
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural, mechanical, spectroscopic and thermodynamic characterization of the copper-uranyl tetrahydroxide mineral vandenbrandeite†
Francisco Colmenero *a,
Jakub Plášilb,
Joaquín Cobosc,
Jiří Sejkorad,
Vicente Timóna,
Jiří Čejkad,
Ana María Fernándezc and
Václav Petříčekb
*a,
Jakub Plášilb,
Joaquín Cobosc,
Jiří Sejkorad,
Vicente Timóna,
Jiří Čejkad,
Ana María Fernándezc and
Václav Petříčekb
aInstituto de Estructura de la Materia (IEM-CSIC), C/Serrano, 113, 28006 Madrid, Spain. E-mail: francisco.colmenero@iem.cfmac.csic.es
bInstitute of Physics ASCR, v.v.i., Na Slovance 2, 182 21, Praha 8, Czech Republic
cCentro de Investigaciones Energéticas, Medioambientales y Tecnológicas (CIEMAT), Avda/ Complutense, 40, 28040 – Madrid, Spain
dMineralogicko-petrologické oddělení, Národní Muzeum, Cirkusová 1740, 193 00 Praha 9, Czech Republic
First published on 9th December 2019
Abstract
The full crystal structure of the copper-uranyl tetrahydroxide mineral (vandenbrandeite), including the positions of the hydrogen atoms, is established by the first time from X-ray diffraction data taken from a natural crystal sample from the Musonoi Mine, Katanga Province, Democratic Republic of Congo. The structure is verified using first-principles solid-state methods. From the optimized structure, the mechanical and dynamical stability of vandenbrandeite is studied and a rich set of mechanical properties are determined. The Raman spectrum is recorded from the natural sample and determined theoretically. Since both spectra have a high-degree of consistence, all spectral bands are rigorously assigned using a theoretical normal-coordinate analysis. Two bands in the Raman spectra, located at 2327 and 1604 cm−1, are recognized as overtones and a band at 1554 cm−1 is identified as a combination band. The fundamental thermodynamic functions of vandenbrandeite are computed as a function of temperature using phonon calculations. These properties, unknown so far, are key-parameters for the performance-assessment of geological repositories for storage of radioactive nuclear waste and for understanding the paragenetic sequence of minerals arising from the corrosion of uranium deposits. The thermodynamic functions are used here to determine the thermodynamic properties of formation of vandenbrandeite in terms of the elements and the Gibbs free-energies and reaction constants for a series of reactions involving vandenbrandeite and a representative subset of the most important secondary phases of spent nuclear fuel. Finally, from the thermodynamic data of these reactions, the relative stability of vandenbrandeite with respect to these phases as a function of temperature and in the presence of hydrogen peroxide is evaluated. Vandenbrandeite is shown to be highly stable under the simultaneous presence of water and hydrogen peroxide.
1 Introduction
The use of copper as an exterior canister corrosion barrier for the disposal of radioactive waste was proposed in 1978 in Sweden.1 The concept has been adopted by the national research programs of nuclear waste management of many countries including those in Sweden (SKB),1–21 Finland (POSIVA),22–27 and Canada (NWMO).28–46 Copper is also considered as an alternative material in Switzerland (NAGRA),47,48 Japan (JNC)49,50 and Korea.51 The fundamental motivations for the choice of composite canisters with a copper external shell are the observation that copper is essentially inert in an anoxic environment, its large thermodynamic stability at the conditions of a deep geological repository (DGR), and the existence of natural analogues for the long-term corrosion of copper nuclear waste containers.7,8,22,29 The disposal models in these countries are similar and comprise an underground repository situated more than four hundred meters below the water table in stable host rock environments which are sealed using a bentonite-based backfill material. Under these conditions, copper provides a very long-lived containment with predicted very long canister lifetimes.9 The corrosion of the exterior copper canister barrier as well as a long series of related issues1–57 have been studied extensively by the Scandinavian, Finnish and Canadian organizations and many other institutions from the European Community.52 An entirely new nuclear facility in which the exterior copper canister concept will be put in practice is being developed to handle the permanent disposal of spent nuclear fuel (SNF) at ONKALO (Olkiluoto site, Finland). Finland was the first country to issue a construction license for a DGR, granted in November 2015. The operation license application will be submitted in 2020, the repository being sealed up to 2120.23–27Although the well-established thermodynamic data of copper lead to the expectation that it will corrode to a very limited extent in a closed system2,3,11–14,19–21,28 and that this kind of canisters will protect the SNF against corrosion in a stable form during very long times,4–6,9,15,16,23,25,42–45 the corrosion processes increase upon oxic and humid conditions and high temperatures and pressures. Furthermore, the experiments carried out in the presence of high radiation fields18,46 show that the dissolution of copper and the rate of formation of oxide layers increase upon radiation significantly, and that aqueous radiation chemistry is not the only process driving the corrosion of copper in these systems. It has been anticipated that, after storage times on the order of thousands of years, the barriers that protect the SNF will be breached and, then, the used fuel will enter in contact with oxygen and water.58 Under high radiation fields producing the radiolysis of water at the surface of SNF, the formation of hydrogen peroxide and other oxidant species is expected.59–64 An oxidative environment has been postulated65 in a layer near the fuel surface (within <50 μm of the fuel surface). Thus, the U(IV) in the matrix of the SNF, mainly composed of uranium dioxide, could be oxidized to U(VI), and dissolve in water forming uranyl groups. The uranyl groups can precipitate forming secondary phases on the spent fuel surface depending on the local conditions of pressure, temperature, pH and redox potential and the concentrations of reactive species present.66–69 The formation of secondary phases can reduce the release and environmental impact of the fission products and heavier actinides contents in the SNF to the biosphere70 because they may be retained in the crystal structures of these phases.
If copper canisters are used, clearly the secondary phases of SNF containing this element will acquire an enormous relevance. In particular, the copper-uranyl tetrahydroxide mineral (Cu(UO2)(OH)4) vandenbrandeite, studied in this work, is one of the most important uranyl minerals containing copper. Other important uranyl and copper containing minerals are the uranyl silicate cuprosklodowskite,71,72 the uranyl carbonates paddlewheelite73 and voglite,74 the uranyl vanadate sengierite,75 the uranyl sulfates johannite,76–78 and pseudojohannite,79 the uranyl phosphates torbernite, metatorbernite80 and ulrichite,81,82 and the uranyl arsenates zeunerite and metazeunerite.80 The knowledge of the structures of uranyl minerals83–85 is crucial to evaluate the possible incorporation of fission products and transuranic elements into their crystal structures.70,86–88 Besides, the study of the crystal structures, spectra and physical and chemical properties of uranyl-containing minerals is fundamental for the study of the corrosion processes occurring in uraninite-bearing ore deposits66,89–91 and for understanding the paragenetic sequence of secondary phases that arises from the corrosion of spent nuclear fuel (SNF) under the final geological disposal conditions.92–98
Vandenbrandeite was collected in 1932 by Schoep99 in the Kalongwe Copper–Cobalt mine from the Katanga Province in Democratic Republic of Congo (DRC). Its name was coined in honor to Pierre Van den Brande, a Belgian geologist who discovered the Kalongwe deposit. However, this mineral was encountered by the first time a year prior to the finding by Schoep, by Thoreau100 in the Shinkolobwe uranium mine also from Katanga Province (DRC), and called it uranolepidite. According to the well-known convention that prior names should be preferred, the name uranolepidite should be used, but the name vandenbrandeite has been adopted as the standard name surely due to the most detailed description of the specimen from the Kalongwe mine. Vandenbrandeite is found as a secondary mineral appearing frequently in the oxidation zones of the hydrothermal uranium ore deposits containing copper. Thus, vandenbrandeite mineral is mainly formed from the oxidation and dissolution/precipitation process of uraninite in contact with primary copper minerals.101,102 Due to his large uranium content, this mineral is extracted from mines for uranium production.
Vandenbrandeite synthetic material was produced by Bignand103 in 1955 from mixtures of copper acetate and uranyl acetate at 140 °C and also by heating schoepite with an excess of copper acetate at 140 °C. The crystal structure of vandenbrandeite was found to be triclinic by Milne and Nuffield104 in 1951 and was precisely specified by Rosenzweig and Ryan in 1977.105 However, the determination of the positions of the hydrogen atoms in the unit-cell of vandenbrandeite has not been possible so far. The infrared spectrum of vandenbrandeite was reported by Povarennykh106 in 1979 and Čejka and Urbanec107 in 1990 but the spectrum was given without interpretation. A detailed investigation on the thermal properties of vandenbrandeite and its infrared spectrum was reported by Čejka in 1994,108 who assigned the bands in the experimental infrared spectrum empirically.
While the crystal structures of the majority of the uranyl minerals are reasonably well-known,71–84 the positions of the hydrogen atoms in their unit cells are unknown in many cases. The solution of this problem is currently being attempted by means of experimental X-ray diffraction techniques72,109–112 and first-principles theoretical methods.113–119 In the specific case of copper-containing uranyl minerals, the full-crystal structure of cuprosklodowkite was recently determined experimentally.72 The full crystal structure of vandenbrandeite, including the positions of the hydrogen atoms within the corresponding unit cell, was first obtained by using the first principles methodology in 2016 (see the ESI of ref. 119). However, it was determined employing small calculation parameters. In the present work, the full crystal structure of vandenbrandeite was determined independently by means of the structure refinement from X-ray diffraction data taken from a natural crystal sample collected from Musonoi Mine (Katanga Province, DRC). The obtained crystal structure was confirmed by means of the first principles solid-state methodology employing accurate calculation parameters, using the previous theoretical structure as initial input.
From the full energy-optimized crystal structure of vandenbrandeite, its elasticity tensor and equation of state120 were computed and used to study the stability of this structure and to determine its mechanical properties using theoretical methods. The values of these properties were predicted since they have not been measured experimentally up to date. The experimental Raman spectrum of vandenbrandeite was recorded from the natural crystal sample from Musonoi Mine (DRC). The corresponding spectrum was also determined using density functional perturbation (DFPT). The computed and experimental spectra were in good agreement and, consequently, all the bands in the Raman spectrum were rigorously assigned to specific vibrational motions using a normal mode analysis of the computed vibrational data.
The fundamental thermodynamic functions of vandenbrandeite (specific heat, entropy, enthalpy and Gibbs free energy) were calculated as a function of temperature using theoretical phonon calculations.121 The resulting functions were utilized to derive the corresponding enthalpies and Gibbs free energies of formation in terms of the elements as a function of temperature. These thermodynamic properties of formation were then merged with those of other important uranyl-containing materials such as dehydrated schoepite (UO2(OH)2), soddyite ((UO2)2(SiO4)·2H2O), rutherfordine (UO2CO3) and gamma uranium trioxide (γ-UO3),121–125 to derive the Gibbs free energies of reaction and the associated reaction constants124,125 of the transformation reactions:
| UO3(cr) + CuO(cr) + 2H2O(l) → Cu(UO2)(OH)4(cr) | (A) |
| UO3·H2O(cr) + CuO(cr) + H2O(l) → Cu(UO2)(OH)4(cr) | (B) |
| UO2CO3(cr) + CuO(cr) + 2H2O(l) → Cu(UO2)(OH)4(cr) + CO2(g) | (C) |
| 1/2(UO2)2(SiO4)·2H2O(cr) + CuO(cr) + H2O(l) → Cu(UO2)(OH)4(cr) + 1/2SiO2(cr) | (D) |
Similarly, the thermodynamic properties of reaction of the reactions of conversion of vandenbrandeite into schoepite,114 [(UO2)8O2(OH)12]·12H2O(cr), and studtite,121 (UO2)O2·4H2O, in the presence of water and hydrogen peroxide and in the presence of high concentrations of hydrogen peroxide, respectively, were obtained by considering the corresponding reactions:
| Cu(UO2)(OH)4(cr) + 1/8H2O(l) + 1/8H2O2(l) → 1/8[(UO2)8O2(OH)12]·12H2O(cr) + CuO(cr) + 1/16O2(g) | (E) |
| Cu(UO2)(OH)4(cr) + 2H2O2(l) → (UO2)O2·4H2O(cr) + CuO(cr) + 1/2O2(g) | (F) |
The temperature dependent thermodynamic properties are key parameters for the performance assessment of radioactive waste repositories because the stability of the secondary phases of the spent nuclear fuel under final geological disposal conditions is highly dependent of the temperature.124,125 The computed thermodynamic parameters together with those derived in previous works114,115,121–125 permitted to determine the thermodynamic stability of vandenbrandeite with respect to a selected set of the most relevant secondary phases of SNF under final geological disposal conditions.
One of the most important motivations of this work is to contribute to the development of nuclear Raman113,117–120 and thermodynamic databases.114,115,121–125,125–129 Raman spectroscopy is one of the main techniques used to identify uranium-containing materials because it does not need a special preparation of the samples, a very small amount of sample is required and it is non-destructive. Thus, this spectroscopic technique minimizes the difficulties in the handling of these materials associated to its radiotoxicity and the costs of the analyses because the samples may be reutilized. Furthermore, the use of the first principles methodology allows for a rigorous assignment of the Raman bands, customarily performed using rough empirical arguments frequently leading to incomplete and even incorrect assignments. The availability of accurate thermodynamic information is indispensable in nuclear sciences because it permits to model the dynamical behavior nuclear materials under diverse geochemical conditions. An accurate thermodynamic database is a prerequisite in calculations supporting the safety performance assessment of radioactive waste disposal systems.128,129 The stabilities and dissolution rates of uranyl minerals are functions of their composition, temperature, and local conditions (mainly pH and redox potential), and their prediction requires the knowledge of the Gibbs free energy, enthalpy, and entropy thermodynamic functions of formation for each phase of interest and their variation with temperature. While the amount of thermodynamic data available at normal conditions is quite large, reliable temperature dependent thermodynamic information is completely lacking for most of the uranyl containing materials.126,127 For the case of vandenbrandeite, there are not thermodynamic data even at room temperature. The development of a more complete nuclear thermodynamic database using the first principles methodology combined with experimental techniques should permit to carry out realistic thermodynamic computations involving a significant number of these materials, providing a profound understanding of uranium mineral paragenesis and of the corrosion processes in uranium deposits and SNF repositories.
