
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A one-pot synthesis of N2,6-diaryl-5,6-dihydro-1,3,5-triazine-2,4-diamines and systematic evaluation of their ability to host ethanol in crystals†
Ahmad Junaidad,
Yee Seng Tanb,
Edward R. T. Tiekink b and
Anton V. Dolzhenko*ac
b and
Anton V. Dolzhenko*ac
aSchool of Pharmacy, Monash University Malaysia, Jalan Lagoon Selatan, Bandar Sunway, Selangor Darul Ehsan 47500, Malaysia. E-mail: anton.dolzhenko@monash.edu
bResearch Centre for Crystalline Materials, School of Science and Technology, 5 Jalan Universiti, Sunway University, Bandar Sunway, Selangor Darul Ehsan 47500, Malaysia
cSchool of Pharmacy and Biomedical Sciences, Curtin Health Innovation Research Institute, Faculty of Health Sciences, Curtin University, GPO Box U1987 Perth, Western Australia 6845, Australia
dDepartment of Chemistry, Prairie View A&M University, P.O. Box 519, Prairie View, Texas 77446, USA
First published on 19th November 2019
Abstract
A convenient one-pot method for the preparation of N2,6-diaryl-5,6-dihydro-1,3,5-triazine-2,4-diamines was developed using a three-component synthesis of 1,6-diaryl-1,6-dihydro-1,3,5-triazine-2,4-diamines followed by their Dimroth rearrangement to the desired products. The prepared compounds crystallized from ethanol as ethanol clathrates (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). X-ray crystallography on several products confirmed the adoption of 5,6-dihydro-tautomer. The thermal analysis and powder X-ray diffraction experiments on selected compounds suggested that thermal desolvation of crystals was irreversible.
:1). X-ray crystallography on several products confirmed the adoption of 5,6-dihydro-tautomer. The thermal analysis and powder X-ray diffraction experiments on selected compounds suggested that thermal desolvation of crystals was irreversible.
Introduction
The first synthesis of N2,6-diphenyl-5,6-dihydro-1,3,5-triazine-2,4-diamine (1a) was reported in 1956 by Modest and Levine.1 They prepared 1a using a piperidine-catalyzed condensation of N-phenylbiguanide with benzaldehyde and alternatively, by a Dimroth rearrangement of 1,6-diphenyl-1,6-dihydro-1,3,5-triazine-2,4-diamine hydrochloride (2a) upon its heating with a base (Scheme 1). | ||
