
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiterpenoids from Isodon rubescens and their nitric oxide production inhibitory activity†
Chumao Wen‡
a,
Su Chen‡a,
Fang Yuana,
Xiangming Liua,
Fajun Songc,
Zhinan Meib,
Xiaofei Yang*a and
Guangzhong Yang *b
*b
aCollege of Biomedical Engineering, South-Central University for Nationalities, Wuhan 430074, P. R. China. E-mail: sunlittlefly@hotmail.com; Fax: +86 27 6784 1196; Tel: +86 27 6784 1196
bSchool of Pharmaceutical Sciences, South-Central University for Nationalities, Wuhan 430074, P. R. China. E-mail: yanggz888@126.com
cCollege of Life Science, South Central University for Nationalities, Wuhan 430074, P. R. China
First published on 9th December 2019
Abstract
Six new ent-kaurane diterpenoids, isodonrubescins A–F (1–6), together with twenty-five known ent-kaurane diterpenoids (7–31), a known ent-atisane diterpenoid (32), and two known ent-abietane diterpenoids (33–34), were isolated from Isodon rubescens. Their structures were established by means of extensive MS and NMR data analysis. Among the all isolates, compound 7 was found in a natural product for the first time, and ent-atisane diterpenoid was discovered from I. rubescens in Hubei Province, P. R. China for the first time. Furthermore, all the isolated compounds were tested for their NO production inhibitory activity in LPS stimulated RAW264.7 cells. Compounds 7–9, 12, 13, 16, and 17 displayed NO production inhibitory activities with IC50 values ranging from 1.36 to 18.25 μM, respectively.
1. Introduction
The genus Isodon, comprising about 150 different species of under-shrubs, sub-undershrubs, or perennial herbs, is a cosmopolitan and important genus of the Lamiaceae family. It is widely distributed in tropical and subtropical Asia. Previous studies have shown that they are rich sources of diterpenoids with diverse structural scaffolds, such as ent-kauranes, ent-abietanes, ent-atisanes, and have a range of biological activities.1–3Isodon rubescens is a perennial herb distributed widely in Henan, Guizhou, Hebei, Jiangxi, Hubei, and some other provinces of P. R. China.4 It has attracted great attention due to the traditional uses in folk medicine for the treatment of respiratory and gastrointestinal bacterial infections, inflammation, and cancer.5–8 Oridonin, an important ent-kaurane from I. rubescens showed the anti-tumor and anti-inflammatory activities. Previous studies have demonstrated that it exhibits anti-tumor effects on human cancer cells, such as HepG2, SGC-7901, MCF-7, mainly by blocking the cell cycle, inducing apoptosis and autophagy of tumor cells, and shows anti-inflammatory effects by inhibiting the expression of inflammatory factors through nuclear factor-kappa B (NF-κB) signal pathway.9,10 In addition, previous investigations on the chemical constituents of I. rubescens collected from different provinces, P. R. China revealed that they contained different structure types of diterpenoids. For example, the chemical constituents of I. rubescens collected from Guizhou Province were mainly 6,7-seco-ent-kaurane diterpenoids, however, 7,20-epoxy-ent-kaurane diterpenoids were main chemical constituents of I. rubescens collected from Henan Province.11,12 Furthermore, the chemical constituents of