This paper is structured as follows. In Section 2, the experimental and first principles solid state methods employed in this work are described. The experimental full crystal structure of vandenbrandeite derived from X-ray diffraction data is also reported. Section 3 contains the main results of this work and the discussion of these results. The results concerning the crystal structure, the hydrogen bonding, the computed X-ray diffraction pattern, and the mechanical, spectroscopic and thermodynamic properties of vandenbrandeite are considered in six separate subsections. Finally, the conclusions of this work are presented in Section 4.
2 Materials and methods
2.1. Experimental
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with a = 5.4597(3) Å, b = 6.1034(3) Å, c = 7.8652(4) Å, α = 78.057(5)°, β = 89.804(4)°, γ = 88.612(4)° (V = 256.341(2) Å3 (Z = 2)). These unit-cell parameters are in good agreement with the values reported by Rosenzweig and Ryan.105 Integration of the diffraction data, including corrections for background, polarization and Lorentz effects and absorption correction was carried out with the CrysAlis RED program.130 The crystal structure of vandenbrandeite was solved by the charge-flipping algorithm using the SHELXT program.131 The structure was then refined using the software Jana2006 with the full-matrix least-squares refinement based on F2.132 The structure solution included the locations of all atoms except hydrogens. The positions of hydrogen atoms were later established from the difference-Fourier maps. The H atoms were refined using a mix of soft constraints on O–H distances, 0.95(4) Å, and with the atomic displacement parameter, Ueq, of each H set to 1.2 times that of the donor O atom.
, with a = 5.4597(3) Å, b = 6.1034(3) Å, c = 7.8652(4) Å, α = 78.057(5)°, β = 89.804(4)°, γ = 88.612(4)° (V = 256.341(2) Å3 (Z = 2)). These unit-cell parameters are in good agreement with the values reported by Rosenzweig and Ryan.105 Integration of the diffraction data, including corrections for background, polarization and Lorentz effects and absorption correction was carried out with the CrysAlis RED program.130 The crystal structure of vandenbrandeite was solved by the charge-flipping algorithm using the SHELXT program.131 The structure was then refined using the software Jana2006 with the full-matrix least-squares refinement based on F2.132 The structure solution included the locations of all atoms except hydrogens. The positions of hydrogen atoms were later established from the difference-Fourier maps. The H atoms were refined using a mix of soft constraints on O–H distances, 0.95(4) Å, and with the atomic displacement parameter, Ueq, of each H set to 1.2 times that of the donor O atom.2.2. Theoretical solid-state methods
The unit-cell parameters of vandenbrandeite and the associated atomic positions were fully optimized by means of the Broyden–Fletcher–Goldfarb–Shanno (BFGS) technique.145 A large plane wave kinetic energy cut-off parameter of ε = 1000 eV and a dense k-mesh146 of 3 × 5 × 4 (30 k-points) were employed. These calculation parameters were selected to produce a well converged crystal structure, energy and properties. The X-ray powder diffraction patterns of vandenbrandeite were obtained147 from the experimental and computed crystal structures using the software REFLEX included in Materials Studio program suite.134
The influence of the Hubbard correction,148 allowing for a correct description of the strong Coulomb repulsion between electrons in f orbitals, was also evaluated. The introduction of this correction, improving significantly the description of uranium containing systems in which uranium exists with IV oxidation state,149–154 seems not to be needed for vandenbrandeite as shown in Section 3. In fact, for a numerous set of materials in which uranium exists with VI oxidation state, the standard DFT description provides a reliable description of their structures and properties.113–125,139–144,155–162
2.2.4.1. Fundamental thermodynamic properties. The fundamental thermodynamic functions of vandenbrandeite were obtained by performing phonon calculations at the optimized structure. The phonon spectra at all the points of the Brillouin zone were calculated by means of the density functional perturbation theory (DFPT) method as second order derivatives of the total energy.174 The phonon dispersion curves and density of states were calculated from the computed phonon spectra and, from them, the fundamental thermodynamic functions (Gibbs free energies, enthalpies, entropies, and specific heats) were obtained in the quasi-harmonic approximation.178,179 The thermodynamic properties of formation and reaction were then evaluated in terms of the calculated fundamental thermodynamic properties using the methods described in the next subsections.121,124,125
2.2.4.2. Thermodynamic properties of formation. The calculated enthalpy and entropy functions, (HT − H298)calc and ScalcT, were employed to obtain the enthalpy and Gibbs free-energy of formation in terms of the elements by means of the equations:121,180
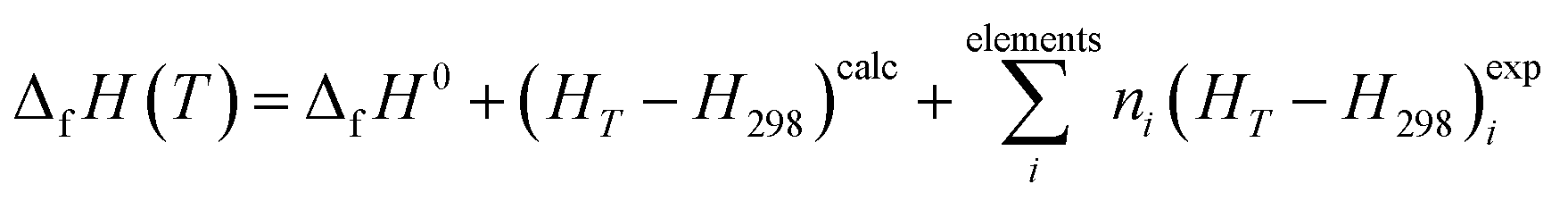 | (1) |
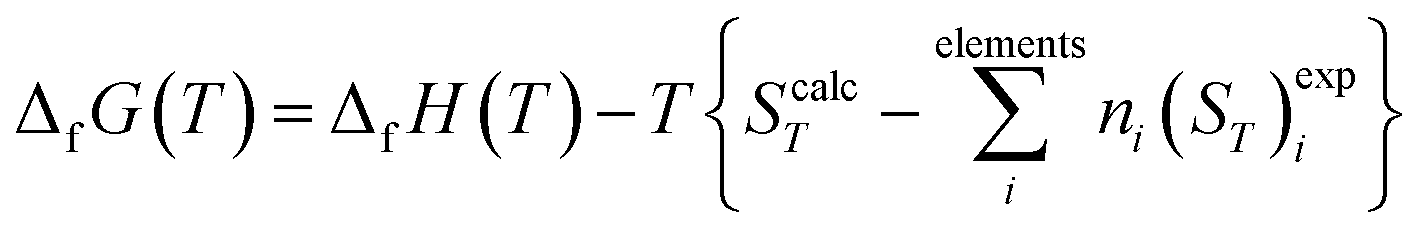 | (2) |
In these relations, ΔfH0 is the enthalpy of formation at the standard state of the material being investigated (temperature of 295.15 K and pressure of 1 bar), and (HT − H298)expi and (ST)expi are the enthalpy and entropy functions of the elements forming part of this material with stoichiometric coefficients ni, respectively. The precise value of ΔfH0 for vandenbrandeite employed in this work will be given below. The thermodynamic functions for copper, oxygen and hydrogen were taken from JANAF (Joint Army-Navy-Air Force) thermochemical tables180 and those for uranium from the book by Barin.181 The reaction constants of the formation reactions were calculated from the corresponding Gibbs free energies of formation employing the relationship:180
ΔG(T) = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnK lnK
| (3) |
2.2.4.3. Thermodynamic properties of reaction. The enthalpies and Gibbs free energies of reaction at the different temperatures were calculated from the computed Gibbs free energy and entropy of formation functions, ΔfG(T)calc and ΔfS(T)calc, by means of the expressions:124,180
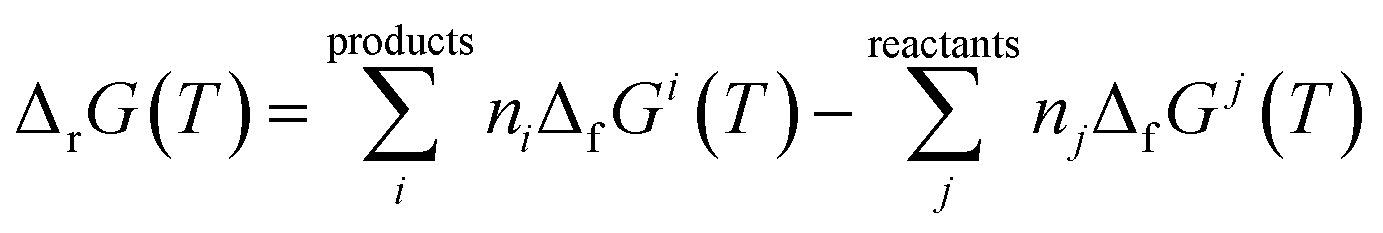 | (4) |
| ΔrH(T) = ΔrG(T) + TΔrS(T) | (5) |
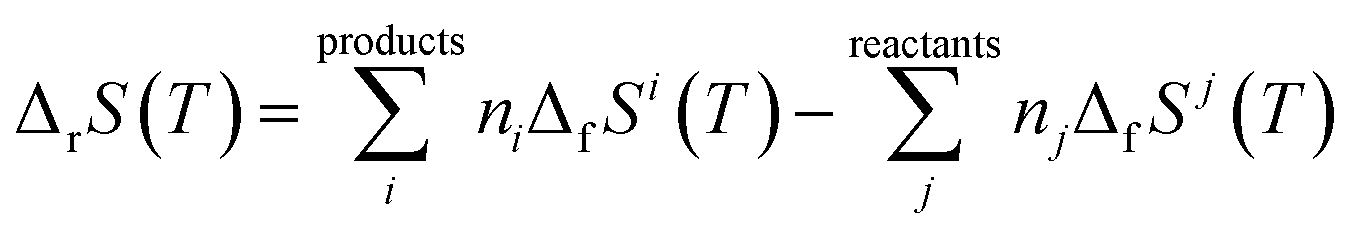 | (6) |
In these equations, ΔfGi(T) and ΔfSi(T) denote the Gibbs free energy and entropy of formation at temperature T of the compound i entering in the reaction with stoichiometric coefficient ni. The thermodynamic functions for CuO(cr), SiO2(cr), H2O(l), CO2(g), and O2(g) were taken from JANAF thermochemical tables180 and those of H2O2(l) from Barin.181 The reaction constants were calculated from the Gibbs free energies of reaction by using the eqn (3).
3 Results and discussion
3.1. Crystal structure
Uranium atom in vandenbrandeite displays pentagonal bipyramidal coordination. Fixing the cutoff for the Cu–O bond-length to 3.2 Å, the copper atom displays distorted octahedral coordination. Although two of the oxygen atoms in the coordination sphere of copper atom have Cu–O distances which are much larger than the remaining four oxygen atoms, the analysis of the computed electron density of vandenbrandeite using Bader's theory182 for the quantum mechanical description of the chemical bond confirmed that they must be included in the coordination structure of copper atom. In vandenbrandeite, the uranium and cooper coordination polyhedra form dimers by sharing an equatorial edge with another polyhedra of the same kind. The main building blocks in the crystal structure of vandenbrandeite are shown in Fig. 1A. The copper dimers share two faces (one from each monomer) with uranium dimers to form uranyl cuprate layers as shown in the Fig. 1B. Finally, the uranyl cuprate layers are linked by sharing the uranium and copper polyhedra apical vertices as shown in the Fig. 1C.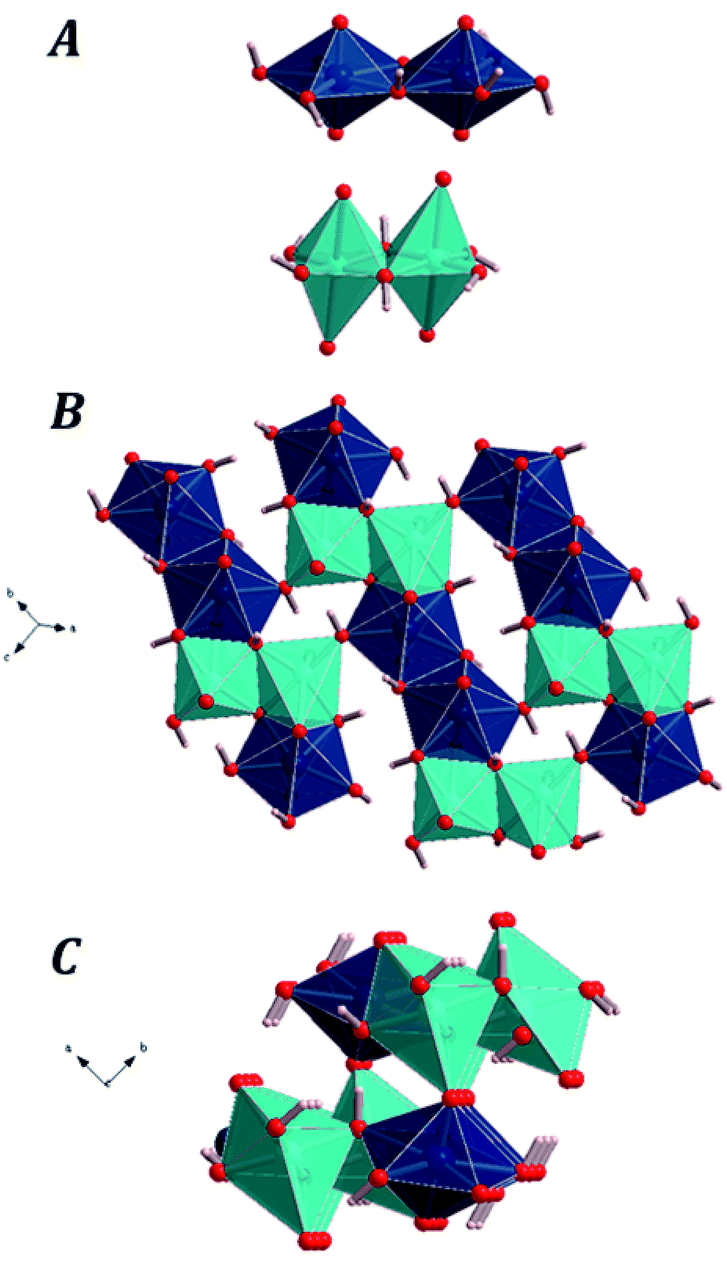 | ||
| Fig. 1 The computed crystal structure of vandenbrandeite: (A) The dimers of the uranyl and copper polyhedra; (B) An uranyl cuprate layer: (C) two connected successive uranyl cuprate layers. Color code: U – dark blue; Cu – clear blue; O – red; H – white. | ||
The unit cell of vandenbrandeite is triclinic, space group P (no. 2). The computed unit-cell parameters, volume and density are given in Table 1 where they are compared with the corresponding experimental data. The differences between the calculated and experimental volumes are only about 0.3% with respect to the present crystal data and 0.8% with respect to those from Rosenzweig and Ryan.105 The dispersion corrections improved significantly the computed crystal structure of vandenbrandeite since, as it can be seen in the first line of Table 1, the computed unit-cell volume obtained using the uncorrected PBE energy-density functional overestimates the experimental volume significantly (2.8%).