| Scheme 1 Reported1 syntheses of N2,6-diphenyl-5,6-dihydro-1,3,5-triazine-2,4-diamine (1a). | ||
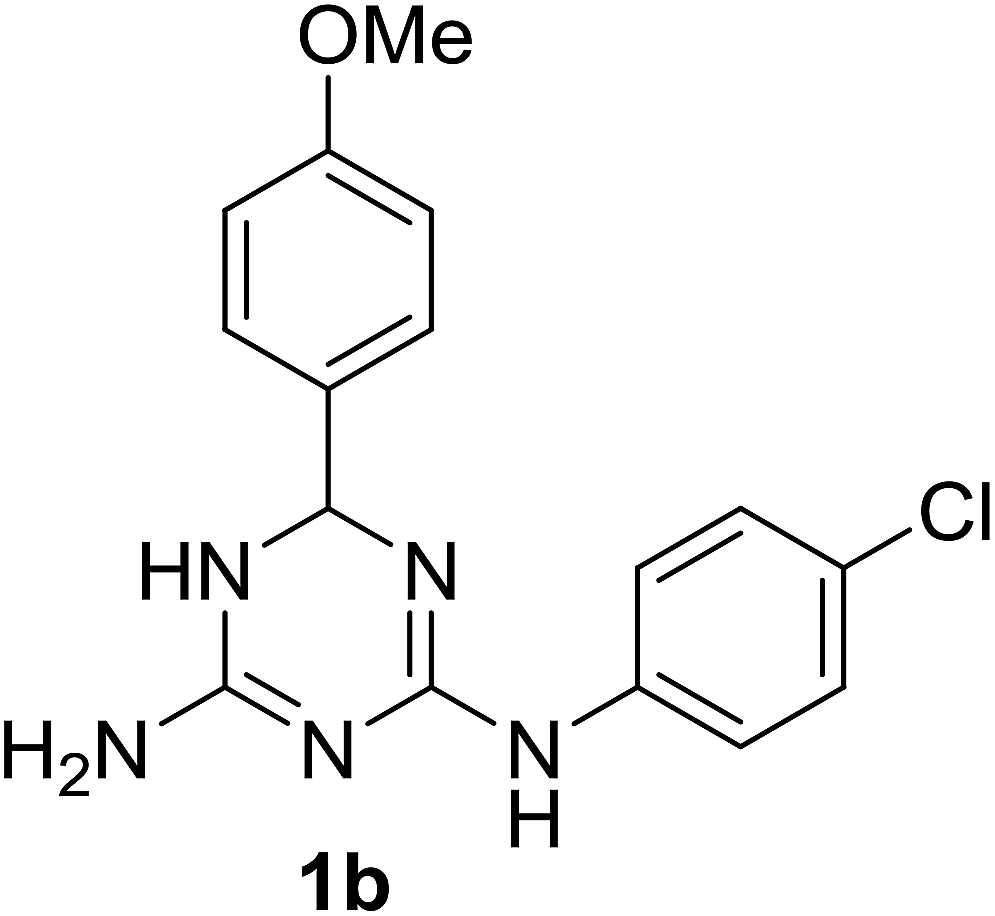
Despite a significant interest in 1,3,5-triazine derivatives as bioactive compounds, particularly due to their anticancer properties,2 since that time no reports on N2,6-diphenyl-5,6-dihydro-1,3,5-triazine-2,4-diamine (1a) and its analogues had appeared until recently. In 2019, a Japanese patent3 mentioned another N2,6-diaryl-5,6-dihydro-1,3,5-triazine-2,4-diamine, compound 1b, as an agent against esophageal squamous cell carcinoma. However, the preparation of this compound was not described. With our interest towards development of new practical methods for the multicomponent synthesis of bioactive 1,3,5-triazines,4,5 we report herein a one-pot synthesis of N2,6-diaryl-5,6-dihydro-1,3,5-triazine-2,4-diamines, including 1a and 1b. The yield of 1a in the rearrangement of 2a described by Modest and Levine1 was not reported, but the fact that 2a could be effectively prepared via a three-component reaction of cyanoguanidine, aniline and benzaldehyde6 prompted us to combine these two reactions in a one-pot procedure.
Modest and Levine reported1 that recrystallization of N2,6-diphenyl-5,6-dihydro-1,3,5-triazine-2,4-diamine (1a) from ethanol resulted in the formation of an ethanol solvate (1:1). In our study, we attempted to explore whether this ability to host ethanol is common for analogues of 1a substituted at the phenyl rings. We also solved crystal structures of several of these ethanol clathrates and assessed their stability.
Results and discussion
The synthesis of N2,6-diaryl-5,6-dihydro-1,3,5-triazine-2,4-diamines (2) was performed using a one-pot methodology combining two steps: (1) a three-component condensation of cyanoguanidine, an aniline and a benzaldehyde in the presence in hydrochloric acid and (2) a base-promoted Dimroth rearrangement of the intermediate 1,6-diaryl-1,6-dihydro-1,3,5-triazine-2,4-diamine hydrochlorides (2) (Scheme 2).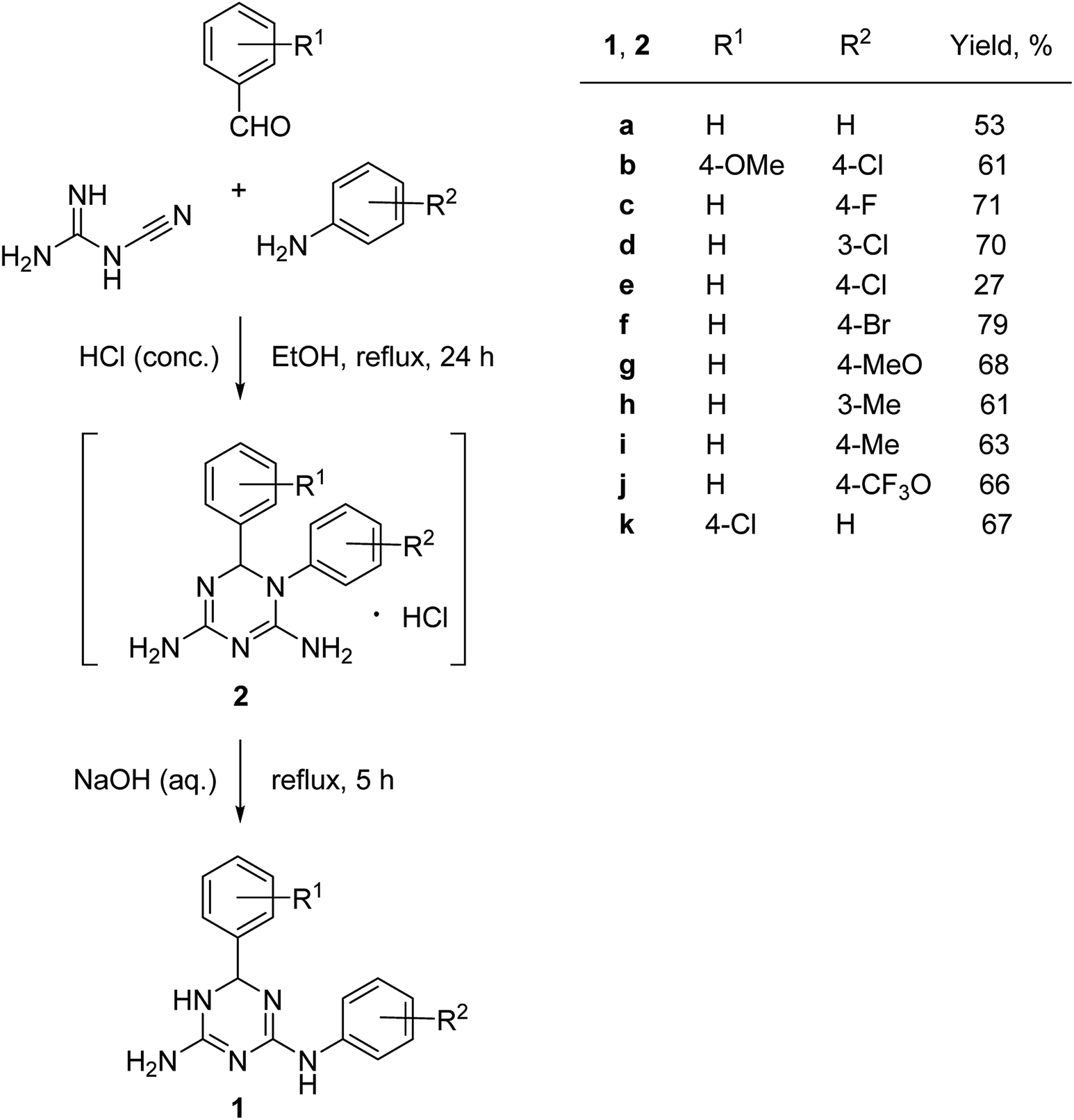 | ||