I. rubescens collected from Hubei Province have not been extensively investigated, only 16 new diterpenoids have been reported, including diterpene alkaloids with an aza-ent-kaurane skeleton.13–17 Therefore, in order to fully understand the active constituents of I. rubescens from different regions, a reinvestigation of I. rubescens collected from Badong county, Hubei Province was undertaken in the hope of discovering diterpenoids with interesting structures and biological activities. As a result, six new diterpenoids (1–6), together with twenty-five known ent-kaurane diterpenoids (7–31), a known ent-atisane diterpenoid (32) and two known ent-abietane diterpenoids (33–34) were isolated from this plant, and it should be noted that compounds 10, and 13–17 have been reported in our previous work.18 Herein we reported the isolation, structural elucidation of six new diterpenoids and biological activities of all isolated compounds (Fig. 1).
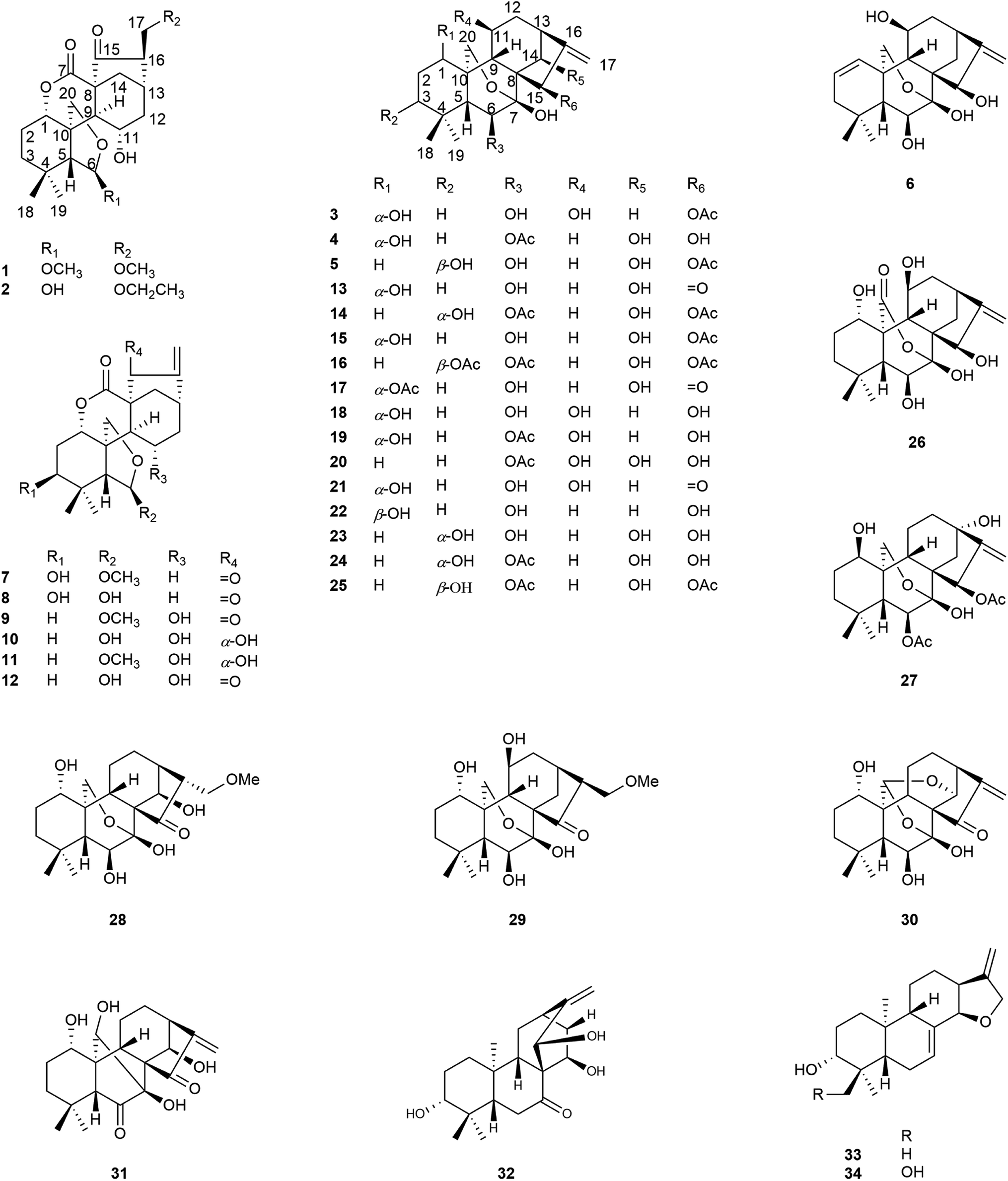 | ||
| Fig. 1 Structures of compounds 1–34. | ||
2. Results and discussion
Compound 1 was obtained as colorless needle crystals, and its molecular formula was determined to be C22H32O7 based on the HR-ESI-MS at m/z 431.20383 [M + Na]+ (calcd for C22H32O7Na, 431.20402), indicating of seven degrees of unsaturation. The 1H NMR spectra (Table 1) of 1 revealed the presence of two singlet methyls [δH 0.96 (s), 0.93 (s)] and two methoxy groups [δH 3.19 (s), 3.15 (s)], one oxygenated methylene [δH 4.32 (d, J = 9.6 Hz), 4.28 (d, J = 9.6 Hz)], three oxygenated methines [δH 4.89 (m), 5.03 (s), 4.46 (m)]. Its 13C NMR and DEPT spectra (Table 2) exhibited 22 carbon signals, including two methoxy groups at δC 54.8 and δC 59.0, two oxygenated methylenes at δC 74.5 and δC 71.8, two oxygenated methines at δC 76.9 and δC 64.8, one hemiacetal group at δC 109.7, one δ-lactone carbonyl group at δC 171.3, one carbonyl group at δC 212.9 and three quaternary carbons at δC 31.7, δC 57.7 and δC 51.2, which implied a 6,7-seco-ent-kaurane skeleton. Detailed analysis of the NMR data of 1 indicated that 1 is structurally related to dayecrystal D.19 The significant difference between them was the change of the chemical shift of C-12 from δC 42.1 in 1 to δC 33.1 in the latter, which was caused by a γ-gauche shielding effect between 16-methoxymethyl group and H-12α. Therefore, it can be deduced that the methoxymethyl group at C-16 in 1 was β-oriented. The location of the methoxymethyl group at C-16 was revealed by the HMBC correlation of OMe (δH 3.15) with C-17 (δC 71.8) and the 1H–1H COSY correlations (Fig. 2) of H2-17 (δH 3.60, δH 3.52) with H-16 (δH 2.67). The ROESY (Fig. 3) correlations of H-16 with H-12α (δH 1.60), of H-12β (δH 2.91) with H-13β and H-11β confirmed the β-orientation of the methoxymethyl group. Consequently, the structure of 1 was assigned as 11α-hydroxy-6β-methoxy-16β-methoxymethyl-6,7-seco-6,20-exoxy-1α,7-olide-ent-kaur-15-one, and named as isodonrubescin A.| No. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 4.89 (m) | 4.92 (dd, 6.9, 10.5) | 4.35 (m) | 3.74 (m) | 1.74–1.86 (m) | 6.38 (dd, 2.4, 10.8) |
| 2 | 1.87 (m) | 1.91 (m) | 1.96 (m) | 1.86 (m) | 2.07 (m); 1.09 (d, 12.6) | 5.73 (ddd, 1.8, 6.0, 10.2) |