| Parameter | a (Å) | b (Å) | c (Å) | α (deg) | β (deg) | γ (deg) | Vol. (Å3) | Dens. (g cm−3) |
|---|---|---|---|---|---|---|---|---|
| PBE | 5.4748 | 6.1774 | 7.9337 | 79.2226 | 90.1392 | 89.6109 | 263.5805 | 5.060 |
| DFT-D2 | 5.4450 | 6.1331 | 7.8348 | 79.1821 | 90.2831 | 89.4481 | 256.9770 | 5.190 |
| DFT-D2+U | 5.4440 | 6.1311 | 7.8343 | 79.1664 | 90.2913 | 89.4393 | 256.8154 | 5.193 |
| Exp [this work] | 5.4597(3) | 6.1034(3) | 7.8652(4) | 78.057(5) | 89.804(4) | 88.612(4) | 256.341(2) | 5.203 |
| Exp.105 | 5.449 | 6.089 | 7.855 | 78.10 | 89.20 | 88.56 | 254.929 | 5.232 |
The Hubbard correction (which was applied with the parameter,152 Ueff = 4.0 eV) does not introduce significant changes to the computed crystal structure as shown in the third row of Table 1. Since the Hubbard correction does not improve the description of vandenbrandeite, it was not used to obtain the properties and spectra of this mineral. The reason of the small impact of this correction is clear from Fig. S.1 of the ESI† where the frontier band functions (the highest occupied and lowest unoccupied one-electron wavefunctions), are displayed. Standard DFT predicts correctly that vandenbrandeite is an insulator with a band gap of 0.72 eV. As can be seen, the highest occupied band function is predominantly composed of contributions localized in d orbitals of copper atoms and p orbitals of oxygen atoms. The lowest unoccupied wavefunction represents mainly a combination of contributions localized in f orbitals of uranium atoms. Since the f orbitals in uranium are unoccupied, the Hubbard correction describing strong correlation between electrons occupying this type of orbitals is not required.
An illustrative set of interatomic distances and angles in vandenbrandeite are given in Tables S.1 and S.2 of the ESI,† respectively. As can be seen, the theoretical and experimental values are in very good agreement. The unit cell of vandenbrandeite contains only one symmetrically independent U atom and one symmetrically independent Cu atom in its unit cell. The uranyl oxygen atoms located in the apical positions of the uranium pentagonal bipyramids have uranium–oxygen distances of 1.786(3) and 1.801(3) Å, which may be compared with the computed values of 1.806 and 1.819 Å, respectively. The average U–O distance for the five equatorial O atoms is 2.38 Å, which hopefully coincides with the theoretical value. Similarly, the experimental and theoretical average Cu–O distances, 2.25 and 2.30 Å agree satisfactorily within 0.05 Å.
The experimental and computed full crystal structures of vandenbrandeite are reported as ESI† in two files of CIF (Crystallographic Information File) type. It should be noticed that in the theoretical structure CIF file the coordinate system axes are permuted with respect to those of the experimental structure in the form a → b′; b → −c′; c → a′. In this coordinate system, the slight triclinic distortion in vandenbrandeite with respect to a monoclinic system is more clearly shown because in this reference system the computed unit-cell parameters become a = 7.8348 Å, b = 5.4450 Å, c = 6.1331 Å, α = 89.4481°, β = 100.8179°, and γ = 90.2831°.
3.2. Hydrogen bonding
There are four inequivalent hydrogen bonds in vandenbrandeite, O5–H10⋯O7, O6–H12⋯O5, O7–H9⋯O3 and O8–H11⋯O4. As shown in Tables S.1 and S.2 of the ESI,† all of them are short and quite nearly linear. Except for the first hydrogen bond O5–H10⋯O7, which is fully contained with a given uranyl-cuprate layer, the remaining hydrogen bonds link two different layers as shown in Fig. 2.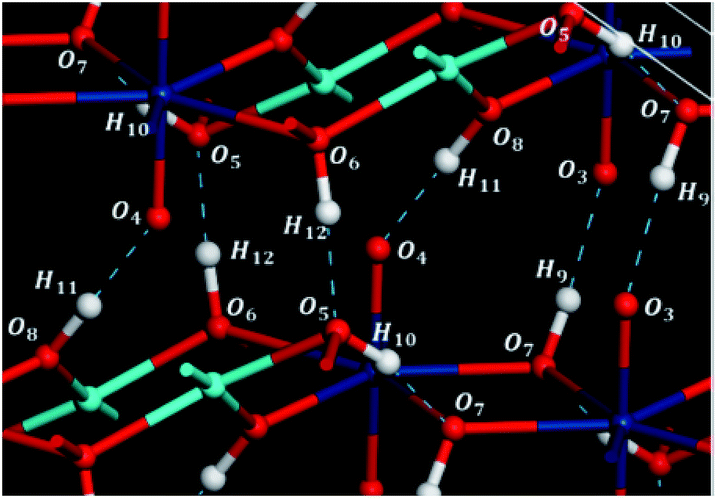 | ||
| Fig. 2 Hydrogen bond structure in vandenbrandeite. Color code: U – dark blue; Cu – clear blue; O – red; H – white. | ||
3.3. X-ray powder diffraction patterns
The software REFLEX included in Materials Studio program suite134 was utilized in order to obtain the X-ray powder diffraction patterns of vandenbrandeite from the theoretical and experimental crystal structures using CuKα radiation (λ = 1.540598 Å). These patterns are compared in Fig. 3 in which a true experimental X-ray powder diffraction pattern of vandenbrandeite183,184 is also included. The last pattern is from a natural sample from Shinkolobwe Mine, Shaba, Democratic Republic of Congo, taken from Record R080114 of RRUFF database.183,184 The agreement of the computed and experimental diffractograms is excellent. A more detailed comparison is provided in Table 2, in which the calculated and experimental positions and intensities of the main reflections are given. As can be observed, the X-ray powder pattern of vandenbrandeite is complex and contains a large number of reflections with intensities larger than 10%. The reflection having the largest difference in the calculated position with respect to experiment is [−121] (2θ ∼ 34.5°), the difference being only about 0.6 degrees.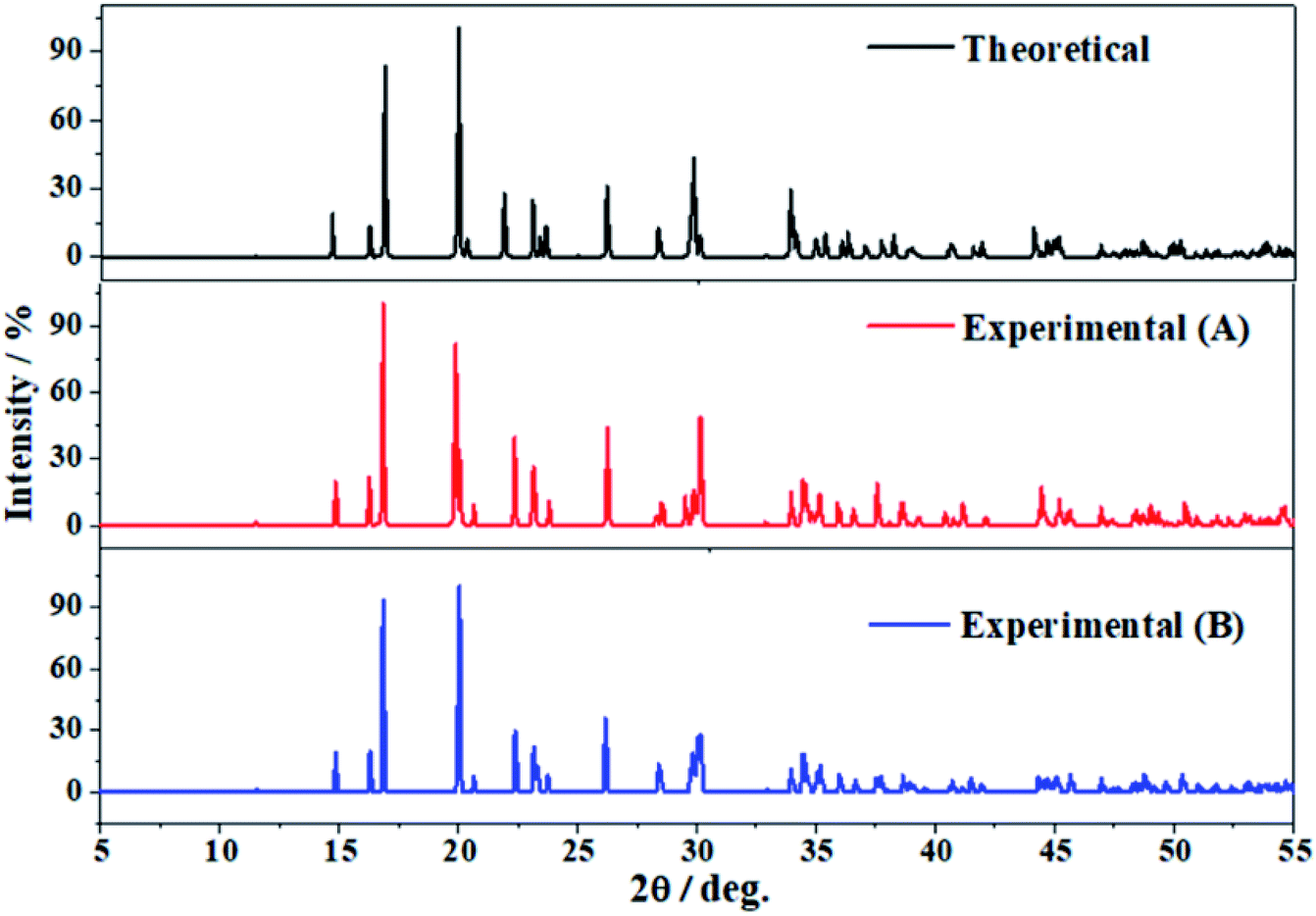 | ||
| Fig. 3 X-ray diffraction powder patterns of vandenbrandeite using CuKα radiation (λ = 1.540598 Å). The upper pattern has been determined from the calculated geometry. The mid pattern was obtained from the experimental geometry reported in this work. Finally the lower diffractogram is an experimental X-ray diffraction pattern from a natural sample from Shinkolobwe Mine, Shaba, Democratic Republic of Congo, taken from Record R080114 of RRUFF database.183,184 | ||
| (a) Calculated | (b) Experimental184 | Δ (2θ) | ||||
|---|---|---|---|---|---|---|
| 2θ (°) | d (Å) | I (%) | [hkl] | 2θ (°) | I (%) | |
| 19.99 | 4.44 | 100.00 | [101] | 20.05 | 100.00 | −0.06 |
| 16.89 | 5.25 | 85.64 | [011] | 16.86 | 93.23 | 0.03 |
| 26.21 | 3.40 | 40.50 | [−1−11] | 26.18 | 35.78 | 0.03 |
| 21.89 | 4.06 | 32.24 | [−110] | 22.39 | 29.65 | −0.50 |
| 29.78 | 3.00 | 35.05 | [0−12] | 30.15 | 28.23 | −0.37 |
| 19.93 | 4.45 | 27.14 | [−101] | 20.08 | 26.84 | −0.15 |
| 29.85 | 2.99 | 28.71 | [−112] | 30.03 | 26.19 | −0.18 |
| 23.10 | 3.85 | 26.14 | [002] | 23.17 | 22.04 | −0.07 |
| 16.27 | 5.44 | 18.58 | [100] | 16.31 | 20.04 | −0.04 |
| 14.69 | 6.02 | 17.31 | [010] | 14.87 | 19.46 | −0.18 |
| 29.73 | 3.00 | 18.71 | [021] | 29.84 | 18.71 | −0.11 |
| 33.91 | 2.64 | 25.76 | [0−21] | 34.44 | 18.64 | −0.53 |
| 33.92 | 2.64 | 13.95 | [−121] | 34.52 | 18.11 | −0.60 |
| 28.34 | 3.15 | 17.34 | [−102] | 28.41 | 13.35 | −0.07 |
| 35.34 | 2.54 | 15.47 | [013] | 35.19 | 12.78 | 0.15 |
| 23.65 | 3.76 | 13.58 | [111] | 23.31 | 12.65 | 0.34 |
| 30.08 | 2.97 | 15.76 | [112] | 29.73 | 11.47 | 0.35 |
| 34.12 | 2.63 | 13.89 | [120] | 33.96 | 10.82 | 0.16 |
| 36.30 | 2.47 | 12.61 | [210] | 35.99 | 8.78 | 0.31 |
| 48.65 | 1.87 | 10.50 | [−203] | 48.78 | 8.56 | −0.13 |
| 50.20 | 1.82 | 9.96 | [−104] | 50.35 | 8.29 | −0.15 |
| 38.21 | 2.35 | 12.22 | [122] | 37.71 | 7.68 | 0.50 |
| 44.91 | 2.02 | 8.08 | [−123] | 45.10 | 7.57 | −0.19 |
| 44.11 | 2.05 | 11.84 | [−1−13] | 44.33 | 7.47 | −0.22 |
3.4. Mechanical properties and stability
The generic necessary and sufficient Born condition185,186 for mechanical stability of a crystal structure is that the stiffness matrix C should be positive definite, i.e. all its eigenvalues should be positive. A numerical diagonalization of the C matrix of vandenbrandeite was carried out. The crystal structure of vandenbrandeite is mechanically stable because all the eigenvalues were found to be positive. Besides, the fulfillment of the dynamical stability condition of the structure was analyzed from phonon calculations. A crystal structure is dynamically stable, if and only if, all its phonon modes have positive frequencies for all wave vectors.186 The calculated phonon density of states is shown in Fig. S.2 of the ESI.† As shown clearly in this figure, there are not negative energy phonons for vandenbrandeite. Consequently, the crystal structure of vandenbrandeite is mechanically and dynamically stable.
