 1H, H-1′′), 5.39 (br s, 1H, H-1′′′), 5.38 (br s, 1H, H-1′′′′) 5.26 (t, J = 3.5 Hz, 1H, H-12), 5.11 (d, J = 12.5 Hz, 1H, CH2-Bn), 5.07 (br s, 1H, H-1′), 4.99 (d, J = 12.5 Hz, 1H, CH2-Bn), 4.50 (dq, J = 9.7, 6.5 Hz, 1H, H-5′′′), 4.44–4.34 (m, 2H, H-5′′′′, H-5′′), 4.32 (dd, J = 8.8, 2.6 Hz, 1H, H-3′), 4.23 (br s, 1H, H-2′), 4.09–3.98 (m, 2H, H-4′, H-5′), 3.14 (dd, J = 10.5, 4.6 Hz, 1H, H-3), 2.27 (d, J = 11.2 Hz, 1H, H-18), 1.53 (d, J = 5.6 Hz, 3H, H-6′), 1.43 (d, J = 6.2 Hz, 3H, H-6′′), 1.36 (d, J = 6.5 Hz, 3H, H-6′′′′), 1.35 (d, J = 6.1 Hz, 3H, H-6′′′), 1.06 (s, 3H, H-27), 0.97 (s, 3H, H-25), 0.95 (s, 3H, H-23), 0.93 (d, J = 6.1 Hz, 3H, H-30), 0.88 (s, 3H, H-24), 0.86 (d, J = 6.3 Hz, 3H, H-29), 0.73 (br d, J = 11.0 Hz, 1H, H-5), 0.65 (s, 3H, H-26). 13C NMR (101 MHz, CDCl3) δ 177.3 (C-28), [166.1, 166, 165.9, 165.5, 165.15, 165.11 (2×), 165.1, 164.6 (CO-Bz)], 138.1 (C-13), 136.4 (C-Bn), [133.3, 133.18, 133.12, 132.88, 132.81 (2×), 132.72, 132.69, 132.4, 130.06 (2×), 130.04 (2×), 129.95 (2×), 129.87 (4×), 129.81 (4×), 129.76 (4×), 129.68 (2×), 129.63, 129.54, 129.51 (2×), 129.4, 129.3, 128.4 (4×), 128.37 (2×), 128.33 (2×), 128.2 (2×), 128.18 (4×), 128.13 (2×), 128.1 (4×), 128 (2×), 127.9 (2×) (CH-Bz, C-Bz, CH-Bn)], 125.7 (C-12), 100.7 (C-1′), 100.6 (C-1′′′), 99.6 (C-1′′), 99.2 (C-1′′′′), 90.0 (C-3), 80.9 (C-4′), 80.7 (C-3′), 79.5 (C-2′), 72.3 (C-4′′), 72.1 (C-4′′′), 72.0 (C-4′′′′), 71.7 (C-2′′), 71.6 (C-2′′′), 71.2 (C-2′′′′), 69.7 (C-3′′′′), 69.6 (C-3′′), 69.5 (C-3′′′), 67.9 (C-5′), 67.8 (C-5′′′), 67.6 (C-5′′), 67.5 (C-5′′′′), 66.0 (C-7), 55.4 (C-5), 52.9 (C-18), 48.1 (C-17), 47.6 (C-9), 42.0 (C-14), 39.6 (C-8), 39.1 (C-19), 39.1 (C-4), 38.8 (C-20), 38.7 (C-1), 36.7 (C-10), 36.7 (C-22), 33.0 (C-7), 30.7 (C-21), 28.5 (C-23), 28.0 (C-15), 25.7 (C-2), 24.3 (C-16), 23.6 (C-27), 23.3 (C-11), 21.2 (C-30), 18.7 (C-6′), 18.3 (C-6), 18.0 (C-6′′′), 17.7 (C-6′′′′), 17.6 (C-6′′), 17.0 (C-29), 17.0 (C-26), 16.8 (C-24), 15.5 (C-25). HRMS calcd for C124H134O28N [M + NH4]+ 2084.90869, found 2084.91553.:1, 2 mL), to which a freshly prepared solution of NaOMe (0.5 M, 2 mL) was added. After overnight stirring, the reaction was neutralized to pH 7 with Dowex G-26 (H+ form) and filtered. The filtrate was concentrated to dryness and purified by reversed-phase flash chromatography SPE (1 g cartridge, H2O:MeOH 50 → 85%), to give 10 (25 mg, 90% over two steps) Rf = 0.18 (26:14:3 CHCl3–MeOH–H2O); [α]20D +36.8 (c 3.9, 1:1, CHCl3–MeOH). 