| 3 | 1.31 (m) | 1.37 (m); 1.32 (m) | 1.51 (m); 1.42 (m) | 1.34 (m) | 3.75 (s) | 1.94 (overlap); 1.77 (dd, 6.0, 16.8) |
| 4 | ||||||
| 5 | 3.48 (s) | 3.27 (s) | 1.74 (d, 5.4) | 1.77 (d, 6.6) | 2.44 (d, 6.6) | 2.16 (d, 6.6) |
| 6 | 5.03 (s) | 5.76 (s) | 4.24 (dd, 5.4, 3.6) | 5.86 (d, 6.6) | 4.33 (dd, 2.4, 6.6) | 4.31 (dd, 4.8, 6.6) |
| 7 | ||||||
| 8 | ||||||
| 9 | 2.65 (m) | 2.93 (d, 10.8) | 2.82 (d, 9.6) | 2.92 (dd, 6.0, 12.9) | 2.84 (overlap) | 2.88 (dd, 1.2, 9.6) |
| 10 | ||||||
| 11 | 4.46 (m) | 4.55 (dd, 8.7, 18.9) | 4.73 (m) | 2.31 (m); 1.91 (m) | 1.54 (m); 1.23 (m) | 4.58 (m) |
| 12 | 2.91 (m); 1.60 (dd, 9.0, 13.8) | 2.95 (m); 1.58 (dd, 9.0, 14.4) | 2.86 (m); 1.91 (m) | 2.42 (m); 1.73 (m) | 2.32 (m); 1.61 (m) | 2.96 (m); 1.91 (overlap) |
| 13 | 2.61 (dd, 4.2, 9.6) | 2.68 (dd, 3.6, 9.6) | 2.72 (dd, 9.6, 3.6) | 2.87 (d, 9.6) | 2.84 (overlap) | 2.76 (dd, 5.4, 10.4) |
| 14 | 2.68 (m); 2.33 (dd, 4.2, 12.0) | 2.72 (d, 12.0); 2.39 (dd, 3.9, 12.3) | 2.22 (d, 12.6); 2.17 (dd, 12.6, 4.2) | 5.15 (overlap) | 5.08 (s) | 2.19 (dd, 4.8, 12.6); 2.01 (d, 12.6) |
| 15 | 6.55 (s) | 5.57 (d, 2.4) | 6.93 (s) | 5.20 (overlap) | ||
| 16 | 2.67 (m) | 2.63 (br t, 5.4) | ||||
| 17 | 3.60 (m); 3.52 (dd, 4.2, 9.0) | 3.61 (m) | 5.29 (s); 5.12 (s) | 5.69 (s); 5.40 (s) | 5.41 (s); 5.28 (s) | 5.51 (s); 5.22 (overlap) |
| 18 | 0.96 (s) | 0.98 (s) | 1.24 (s) | 0.92 (s) | 1.57 (s) | 1.20 (s) |
| 19 | 0.93 (s) | 0.98 (s) | 1.21 (s) | 1.22 (s) | 1.21 (s) | 1.12 (s) |
| 20 | 4.32 (d, 9.6); 4.28 (d, 9.6) | 4.42 (d, 9.0); 4.30 (d, 9.0) | 4.81 (d, 9.6); 4.50 (d, 9.6) | 4.86 (d, 9.6); 4.46 (d, 9.6) | 4.36 (d, 9.6); 4.05 (d, 9.6) | 4.36 (d, 9.6); 4.16 (dd, 1.2, 9.6) |
| OAc | 2.20 (s) | 2.21 (s) | 2.28 (s) | |||
| OMe | 3.19 (s) | |||||
| OMe | 3.15 (s) | |||||
| OCH2CH3 | 3.32 (m); 1.07 (t, 7.2) | |||||
| HO-1 | 6.75 (d, 4.2) | 5.97 (d, 4.2) | ||||
| HO-3 | 6.00 (s) | |||||
| HO-6 | 9.11 (s) | 6.25 (d, 3.0) | 5.91 (s) | 8.17 (d, 4.2) | ||
| HO-7 | 7.91 (s) | 8.31 (s) | 8.01 (s) | 8.07 (s) | ||
| HO-11 | 5.75 (s) | 7.24 (overlap) | 7.10 (d, 6.0) | 5.77 (br s) | ||
| HO-14 | 8.01 (s) | 8.06 (s) | ||||
| HO-15 | 4.40 (d, 3.0) | 6.84 (d, 2.4) |
| No. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 76.9(d) | 77.1(d) | 74.2(d) | 73.5(d) | 26.4(t) | 130.2(d) |
| 2 | 24.2(t) | 24.4(t) | 28.8(t) | 30.7(t) | 24.6(t) | 125.3(d) |
| 3 | 37.1(t) | 37.3(t) | 40.5(t) | 39.1(t) | 74.8(d) | 41.4(t) |
| 4 | 31.7(s) | 31.9(s) | 34.5(s) | 34.2(s) | 38.7(s) | 32.7(s) |
| 5 | 53.1(d) | 54.2(d) | 57.3(d) | 55.5(d) | 50.3(d) | 57.5(d) |
| 6 | 109.7(d) | 102.5(d) | 74.5(d) | 75.3(d) | 73.5(d) | 74.3(d) |
| 7 | 171.3(s) | 171.4(s) | 97.2(s) | 98.8(s) | 100.1(s) | 97.6(s) |
| 8 | 57.7(s) | 57.4(s) | 53.1(s) | 53.8(s) | 53.0(s) | 53.8(s) |
| 9 | 52.6(d) | 52.7(d) | 52.1(d) | 45.7(d) | 46.5(d) | 50.8(d) |
| 10 | 51.2(s) | 51.3(s) | 42.9(s) | 41.7(s) | 35.9(s) | 39.4(s) |
| 11 | 64.8(d) | 63.9(d) | 63.6(d) | 18.8(t) | 15.4(t) | 62.8(d) |
| 12 | 42.1(t) | 41.9(t) | 41.4(t) | 33.0(t) | 32.4(t) | 45.7(t) |
| 13 | 32.2(d) | 32.1(d) | 37.7(d) | 46.2(d) | 46.1(d) | 37.2(d) |
| 14 | 34.3(t) | 34.1(t) | 28.9(t) | 76.4(d) | 76.4(d) | 28.0(t) |
| 15 | 212.9(s) | 213.1(s) | 75.3(d) | 73.4(d) | 74.2(d) | 75.9(d) |
| 16 | 58.5(d) | 58.8(d) | 160.7(s) | 161.5(s) | 160.0(s) | 161.7(s) |
| 17 | 71.8(t) | 69.5(t) | 107.7(t) | 110.2(t) | 110.6(t) | 107.9(t) |