| Property | Value | |
|---|---|---|
| B | Bulk modulus | 36.97 |
| G | Shear modulus | 22.12 |
| E | Young modulus | 55.33 |
| ν | Poisson ratio | 0.25 |
| D | Ductility index | 1.67 |
| H | Hardness index | 3.71 |
| AU | Universal anisotropy index | 0.84 |
|
||
| EOS | ||
| B | Bulk modulus | 39.48 ± 0.74 |
| B′ | Bulk modulus first derivative | 4.55 ± 0.75 |
| B′′ | Bulk modulus second derivative | 0.27 ± 0.28 |
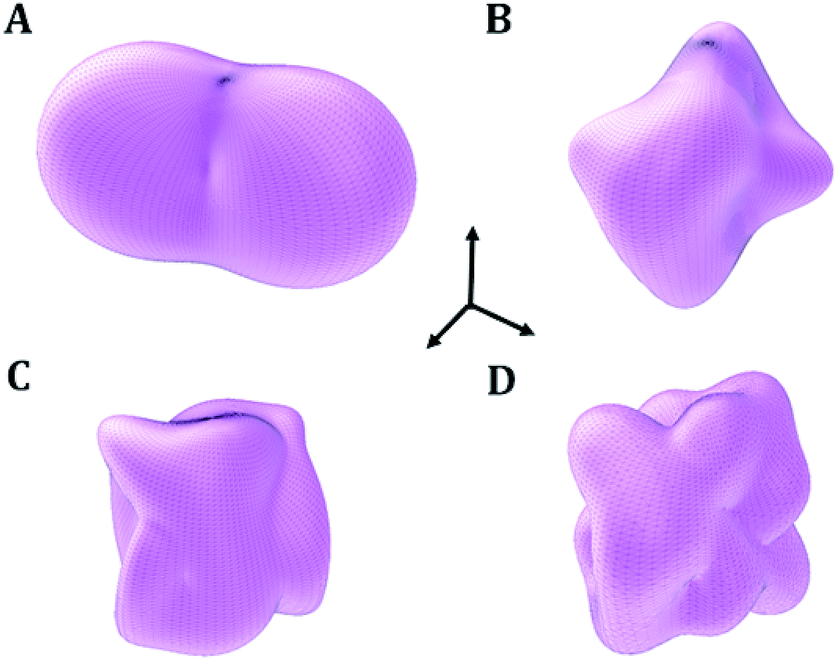 | ||
| Fig. 4 Elastic properties of vandenbrandeite mineral as a function of the orientation of the applied strain: (A) compressibility; (B) Young modulus; (C) shear modulus; (D) Poisson ratio. The maximum values of the compressibility, Young modulus, shear modulus and Poisson's ratio are 15.46 TPa−1, 85.21 GPa, 35.22 GPa and 0.56, respectively. | ||
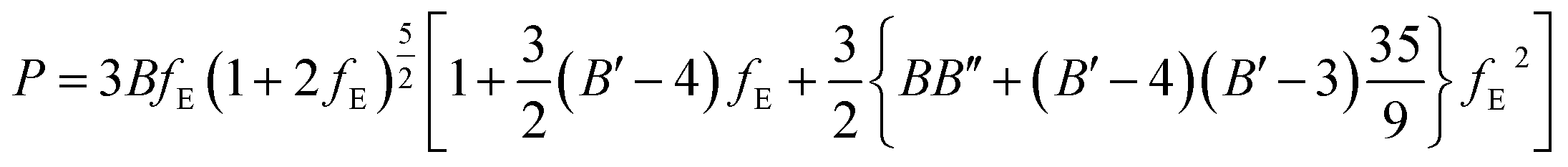 | (7) |
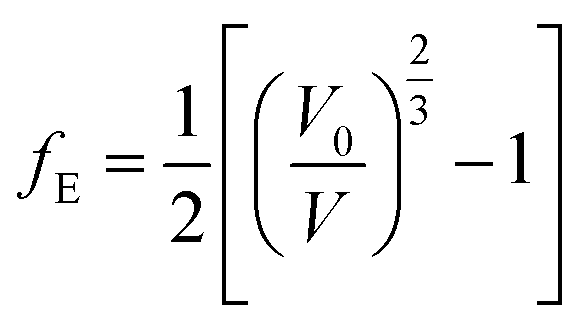 | (8) |
The fitted EOS parameters are the bulk modulus and its first two derivatives with respect to pressure. The values found are given in Table 3. The calculated bulk modulus, B = 39.48 ± 0.74 GPa, is in excellent agreement with the value derived from the computed elastic constants, B = 36.97 ± 0.27 GPa.
3.5. Raman spectroscopic characterization
The Raman spectrum of vandenbrandeite was recorded from a natural crystal sample from Musonoi Mine, Katanga Province (DRC) and determined using the first principles methodology. The experimental and theoretical spectra are compared in Fig. 5. The theoretical calculations reproduced satisfactorily the observed Raman spectrum. The band wavenumbers of both spectra along with the corresponding calculated intensities and assignments are given in Table 4. A series of pictures of the atomic motions in some Raman active vibrational modes are provided in Fig. S.4 of the ESI.† The Raman spectrum is analyzed in five different wavenumber regions: (i) OH stretching vibrations region from 3600 to 2700 cm−1 (Fig. 5A); (ii) the region from 2700 to 1300 cm−1 (Fig. 5E); (iii) the region from 1300 to 900 cm−1 (Fig. 5B); (iv) the region from 900 to 600 cm−1 (Fig. 5C); and (v) the region from 600 to 40 cm−1 (Fig. 5D). Since there are not theoretical bands in the region from 2700 to 1300 cm−1, it will be described separately after the analysis of the four main regions.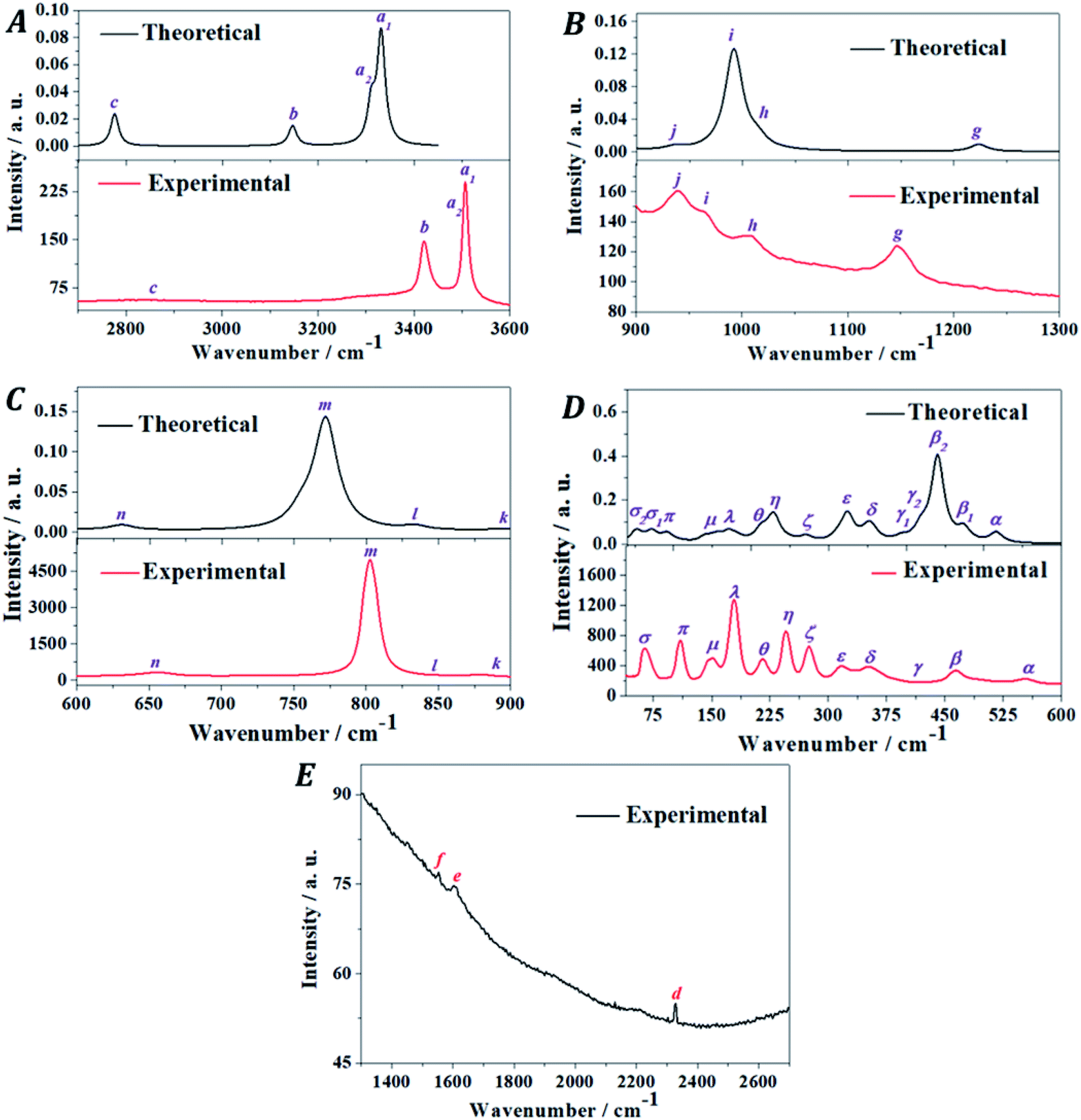 | ||
| Fig. 5 Experimental and theoretical Raman spectra of vandenbrandeite: (A) region: 3600–2700 cm−1; (B) region: 1300–900 cm−1; (C) region: 900–600 cm−1; (D) region: 600–40 cm−1; (E) region: 2700–1300 cm−1. | ||
| Band name | Exp. shift (cm−1) | Calc. shift (cm−1) | Int.[Å4 amu−1] | Assignment |
|---|---|---|---|---|
| OH stretching region (3600–2700 cm−1) | ||||
| a | 3510 | 3331 | 18628.64 |
ν(OH) |
| 3505 | 3311 | 6538.74 | ν(OH) | |
| b | 3419 | 3146 | 3075.31 | ν(OH) |
| c | 2827 | 2775 | 3938.92 | ν(OH) |
|
||||
| 2700–1300 cm−1 region | ||||
| d | 2327 | — | — | 2v1 = 2 × 1153 = 2306[2g] |
| e | 1604 | — | — | 2v1 = 2 × 803 = 1606[2m] |
| f | 1554 | v1 + v2 = 884 + 656 = 1540[n + k] | ||
|
||||
| 1300–900 cm−1 region | ||||
| g | 1153 | 1224 | 437.36 | δ(UOH) |
| h | 1008 | 1014 | 442.42 | δ(UOH) |
| i | 967 | 991 | 3847.72 | δ(UOH) |
| j | 941 | 936 | 143.12 | δ(UOH) |
|
||||
| 900–600 cm−1 region | ||||
| k | 884 | 894 | 53.03 | δ(UOH) |
| l | 844 | 834 | 155.87 | νa(UO2+2) + δ(UOH) |
| m | 803 | 772 | 3857.85 | νs(UO2+2) + δ(UOH) |
| n | 656 | 631 | 136.88 | δ(UOH) |
|
||||
| 600–40 cm−1 region | ||||
| α | 554 | 516 | 758.66 | ν(CuO) + δ(UOH) |
| β | 492 | 474 | 822.57 | δ(UOCu) + δ(UOH) |
| 463 | 440 | 5199.11 | δ(UOCu) + T(OH) | |
| γ | 409 | 419 | 706.75 | δ(UOCu) + T(OH) |
| 394 | 199.57 | ν(OH) + δ(UOH) | ||
| δ | 362 | 355 | 593.06 | δ(UOCu) + T(OH) |
| 351 | 348 | 275.18 | ν(UOeq) + T(OH) | |
| ε | 331 | 324 | 1071.66 | δ(UOCu) + T(OH) |
| 316 | 312 | 170.86 | T(OH) | |
| ζ | 276 | 271 | 167.95 | γ(UO22+) + δ(UOCu) + T(OH) |
| η | 246 | 249 | 34.95 | δ(UO22+) + δ(UOH) |
| 232 | 144.75 | γ(UO22+) + δ(UOCu) + δ(UOH) | ||
| 228 | 481.00 | δ(UO22+) + δ(UOCu) + δ(UOH) | ||
| θ | 216 | 213 | 235.64 | γ(UO22+) + T(OH) |
| λ | 179 | 171 | 130.56 | T(Cu) + ρ(UO22+) + δ(UOCu) + δ(UOH) |
| μ | 152 | 155 | 73.65 | T(Cu) + ρ(UO22+) + T(OH) |
| 144 | 141 | 55.96 | T(Cu) + ρ(UO22+) + T(OH) | |
| π | 110 | 92 | 40.54 | T(Cu) + T(UO22+) + δ(UOCu) + δ(UOH) |
| σ | 74 | 72 | 31.65 | T(Cu) + T(UO22+) + δ(UOCu) + δ(UOH) |
| 65 | 52 | 19.83 | T(Cu) + T(UO22+) + δ(UOCu) + δ(UOH) | |
The band ξ, located at 276 cm−1, is calculated to be at 271 cm−1 and assigned to uranyl deformations, UOCu bending vibrations and hydroxyl translations. Band η, at 246 cm−1, corresponds to the three close theoretical bands at 249 and 232 and 228 cm−1. The first of these bands is assigned to uranyl deformations and UOH bending vibrations. The second one is ascribed to uranyl deformations and UOCu and UOH bending vibrations. The assignment of the third sub-band is the same as that of the 232 cm−1 band but uranyl deformations are replaced by uranyl bending vibrations. The band θ, at 216 cm−1, is very well reproduced theoretically at 2123 cm−1 and assigned to uranyl deformations and hydroxyl translations.