1H NMR (400 MHz, CD3OD/CDCl3 1:1) δ 5.24 (br s, 1H, H-12), 4.98 (s, 1H, H-1′′), 4.92 (s, 1H, H– H-1′′′′), 4.86 (s, 1H, H-1′′′), 4.85 (s, 1H, H-1′), 3.98 (dd, J = 9.8, 2.8 Hz, 1H, H-3′), 3.94–3.87 (m, 3H, H-2′′′′, H-2′′′, H-2′), 3.85–3.57 (m, 9H, H-2′′, H-5′, H-5′′, H-3′′′′, H-5′′′, H-5′′′′, H-3′′, H-3′′′, H-4′), 3.47–3.32 (m, 3H, H-4′′′, H-4′′, H-4′′′′), 3.11 (t, J = 8.4 Hz, 1H, H-3), 2.20 (br d, J = 11.2 Hz, 1H, H-18), 1.28 (m, 12H, H-6′, H-6′′, H-6′′′, H-6′′′′), 1.11 (s, 3H, H-27), 0.96 (m, 9H, H-30, H-25, H-23), 0.88 (d, J = 6.4 Hz, 3H, H-29), 0.84 (s, 3H, H-26), 0.81 (s, 3H, H-24). 13C NMR (101 MHz, CD3OD/CDCl3 1:1) δ 181.5 (C-28), 138.9 (C-13), 126.0 (C-12), 103.6 (C-1′′′), 102.9 (C-1′′), 102.9 (C-1′′′′), 101.9 (C-1′), 90.0 (C-3), 80.2 (C-3′), 80.2 (C-4′), 79.1 (C-2′), 73.2 (C-4′′′′), 73.1 (C-4′′′), 72.9 (C-4′′), 71.8 (C-3′′), 71.7 (C-3′′′), 71.6 (C-2′′), 71.6 (C-3′′′′), 71.3 (C-2′′′′), 71.3 (C-2′′′), 69.9 (C-5′′), 69.9 (C-5′′′′), 69.6 (C-5′′′), 68.3 (C-5′), 55.9 (C-5), 53.6 (C-18), 48.3 (C-17), 48.2 (C-9), 42.7 (C-14), 40.1 (C-8), 39.7 (C-19), 39.6 (C-20), 39.6 (C-4), 39.1 (C-1), 37.5 (C-22), 37.3 (C-10), 33.6 (C-7), 31.2 (C-21), 28.7 (C-23), 28.6 (C-15), 26.1 (C-2), 24.8 (C-16), 23.9 (C-27), 23.8 (C-11), 21.4 (C-30), 18.8 (C-6), 18.3 (C-6′′), 17.8 (C-6′′′′), 17.7 (C-6′), 17.5 (C-6′′′), 17.4 (C-26), 17.4 (C-29), 16.8 (C-24), 15.8 (C-25). HRMS calcd for C54H88O19Na [M + Na]+ 1063.5812, found 1063.5845.Authors contribution
A. P., B. S., and C. G. designed the experiments. B. S. and J. L. performed the experiments. B. S. and S. L. analyzed the NMR data. B. S., C. G., and J. L. analyzed the data and wrote the paper. A. P. and J. L. secured funding. A. P., J. L., and C. G. directed the students.Conflicts of interest
The authors have no conflict of interest to declare.Acknowledgements
This work was supported by operating grants 311906 and 326086 from the Canadian Institutes of Health Research (CIHR, to A. P. and J. L.) and by a Discovery grant from the Natural Sciences and Engineering Research Council of Canada (NSERC) under award number RGPIN-2016-04950 (to C. G.). C. G. holds a Fonds de recherche du Québec – Santé (FRQS) Research Scholars Junior 2 Career Award. We thank Karl Lalancette and Catherine Dussault for the biological assays.References
- C. Gauthier, J. Legault, M. Piochon-Gauthier and A. Pichette, Phytochem. Rev., 2011, 10, 521–544 CrossRef CAS.