| Scheme 2 One-pot synthesis N2,6-diaryl-5,6-dihydro-1,3,5-triazine-2,4-diamines (1). | ||
Variations of substituents on aniline (1c–j) benzaldehyde (1k), and their combinations (1b) were tolerated in the reaction. The structure of the prepared compounds 1 was confirmed by NMR spectroscopic data. The signals of the sp3-hydridized carbon atom in 13C NMR spectra at 67.5–68.4 ppm and the corresponding proton in 1H NMR spectra at 5.70–5.77 ppm confirmed formation of the dihydrotriazine ring. We found that it was critical for the success of the reaction to maintain pH at 10–11. Further increase of the pH resulted in the partial dehydrogenation of the product and aromatization of the dihydrotriazine in a process similar to the one we reported earlier.5
The products were isolated as a racemic mixture of R- and S-stereoisomers in the form of ethanol solvates after recrystallization from this solvent. The integration of signals at 1.06, 3.44–3.45, and 4.34–4.37 ppm, attributed to ethanol, in the 1H NMR spectra of samples dried in air at room temperature indicated that all prepared solvates of N2,6-diaryl-5,6-dihydro-1,3,5-triazine-2,4-diamines (1) contained one molecule of ethanol per heterocycle molecule. This was further confirmed by X-ray crystallographic studies as described below. We did not observe formation of stable clathrates with methanol and desolvated compounds 1 can be obtained by the recrystallization from this solvent. The NMR spectra of a representative desolvated sample of 1a can be found in ESI.†
In principle, three tautomeric forms of the prepared compounds are possible due to annular prototropic tautomerism in the dihydrotriazine ring (Scheme 3). In the 1H NMR spectra of compounds 1, signals of the annular NH and the NH link between the triazine and benzene rings were very broad and not always detectable. This can be attributed to the tautomerism, proton exchange with ethanol, or both processes taking place together. X-ray crystallography (vide infra) revealed that the compounds crystallized as the 5,6-dihydro-tautomer, A.