| 18 | 33.1(q) | 33.3(q) | 33.8(q) | 32.0(q) | 29.8(q) | 31.4(q) |
| 19 | 23.6(q) | 23.4(q) | 23.3(q) | 22.2(q) | 23.8(q) | 22.7(q) |
| 20 | 74.5(t) | 74.0(t) | 64.7(t) | 64.1(t) | 67.0(t) | 66.8(t) |
| OAc | 171.4(s) | 169.6(s) | 171.6(s) | |||
| 22.3(q) | 21.7(q) | 22.5(q) | ||||
| OMe | 54.8(q) | |||||
| OMe | 59.0(q) | |||||
| OCH2CH3 | 66.9(t) | |||||
| 15.5(q) |
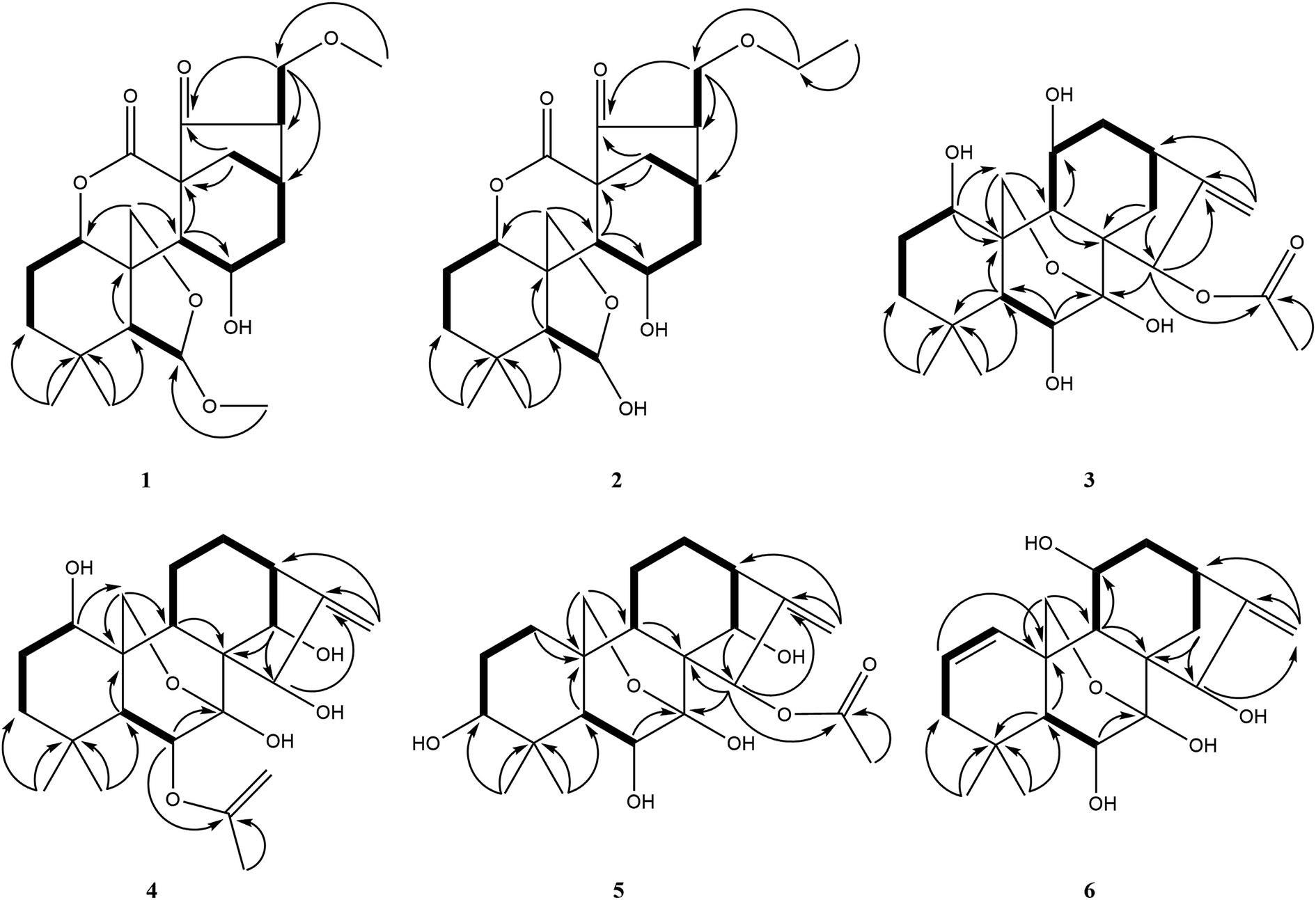 | ||
| Fig. 2 Key HMBC and 1H–1H COSY correlations of compounds 1–6. | ||
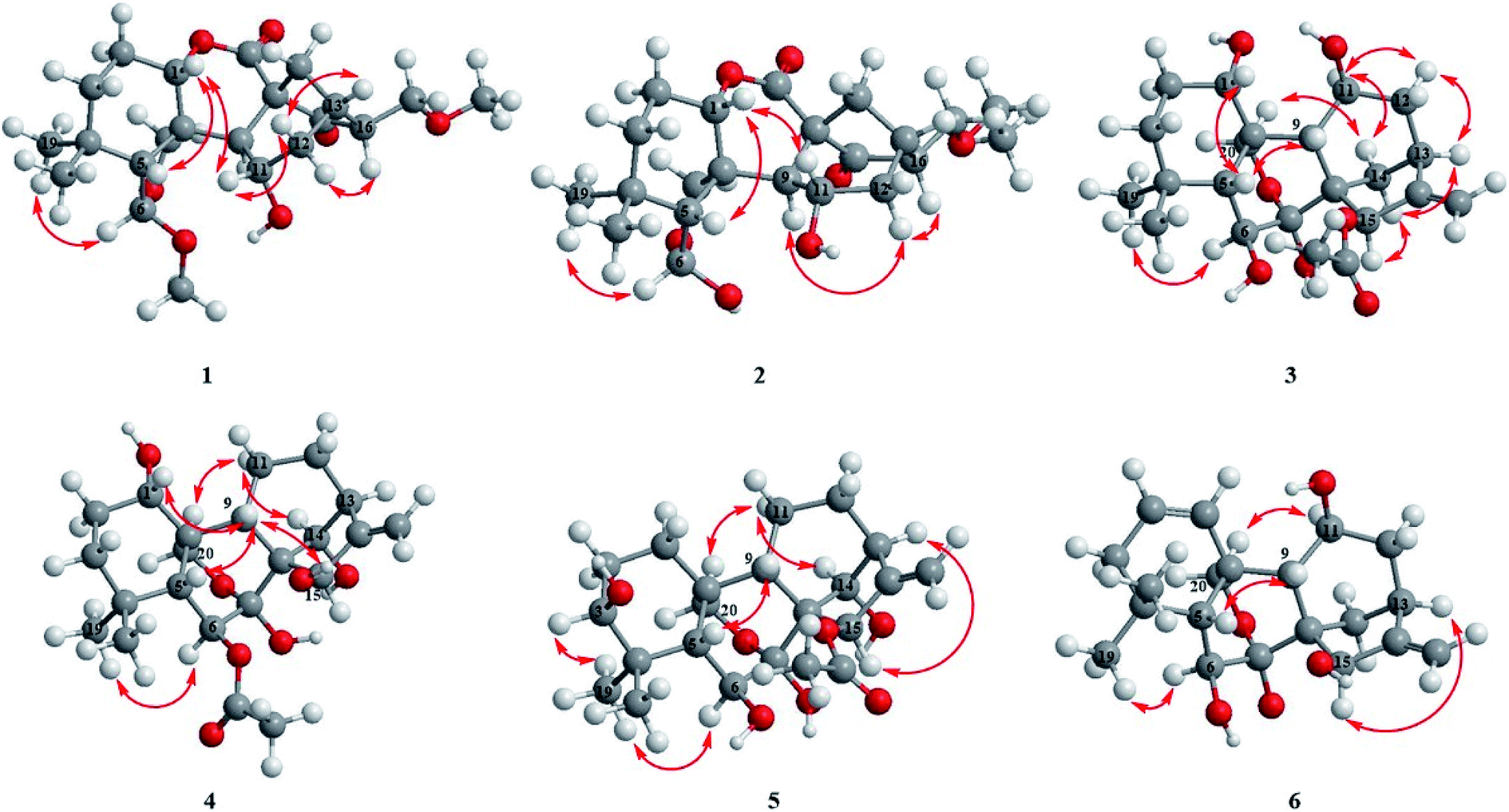 | ||
| Fig. 3 Key ROESY correlations of compounds 1–6. | ||
Compound 2 was isolated as colorless crystals (MeOH), and its molecular formula was the same as 1, as established to be C22H32O7 by HR-ESI-MS at m/z 431.20407 [M + Na]+ (calcd for C22H32O7Na, 431.20402) and 13C NMR data. A comparison of the NMR data of 2 (Tables 1 and 2) with those of 1 suggested that compound 2 had a 6,7-seco-ent-kaurane skeleton as 1, except for the disappearance of two methoxyl signal and the presence of an additional ethoxyl signal. The location of the ethoxyl group at C-17 was revealed by the HMBC correlations of H2-17 (δH 3.61) with the carbon (δC 66.9) of the ethoxyl group. In addition, the chemical shift of C-6 was shifted upfield from δC 109.7 in 1 to δC 102.5 in 2 due to the change of the substituent at C-6 from a methoxyl group in 1 to a hydroxyl group in 2. The relative stereochemistry of 2 was consistent with those of 1 and was ensured by the ROESY correlations (Fig. 3). Accordingly, the structure of compound 2 was established as 6β,11α-dihydroxy-16β-ethoxymethyl-6,7-seco-6,20-exoxy-1α,7-olide-ent-kaur-15-one and given the name isodonrubescin B.
Compound 3 was exhibited to have the molecular formula C22H32O7 by HR-ESI-MS (m/z 431.20288 [M + Na]+, calcd 431.20402). The 1H-NMR spectra (Table 1) of 3 established the existence of three single methyls [δH 1.24 (s), 1.21 (s), 2.20 (s)], one olefinic methylene [δH 5.29 (s), 5.12 (s)], one oxygenated methylene [δH 4.81 (d, J = 9.6 Hz), 4.50 (d, J = 9.6 Hz)], four oxygenated methines [δH 4.35 (m), 4.24 (dd, J = 5.4, 3.6 Hz), 4.73 (m), 6.55 (s)]. The methyl at δH 2.20 (3H, s) and the carbonyl group at δC 171.4 in the NMR spectrum suggested the presence of an acetoxyl group in 3. Apart from the acetoxyl group, there were 20 carbon resonances, consisting of two methyls, six methylenes (one oxygenated carbon at δC 64.7 and one olefinic carbon at δC 107.7), seven methines (four oxygenated carbons at δC 74.2, δC 74.5, δC 63.6 and δC 75.3, respectively), and five quaternary carbons (one hemiacetal group at δC 97.2 and one olefinic carbon at δC 160.7). The above-mentioned data suggested compound 3 to be a 7,20-epoxy-ent-kaurane diterpenoid. Comparison of the NMR date of 3 with those of hebeirubescensin K20 indicated that their structures were closely related. The only structural difference between them was that the hydroxyl group at C-15 in the latter was replaced by an acetoxyl group in 3, which can be deduced by the change of the chemical shift of H-15 from δH 5.06 in the latter to δH 6.55 in 3 and was further confirmed by the HMBC correlations (Fig. 2) from H-15 to C-16 (δC 160.7) and OAc (δC 171.4). The remaining structure was corroborated by the HMBC experiment.