All the bands with lower wavenumbers in the Raman spectrum of vandenbrandeite (λ, μ, π and σ) contain contributions from copper atom translations. The band λ at 179 cm−1 appears in the computed spectrum at 171 cm−1 and must be assigned to uranyl rotations, UOCu bending vibrations and cooper and hydroxyl translations. The band μ is composed of two sub-bands at 152 and 144 cm−1 which are reproduced theoretically at 155 and 141 cm−1. Both bands are ascribed to uranyl rotations and cooper and hydroxyl translations. The band π, at 110 cm−1, is calculated to be at 92 cm−1 and it is assigned to cooper, uranyl and hydroxyl translations and UOCu bending vibrations. Finally the band σ, contains two contributing sub-bands at 74 and 65 cm−1 which are reproduced theoretically at 72 and 52 cm−1, respectively. The two components of band σ have the same assignment as band π.
The deformation (γ), bending (δ) and rotation (ρ) motions of the nearly linear uranyl cation appear in the assignments of the different bands of the low wavenumber region of the Raman spectrum of vandenbrandeite. Therefore, this spectral region should not be referred to as the uranyl bending region as it has been frequently denoted. The same was observed in the Raman spectrum of other uranyl minerals. The distinction between these motions was very clearly explained in a previous work.117 The bending and rotation motions involve displacements of the uranyl oxygens out of the uranyl axis in the same and opposite directions, respectively. In the deformation motion, each oxygen displaces in a different direction.
It must be noticed that while the distribution of intensities in the 600–40 cm−1 region of the computed and experimental Raman spectra (Fig. 5D) is quite different, the matching of the wavenumbers of the peaks of both spectra is very good and faultless and, therefore, the assignment of the bands of this region should be reliable.
3.6. Thermodynamics
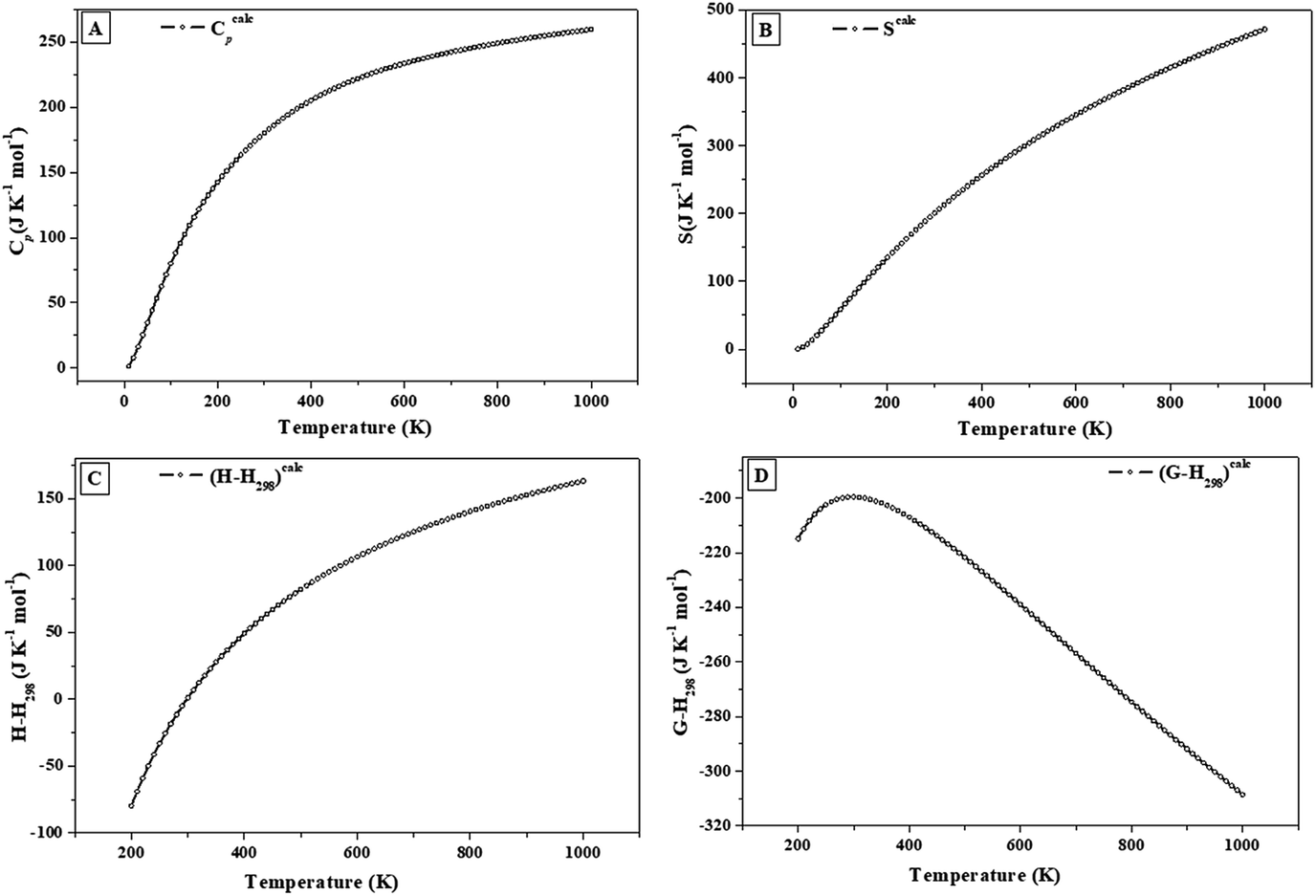 | ||
| Fig. 6 Calculated heat capacity, entropy, enthalpy and free energy functions of vandenbrandeite: (A) isobaric heat capacity; (B) entropy; (C) enthalpy; (D) Gibbs free energy. | ||
The calculated specific heat and entropy of vandenbrandeite at 298.15 K are 179.8 and 199.6 J K−1 mol−1, respectively. An accuracy of about 3–5% may be expected for the calculated specific heat and entropy at room temperature. The expected accuracy was estimated in a previous work125 from the analysis of the results obtained for uranyl-containing materials using the same methods as in this work in the cases in which the corresponding experimental values were available. The fundamental thermodynamic properties of several non-uranyl containing materials, as natroxalate and oxammite minerals and solid squaric acid,194–196 were also determined utilizing the same methods as in the present work and the results were found to be within the above mentioned expected accuracy. The dependence of thermodynamic properties on the temperature is also expected to be accurately reproduced. For example, the computed specific heats and entropies of γ-UO3 were in excellent agreement with their experimental counterparts in the temperature range from 0 to 1000 K.123,124 The differences of the calculated and experimental values of Cp and S were 3.9% and 1.8% at 100 K and 6.1% and 3.6% at 1000 K, respectively.
K) of vandenbrandeite as a function of temperature. The values of ΔfH and ΔfG are in units of kJ mol−1
| T (K) | ΔfH | ΔfG | logK |
|---|---|---|---|
| 298.15 | −1998.10 | −1771.30 | 310.32 |
| 300 | −1997.35 | −1769.11 | 308.02 |
| 350 | −1980.70 | −1713.77 | 255.76 |
| 400 | −1969.30 | −1664.10 | 217.30 |
| 450 | −1961.70 | −1618.60 | 187.88 |
| 500 | −1956.98 | −1576.30 | 164.67 |
| 540 | −1952.69 | −1544.11 | 149.36 |
| 600 | −1953.96 | −1498.79 | 130.48 |
| 650 | −1954.89 | −1462.74 | 117.54 |
| 700 | −1957.11 | −1428.09 | 106.56 |
| 750 | −1960.41 | −1394.62 | 97.13 |
| 800 | −1964.66 | −1362.18 | 88.94 |
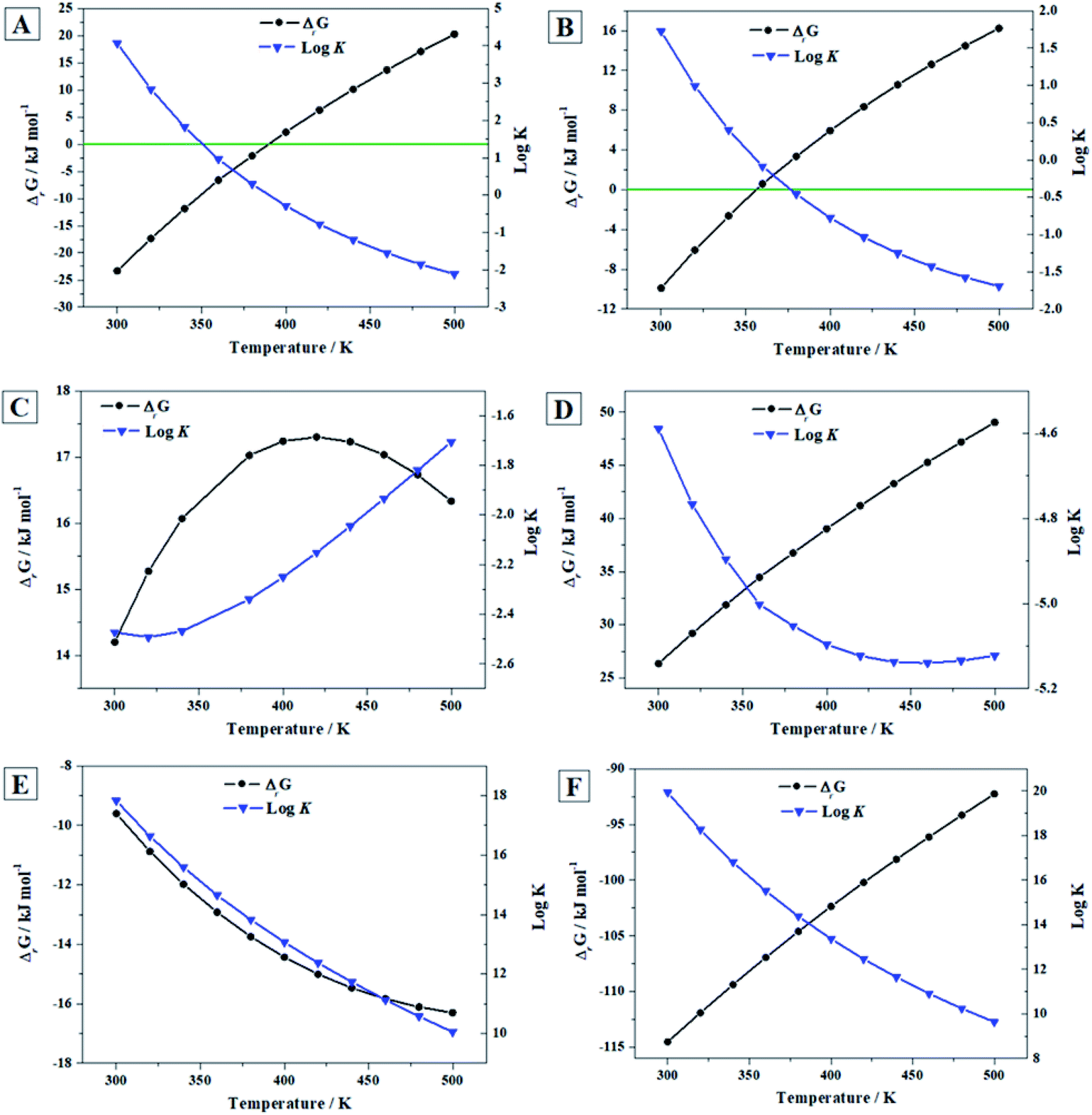 | ||
| Fig. 7 Calculated free energies of reaction and associated reaction constants of reactions (A)–(F) as a function of temperature. | ||
From the results (see Fig. 7A), it follows that vandenbrandeite becomes unstable with respect to the corresponding oxides approximately at 116 °C (390 K). The calculations predict (Fig. 7B) that dehydrated schoepite will transform, in the presence of copper oxide, into vandenbrandeite up to 83 °C (356 K). As shown in Fig. 7C, under the presence of carbon dioxide, vandenbrandeite should transform into rutherfordine within the full range of temperatures considered (300 to 500 K). Similarly (Fig. 7D), from 300 to 500 K, vandenbrandeite should transform into soddyite in the presence of silicon dioxide (quartz). If both carbon dioxide and silicon dioxide are simultaneously present, vandenbrandeite should transform into soddyite because the Gibbs free energy of reaction (D) is larger than that of reaction (C) at all the temperatures.
These thermodynamic relations, being very useful and rigorous, must be interpreted and used with caution. A full evaluation and understanding of the number and relative amount of the secondary phases of spent nuclear fuel present at the conditions of a final geological disposal over time requires the realization of complete thermodynamic computations124,125,201–204 employing thermochemical data for a significant number of materials, including the most important secondary phases, amorphous phases and aqueous species, at a wide range of temperature and pressure conditions. For example, the presence of oxidant species as hydrogen peroxide (see next section) and their concentration alters drastically the relative stability of the secondary phases of the SNF.