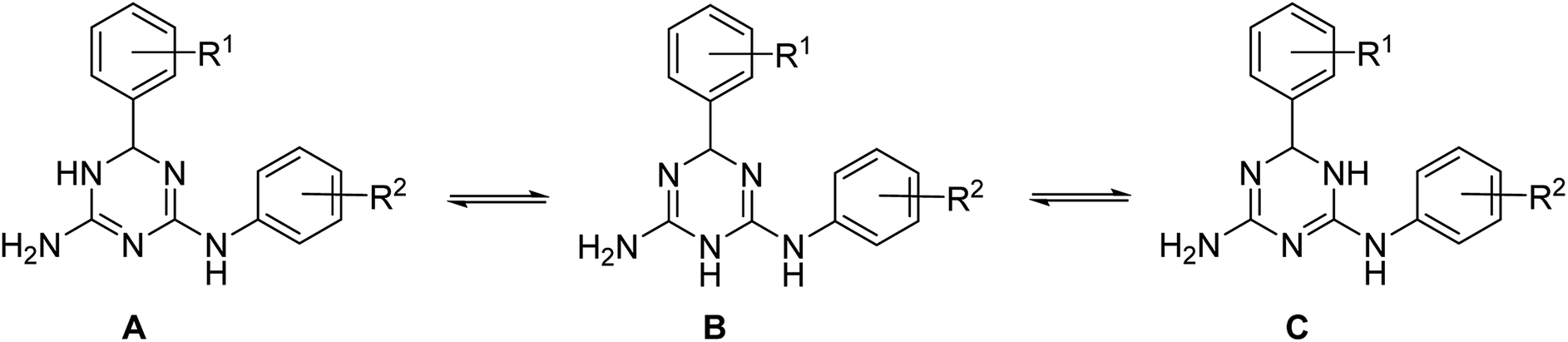 | ||
| Scheme 3 Annular tautomerism in N2,6-diaryl-X,6-dihydro-1,3,5-triazine-2,4-diamines. | ||
The molecular structures of 1b, 1e and 1f were established by single crystal X-ray crystallography as their ethanol solvates (1:1) and found to be isostructural. The discussion will focus on 1e·EtOH with details of 1b·EtOH and 1f·EtOH available in the ESI. Crystals of 1e·EtOH adopt the non-centrosymmetric space group P21 with two independent molecules each of 1e and ethanol comprising the asymmetric unit of 1e·EtOH, Fig. 1; the molecular structure diagrams for 1b·EtOH and 1f·EtOH are given in ESI Fig. S1 and S2,† respectively.
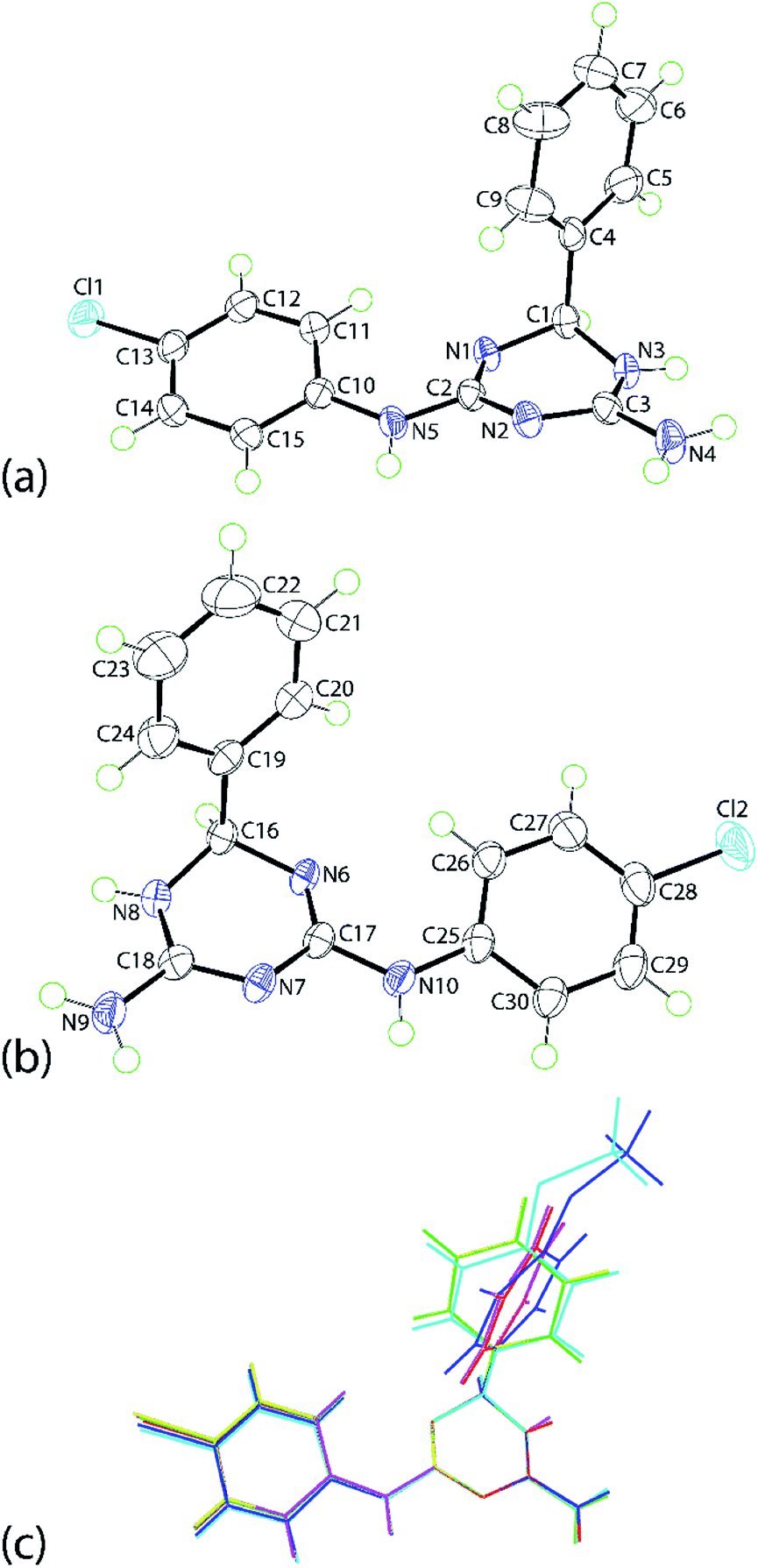 | ||
| Fig. 1 The molecular structures of the two independent organic molecules comprising the asymmetric unit of 1e, showing atom labelling scheme and displacement ellipsoids at the 70% probability level: (a) N1-containing molecule and (b) N6-containing molecule. (c) An overlay diagram of 1b (N1-molecule, blue image), 1b (inverted N6-molecule, aqua), 1e (N1-molecule, red), 1e (inverted N6-molecule, green), 1f (inverted N1-molecule, pink) and 1f (N6-molecule, yellow). The molecules have been overlapped so the N1, N2 and N3 atoms are coincident. Solvent ethanol molecules are omitted. | ||
The independent molecules of 1e are related by a non-crystallographic centre of inversion with the configurations at the C1 and C16 atoms being R and S, respectively; this therefore, an example of kyptoracemic behaviour.7 Key geometric parameters are collated in Table 1 for 1e·EtOH and ESI Table S1† for 1b·EtOH and 1f·EtOH. The parameters describing chemically equivalent bonds and angles follow the same trends across the series. At least three key parameters point to the adoption of 5,6-dihydro-tautomer, A, shown in Scheme 3. These are the short C2–N1 bond, consistent with significant double bond character, the wider angle subtended at