The relative configuration of 3 was revealed by analysis of the ROESY spectrum (Fig. 3), in which the correlations of H-6/H3-19α (δH 1.21), H-11/H-12α (δH 2.86)/H-13α (δH 2.72), H-15/H-14β (δH 2.17)/H-13α were clearly observed, indicating that HO-6, HO-11, and AcO-15 were β-orientation. Correlations of H-1/H-5β assigned HO-1 to be α-oriented. Thus. Compound 3 was determined as 1α,6β,11β-trihydroxy-15β-acetoxy-7,20-exoxy-ent-kaur-16-ene, and named as isodonrubescin C.
Compound 4 had the same molecular formula C22H32O7 as that of 3, which was established by HR-ESI-MS at m/z 431.20404 [M + Na]+ (calcd for C22H32O7Na, 431.20402). Its 1H and 13C NMR spectra (Tables 1 and 2) showed that compound 4 possessed the same 7,20-epoxy-ent-kaurane skeleton as that of 3. A comparision of the NMR data of 4 (Tables 1 and 2) with those of enmenol21 disclosed that 4 was a 6-acetyl derivative of enmenol. The key HMBC correlation (Fig. 2) from H-6 (δH 5.86) to OAc (δC 169.6) in 4 confirmed this conclusion. Furthermore, the 1H–1H COSY correlations (Fig. 2) of H-1 (δH 3.74) with H2-2 (δH 1.86), of H-14 (δH 5.15) with H-13 (δH 2.87) and the HMBC correlations of H-15 (δH 5.57) with C-16 (δC 161.5) and C-17 (δC 110.2) indicated that three hydroxyl groups were located at C-1, C-14 and C-15 respectively. The relative configuration of 4 was assigned by the ROESY correlations (Fig. 3) of H-1/H-9β (δH 2.92), H-6/H3-19α (δH 1.22), HO-15 (4.40)/H-9β (2.92), which revealed the α-orientation of HO-1 and the β-orientation of AcO-6, HO-14, HO-15. Therefore, the structure of 4 was elucidated as 1α,14β,15β-trihydroxy-6β-acetoxy-7,20-exoxy-ent-kaur-16-ene, and given the name isodonrubescin D.
Compound 5 was obtained as a white amorphous powder with a molecular formula of C22H32O7 as assigned by HR-ESI-MS (m/z 431.20380 [M + Na]+, calcd 431.20402). Its 1H and 13C NMR data (Tables 1 and 2) resembled those of hikiokoshins G,22 suggesting that 5 had the same carbon skeleton as that of hikiokoshins G. The difference between them was that hikiokoshins G had two acetoxyl groups while compound 5 only possessed one acetoxyl group, and in the HMBC spectrum of 5 (Fig. 2), the cross-peak of H-15 with OAc (δC 171.6) indicated that the acetoxyl group was located at C-15. Thus, 5 was a 6-deacetyl derivative of hikiokoshins G, this conclusion was further supported by the change of the chemical shift of H-6 from δH 5.98 in hikiokoshins G to δH 4.33 in 5. The relative stereochemistry of 5 was consistent with those of hikiokoshins G, and was confirmed by the ROESY analysis (Fig. 3). Accordingly, compound 5 was established as 3β,6β,14β-trihydroxy-15β-acetoxy-7,20-exoxy-ent-kaur-16-ene, and named as isodonrubescin E.
Compound 6 had the molecular formula of C20H28O5 as determined by its HR-ESI-MS (m/z 349.20029 [M + H]+, calcd 349.20095) and 13C NMR data, indicating seven degrees of unsaturation. The 1H NMR and 13C NMR spectra (Tables 1 and Table 2) of 6 implied that compound 6 was a 7,20-epoxy-ent-kaurane diterpenoid. However, unlike the normal type of 7,20-epoxy-ent-kaurane diterpenoids, such as compound 3–5, a cis double bond signal [δH 6.38 (dd, J = 2.4, 10.8 Hz), 5.73 (ddd, J = 1.8, 6.0, 10.2 Hz); δC 130.2, 125.3] was presented in the NMR spectra of 6, and the double bond was assigned to C-1 and C-2 by the key 1H–1H COSY correlations of 6 (Fig. 2) from H-1 (δH 6.38) to H-2 (δH 5.73), from H-2 to H-3 (δH 1.94) and the key HMBC correlations (Fig. 2) from H3-18 (δH 1.20) to C-3 (δC 41.4), from H-2 to C-10 (δC 39.4). The remaining three hydroxyl groups were respectively assigned to C-6, C-11 and C-15 by interpretation of the 1H–1H COSY and HMBC correlations. The relative configuration of 6 was determined by the ROESY correlations (Fig. 3) of H-6 (δH 4.31)/H3-19α (δH 1.12), H-11 (δH 4.58)/H-20 (δH 4.36) and H-15 (δH 5.20)/H-13α (δH 2.76), which suggested the β-orientation of HO-6, HO-11 and HO-15. Consequently, the structure of 6 was assigned as 6β,11β,15β-trihydroxy-7,20-exoxy-ent-kaur-1,16-diene, and given the name isodonrubescin F.