Since the Gibbs free energies and enthalpies of reaction (E) are negative everywhere (see Fig. 7E), the reaction of conversion of vandenbrandeite into schoepite under the presence of water and hydrogen peroxide is spontaneous and exothermic from 300 to 500 K. This is due to the great thermodynamic stability of schoepite at these conditions.115 The same is true for reaction (F) as shown in Fig. 7F. Hence, vandenbrandeite phase will be converted into studtite under high hydrogen peroxide concentrations. Although vandenbrandeite, as it occurred with schoepite, metaschoepite and becquerelite uranyl oxide hydrate phases,115,116 is largely stabilized under high concentrations of hydrogen peroxide, its stabilization under these conditions is not as large as that of studtite phase.124 Kubatko et al.205 and Forbes et al.206 observed experimentally that becquerelite, dehydrated schoepite and soddyite transform readily into studtite in the presence of high concentrations of hydrogen peroxide. The transformation of uranium trioxide, rutherfordine, metastudtite, schoepite and metaschoepite and becquerelite into studtite was also predicted in previous works.115,116,124,125 Vandenbrandeite should also transform into studtite under these conditions.
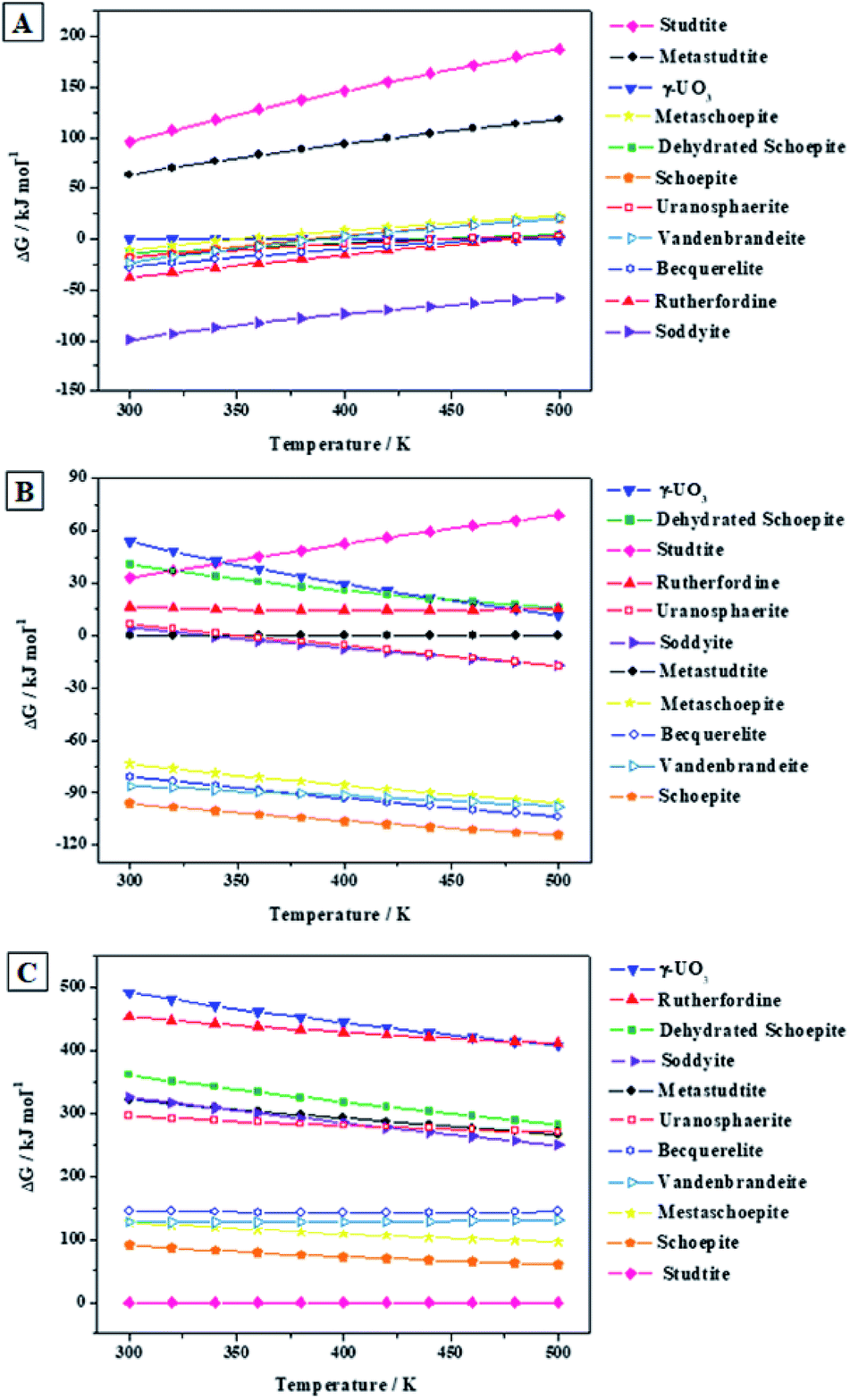 | ||
| Fig. 8 Relative thermodynamic stability of vandenbrandeite with respect to other secondary phases of SNF: (A) under the absence of hydrogen peroxide; (B) under the presence of water and hydrogen peroxide; (C) under high concentrations of hydrogen peroxide. The labels of the different phases are ordered vertically according to their relative stability at 300 K. | ||
Fig. 8A shows that, in the absence of hydrogen peroxide, soddyite is the most stable phase and that rutherfordine and becquerelite (the second and third most stable phases at 300 K) are also more stable than vandenbrandeite. Thus, at hydrogen peroxide free conditions, vandenbrandeite should be replaced by other mineral phases in the presence of, for example, silicate or carbonate ions. The uranyl oxide hydrate phases (Fig. 8B) are stabilized by the presence of water and hydrogen peroxide. The same appears to occur with the uranyl-copper tetrahydroxide phase. As shown in Fig. 8B, under the presence of water and hydrogen peroxide, vandenbrandeite becomes the second most stable phase after schoepite (becquerelite becomes more stable than vandenbrandeite at 373 K). Fig. 8B shows that in the presence of water and hydrogen peroxide, there is a large stability gap between schoepite, vandenbrandeite, metaschoepite and becquerelite minerals and the remaining ones. Finally, in the presence of high hydrogen peroxide concentrations (Fig. 8C), the stability of vandenbrandeite also increases as it also occurs to studtite, schoepite, metaschoepite and becquerelite.114,115,124 Under high concentrations of hydrogen peroxide, studtite becomes by far the most stable phase. Vandenbrandeite becomes the fourth most stable phase among those considered in this work and again there is a large stability gap between studtite, schoepite, metaschoepite, vandenbrandeite and becquerelite and the remaining phases under study.
4 Conclusions
A complete characterization of the crystal structure, the Raman spectrum and the mechanical and thermodynamic properties of the copper-uranyl tetrahydroxide mineral vandenbrandeite has been achieved by means of the X-ray diffraction and infrared and Raman spectroscopy techniques combined with first principles solid-state methods. The full crystal structure of vandenbrandeite, including the positions of the hydrogen atoms is obtained by the first time from X-ray diffraction data by structure refinement. Furthermore, the computed crystal structure is confirmed theoretically since the computed unit-cell parameters, interatomic distances and angles and X-ray diffraction pattern of vandenbrandeite are in excellent agreement with the experimental data. The availability of the full energy-optimized crystal structure of vandenbrandeite made possible the computation of its mechanical properties, Raman spectrum and thermodynamic properties using the first-principles methodology. We conclude that the first-principles methods provided a faithful description of vandenbrandeite crystal structure and spectra at the nanometer scale and allowed to determine a significant amount of relevant material properties whose accurate determination by means of experimental methods could be extremely difficult.From the computed elasticity matrix, equation of state and phonon spectra, the mechanical and dynamical stability of the crystal structure of vandenbrandeite were stablished and a rich set of relevant elastic properties were derived. This set comprises the bulk, shear and Young moduli, ductility, hardness and universal anisotropy indices and the derivatives of the bulk modulus with respect to pressure. The Raman spectrum of vandenbrandeite was recorded from a natural crystal sample and was also determined using density functional perturbation theory. The computed and experimental spectra have a very high degree of consistence and, therefore, all the Raman bands were rigorously assigned using a normal coordinate analysis of the theoretical vibrational results. The Raman spectrum within the spectral region from 1300 to 2700 cm−1 was shown to contain three low intensity bands. Two of these bands were recognized as overtones and the other one as a combination band.
From accurate phonon calculations, the fundamental thermodynamic properties of vandenbrandeite were evaluated. These thermodynamic properties were then used to determine the thermodynamic properties of formation of vandenbrandeite in terms of the elements as a function of temperature. In turn, the thermodynamic functions of formation were employed in order to obtain the Gibbs free energies of reaction and the associated reaction constants of a series of reactions involving vandenbrandeite and a representative subset of the most important secondary phases of spent nuclear fuel. The relative stability of vandenbrandeite with respect to these phases as a function of temperature and under different concentrations of hydrogen peroxide was reported. It was found that vandenbrandeite is strongly stabilized under the presence of hydrogen peroxide and that, under the simultaneous presence of water and hydrogen peroxide, becomes the second most stable phase among those considered in this work after schoepite.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The supercomputer time provided by the CTI-CSIC center is greatly acknowledged. This work has been carried out in the context of a CSIC-CIEMAT collaboration agreement: “Caracterización experimental y teórica de fases secundarias y óxidos de uranio formados en condiciones de almacenamiento de combustible nuclear”. JP acknowledges the support through the project no. LO1603 of the Ministry of Education, Youth and Sports National Sustainability Program I of the Czech Republic. JS and JČ were supported by the Ministry of Culture of the Czech Republic (long-term project DKRVO 2019-2023/1.II.a and 1.II.b; National Museum, 00023272). VT was supported by the Ministry of Science, Innovation and Universities within the Project FIS2016-77726-C3-1-P. VP thanks the support from the Czech Science Foundation (project 18-10504S).References
- Swedish Corrosion Institute, Copper as canister material for unreprocessed nuclear waste - evaluation with respect to corrosion, Report KBS-TR-90, Swedish Nuclear Fuel Supply Company, Stockholm, 1978 Search PubMed.
- G. Hultquist, Corros. Sci., 1986, 26, 173 CrossRef CAS.
- T. E. Eriksen, P. Ndalgmba and I. Grenthe, Corros. Sci., 1989, 29, 1241 CrossRef CAS.
- P. Wersin, K. Spahiu and J. Bruno, Kinetic modelling of bentonite-canister interaction. Long-term predictions of copper canister corrosion under oxic and anoxic conditions, Technical Report SKB-TR-94-25, Swedish Nuclear Fuel and Waste Management Company, Stockholm, 1994 Search PubMed.
- R. Sjoblom, H. P. Hermansson and O. Amcoff, Mater. Res. Soc. Symp. Proc., 1995, 353, 687 CrossRef CAS.
- H. P. Hermansson and S. Eriksson, Corrosion of the Copper Canister in the Repository Environment, SKI Report 99:52, Swedish Nuclear Power Inspectorate (SKI), Stockholm, 1999 Search PubMed.
- A. E. Mildowski, M. T. Styles and V. L. Hards, A natural analogue for copper waste canisters: the copper-uranium mineralised concretions in the Permian mudrocks of south Devon, United Kingdom, Technical Report SKB-TR-00-11, Swedish Nuclear Fuel and Waste Management Company, Stockholm, 2000 Search PubMed.
- A. E. Mildowski, M. T. Styles, M. S. A. Horstwood and S. J. Kemp, Alteration of uraniferous and native copper concretions in the Permian mudrocks of south Devon, United Kingdom, Technical Report SKB-TR-02-09, Swedish Nuclear Fuel and Waste Management Company, Stockholm, 2002 Search PubMed.
- F. King, L. Ahonen, C. Taxén, U. Vuorinen and L. Werme, Copper corrosion under expected conditions in a deep geologic repository, Technical Report SKB-TR-01-23, Swedish Nuclear Fuel and Waste Management Company, Stockholm, 2001 Search PubMed.
- R. Gubner and U. Andersson, Corrosion resistance of copper canister weld material, Technical Report SKB-TR-07-07, Swedish Nuclear Fuel and Waste Management Company, Stockholm, 2007 Search PubMed.
- P. Szakálos, G. Hultquist and G. Wikmark, Electrochem. Solid-State Lett., 2007, 10, C63 CrossRef.
- G. Hultquist, P. Szakálos, M. J. Graham, A. B. Belonoshko, G. I. Sproule, L. Gråsjö, P. Dorogokupets, B. Danilov, T. Aastrup, G. Wikmark, G. K. Chuah, J. C. Eriksson and A. Rosengren, Catal. Lett., 2009, 132, 311 CrossRef CAS.
- G. Hultquist, M. J. Graham, P. Szakálos, G. I. Sproule, A. Rosengren and L. Gråsjö, Corros. Sci., 2011, 53, 310 CrossRef CAS.
- B. Rosborg and L. Werme, J. Nucl. Mater., 2008, 379, 142 CrossRef CAS.
- B. Rosborg, Recorded corrosion rates on copper electrodes in the Prototype Repository at the Äspö HRL, Report SKB-R-13-13, Swedish Nuclear Fuel and Waste Management Company, Stockholm, 2013 Search PubMed.
- P. Wersin, LOT A2 test parcel. Compilation of copper data in the LOT A2 test parcel, Report TR-13-17, Swedish Nuclear Fuel and Waste Management Company, Stockholm, 2013 Search PubMed.
- S. Kärnbränslehantering, Design, production and initial state of the canister, Technical Report SKB-TR-10-14, Swedish Nuclear Fuel and Waste Management Company, Stockholm, 2014 Search PubMed.
- Å. Björkbacka, S. Hosseinpour, M. Johnson, C. Leygraf and M. Jonsson, Radiat. Phys. Chem., 2013, 92, 80 CrossRef.
- G. Hultquist, Corros. Sci., 2015, 93, 327 CrossRef CAS.
- M. Ottossona, M. Bomana, P. Berasteguia, Y. Anderssona, M. Hahlina, M. Korvelab and R. Bergera, Corros. Sci., 2017, 122, 53 CrossRef.
- A. Hedin, A. J. Johansson, C. Lilja, M. Boman, P. Berastegui, R. Berger and M. Ottosson, Corros. Sci., 2018, 137, 1 CrossRef CAS.