the N3 atom compared with those at the N1 and N2 atoms, consistent with protonation at N3, and the participation of the N3-bound proton in significant hydrogen bonding interactions with the N7 atom of the second independent molecule and conversely, the N8-bound proton to the N2 atom. Based on the closeness of the C3–N2 and C3–N3 bond lengths, there is some evidence of delocalization of the π-electron density over these atoms.
| C1–N1 | 1.449(3) | C16–N6 | 1.458(3) |
| C1–N3 | 1.469(3) | C16–N8 | 1.456(3) |
| C2–N1 | 1.305(3) | C17–N6 | 1.300(3) |
| C2–N2 | 1.383(3) | C17–N7 | 1.378(3) |
| C3–N2 | 1.337(3) | C18–N7 | 1.330(3) |
| C3–N3 | 1.329(3) | C18–N8 | 1.339(3) |
| C1–N1–C2 | 115.67(19) | C16–N6–C18 | 114.63(19) |
| C2–N2–C3 | 114.0(2) | C17–N7–C18 | 114.4(2) |
| C1–N3–C3 | 118.5(2) | C16–N8–C18 | 118.1(2) |
| N1–C1–N3 | 110.70(19) | N6–C16–N8 | 110.92(19) |
| N1–C2–N2 | 127.6(2) | N6–C17–N7 | 127.5(2) |
| N2–C3–N3 | 122.9(2) | N7–C18–N8 | 122.6(2) |
The N1-triazine ring adopts an envelope conformation whereby the C1 atom lies 0.442(3) Å out of the plane defined by the five remaining atoms, r.m.s. deviation = 0.033 Å; the equivalent parameters for the N6-ring/C16 atom are 0.471(3) and 0.034 Å, respectively. There are some conformational differences evident between the two independent molecules as highlighted in the overlay diagram of Fig. 1(c). These relate primarily to the relative orientation of the phenyl rings. The dihedral angles between the best plane through the triazine ring and the phenyl and chlorophenyl rings are 89.59(8) and 17.74(12)°, respectively for the N1-molecule; the equivalent angles for the N6-molecule are 72.76(9) and 19.24(14)°, respectively. For the N1- and N6-molecules, the dihedral angle between the peripheral rings are 77.95(8) and 57.86(9)°, respectively. Very similar trends are evident in the molecules comprising the methoxy (1b) and bromo (1f) analogues; see ESI Table S2† for data.