- M. Ali-Seyed, I. Jantan, K. Vijayaraghavan and S. N. A. Bukhari, Chem. Biol. Drug Des., 2016, 87, 517–536 CrossRef CAS PubMed.
- M. Grymel, M. Zawojak and J. Adamek, J. Nat. Prod., 2019, 82, 1719–1730 CrossRef CAS PubMed.
- J. L. C. Sousa, C. S. R. Freire, A. J. D. Silvestre and A. M. S. Silva, Molecules, 2019, 24, E355 CrossRef PubMed.
- H. Chen, Y. Gao, A. Wang, X. Zhou, Y. Zheng and J. Zhou, Eur. J. Med. Chem., 2015, 92, 648–655 CrossRef CAS PubMed.
- D. Kashyap, H. S. Tuli and A. K. Sharma, Life Sci., 2016, 146, 201–213 CrossRef CAS PubMed.
- H. J. Jeong, H. B. Chai, S. Y. Park and D. S. H. L. Kim, Bioorg. Med. Chem. Lett., 1999, 9, 1201–1204 CrossRef CAS PubMed.
- M. Kvasnica, J. Sarek, E. Klinotova, P. Dzubak and M. Hajduch, Bioorg. Med. Chem., 2005, 13, 3447–3454 CrossRef CAS PubMed.
- H. Kommera, G. N. Kaluderović, S. Dittrich, J. Kalbitz, B. Dräger, T. Mueller and R. Paschke, Bioorg. Med. Chem. Lett., 2010, 20, 3409–3412 CrossRef CAS PubMed.
- C. Suresh, H. Zhao, A. Gumbs, C. S. Chetty and H. S. Bose, Bioorg. Med. Chem. Lett., 2012, 22, 1734–1738 CrossRef CAS PubMed.
- Q. Michaudel, G. Journot, A. Regueiro-Ren, A. Goswami, Z. Guo, T. P. Tully, L. Zou, R. O. Ramabhadran, K. N. Houk and P. S. Baran, Angew. Chem., Int. Ed., 2014, 53, 12091–12096 CrossRef CAS PubMed.
- C. Gauthier, J. Legault, K. Girard-Lalancette, V. Mshvildadze and A. Pichette, Bioorg. Med. Chem., 2009, 17, 2002–2008 CrossRef CAS PubMed.
- C. Gauthier, J. Legault, S. Lavoie, S. Rondeau, S. Tremblay and A. Pichette, Tetrahedron, 2008, 64, 7386–7399 CrossRef CAS.
- C. Gauthier, J. Legault, S. Lavoie, S. Rondeau, S. Tremblay and A. Pichette, J. Nat. Prod., 2009, 72, 72–81 CrossRef CAS PubMed.
- C. Gauthier, J. Legault, M. Lebrun, P. Dufour and A. Pichette, Bioorg. Med. Chem., 2006, 14, 6713–6725 CrossRef CAS PubMed.
- C. Gauthier, J. Legault and A. Pichette, Mini-Rev. Org. Chem., 2009, 6, 321–344 CrossRef CAS.
- C. Gauthier, J. Legault, M. Piochon, S. Lavoie, S. Tremblay and A. Pichette, Bioorg. Med. Chem., 2009, 19, 2310–2314 CrossRef CAS PubMed.
- C. Gauthier, J. Legault, S. Rondeau and A. Pichette, Tetrahedron Lett., 2009, 50, 988–991 CrossRef CAS.
- Y. Li, J. Sun and B. Yu, Org. Lett., 2011, 13, 5508–5511 CrossRef CAS PubMed.
- P. Cmoch, Z. Pakulski, J. Swaczynová and M. Strnad, Carbohydr. Res., 2008, 343, 995–1003 CrossRef CAS PubMed.