- N. Marcos, Mater. Res. Soc. Symp. Proc., 1997, 465, 1153 Search PubMed.
- F. King and P. Wersin, Review of Supercontainer Copper Shell-Bentonite Interactions and Possible Effects on Buffer Performance for the KBS-3H Design, Posiva Working Report 2013-03, Posiva, Olkiluoko, 2014 Search PubMed.
- P. Wersin, P. A. Epping, M. Pekala, P. Pitkänen and M. Snellman, Procedia Earth Planet. Sci., 2017, 17, 722 CrossRef.
- P. Alt-Epping, M. Pekala, P. Wersin and P. Pitkänen, in Proceedings of the 7th International Conference on Clay in Natural and Engineered Barriers for Radioactive Waste Confinement, Davos, Switzerland, 2017, p. 50 Search PubMed.
- P. Wersin, M. Pękala, P. Alt-Epping, P. Pitkänen and V. Cloet, in Proceedings of the 16th International High-Level Radioactive Waste Management (IHLRWM 2017), Charlotte, NC, 2017, p. 30 Search PubMed.
- M. Pekala, P. Alt-Epping and P. Wersin, 3D and 1D Dual-Porosity Reactive Transport Simulations - Model Improvements, Sensitivity Analyses, and Results from the Integrated Sulfide Project Inter-Model Comparison Exercise, Posiva Working Report 2018-31, Posiva, Olkiluoko, 2019 Search PubMed.
- F. King and C. D. Litke, The corrosion of copper in synthetic groundwater at 150 °C. Part I. The results of short term electrochemical tests, Technical Record TR-428, Atomic Energy of Canada Ltd. Toronto, 1987 Search PubMed.
- F. King, Appl. Geochem., 1995, 10, 477 CrossRef CAS.
- F. King, C. D. Litke, M. J. Quinn and D. M. LeNeveu, Corros. Sci., 1995, 37, 833 CrossRef CAS.
- K. Worgan, M. Apted and R. Sjoblom, Mater. Res. Soc. Symp. Proc., 1995, 353, 895 Search PubMed.
- F. King and M. Kolar, Theory manual for the copper corrosion model for stress corrosion cracking of used fuel disposal containers CCM-SCC.0, Nuclear Waste Management Division Report 06819-REP-01300-10095-R00, Ontario Power Generation, Toronto, Ontario, 2004 Search PubMed.
- F. King and M. Kolar, Simulation of the consumption of oxygen in long-term in situ experiments and in the third case study repository using the copper corrosion model CCM-UC.1.1, Nuclear Waste Management Division Report 06819-REP-01300-10084-R00, Ontario Power Generation, Toronto, Ontario, 2006 Search PubMed.
- F. King, Review and gap analysis of the corrosion of copper containers under unsaturated conditions, Nuclear Waste Management Division Report 06819-REP-01300-10124-R00, Ontario Power Generation, Toronto, Ontario, 2006 Search PubMed.
- J. M. Smith, The corrosion and electrochemistry of copper in aqueous, anoxic sulphide solutions, PhD Thesis, The University of Western Ontario, Canada, 2007.
- J. Smith, Z. Qin, F. King, L. Werme and D. W. Shoesmith, Corrosion, 2007, 63, 135 CrossRef CAS.
- B. M. Ikeda and C. D. Litke, Stress corrosion cracking of copper in nitrite/chloride mixtures at elevated temperatures, Technical Report NWMO TR-2007-04, Nuclear Waste Management Organization, Toronto, Ontario, 2007 Search PubMed.
- F. King, M. Kolar and P. Maak, J. Nucl. Mater., 2008, 379, 133 CrossRef CAS.
- F. King, Corrosion, 2009, 65, 233 CrossRef CAS.
- G. M. Kwong, Status of corrosion studies for copper used fuel containers under low salinity conditions, NWMO Technical Report NWMO-TR-2011-14, Nuclear Waste Management Organization, Toronto, Canada, 2011 Search PubMed.
- J. Chen, Z. Qin and D. W. Shoesmith, Long-term corrosion of copper in a dilute anaerobic sulfide solution, Electrochim. Acta, 2011, 56, 7854 CrossRef CAS.
- F. King, C. Lilja and M. Vähänen, J. Nucl. Mater., 2013, 438, 228 CrossRef CAS.
- P. G. Keech, P. Vo, S. Ramamurthy, J. Chen, R. Jacklin and D. W. Shoesmith, Corros. Eng., Sci. Technol., 2014, 49, 425 CrossRef CAS.
- C. H. Boyle and S. A. Meguid, Nucl. Eng. Des., 2015, 293, 403 CrossRef CAS.
- T. Standish, J. Chen, R. Jacklin, P. Jakupi, S. Ramamurthy, D. Zagidulin, P. Keech and D. W. Shoesmith, Electrochim. Acta, 2016, 211, 331 CrossRef CAS.
- B. Ibrahim, D. Zagidulin, M. Behazin, S. Ramamurthy, J. C. Wren and D. W. Shoesmith, Corros. Sci., 2018, 141, 53 CrossRef CAS.
- J. P. Simpson, Experiments on container materials for Swiss high-level waste disposal projects Part II, Nagra Technical Report 84-01, National Cooperative for the Disposal of Radioactive Waste, Wettingen, 1984 Search PubMed.
- L. H. Johnson and F. King, Canister options for the disposal of spent fuel, NAGRA Technical Report 02-11, National Cooperative for the Disposal of Radioactive Waste, Wettingen, 2003 Search PubMed.
- JNC, Second Progress Report in Research and Development for the Geological Disposal of HLW in Japan, Supporting Report 2, Repository Design and Engineering Technology, Japan Nuclear Cycle Development Institute, 2000 Search PubMed.
- N. Taniguchi and M. Kawasaki, J. Nucl. Mater., 2008, 379, 154 CrossRef CAS.
- H. J. Choi, M. Lee and J. Y. Lee, Nucl. Eng. Des., 2010, 240, 2714 CrossRef CAS.
- B. Kursten, E. Smailos, I. Azkarate, L. Werme, N. R. Smart and G. Santarini, State-of-the-art document on the COrrosion BEhaviour of COntainer MAterials, COBECOMA project Final Report, 5th Euratom Framework Programme, Contract No. FIKW-CT-20014-20138, European Commission, 2004 Search PubMed.
- D. Landolt, R. H. Muller and C. W. Tobias, J. Electrochem. Soc., 1969, 116, 1385 CrossRef.
- A. L. Bacarella and J. C. Griess, J. Electrochem. Soc., 1973, 120, 459 CrossRef CAS.
- S. B. Adeloju and Y. Y. Duan, Br. Corros. J., 1994, 29, 309 CrossRef CAS.
- A. Fateh, M. Aliofkhazraei and A. R. Rezvanian, Arabian J. Chem., 2017 DOI:10.2016/j.arabjc.2017.05.021.
- S. Li, M. T. Teague, G. L. Doll, E. J. Schindelholz and H. Conga, Corros. Sci., 2018, 141, 243 CrossRef CAS.
- R. C. Ewing, Nat. Mater., 2015, 14, 252 CrossRef CAS PubMed.
- R. Wang and Y. B. Katayama, Nucl. Chem. Waste Manage., 1982, 3, 83 CrossRef CAS.
- D. W. Shoesmith and S. Sunder, J. Nucl. Mater., 1992, 190, 20 CrossRef CAS.
- S. Sunder, D. W. Shoesmith, H. Christensen and N. H. Miller, J. Nucl. Mater., 1992, 190, 78 CrossRef CAS.
- D. W. Shoesmith, J. Nucl. Mater., 2000, 282, 1 CrossRef CAS.
- G. Sattonnay, C. Ardois, C. Corbel, J. F. Lucchini, M. F. Barthe, F. Garrido and D. Gosset, J. Nucl. Mater., 2001, 288, 11 CrossRef CAS.
- O. Roth and M. Jonsson, Cent. Eur. J. Chem., 2008, 6, 1 CAS.
- H. Christensen and S. Sunder, J. Nucl. Mater., 1996, 238, 70 CrossRef CAS.
- J. Plášil, J. Geosci., 2014, 59, 99 CrossRef.
- R. J. Finch and T. Murakami, Rev. Mineral. Geochem., 1999, 38, 91 CAS.
- I. Grenthe, J. Drozdzynski, T. Fujino, E. C. Buck, T. E. Albrecht-Schmitt and S. F. Wolf, in The Chemistry of Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J. Fuger, Springer Science and Business Media, Berlin, 2006, ch. V, vol. I; pp. 253–638 Search PubMed.
- S. V. Krivovichev and J. Plášil, in Uranium: From Cradle to Grave, ed. P. C. Burns and G. E. Sigmon, Mineralogical Association of Canada, Winnipeg, MB, Canada, 2013, short course 43, pp. 15–119 Search PubMed.
- P. C. Burns, R. C. Ewing and M. L. Miller, J. Nucl. Mater., 1997, 245, 1 CrossRef CAS.
- A. Rosenzweig and R. R. Ryan, Am. Mineral., 1975, 60, 448 CAS.
- J. Plášil, Minerals, 2018, 8, 551 CrossRef.
- P. Piret, J. Appl. Crystallogr., 1979, 12, 616 CrossRef CAS.
- T. A. Olds, J. Plášil, A. R. Kampf, F. Dal Bo and P. C. Burns, Minerals, 2018, 8, 511 CrossRef CAS.
- P. Piret, J. P. Declercq and D. Wauters-Stoop, Bull. Mineral., 1980, 103, 176 CAS.
- J. Čejka, Z. Urbanec, J. Čejka Jr and Z. Mrázek, Neues Jahrb. Mineral., Abh., 1988, 159, 297 Search PubMed.
- P. Ondruš, F. Veselovský, R. Skála, I. Císařová, J. Hloušek, J. Frýda, Vavřín, J. Čejka and A. Gabašová, J. Czech Geol. Soc., 1997, 42, 7 Search PubMed.
- P. Ondruš, F. Veselovský, A. Gabašová, J. Hloušek and V. Šrein, J. Czech Geol. Soc., 2003, 48, 149 Search PubMed.
- J. Plášil, K. Fejfarová, K. S. Wallwork, M. Dušek, R. Škoda, J. Sejkora, J. Čejka, F. Veselovský, J. Hloušek, N. Meisser and J. Brugger, Am. Mineral., 2012, 97, 1796 CrossRef.
- A. J. Locock and P. C. Burns, Can. Mineral., 2003, 41, 489 CrossRef CAS.
- W. D. Birch, W. D. Mumme and E. R. Segnit, Aust. Mineral., 1988, 3, 125 Search PubMed.
- U. Kolitsch and G. Giester, Mineral. Mag., 2001, 65, 717 CrossRef CAS.
- P. C. Burns, R. C. Ewing and F. C. Hawthorne, Can. Mineral., 1997, 35, 1551 CAS.
- P. C. Burns, Rev. Mineral. Geochem., 1999, 38, 23 CAS.
- P. C. Burns, Can. Mineral., 2005, 43, 1839 CrossRef CAS.
- P. C. Burns, J. Nucl. Mater., 1999, 265, 218 CrossRef CAS.
- P. C. Burns, K. M. Deely and S. Skanthakumar, Radiochim. Acta, 2004, 92, 151 CAS.
- A. L. Klingensmith and P. C. Burns, Am. Mineral., 2007, 92, 1946 CrossRef CAS.
- C. Frondel, Am. Mineral., 1956, 41, 539 CAS.
- R. M. Garrels and C. L. Christ, US Geol. Surv. Prof. Pap., 1959, 320, 81 CAS.
- R. J. Finch and R. C. Ewing, J. Nucl. Mater., 1992, 190, 133 CrossRef CAS.
- R. J. Finch and R. C. Ewing, Uraninite alteration in an oxidizing environment and its relevance to the disposal of spent nuclear fuel, SKB Technical Report 91–15, Swedish Nuclear Fuel and Waste Management Co., Stockholm, Sweden, 1994 Search PubMed.
- R. S. Forsyth and L. O. Werme, J. Nucl. Mater., 1992, 190, 3 CrossRef CAS.
- E. C. Pearcy, J. D. Prikryl, W. M. Murphy and B. W. Leslie, Appl. Geochem., 1994, 9, 713 CrossRef CAS.
- D. J. Wronkiewicz, J. K. Bates, T. J. Gerding, E. Veleckis and B. S. Tani, J. Nucl. Mater., 1992, 190, 107 CrossRef CAS.
- D. J. Wronkiewicz, J. K. Bates, S. F. Wolf and E. C. Buck, J. Nucl. Mater., 1996, 238, 78 CrossRef CAS.
- J. Bruno, I. Casas, E. Cera, R. C. Ewing, R. J. Finch and L. O. Werme, Mater. Res. Soc. Symp. Proc., 1994, 353, 633 CrossRef.
- P. A. Finn, J. C. Hoh, S. F. Wolf, S. A. Slater and J. K. Bates, Radiochim. Acta, 1996, 74, 65 CAS.
- A. Schoep, Ann. Mus. Congo Belge, 1932, 1, 22 Search PubMed.
- J. Thoreau, Ann. Soc. Geol. Belge, 1931, 55, C3 Search PubMed.
- C. Frondel, U.S. Geol. Surv. Bull., 1958, 1064, 1 Search PubMed.
- G. Gauthier, A. François, M. Deliens and P. Piret, Mineral. Rec., 1989, 20, 265 CAS.
- C. Bignand, Bull. Soc. Fr. Mineral. Cristallogr., 1955, 78, 1 CAS.
- I. H. Milne and E. W. Nuffield, Am. Mineral., 1951, 36, 394 CAS.
- A. Rosenzweig and R. R. Ryan, Cryst. Struct. Commun., 1977, 6, 53 CAS.
- A. S. Povarennykh, Konstitut. Svoy. Mineral., 1979, 13, 78 CAS.
- J. Čejka and Z. Urbanec, Trans. Czech. Acad. Sci., Math. Natur. Sci. Ser., 98, 1–93 Search PubMed.