An interesting feature of the crystal structure determinations of 1b·EtOH, 1e·EtOH and 1f·EtOH is that each has occluded solvent in their structures so the ratio of organic molecule to ethanol was 1:1. A description of the molecular packing of 1e·EtOH ensues; details of the intermolecular interactions are gathered in Table 2. Fig. 2(a) shows a view of the molecular packing for 1e·EtOH showing only contacts occurring between the organic molecules, i.e. 1e only. The two 1e molecules in the asymmetric unit are connected into helical supramolecular chains via ring amine-N–H⋯N(imine) hydrogen-bonding. The chains are aligned along the b-axis direction, being propagated by screw (21) symmetry. The connections between chains along the c-axis are of the type primary amine-N–H⋯π(chlorophenyl), again occurring between the different 1e molecules of the asymmetric unit. Links between layers along the a-axis are of the type phenyl-C–H⋯π(phenyl). Here, the donor and acceptor molecules are the N1- and N6-containing molecules, respectively. As there is no reciprocal contact, i.e. having the N6- and N- molecules as the donor and acceptor, this interaction distinguishes the two independent molecules. Each of the independent ethanol molecules participates in analogous hydrogen-bonding with the 1e molecules defining the host structure. The donor interactions are of the type ethanol–O–H⋯N6(triazine), where the nitrogen acceptor is adjacent to the methine-carbon atom. The ethanol molecules each accept two hydrogen bonds, one from each of the exocyclic amines.
| A | H | B | A–H | H⋯B | A⋯B | A–H⋯B | Symmetry operation |
|---|---|---|---|---|---|---|---|
| N3 | H3n | N7 | 0.87(2) | 2.11(2) | 2.931(3) | 156(2) | 1 − x, 1/2 + y, 1 − z |
| N8 | H8n | N2 | 0.87(2) | 2.12(2) | 2.939(3) | 156(2) | 1 − x, 1/2 + y, 1 − z |
| N4 | H4na | Cg(C10–C15) | 0.88(2) | 2.60(3) | 135(2) | 3.279(2) | x, y, z |
| N9 | H9na | Cg(C25–C30) | 0.87(2) | 2.63(4) | 128(3) | 3.236(3) | x, y, 1 + z |
| C22 | H22 | Cg(C4–C9) | 0.95 | 2.71 | 3.577(3) | 152 | 1 + x, y, 1 + z |
| O1 | H1o | N6 | 0.84(2) | 1.88(2) | 2.706(3) | 169(3) | x, y, z |
| O2 | H2o | N1 | 0.84(2) | 1.89(2) | 2.726(3) | 172(3) | x, y, 1 + z |
| N4 | H4nb | O1 | 0.88(3) | 1.99(3) | 2.865(3) | 176(3) | x, y, z |
| N10 | H10n | O1 | 0.88(2) | 2.05(3) | 2.913(3) | 165(2) | 1 − x, −1/2 + y, 1 − z |
| N5 | H5n | O2 | 0.88(2) | 2.04(2) | 2.907(3) | 169(2) | 1 − x, −1/2 + y, 1 − z |
| N9 | H9nb | O2 | 0.88(3) | 2.00(3) | 2.874(3) | 177(3) | x, y, z |
 | ||
| Fig. 2 Molecular packing in the crystal of 1e·EtOH: (a) a view of the unit cell contents in projection down the b-axis. The N–H⋯N, N–H⋯π and C–H⋯π interactions are shown as blue, purple and pink dashed lines, respectively. (b) A side-on view of a supramolecular layer highlighting the encapsulation of the solvent ethanol molecules and (c) a plan view of the layer shown in (b). Hydrogen-bonding interactions involving ethanol are indicated by orange dashed lines. For (b) and (c), non-participating hydrogen atoms are omitted for reasons of clarity. | ||
Subtle differences in the molecular packing are apparent in the crystal of 1b·EtOH; see ESI Table S3 and Fig. S3.† Crucially, the intra-layer O–H⋯N, N–H⋯O and N–H⋯N hydrogen-bonding remains the same. However, the connections between layers are chlorophenyl-C–H⋯O(methoxy) interactions. Additional intra-layer methoxyphenyl-C–H⋯π(chlorophenyl) and ethanol–methylene–C–H⋯Cl interactions are noted, which contribute to the stability of the layer. The molecular packing of the bromo analogue of 1e·EtOH, i.e. 1f·EtOH, mimics that described above with the addition of inter-layer phenyl-C–H⋯Br contacts; see ESI Table S4 and Fig. S4.†
Additional investigations were conducted on the three crystallographically characterised compounds, i.e. simultaneous thermal analysis (STA) and powder X-ray diffraction (PXRD) studies. The original STA traces and data are given in ESI Fig. S5.† For 1b·EtOH, the results of the thermogravimetric analyses showed a single-step between 90 and 162 °C corresponding to the loss of one ethanol molecule per molecule of 1b. Similar features were noted for each of 1e·EtOH and 1f·EtOH although the onset temperatures were higher and the process was completed at lower temperatures. For 1e·EtOH and 1f·EtOH, a second exothermic event corresponding to melting was also observed. This was not seen in the STA of 1b·EtOH and visual inspection showed the sample to be a paste after desolvation in contrast to the powders generated after the desolvation of the other samples. In order to ascertain whether desolvation altered the crystal structures of 1e·EtOH and 1f·EtOH, PXRD measurements were conducted. As seen from ESI Fig. S6,† the desolvated forms did not retain the original crystal structures. Attempts at re-solvation of 1e and 1f were performed. Thus, powders were placed in a sample vial which was in turn placed in a larger container containing ethanol, capped and allow to stand overnight. Subsequent PXRD measurements showed no evidence of re-solvation.
Conclusions
An efficient one-pot protocol for the synthesis of N2,6-diaryl-5,6-dihydro-1,3,5-triazine-2,4-diamines (1) from simple, readily available reagents i.e. cyanoguanidine, anilines and benzaldehydes was developed. After the initial three-component condensation of these building blocks, the intermediate 1,6-diaryl-1,6-dihydro-1,3,5-triazine-2,4-diamines (2) were isomerized to the targeted products via the base-promoted Dimroth rearrangement. The X-ray crystallographic study established the tautomeric form as the 5,6-dihydro-tautomer. It was found that N2,6-diaryl-5,6-dihydro-1,3,5-triazine-2,4-diamines (1) crystallised from ethanol trapping one molecule of this solvent per molecule of 1 in the crystals, giving 1·EtOH. The STA and PXRD data for selected compounds suggested that desolvation irreversibly change crystal structure.Experimental
General information
All the chemicals and reagents were purchased from commercial suppliers (Sigma-Aldrich, Merck, and Alfa-Aesar) and were used without additional purification. Melting points (uncorrected) were determined on a Stuart™ SMP40 automatic melting point apparatus. 1H and 13C NMR spectra were recorded on a Bruker Fourier NMR spectrometer (300 MHz) using DMSO-d6 as a solvent and TMS as an internal reference. The chemical shifts are reported in parts per million (ppm, δ) and coupling constants are reported in Hertz with the splitting pattern described as singlet (s), broad signal (brs), doublet (d), triplet (t), quartet (q), doublet of doublets (dd), double of doublet of doublets (ddd), or multiplet (m). Thermogravimetric analyses were performed on a PerkinElmer STA 6000 Thermogravimetric Analyzer in the range 25–600 °C at the rate of 10 °C min−1 under a nitrogen purge. The data was manipulated by PerkinElmer thermal analysis proprietary software Pyris®. Powder X-ray diffraction (PXRD) patterns were measured on a Rigaku SmartLab X-ray diffractometer using Cu-Kα (λ = 1.5406 Å) radiation in the 2θ range 10 to 60° with step size 0.01°.General procedure for the preparation of N2,6-diaryl-5,6-dihydro-1,3,5-triazine-2,4-diamines (1)
To a solution of cyanoguanidine (0.84 g, 10 mmol), a (un)substituted benzaldehyde (10 mmol) and a (un)substituted aniline (10 mmol) in EtOH (5 mL), conc. HCl (0.84 mL, 10 mmol) was added and the reaction mixture was heated under reflux for 24 h. The reaction mixture was cooled to rt and diluted with 50% aq. EtOH (5 mL). Then, aq. NaOH solution (5 N) was added drop-wise to the solution until the pH increased to 10–11. The mixture was heated under reflux for another 5 h. After cooling at 0 °C, the resulting precipitate was filtered, washed with water and recrystallized from EtOH.X-ray crystallographic analysis
X-ray intensity data for 1b·EtOH, 1e·EtOH and 1f·EtOH were measured at T = 100 K on Rigaku/Oxford Diffraction XtaLAB Synergy diffractometer (Dualflex, AtlasS2) fitted with Cu-Kα radiation (λ = 1.54178 Å) so that θmax = 67.1°. Data reduction, including absorption correction, was accomplished with CrysAlisPro.8 The structures were solved by direct-methods9 and refined (anisotropic displacement parameters and C-bound H atoms in the riding model approximation) on F2.10 The O- and N-bound H atoms were refined with O–H and N–H constrained to 0.84 ± 0.01 and 0.88 ± 0.01 Å, respectively, and with Uiso(H) = 1.2Ueq(O, N). A weighting scheme of the form w = 1/[σ2(Fo2) + (aP)2 + bP] where P = (Fo2 + 2Fc2)/3 was introduced in each case. The absolute structures were determined based on differences in Friedel pairs included in the respective data sets. In 1b·EtOH, two reflections, i.e. (3–5 8) and (3 5 8), were omitted from the final cycles of refinement owing to poor agreement. The molecular structure diagrams were generated with ORTEP for Windows11 with 50% displacement ellipsoids, and the packing diagrams were drawn with DIAMOND.12 Additional data analysis was made with PLATON.13 Crystal data and refinement details are given in Table 3.| Compound | 1b·EtOH | 1e·EtOH | 1f·EtOH |
|---|---|---|---|
| Formula | C16H16ClN5O·C2H6O | C15H14ClN5·C2H6O | C15H14BrN5·C2H6O |
| Molecular weight | 375.85 | 345.83 | 390.29 |
| Crystal size/mm3 | 0.04 × 0.07 × 0.13 | 0.03 × 0.06 × 0.06 | 0.02 × 0.08 × 0.11 |
| Colour | Colourless | Colourless | Colourless |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21 | P21 | P21 |
| a/Å | 13.2045(2) | 12.6684(2) | 12.7568(4) |
| b/Å | 9.7903(2) | 9.81230(10) | 9.7768(3) |
| c/Å | 14.8676(2) | 15.0780(2) | 15.1645(4) |
| β/° | 93.816(2) | 109.8020(10) | 109.622(3) |
| V/Å3 | 1917.76(6) | 1763.46(4) | 1781.49(10) |
| Z | 4 | 4 | 4 |
| Dc/g cm−3 | 1.302 | 1.303 | 1.455 |
| μ/mm−1 | 1.949 | 2.030 | 3.258 |
| Measured data | 24367 |
42215 |
22035 |
| θ range/° | 3.0–67.1 | 3.1–67.1 | 3.1–67.1 |
| Unique data | 6693 | 6286 | 5907 |
| Observed data (I ≥ 2.0σ(I)) | 6456 | 6062 | 5554 |
| No. parameters | 503 | 465 | 465 |
| R, obs. data; all data | 0.053; 0.145 | 0.029; 0.072 | 0.037; 0.093 |
| a; b in weighting scheme | 0.071; 3.036 | 0.041; 0.308 | 0.058; 0.019 |
| Rw, obs. data; all data | 0.055; 0.146 | 0.031; 0.073 | 0.040; 0.096 |
| Range of residual electron | |||
| Density peaks/eÅ−3 | −0.46 to 0.60 | −0.27 to 0.18 | −0.74 to 0.74 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Ministry of Higher Education, Malaysia under Fundamental Research Grant Scheme, grant number FRGS/1/2015/SG01/MUSM/03/1. The authors thank Sunway University Sdn Bhd (grant number STR-RCTR-RCCM-001-2019) for support of crystallographic studies.References
- E. J. Modest and P. Levine, J. Org. Chem., 1956, 21, 14 CrossRef CAS.
- S. Cascioferro, B. Parrino, V. Spano, A. Carbone, A. Montalbano, P. Barraja, P. Diana and G. Cirrincione, Eur. J. Med. Chem., 2017, 142, 523 CrossRef CAS PubMed.
- S. Tsuchiya, H. Murayama and Y. Sugimoto, Japan Patent, JP 2019064976, 2019.
- F. P. L. Lim, K. C. Tan, G. Luna, E. R. T. Tiekink and A. V. Dolzhenko, Tetrahedron, 2019, 75, 2322 CrossRef CAS; F. P. L. Lim, L. Y. Tan, E. R. T. Tiekink and A. V. Dolzhenko, RSC Adv., 2018, 8, 21495 RSC; F. P. L. Lim, N. R. Halcovitch, E. R. T. Tiekink and A. V. Dolzhenko, Tetrahedron, 2018, 74, 1868 CrossRef; F. P. L. Lim, S. T. Low, E. L. K. Ho, N. R. Halcovitch, E. R. T. Tiekink and A. V. Dolzhenko, RSC Adv., 2017, 7, 51062 RSC; F. P. L. Lim, G. Luna and A. V. Dolzhenko, Tetrahedron Lett., 2015, 56, 7016 CrossRef; F. P. L. Lim, G. Luna and A. V. Dolzhenko, Tetrahedron Lett., 2015, 56, 521 CrossRef; F. P. L. Lim, G. Luna and A. V. Dolzhenko, Tetrahedron Lett., 2014, 55, 5159 CrossRef; S. A. Kalinina, D. V. Kalinin and A. V. Dolzhenko, Tetrahedron Lett., 2013, 54, 5537 CrossRef; A. V. Dolzhenko, S. A. Kalinina and D. V. Kalinin, RSC Adv., 2013, 3, 15850 RSC.
- A. Junaid, F. P. L. Lim, E. R. T. Tiekink and A. V. Dolzhenko, ACS Comb. Sci., 2019, 21, 548 CrossRef CAS.
- E. J. Modest, J. Org. Chem., 1956, 21, 1 CrossRef CAS.
- L. Fábián and C. P. Brock, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 94 CrossRef.
- Rigaku Oxford Diffraction, CrysAlis PRO, Yarnton, Oxfordshire, England, 2017 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. B: Struct. Sci., 2015, 71, 3 CrossRef.
- K. Brandenburg, Diamond, Crystal Impact GbR, Bonn, Germany, 2006 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1961317–1961319. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra08795h |
| This journal is © The Royal Society of Chemistry 2019 |