The other twenty-eight known diterpenoids (7–34) were identified by comparison of their NMR data with those reported in the literature. As a result, they were identified to be 3β-hydroxy-6β-methoxy-6,7-seco-6,20-epoxy-1α,7-olide-ent-kaur-16-en-15-one (7),23 enmein (8),20 rabdosin A (9),19 epinodosinol (10),18 isojaponin A (11),24 epinodosin (12),25 oridonin (13),18 hubeirubesin K (14),18 neolaxiflorin U (15),18 hubeirubesin I (16),18 lasiokaurin (17),18 hebeirubescensin K (18),20 maoyecrystal F (19),24 rabdoternin D (20),26 lasiodonin (21),27 enmelol (22),28 rabdonervosin G (23),29 rabdonervosin D (24),29 hikiokoshins G (25),22 isodonhenrin E (26),30 maoyecrystal L (27),20 dayecrystal B (28),31 lushanrubescensin F (29),32 ponicidin (30),33 rubescensin D (31),34 isorosthornin D (32),35 isoadenolin M (33),36 rubescensin J (34).37
In addition, all the isolated compounds were assessed for their inhibitory activity against NO production in LPS stimulated RAW264.7 cells with dexamethasone as a positive control (IC50 = 9.58 μM). The cell viability of the tested compounds was firstly measured using CCK-8 assay to determine whether the NO production inhibitory activities were induced by the cytotoxicity. As a result, compounds 7, 9, 13, 16, and 17 exhibited obvious NO production inhibitory effects with IC50 values of 3.97, 2.25, 6.51, 1.48 and 1.36 μM, respectively. Compounds 8 and 12 displayed mild NO production inhibitory effects with IC50 values of 17.43 and 18.25 μM, respectively, while the rest of the tested compounds had no obvious NO production inhibitory activity (IC50 > 20 μM). In the present study, the 6,7-seco-ent-kaurane diterpenoids, such as 7–9 and 12 which possessed an α,β-unsaturated ketone moiety, exhibited NO production inhibitory effects, the result indicated that α,β-unsaturated ketone moiety was an essential pharmacophore. However, this conclusion did not fully be applied to 7,20-epoxy-ent-kaurane diterpenoids. For compounds 13, 17, 21 and 30, they shared an α,β-unsaturated ketone moiety, but compound 21 and 30 did not show the activity. This could be caused by the lack of HO-14β in 21 and 30. Additionally, compound 16 without an α,β-unsaturated ketone moiety also exhibited obvious NO production inhibitory effects. This result further demonstrated that the α,β-unsaturated ketone moiety was not absolutely essential active center for the activity. Besides, it was interesting that compound 25 was a 3-deacetyl derivative of 16, but it did not show NO production inhibitory effect, the result suggested that 3β-OAc might played an important role in the NO production inhibitory activity.
3. Experimental
3.1 General experimental procedures
Optical rotations were measured with an Autopol IV polarimeter (Rudolph Research Analytical, Hackettstown, NJ, USA). UV spectra were recorded on a UH5300 UV-VIS Double Beam spectrophotometer (Hitachi Co., Tokyo, Japan). NMR spectra were obtained on a Bruker AVANCE IIITM 600 MHz spectrometer (Bruker, Ettlingen, Germany) in C5D5N with tetramethylsilane (TMS) as an internal reference standard. Chemical shifts (δ) have been given in ppm and the coupling constants (J) have been expressed in Hz. High-resolution electrospray mass spectroscopy was conducted on a Thermo Scientific Q Exactive Orbitrap LC-MS/MS System (HR-ESI-MS) (Thermo Scientific, Waltham, MA, USA). High-performance liquid chromatography (HPLC) was performed on an Ultimate 3000 HPLC system (Dionex Co., Sunnyvale, CA, USA) equipped with an Ultimate 3000 pump and Ultimate 3000 Variable Wavelength detector, as well as a semi-preparative YMC-Pack ODS-A column (250 × 10 mm, 5 μm), column chromatography (CC) was conducted with silica gel (200–300 mesh and 300–400 mesh, Qingdao Haiyang Chemical Industry Co., Ltd., Qingdao, China). Chromatographic grade acetonitrile was purchased from Chang Tech Enterprise Co., Ltd (Taiwan, China). RAW264.7 murine macrophages were purchased from the cell bank of Chinese Academy of Sciences (Shanghai, China). Dexamethasone and lipopolysaccharides (LPS) were purchased from Sigma Chemical Co. Ltd. (St. Louis, MO, USA). Cell Counting Kit (CCK-8) was purchased from Beyotime Biotechnology (Shanghai, China). Dulbecco modified Eagle medium (DMEM) and penicillin-streptomycin solution were purchased from GE Healthcare Life Sciences (Logan, UT, USA). Fetal bovine serum (FBS) was purchased from Gibco, Life Technologies (Grand Island, NY, USA). Reagent grade dimethyl sulfoxide (DMSO) was purchased from Vetec, Sigma Chemical Co. (St. Louis, MO, USA). The absorbance was read on a Multiskan GO microplate reader (Thermo Fisher Scientific Inc. Waltham, MA, USA).3.2 Plant material
Isodon rubescens were collected from Badong county, Hubei Province and identified by Prof. Fajun Song, College of Life Science, South Central University for Nationalities. The voucher specimen (2016101201) was deposited in the herbarium of School of Pharmaceutical Sciences, South Central University for Nationalities.3.3 Extraction and isolation
The air-dried and powdered parts of I. rubescens (11.2 kg) were extracted with 95% EtOH (25 L × 3, each 24 h) at room temperature. The extract was filtered and evaporated to afford a crude extract (1.1 kg), which was partitioned successively with petroleum ether (P. E.) and EtOAc. The EtOAc extract (556 g) was subjected to column chromatography on a silica gel column eluting with the gradient of CHCl3–acetone (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 9:1, 8:2, 7:3, 6:4, 1:1, 3:7, 0:10) to yield eight fractions (Fr. A–Fr. H). Fr. D (123.7 g) was separated by silica gel CC (CH2Cl2–EtOAc, 10:1, 8:2, 6:4, 1:1) into fractions D1–D10. Fr. D6 was subjected to RP-18 CC (MeOH–H2O, 3:7, 5:5, 7:3, 0:10) to obtain eleven fractions (Fr. D6A–Fr. D6K). Fr. D6D was purified by semi-preparative HPLC (MeOH–H2O 33:67) to afford compounds 2 (15 mg, tR 20.8 min) and 25 (25 mg, tR 15.1 min).
:0, 9:1, 8:2, 7:3, 6:4, 1:1, 3:7, 0:10) to yield eight fractions (Fr. A–Fr. H). Fr. D (123.7 g) was separated by silica gel CC (CH2Cl2–EtOAc, 10:1, 8:2, 6:4, 1:1) into fractions D1–D10. Fr. D6 was subjected to RP-18 CC (MeOH–H2O, 3:7, 5:5, 7:3, 0:10) to obtain eleven fractions (Fr. D6A–Fr. D6K). Fr. D6D was purified by semi-preparative HPLC (MeOH–H2O 33:67) to afford compounds 2 (15 mg, tR 20.8 min) and 25 (25 mg, tR 15.1 min).
Fr. E (56.3 g) was separated on RP-18 CC into six fractions (Fr. E1–Fr. E6) by eluting with MeOH–H2O (3:7, 5:5, 7:3, 0:10). Fr. E2 and Fr. E4 was purified by recrystallizing in MeOH to afford compounds 8 (735 mg). Fr. E3 was firstly purified by a silica gel column (eluted with CH2Cl2–MeOH, 100:1, 50:1, 25:1, 15:1, 12:1 gradient) to yield nine fractions Fr. E3A–Fr. E3I. Fr. E3C was purified by recrystallizing in MeOH to afford compound 12 (8 mg), then Fr. E3A was subjected to silica gel CC (petroleum ether–EtOAc, 9:1, 8:2, 7:3 gradient) to obtain fractions E3A1–E3A8. Fr. E3A8 was finally purified by semi-preparative HPLC (MeOH–H2O 43:57) to afford compounds 1 (10 mg, tR 16.3 min) and 9 (18 mg, tR 17.6 min). Similarly, compound 7 (5 mg, tR 15.8 min) was obtained from Fr. E3B by semi-preparative HPLC (MeOH–H2O, 40:60). Fr. E3F was successively chromatographed over silica gel CC (CH2Cl2–MeOH, 50:1, 25:1, 12:1) and semi-preparative HPLC to yield compounds 6 (1 mg, MeOH–H2O, 36:64, tR 41.8 min), 26 (9 mg, MeOH–H2O, 36:64, tR 43.3 min), 18 (25 mg, MeOH–H2O, 45:55, tR 22.4 min), 19 (19 mg, MeOH–H2O, 47:53, tR 22.3 min) and 32 (5 mg, MeOH–H2O, 36:64, tR 46.3 min). Fr. E6 was similarly purified with semi-preparative HPLC to yield compounds 33 (1.5 mg, CH3CN–H2O, 77:23, tR 14.2 min) and 34 (7 mg, MeOH–H2O, 79:21, tR 15.5 min).