- J. Čejka, Neues Jahrb. Mineral., Abh., 1994, H3, 112 Search PubMed.
- P. Škácha, J. Plášil, J. Sejkora, J. Čejka, R. Škoda and N. Meisser, Bull. Mineral. Petrolog. Odd. Nár. Muz., 2014, 22, 240 Search PubMed.
- J. Plášil, Eur. J. Mineral., 2018, 30, 253 CrossRef.
- J. Plášil, Z. Kristallogr., 2019, 234, 733–738 Search PubMed.
- S. Ghazisaeed, B. Kiefer and J. Plášil, RSC Adv., 2019, 9, 10058 RSC.
- F. Colmenero, J. Cobos and V. Timón, Inorg. Chem., 2018, 57, 4470 CrossRef CAS PubMed.
- F. Colmenero, A. M. Fernández, J. Cobos and V. Timón, ACS Earth Space Chem., 2019, 3, 17 CrossRef CAS.
- F. Colmenero, A. M. Fernández, J. Cobos and V. Timón, RSC Adv., 2019, 8, 24599 RSC.
- F. Colmenero, J. Cobos and V. Timón, J. Phys.: Condens. Matter, 2019, 31, 175701 CrossRef CAS.
- F. Colmenero, J. Plášil, J. Cobos, J. Sejkora, V. Timón, J. Čejka and L. J. Bonales, RSC Adv., 2019, 9, 15323 RSC.
- F. Colmenero, J. Plášil and J. Sejkora, Dalton Trans., 2019, 48, 16722 RSC.
- L. J. Bonales, F. Colmenero, J. Cobos and V. Timón, Phys. Chem. Chem. Phys., 2016, 18, 16575 RSC.
- F. Colmenero, in Minerals, ed. K. S. Essa, InTechOpen, London, 2018, ISBN: 978-953-51-6784-6 Search PubMed.
- F. Colmenero, A. M. Fernández, J. Cobos and V. Timón, J. Phys. Chem. C, 2018, 122, 5254 CrossRef CAS.
- F. Colmenero, L. J. Bonales, J. Cobos and V. Timón, J. Phys. Chem. C, 2017, 121, 5994 CrossRef CAS.
- F. Colmenero, L. J. Bonales, J. Cobos and V. Timón, J. Phys. Chem. C, 2017, 121, 14507 CrossRef CAS.
- F. Colmenero, A. M. Fernández, J. Cobos and V. Timón, J. Phys. Chem. C, 2018, 122, 5268 CrossRef CAS.
- F. Colmenero, in Density Functional Theory, ed. D. Glossman-Mitnik, InTechOpen, London, 2018, ISBN: 978-953-51-7020-4 Search PubMed.
- D. Langmuir, Aqueous Environmental Geochemistry, Prentice-Hall, New York, 1997, pp. 486–557 Search PubMed.
- I. Grenthe, J. Fuger, R. J. M. Konings, R. J. Lemire, A. B. Muller, C. Nguyen-Trung and H. Wanner, Chemical Thermodynamics of Uranium, Nuclear Energy Agency Organisation for Economic Co-Operation and Development, OECD, Issy-les-Moulineaux, France, 2004 Search PubMed.
- NEA Data Bank, Thermochemical Database (TDB), https://www.oecd-nea.org/dbtdb/, accessed Nov. 22, 2019 Search PubMed.
- Thermo-Chimie database (Consortium Andra-Ondraf/Niras-RWM), http://www.thermochimie-tdb.com/, accessed Nov. 22, 2019 Search PubMed.
- Rigaku Oxford Diffraction, CrysAlisCCD, CrysAlisRED and CrysAlis PRO. Oxford Diffraction Ltd, Yarnton, Oxfordshire, UK, 2019 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2015, 71, 3 CrossRef PubMed.
- Crystallographic Computing System for Standard and Modulated Structures, Jana2006, http://jana.fzu.cz/, accessed Sept. 15, 2019 Search PubMed.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567 CAS.
- MaterialsStudio, http://3dsbiovia.com/products/collabora-tive-science/biovia-materials-studio/, accessed Sept. 15, 2019 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS.
- M. C. Payne, M. P. Teter, D. C. Ailan, A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045 CrossRef CAS.
- N. Troullier and J. L. Martins, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 1993 CrossRef CAS PubMed.
- F. Colmenero, Characterization of Secondary Phases of Spent Nuclear Fuel under Final Geological Disposal Conditions: Experimental and Theoretical Studies, PhD Thesis, Universidad Autónoma de Madrid, 2017 DOI:10.13140/RG.2.2.10526.43843.
- F. Colmenero, L. J. Bonales, J. Cobos and V. Timón, Spectrochim. Acta, Part A, 2017, 174, 245 CrossRef CAS PubMed.
- F. Colmenero, L. J. Bonales, J. Cobos and V. Timón, J. Solid State Chem., 2017, 253, 249 CrossRef CAS.
- F. Colmenero, L. J. Bonales, J. Cobos and V. Timón, Clay Miner., 2018, 53, 377 CrossRef CAS.
- F. Colmenero, J. Cobos and V. Timón, Theor. Chem. Acc., 2019, 138, 43 Search PubMed.
- F. Colmenero, Appl. Sci., 2018, 8, 2281 Search PubMed.
- B. G. Pfrommer, M. Cote, M. S. G. Louie and M. L. Cohen, J. Comput. Phys., 1997, 131, 233 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- R. T. Downs, K. L. Bartelmehs, G. V. Gibbs and M. B. Boisen, Am. Mineral., 1993, 78, 1104 CAS.
- M. Coccioni, in Correlated Electrons: From Models to Materials Modeling and Simulation, ed. E. Pavarini, E. Koch, F. Anders and M. Jarrell, Forschungszentrum Jülich, Berlin, 2012, vol. 2, ch. 4 Search PubMed.
- S. L. Dudarev, D. Nguyen Manh and A. P. Sutton, Philos. Mag. B, 1997, 75, 613 CrossRef CAS.
- J. P. Crocombette, F. Jollet, L. T. Nga and T. Petit, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 104107 CrossRef.
- P. Nerikar, T. Watanabe, J. S. Tulenko, S. R. Phillpot and S. B. Sinnott, J. Nucl. Mater., 2009, 384, 61 CrossRef CAS.
- P. F. Weck, E. Kim, C. F. Jové-Colón and D. C. Sassani, Dalton Trans., 2013, 42, 4570 RSC.
- D. A. Andersson, G. Baldinozzi, L. Desgranges, D. R. Conradson and S. D. Conradson, Inorg. Chem., 2013, 52, 2769 CrossRef CAS.
- G. Beridze and P. M. Kowalski, J. Phys. Chem. A, 2014, 118, 11797 CrossRef CAS.
- P. F. Weck and E. Kim, Dalton Trans., 2004, 43, 17191 RSC.
- P. F. Weck, E. Kim and E. C. Buck, RSC Adv., 2015, 5, 79090 RSC.
- P. F. Weck and E. Kim, J. Phys. Chem. C, 2016, 120, 16553 CrossRef CAS.
- D. C. Sassani, C. F. Jové-Colón, P. F. Weck, J. L. Jerden, K. E. Frey, T. Cruse, W. L. Ebert, E. C. Buck, R. S. Wittman, Fuel Cycle Research and Development Report FCRD-UFD-2013–000404, Sandia National Laboratories, Albuquerque, 2013 Search PubMed.
- T. M. Alam, Z. Liao, M. Nyman and J. Yates, J. Phys. Chem. C, 2016, 120, 10675 CrossRef CAS.
- N. Kalashnyk, D. L. Perry, F. Massuyeau and E. Faulques, J. Phys. Chem. C, 2018, 122, 7410 CrossRef CAS.
- S. Ostanin and P. Zeller, J. Phys.: Condens. Matter, 2007, 19, 246108 CrossRef CAS PubMed.
- S. Ostanin and P. Zeller, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 073101 CrossRef.
- R. Yu, J. Zhu and H. Ye, Comput. Phys. Commun., 2010, 181, 671 CrossRef CAS.
- J. F. Nye, Physical Properties of Crystals, Clarendon, Oxford, 1976 Search PubMed.
- F. Colmenero, Mater. Res. Express, 2019, 6, 045610 (Mater. Res. Express, 2019, 6, 069401) CrossRef.
- F. Colmenero, Phys. Chem. Chem. Phys., 2019, 21, 2673 RSC.
- F. Colmenero, Mater. Lett., 2019, 245, 25 CrossRef CAS.
- F. Colmenero, Adv. Theory Simul., 2019, 2, 1900040 CrossRef.
- F. Colmenero and V. Timón, J. Mater. Sci., 2020, 55, 218 CrossRef CAS.
- F. Birch, Phys. Rev., 1947, 71, 809 CrossRef CAS.
- R. J. Angel, Rev. Mineral. Geochem., 2000, 41, 35 CrossRef.
- EOSFIT 5.2 software; http://programming.ccp14.ac.uk/ccp/web-mirrors/ross-angel/crystal/software.html, accessed Sept. 15, 2019 Search PubMed.
- A. Marmier, Z. A. D. Lethbridge, R. I. Walton, C. W. Smith, S. C. Parker and K. E. Evans, Comput. Phys. Commun., 2010, 181, 2102 CrossRef CAS.
- S. Baroni, S. de Gironcoli and A. Dal Corso, Rev. Mod. Phys., 2001, 73, 515 CrossRef CAS.
- X. Gonze and C. Lee, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 10355 CrossRef CAS.
- K. Refson, P. R. Tulip and S. J. Clark, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 155114 CrossRef.
- W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- C. Lee and X. Gonze, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 8610 CrossRef CAS PubMed.
- A. A. Maradudin, E. M. Montroll, G. H. Weiss and I. P. Ivatova, Solid State Physics, ed. E. H. Ehrenreich, F. Seitz and D. Turnbull, Academic, New York, 2nd edn, 1971 Search PubMed.
- M. W. Chase, C. A. Davies, J. R. Downey, D. J. Frurip, R. A. McDonald and A. N. Syverud, J. Phys. Chem. Ref. Data, 1985, 14(suppl. 1), 1 CAS.
- I. Barin, Thermochemical Data of Pure Substances, VCH, Weinheim, 3rd edn, 1995 Search PubMed.
- R. F. W. Bader, J. Hernández-Trujillo and F. Cortés-Guzman, J. Comput. Chem., 2006, 28, 4 CrossRef PubMed.
- B. Lafuente, R. T. Downs, H. Yang and N. Stone, in Highlights in Mineralogical Crystallography, ed. T. Armbruster, R. M. Danisi and W. De Gruyter, Berlin, Germany, 2015, pp. 1–30 Search PubMed.
- RRUFF database, http://rruff.info/vandenbrandeite, Record R080114, accessed Sept. 15, 2019 Search PubMed.
- M. Born, Math. Proc. Cambridge Philos. Soc., 1940, 36, 160 CrossRef CAS.
- F. Mouhat and F. X. Coudert, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 224104 CrossRef.
- W. Voigt, Lehrbuch der Kristallphysik, Teubner, Leipzig, 1962 Search PubMed.
- A. Reuss, Z. Angew. Math. Mech., 1929, 9, 49 CrossRef CAS.
- R. Hill, Proc. Phys. Soc., London, Sect. A, 1952, 65, 349 CrossRef.
- S. F. Pugh, Philos. Mag., 1954, 45, 823 CAS.
- Y. Bouhadda, S. Djella, M. Bououdina, Y. Fenineche and Y. Boudouma, J. Alloys Compd., 2012, 534, 20 CrossRef CAS.
- H. Niu, P. Wei, Y. Sun, C. X. Chen, C. Franchini, D. Li and Y. Li Y, Appl. Phys. Lett., 2011, 99, 031901 CrossRef.
- S. I. Ranganathan and M. Ostoja-Starzewski, Phys. Rev. Lett., 2008, 101, 055504 CrossRef PubMed.
- F. Colmenero and V. Timon, J. Solid State Chem., 2018, 263, 131 CrossRef CAS.
- F. Colmenero, J. Phys. Chem. Solids, 2019, 125, 31 CrossRef CAS.
- F. Colmenero and R. Escribano, J. Phys. Chem. A, 2019, 123, 424 CrossRef PubMed.
- Y. Tardy and R. M. Garrels, Geochim. Cosmochim. Acta, 1976, 41, 1051 CrossRef.
- R. J. Finch, Mater. Res. Soc. Symp. Proc., 1997, 465, 1185 CrossRef CAS.
- S. B. Clark, R. C. Ewing and J. C. Schaumloffel, J. Alloys Compd., 1998, 271–273, 189 CrossRef CAS.
- F. Chen, R. C. Ewing and S. B. Clark, Am. Mineral., 1999, 84, 650 CrossRef CAS.
- B. George, L. P. Brown, C. H. Farmer, P. Buthod and F. S. Manning, Ind. Eng. Chem. Process Des. Dev., 1976, 15, 332 CrossRef.
- W. R. Smith and R. W. Missen, Chemical Reaction Equilibrium Analysis: Theory and Algorithms, Wiley-Interscience, New York, 1982 Search PubMed.
- C. E. Harvie, J. P. Greenberg and J. H. Weare, Geochim. Cosmochim. Acta, 1987, 51, 1045 CrossRef CAS.
- M. H. A. Piro, Calphad, 2017, 58, 115 CrossRef CAS.
- K. A. Kubatko, D. Unruh and P. C. Burns, Mater. Res. Soc. Symp. Proc., 2006, 893, 423 CAS.
- T. Z. Forbes, P. Horan, T. Devine, D. McInnis and P. C. Burns, Am. Mineral., 2011, 96, 202 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: (a) Interatomic distances and angles in vandenbrandeite; (b) frontier band functions in vandenbrandeite; (c) pictures of the atomic motions in the Raman active vibrational normal modes of vandenbrandeite; (d) computed fundamental thermodynamic properties of vandenbrandeite as a function of temperature; (e) experimental and theoretical positions of the atoms in the unit cell of vandenbrandeite. CCDC 1952205 and 1946879. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra09047a |
| This journal is © The Royal Society of Chemistry 2019 |