- K. Kuczynska, P. Cmoch, L. Rárová, J. Oklešťková, A. Korda, Z. Pakulski and M. Strnad, Carbohydr. Res., 2016, 423, 49–69 CrossRef CAS PubMed.
- K. Sidoryk, L. Rárová, J. Oklešťková, Z. Pakulski, M. Strnad, P. Cmoch and R. Luboradzki, Org. Biomol. Chem., 2016, 14, 10238–10248 RSC.
- A. Korda, Z. Pakulski, P. Cmoch, K. Gwardiak and R. Karczewski, Carbohydr. Res., 2018, 461, 32–37 CrossRef CAS PubMed.
- M. Mihoub, A. Pichette, B. Sylla, C. Gauthier and J. Legault, PLoS One, 2018, 13, e0193386 CrossRef PubMed.
- P. Tomsik, T. Soukup, E. Cermakova, S. Micuda, M. Niang, L. Sucha and M. Rezacova, Cent. Eur. J. Biol., 2011, 6, 1–9 CAS.
- A. Martín, T. D. Butters and G. W. J. Fieet, Chem. Commun., 1998, 2119–2120 RSC.
- L. Moreno Y Banuls, A. Katz, W. Miklos, A. Cimmino, D. M. Tal, E. Ainbinder, M. Zehl, E. Urban, A. Evidente, B. Kopp, W. Berger, O. Feron, S. Karlish and R. Kiss, Mol. Cancer, 2013, 12 Search PubMed.
- H. Y. L. Wang, W. Xin, M. Zhou, T. A. Stueckle, Y. Rojanasakul and G. A. O'Doherty, ACS Med. Chem. Lett., 2011, 2, 73–78 CrossRef CAS PubMed.
- H. Y. L. Wang, Y. Rojanasakul and G. A. O'Doherty, ACS Med. Chem. Lett., 2011, 2, 264–269 CrossRef CAS PubMed.
- Y. Wang, J. Gao, G. Gu, G. Li, C. Cui, B. Sun and H. Lou, ChemBioChem, 2011, 12, 2418–2420 CrossRef CAS PubMed.
- Y. L. Ku, G. V. Rao, C. H. Chen, C. Wu, J. H. Guh and S. S. Lee, Helv. Chim. Acta, 2003, 86, 697–702 CrossRef CAS.
- D. Thibeault, C. Gauthier, J. Legault, J. Bouchard, P. Dufour and A. Pichette, Bioorg. Med. Chem., 2007, 15, 6144–6157 CrossRef CAS PubMed.
- M. Tamigney Kenfack, M. Mazur, T. Nualnoi, T. L. Shaffer, A. Ngassimou, Y. Blériot, J. Marrot, R. Marchetti, K. Sintiprungrat, N. Chantratita, A. Silipo, A. Molinaro, D. P. AuCoin, M. N. Burtnick, P. J. Brett and C. Gauthier, Nat. Commun., 2017, 8, 115 CrossRef PubMed.
- S. O. Bajaj, E. U. Sharif, N. G. Akhmedoz and G. A. O'Doherty, Chem. Sci., 2014, 5, 2230–2234 RSC.
- R. R. Schmidt and A. Toepfer, Tetrahedron Lett., 1991, 32, 3353–3356 CrossRef CAS.
- Y. Zhang, Y. Li, T. Guo, H. Guan, J. Shi, Q. Yu and B. Yu, Carbohydr. Res., 2005, 340, 1453–1459 CrossRef CAS PubMed.
- S. C. B. Gnoatto, A. Dassonville-Klimpt, S. Da Nascimento, P. Galéra, K. Boumediene, G. Gosmann, P. Sonnet and S. Moslemi, Eur. J. Med. Chem., 2008, 43, 1865–1877 CrossRef CAS PubMed.
- R. Rago, J. Mitchen and G. Wilding, Anal. Biochem., 1990, 191, 31–34 CrossRef CAS PubMed.
- S. J. Green, M. S. Meltzer, J. B. Hibbs Jr and C. A. Nacy, J. Immunol., 1990, 144, 278–283 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09389c |
| This journal is © The Royal Society of Chemistry 2019 |