Fr. F (51.3 g) was separated over RP-18 CC (MeOH–H2O, 3:7, 5:5, 7:3, 0:10 gradient) into five fractions (Fr. F1–Fr. F5), Fr. F2 and Fr. F3 was separated over repeatedly chromatographed by silica gel column, and then further purified by semi-preparative HPLC to afford compounds 3 (46 mg, MeOH–H2O, 49:51, tR 23.9 min), 4 (5.0 mg, MeOH–H2O, 35:65, tR 12.2 min), 5 (8.0 mg, MeOH–H2O, 25:75, tR 19.5 min), 11 (3.0 mg, MeOH–H2O, 60:40, tR 12.9 min), 20 (13 mg, CH3CN–H2O, 35:65, tR 11.7 min), 21 (23 mg, CH3CN–H2O, tR 7.7 min), 22 (2.5 mg, CH3CN–H2O, 25:75, tR 11.7 min), 23 (9 mg, MeOH–H2O, 45:55, tR 11.4 min), 24 (12 mg, MeOH–H2O, 35:65, tR 34.7 min), 27 (6.0 mg, MeOH–H2O, 40:60, tR 17.0 min), 28 (12 mg, CH3CN–H2O, 35:65, tR 8.8 min), 29 (23 mg, CH3CN–H2O, 35:65, tR 6.8 min), 30 (1.7 mg, MeOH–H2O, 35:65, tR 26.9 min), 31 (3 mg, CH3CN–H2O, 35:65, tR 10.8 min).
3.4 Spectroscopic data
Isodonrubescin A (1): colorless needle crystals (MeOH); [α]D = −107.8° (c 0.10, MeOH); UV (MeOH) λmax (logε): 215 (2.48), 295 (1.58) nm; 1H and 13C NMR data see Tables 1 and 2; HR-ESI-MS m/z 431.20383 [M + Na]+ (calcd for C22H32O7 Na, 431.20402).
Isodonrubescin B (2): colorless crystals (MeOH); [α]D = −47.6° (c 0.01, MeOH); UV (MeOH) λmax (logε): 205 (2.92) nm; 1H and 13C NMR data see Tables 1 and 2; HR-ESI-MS m/z 431.20407 [M + Na]+ (calcd for C22H32O7Na, 431.20402).
Isodonrubescin C (3): colorless crystals (MeOH); [α]D = −101.0° (c 0.02, MeOH); UV (MeOH) λmax (logε): 205 (3.21), 250 (2.43) nm; 1H and 13C NMR data see Tables 1 and 2; HR-ESI-MS m/z 431.20288 [M + Na]+ (calcd for C22H32O7Na, 431.20402).
Isodonrubescin D (4): white amorphous powder; [α]D = +6.2° (c 0.03, MeOH); UV (MeOH) λmax (logε): 210 (3.24), 250 (2.29) nm; 1H and 13C NMR data see Tables 1 and 2; HR-ESI-MS m/z 431.20404 [M + Na]+ (calcd for C22H32O7Na, 431.20402).
Isodonrubescin E (5): white amorphous powder; [α]D = +9.1° (c 0.02, MeOH); UV (MeOH) λmax (logε): 205 (3.29), 250 (2.86) nm; 1H and 13C NMR data see Tables 1 and 2; HR-ESI-MS m/z 431.20380 [M + Na]+ (calcd for C22H32O7Na, 431.20402).
Isodonrubescin F (6): white amorphous powder; [α]D = +12.2° (c 0.02, MeOH); UV (MeOH) λmax (logε): 210 (3.38) nm; 1H and 13C NMR data see Tables 1 and 2; HR-ESI-MS m/z 349.20029 [M + H]+ (calcd for C20H29O5, 349.20095).
3.5 NO production measurement and cell viability assay
The NO production and cell viability were determined by the Griess reaction and CCK-8 method respectively, which have been described in our previous paper.384. Conclusions
In this study, six previously undescribed ent-kaurane diterpenoids, including two 6,7-seco-ent-kaurane diterpenoids (1–2), four 7,20-epoxy-ent-kaurane diterpenoids (3–6), together with twenty-five known ent-kaurane diterpenoids (7–31), a known ent-atisane diterpenoids (32), and two known ent-abietane diterpenoids (33–34) were isolated from I. rubescens collected from Badong county of Hubei Province, P. R. China. It was noteworthy that compounds 7 was isolated as a natural product for the first time, and ent-atisane diterpenoid was found from I. rubescens in Hubei Province for the first time. Among all the isolated compounds, 7,20-epoxy-ent-kaurane diterpenoids and 6,7-seco-ent-kaurane diterpenoids were the main chemical constituents of I. rubescens collected from Hubei Province, which contained two types of diterpenoids isolated from I. rubescens collected from Henan and Guizhou Province, this may be related to the geographical location of Hubei Province lying between Henan and Guizhou Province. Moreover, all the isolated compounds were evaluated for their inhibitory effect against LPS induced nitric oxide production in RAW 264.7 macrophages. Compounds 7–9, 12, 13, 16, and 17 displayed obvious NO production inhibitory effects. In conclusion, those results have further facilitated our understanding of the active constituents of I. rubescens from Badong region and the potential bioactive constituents of I. rubescens accounting for the application as anti-inflammatory agents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Major New Drugs Innovation and Development (2017ZX09301060), the National Key Research and Development Program of China (2018YFC1708004), the Special Fund for Basic Scientific Research of Central Colleges, South-Central University for Nationalities (CZP18004 and CZP17076) and the National Science Foundation of China (31600272).References
- H. D. Sun, S. X. Huang and Q. B. Han, Nat. Prod. Rep., 2006, 23, 673–698 RSC.
- M. Liu, W. G. Wang, H. D. Sun and J. X. Pu, Nat. Prod. Rep., 2017, 34, 1090–1140 RSC.
- A. Ohsaki, M. Ozawa, K. Komiyama, A. Kishida and T. Isobe, Nat. Prod. Commun., 2012, 7, 977–978 CrossRef CAS PubMed.
- X. M. Wang, C. X. Xie, S. L. Chen, H. Yao and J. P. Han, J. Anhui Agric. Sci., 2008, 36, 13677–13680 Search PubMed.
- L. L. Tian, D. L. Sheng, Q. H. Li, C. X. Guo and G. F. Zhu, Pharm. Biol., 2019, 57, 632–640 CrossRef CAS PubMed.
- F. L. Zhao, M. C. Sun, W. J. Zhang, C. L. Jiang, J. T. Teng, W. Sheng, M. Z. Li, A. M. Zhang, Y. B. Duan and J. P. Xue, BMC Plant Biol., 2018, 18, 272 CrossRef CAS PubMed.
- H. D. Sun, Z. W. Lin, F. D. Niu, L. Z. Lin, H. B. Chai, J. M. Pezzuto and G. A. Cordell, Phytochemistry, 1995, 38, 437–442 CrossRef CAS PubMed.
- N. S. Bai, K. He, Z. Zhou, M. L. Tsai, L. Zhang, Z. Quan, X. Shao, M. H. Pan and C. T. Ho, Planta Med., 2010, 76, 140–145 CrossRef CAS PubMed.
- M. Hu, S. T. Xu and J. Y. Xu, Pharmaceutical and Clinical Research, 2017, 25, 425–430 Search PubMed.
- W. C. Huang, M. C. Huang, H. Ouyang, J. W. Peng and J. Liang, Eur. J. Pharmacol., 2018, 826, 133–139 CrossRef CAS PubMed.
- Q. B. Han, Q. S. Zhao, S. H. Li, L. Y. Peng and H. D. Sun, Acta Chim. Sin., 2003, 61, 1077–1082 CAS.
- Q. B. Han, R. T. Li, M. L. Li, Y. K. Mou, Q. E. Tian, S. W. Li and H. D. Sun, J. Asian Nat. Prod. Res., 2005, 7, 31–36 CrossRef CAS PubMed.
- X. Liu, Y. B. Xue, K. Dong, X. N. Li, Y. Li, J. X. Pu, J. Z. Wu and H. D. Sun, Chin. J. Nat. Med., 2012, 10, 464–470 CAS.
- X. Liu, R. Zhan, W. G. Wang, X. Du, X. N. Li, J. H. Yang, P. Zhang, Y. Li, J. X. Pu, J. Z. Wu and H. D. Sun, Chem. Pharm. Bull., 2013, 61, 90–95 CrossRef CAS PubMed.
- X. Liu, W. G. Wang, X. Du, X. N. Li, L. M. Kong, Y. Li, J. X. Pu, J. Z. Wu and H. D. Sun, Fitoterapia, 2012, 83, 1451–1455 CrossRef CAS PubMed.
- X. Liu, J. Yang, W. G. Wang, Y. Li, J. Z. Wu, J. X. Pu and H. D. Sun, J. Nat. Prod., 2015, 78, 196–201 CrossRef CAS PubMed.
- X. Liu, R. Zhan, W. G. Wang, X. Du, Y. Li, P. Zhang, J. X. Pu, J. Z. Wu and H. D. Sun, Arch. Pharmacal Res., 2012, 35, 2147–2151 CrossRef CAS PubMed.
- J. W. Shu, F. Yuan, C. M. Wen and G. Z. Yang, J. Green. Sci. Technol., 2017, 18, 216–218 Search PubMed.
- J. X. Zhang, Z. A. He, Z. Y. Chen, Y. X. Wang, S. P. Bai and H. D. Sun, J. Asian Nat. Prod. Res., 2009, 11, 693–697 CrossRef PubMed.
- X. M. Di and F. L. Yan, Anhui Med. Pharm. J., 2013, 17, 1470–1472 CAS.
- S. Mori, K. Shudo, T. Ageta, T. Koizumi and T. Okamoto, Chem. Pharm. Bull., 1970, 18, 871–883 CrossRef CAS.
- N. Tanaka, E. Tsuji, K. Sakai, T. Gonoi and J. Kobayashi, Phytochemistry, 2014, 102, 205–210 CrossRef CAS PubMed.
- E. Fujita, T. Fujita, Y. Okada, S. Nakamura and M. Shibuya, Chem. Pharm. Bull., 1972, 20, 2377–2383 CrossRef CAS.
- F. L. Yan, L. Q. Guo, S. P. Bai and H. D. Sun, J. Chin. Chem. Soc., 2008, 55, 933–936 CrossRef CAS.
- L. B. Yang, S. X. Huang, J. X. Pu and L. M. Li, Res. Pract. Chin. Med., 2013, 20, 25–29 Search PubMed.
- Y. Takeda, K. I. Takeda, T. Fujita, H. D. Sun and Y. Minami, Phytochemistry, 1994, 35, 1513–1516 CrossRef CAS.
- W. Li, B. Li and Y. Chen, Phytochemistry, 1998, 49, 2433–2435 CrossRef CAS.
- S. Mori, T. Koizumi, K. Shudo and T. Okamoto, Chem. Pharm. Bull., 1970, 18, 884–889 CrossRef CAS.
- Y. H. Gao, Z. X. Wei, Y. Cheng, T. H. Wang, L. Ni, X. H. Zhou and L. Y. Zhang, Chem. Biodiversity, 2013, 10, 1487–1493 CrossRef CAS PubMed.
- Z. Hu, R. Zhang, X. Du, J. Su, X. N. Li and J. H. Yang, Chem. Pharm. Bull., 2011, 59, 1562–1566 CrossRef CAS PubMed.
- H. B. Zhang, J. X. Pu, Y. Y. Wang, F. He, Y. Zhao, X. N. Li, X. Luo, W. L. Xiao, Y. Li and H. D. Sun, Chem. Pharm. Bull., 2010, 58, 56–60 CrossRef CAS PubMed.
- Q. B. Han, M. L. Li, S. H. Li, Y. K. Mou, Z. W. Lin and H. D. Sun, Chem. Pharm. Bull., 2003, 51, 790–793 CrossRef CAS PubMed.
- F. Yin, J. Y. Liang and J. Liu, J. China Pharm. Univ., 2003, 34, 302–304 CAS.
- H. D. Sun, Q. Zhou, T. Fujita, Y. Takeda, Y. Minami, T. Maronaka, Z. W. Lin and X. Y. Shen, Phytochemistry, 1992, 31, 1418–1419 CrossRef CAS.
- R. Zhan, X. N. Li, X. Du, W. G. Wang, K. Dong, J. Su, Y. Li, J. X. Pu and H. D. Sun, Fitoterapia, 2013, 88, 76–81 CrossRef CAS PubMed.
- W. Zhao, J. X. Pu, X. Du, J. Su, X. N. Li, J. H. Yang, Y. B. Xue, Y. Li and H. D. Sun, Chin. J. Nat. Med., 2011, 9, 253–258 CAS.
- J. Wan, H. Y. Jiang, J. W. Tang, X. R. Li, X. Du, Y. Li, H. D. Sun and J. X. Pu, Molecules, 2017, 22, 309–318 CrossRef PubMed.
- H. D. Teng, Y. S. Ren, Z. Y. Ma, X. Tan, J. Xu, Y. Chen and G. Z. Yang, Fitoterapia, 2019, 137, 104–245 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08831h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |