
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical study of substituent effects on electride characteristics and the nonlinear optical properties of Li@calix[4]pyrrole†
Hui Wenga,
Yunyang Tenga,
Qi Shenga,
Zhongjun Zhou *ab,
Xuri Huanga,
Zhiru Lia and
Tao Zhangb
*ab,
Xuri Huanga,
Zhiru Lia and
Tao Zhangb
aInstitute of Theoretical Chemistry, Jilin University, Changchun, 130023, People's Republic of China. E-mail: zjzhou@jlu.edu.cn
bCollege of Physics, Jilin University, Changchun, 130012, People's Republic of China
First published on 21st November 2019
Abstract
Electrides, a novel kind of ionic compound in which electrons serve as anions, have been proposed as potential second-order nonlinear optical (NLO) materials. In this work, the substituent effects on the electride characteristics and the NLO behaviour of Li@calix[4]pyrrole with an electride-like structure were studied theoretically. The results show that electron-donating and electron-withdrawing groups can effectively increase and decrease the first hyperpolarizability (β0), respectively, without affecting the electride characteristics (electron population). More interestingly, lithiation in which four H atoms bonded to N atoms are substituted by four Li atoms within the core structure of Li@calix[4]pyrrole remarkably improves the electride characteristics, with a large electron population of 0.74 e (1.02 e) at the NNA (ELF) basins, making this structure perhaps the first formal molecular electride with almost one electron isolated from the rest of the molecules. Furthermore, a relationship between the electride characteristics and the NLO properties is found: the more delocalization the excess electron of the electride experiences, the larger the β0 value is. The present investigation may provide useful information for exploring high-performance second-order nonlinear optical materials based on organic electrides.
1. Introduction
During the past few decades, nonlinear optic (NLO) materials have made rapid progress in terms of both theoretical research and experimentation and have become an important component in the fields of optics and electricity.1–20 With the research, design and improvement of new nonlinear optical materials, various methods for increasing nonlinear optical responses have been proposed.21–31Electrides are a novel kind of ionic compound in which electrons serve as the anions.32–35 With such a unique structure, electrides show great potential in various applications, such as superconductivity,36,37 electronics,38–40 and catalysis.41–43 Recently, theoretical investigations by Li et al. have shown that systems in which the excess electrons are diffuse exhibit great NLO responses. Thereafter, a series of inorganic and organic molecular electrides were theoretically designed and studied by Li and co-workers, and the electride characteristics of these materials were identified by the highest occupied molecular orbitals.44–50 Among these materials, Li@calix[4]pyrrole is the most representative electride with a large NLO response.51 More recently, Luis and Matito proposed four simultaneous criteria as necessary means to distinguish molecular electrides from similar species, namely, the existence of a non-nuclear attractor (NNA), an ELF basin, negative values of the Laplacian of the electron density, and NLO properties, which are very meaningful for identifying new molecular electrides.52–54 According to these criteria, Li@calix[4]pyrrole is accurately classified as an electride-like molecule because the electron number of NNA and ELF basins is far less than one electron. To the best of our knowledge, there is no formal molecular electride meeting all the above requirements except Li2@TCNQ and Na2@TCNQ, in which the electride characteristics are considered to derive from Li2 and Na2. How do we construct a formal electride? Recently, the effects of substituents on the alkalide characteristics and NLO properties of Li+calix[4]pyrroleLi−, as an analogue of the electride of calix[4]pyrrole, have been reported theoretically. The authors demonstrate that electron-donating substituents can improve the alkalide characteristics of Li+(calix[4]pyrrole)Li−.55 Then, can we obtain an formal electride with an isolated electron by means of the substituent effects based on Li@calix[4]pyrrole? With this question, we start this work. In the structure of calix[4]pyrrole, many different kinds of H atoms exist, such as –CH, –CH2, and –NH, which can be substituted by electron-donating or electron-withdrawing groups. Many derivatives of calix[4]pyrrole with electron-donating or electron-withdrawing groups, such as methoxy (–OCH3) and fluorine (–F), have been synthesized, providing abundant structures for our present work.56 In addition to the conventional push–pull groups, lithiation by which H atoms in –NH groups are substituted by additional Li atoms is also used. In this work, we will pay attention to not only the substituent effect on the electride characteristic but also the substituent effects on the NLO properties, as well as the relationship between the electride characteristics and the NLO properties. The present investigation may provide useful information for exploring high-performance second-order nonlinear optical materials based on organic electrides.
2. Computational methods
The optimized geometric structures were obtained using the B3LYP method and 6-31+G(d) basis set in conjunction with the same thresholds as the article published by Luis and Matito (see ESI†).52 Frequency calculations were performed to confirm whether the obtained structures are real minima at the same level of theory.For a large system, the DFT57,58 method is a good choice for calculating the first hyperpolarizability (β0) because it considers a balance between the efficiency and the computational cost. However, different functionals may yield different results for β0, and thus, testing different functionals is necessary. Here, different functionals were used to calculate the β0 value of Li@calix[4]pyrrole with a medium size. In this testing, CAM-B3LYP59,60 was chosen to calculate the β0 and polarizability (α0) value for all the systems in this work because the β0 value (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 876 au) of CAM-B3LYP is closest to that of MP2 (ref. 61) (11853 au) with the same mixed basis set where the 6-311++G(3df,3pd) basis set is employed for N, Li and the four H atoms of –NH groups and the 6-31+g(d) basis set is used for other atoms. The results are shown in the ESI (Table S1†). The finite-field (FF) approach62–64 has been applied to evaluate the tensor components of the static hyperpolarizabilities. It simply requires the evaluation of the system energy for different amplitudes of the applied external electric field and can therefore be applied to a broad range of methods. When a system is in a weak and stable applied electric field, its energy can be written as:
876 au) of CAM-B3LYP is closest to that of MP2 (ref. 61) (11853 au) with the same mixed basis set where the 6-311++G(3df,3pd) basis set is employed for N, Li and the four H atoms of –NH groups and the 6-31+g(d) basis set is used for other atoms. The results are shown in the ESI (Table S1†). The finite-field (FF) approach62–64 has been applied to evaluate the tensor components of the static hyperpolarizabilities. It simply requires the evaluation of the system energy for different amplitudes of the applied external electric field and can therefore be applied to a broad range of methods. When a system is in a weak and stable applied electric field, its energy can be written as:
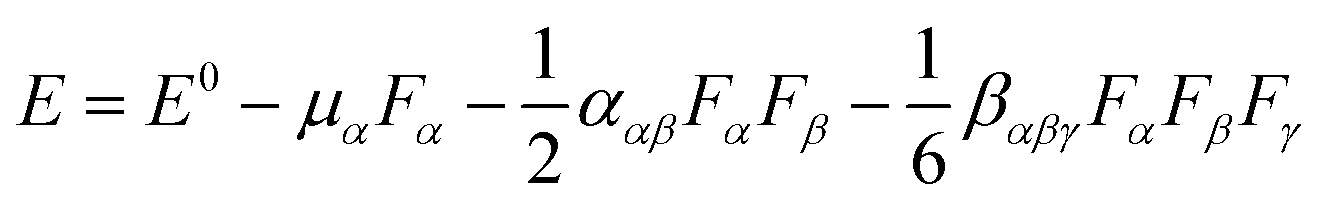
Topological analysis of electron density was performed using the MP2 method since Timerghazin and Peslherbe have shown that the characterization of excess electrons is sensitive to electron correlation and large basis sets.71 The topological analysis of atoms in molecules (AIM)72,73 and the electron localization function (ELF)74–76 were conducted by using the free and open source Multiwfn program.77
We used the following equations to calculate β0, α0 and VIE.
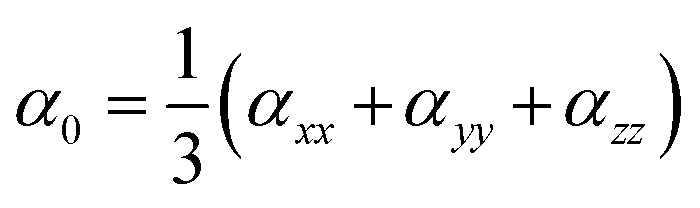
| β0 = (βx2 + βy2 + βz2)1/2 |
| βi = βiii + βijj + βikk, i, j, k = x, y, z |
| VIE = E[M+] − E[M] |
All calculations are performed using the Gaussian 09 program package.78
3. Results and discussion
3.1 Topological analysis of electron density
All optimized structures without imaginary frequencies are shown in Fig. 1. To facilitate the discussion, Li@calix[4]pyrrole is denoted as 1. All other structures are constructed based on this structure of 1. When eight H atoms at the β-positions of 1 are replaced with methoxy (–OCH3) and fluorine (–F) groups, octamethoxy-1 and octafluorine-1 are obtained, respectively. In addition, four H atoms within NH– groups of 1 are substituted by four Li atoms to form tetra-lithium-1. Except for octamethoxy-1, which has C1 symmetry, these complexes all have C4v symmetry.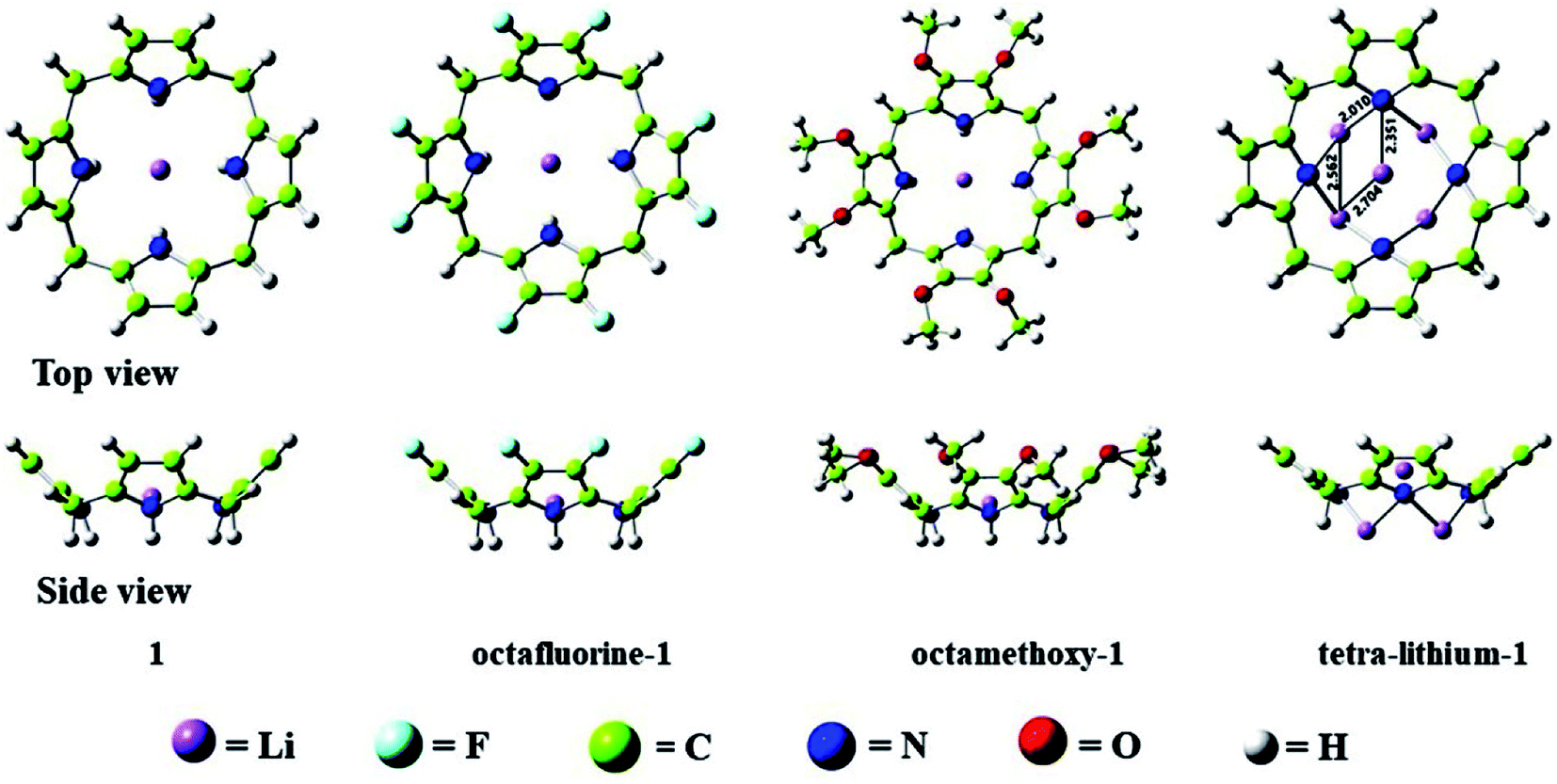 | ||
| Fig. 1 Optimized structures of 1, octafluorine-1, octamethoxy-1, and tetra-lithium-1. | ||
079 bohr3). Octafluorine-1, with electron-withdrawing substituents, has the smallest integral volume, while octamethoxy-1, with electron-donating substituents, has the largest integral volume, which indicates that the excess electron of octamethoxy-1 is more diffuse than that of octafluorine-1. The above analysis shows that the introduction of electron-donating or electron-withdrawing substituents does not change the electron population of electrides but significantly affects the diffusion of the excess electron, which may be an effective way to change the NLO properties while maintaining the electride characteristics. The NLO properties will be discussed in the following section.
| Chemical models | AIM | ELF | |||||
|---|---|---|---|---|---|---|---|
| CP | ρ(rc) | ∇2ρ(rc) | N(pop) | CP | N(Ω) | Basin volume | |
| 1 | NNA | 0.006 | −0.004 | 0.47 | C(NNA) | 0.91 | 8763 |
| Octafluorine-1 | NNA | 0.007 | −0.004 | 0.45 | C(NNA) | 0.91 | 7976 |
| Octamethoxy-1 | NNA | 0.005 | −0.002 | 0.47 | C(NNA) | 0.91 | 13079 |
| Tetra-lithium-1 | NNA | 0.010 | −0.009 | 0.74 | C(NNA) | 1.02 | 5353 |
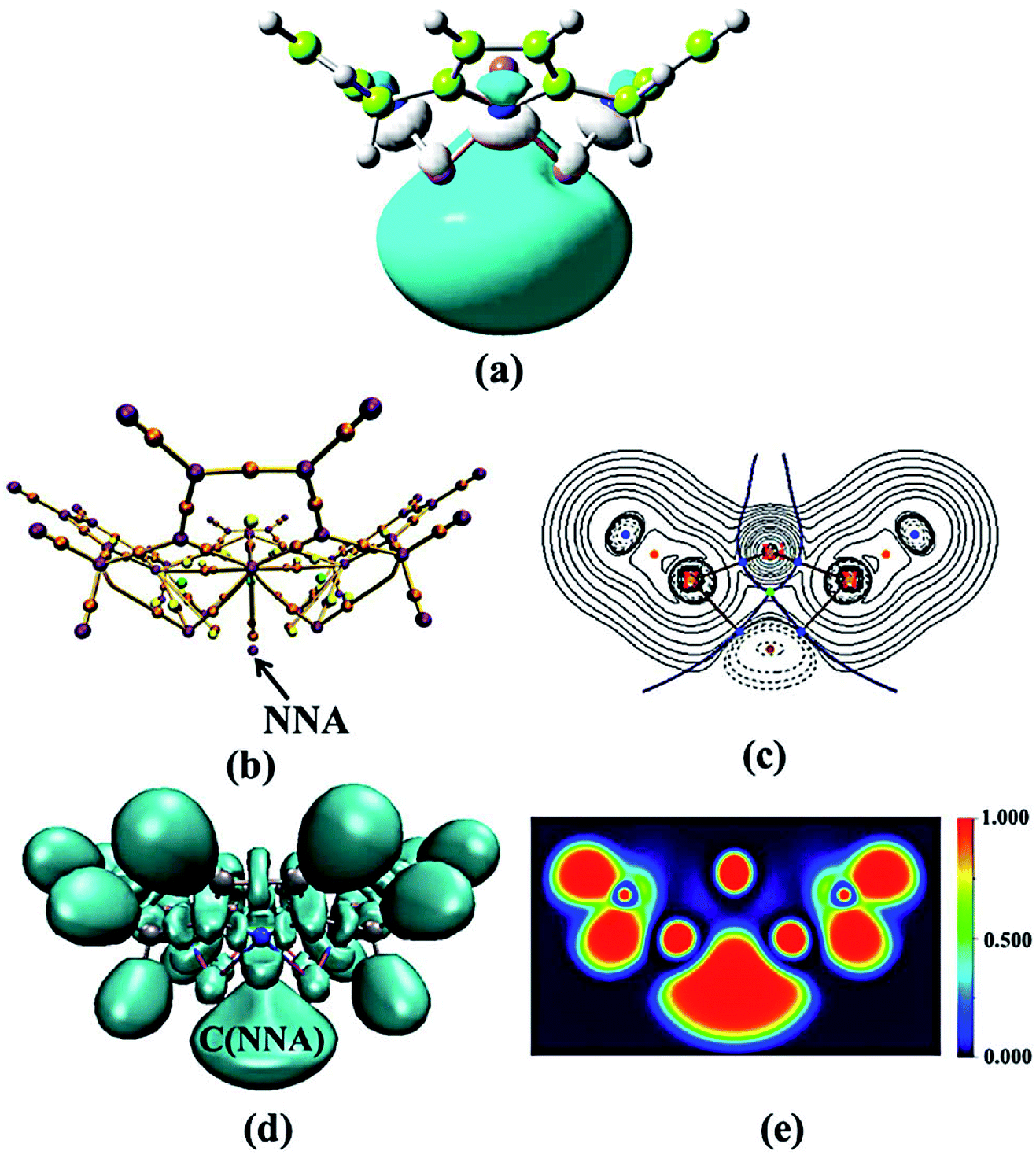 | ||
| Fig. 2 (a) HOMO (isovalue = 0.02 au). (b) Non-nuclear attractor (NNA). (c) Two-dimensional (2D) plot of ∇2ρ(rc). (d) ELF isosurface image at a 0.800 value. (e) The colour-filled map of ELF for tetra-lithium-1. | ||
3.2 Nonlinear optical properties
The NLO properties of 1, octafluorine-1 and octamethoxy-1 have been calculated at the CAM-B3LYP level of theory, and the results are collected in Table 2. Unlike the electron population, the first hyperpolarizabilities (β0) are very significantly affected by the substituents. When eight electron-withdrawing substituents (–F) is used, the β0 value decreases from 11876 au (1) to 8977 au (octafluorine-1). However, when eight electron-donating substituents (–OCH3) is used, the β0 value is considerably increased from 11876 au (1) to 20562 au (octamethoxy-1). For systems with electride or alkalide characteristics, a two-level expression is often used to explain the β0 value.79,80where ΔE, f0 and Δμ are transition energy, oscillator strength and the difference in dipole moments between the ground state and crucial excited state. In the two-level expression, the third power of the transition energy (ΔE) is inversely proportional to the β0 value; thus, the ΔE value is the decisive factor in the first hyperpolarizability. For the polarizabilities, from Table 2, the order of the α0 values is 383 (octafluorine-1) < 390 (1) < 581 au (octamethoxy-1). The properties of the related excited state are calculated using the time-dependent CAM-B3LYP method, and the results are listed in Table 2. From Table 2, the order of ΔE is 1.369 (octamethoxy-1) < 1.599 (1) < 1.603 eV (octafluorine-1). The ΔE value is the difference in energy between the ground state and the excited state with a large oscillator. After Franck–Condon excitation, octafluorine-1, 1, and octamethoxy-1 are excited into S2, S2, and S1, respectively. All the electronic configurations in these excited states involved the excitation of the highest occupied molecular orbital (HOMO), which is the orbital containing the excess electron. When electron-donating substituents (–OCH3) are used, the energy level of the HOMO is increased from −3.35 (1) to −3.03 eV (octamethoxy-1), which will lead to a small ΔE. When electron-withdrawing substituents (–F) are used, the energy level of the HOMO is decreased from −3.35 (1) to −4.01 eV (octafluorine-1), which leads to a large ΔE. Moreover, the order of VIE is octafluorine-1 (4.38 eV) > 1 (3.71 eV) > octamethoxy-1 (3.32 eV), consistent with the order of the HOMO energy levels. From the above analysis, we can conclude that the electron-withdrawing or electron-donating substituents cannot affect the electride characteristics but significantly affect the NLO behaviour.
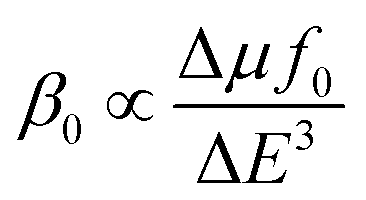
| Molecules | β0 | α0 | Transition | ΔE | f0 | Configuration | Ci | HOMO | VIE | q(Li+) |
|---|---|---|---|---|---|---|---|---|---|---|
| a The values outside parentheses were calculated at the CAM-B3LYP/6-31+g(d)/6-311++G(3df,3pd) level of theory. The values within parentheses were obtained at the MP2/6-31+g(d)/6-311++g(3df,3pd) level of theory. | ||||||||||
| Octafluorine-1 | 8977 | 383 | S0 → S2 | 1.603 | 0.157 | H → L+1 | 0.86 | −4.01 | 4.38 (4.26) | 0.481 (0.361) |
| H → L+4 | −0.45 | |||||||||
| 1 | 11876 |
390 | S0 → S2 | 1.599 | 0.169 | H → L+1 | 0.70 | −3.35 | 3.71 (3.62) | 0.477 (0.372) |
| H → L+4 | 0.68 | |||||||||
| Octamethoxy-1 | 20562 |
581 | S0 → S1 | 1.369 | 0.152 | H → L | 0.72 | −3.03 | 3.32 (3.23) | 0.491 (0.356) |
| H → L+5 | −0.62 | |||||||||
| Tetra-lithium-1 | 5097 | 328 | S0 → S1 | 2.571 | 0.163 | H → L | 0.91 | −5.48 | 5.91 (5.77) | 0.915 (0.921) |
| H → L+4 | −0.28 | |||||||||
Of the above three molecules, tetra-lithium-1 has the smallest β0 and α0 value of 5097 and 328 au and the largest transition energy of 2.571 eV. The electronic configuration of tetra-lithium-1 is the HOMO → LUMO excitation. Due to lithiation, the energy level of the HOMO is considerably decreased from −3.35 eV (1) to −5.48 eV (tetra-lithium-1), leading to a large ΔE. As usual, the VIE value of the molecular electride is smaller than that of alkali atoms, ranging from 5.4 to 3.9 eV (Li–Cs),81,82 and thus, sometimes, these molecules are called superalkalides. For example, the VIE values of octafluorine-1, 1, and octamethoxy-1 lie between 3.32 and 4.38 eV, which is less than that of Li. However, tetra-lithium-1 has a very large VIE value of 5.91 eV at the CAM-B3LYP level of theory, which is greater than that of Li. From the above analysis, the following relationship is obtained: the more obvious the electride characteristic (the electron population), the larger the VIE value is. The large VIE value of tetra-lithium-1 may be attributed to the orbital of excess electron lowered by lithiation.
4. Conclusion
The effects of multiple substitutions on the electride characteristics and the second-order NLO properties (β0) of Li@calix[4]pyrrole (1) and its analogues (octafluorine-1, octamethoxy-1 and tetra-lithium-1) were theoretically studied at the CAM-B3LYP and MP2 levels of theory. Some conclusions are summarized as follows:(1) Electron-donating and electron-withdrawing groups can effectively increase and decrease the first hyperpolarizability (β0), respectively, but do not damage the electride characteristics (electron population). For example, when all the H atoms at the β-positions of 1 were substituted by –F groups, the β0 value was decreased from 11876 (1) to 8977 au (octafluorine-1), but when all the H atoms at the β-positions of 1 were substituted by –OCH3 groups, the β0 value was substantially increased from 11876 (1) to 20562 au (octamethoxy-1). However, the electride characteristics were not changed, resulting in a nearly identical electron population (approximately 0.47 e).
(2) A different molecular electride, tetra-lithium-1, was successfully theoretically designed by lithiation in which four H atoms on N atoms were substituted by four Li atoms. Due to the large electron numbers of 0.74 and 1.02 e at the NNA and ELF basins, respectively, tetra-lithium-1 may be the first formal molecular electride with one isolated excess electron serving as an anion, except for Li2@TCNQ and Na2@TCNQ, in which the electride characteristics are considered to derive from Li2 and Na2. Furthermore, the vertical ionization energy of tetra-lithium-1 was 5.91 eV, which is larger than those (5.4–3.9 eV) of alkali atoms from Li to Cs, indicating its higher stability. In addition, enhancement of the electride characteristics was accompanied by a decrease in the NLO properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the China Postdoctoral Science Foundation (2015M571355).References
- B. J. Coe, Acc. Chem. Res., 2006, 39, 383–393 CrossRef CAS PubMed.
- J. A. Delaire and K. Nakatani, Chem. Rev., 2000, 100, 1817–1846 CrossRef CAS PubMed.
- S. H. Lee, J. R. Park, M. Y. Jeong, H. M. Kim, S. Li, J. Song, S. Ham, S. J. Jeon and B. R. Cho, ChemPhysChem, 2006, 7, 206–212 CrossRef CAS PubMed.
- C. G. Liu, W. Guan, P. Song, L. K. Yan and Z. M. Su, Inorg. Chem., 2009, 48, 6548–6554 CrossRef CAS PubMed.
- F. Meyers, S. Marder, B. Pierce and J. Bredas, J. Am. Chem. Soc., 1994, 116, 10703–10714 CrossRef CAS.
- S. Muhammad, H.-L. Xu, R.-L. Zhong, Z.-M. Su, A. G. Al-Sehemi and A. Irfan, J. Mater. Chem. C, 2013, 1, 5439 RSC.
- P. C. Ray, Chem. Rev., 2010, 110, 5332–5365 CrossRef CAS PubMed.
- M. Schulz, S. Tretiak, V. Chernyak and S. Mukamel, J. Am. Chem. Soc., 2000, 122, 452–459 CrossRef CAS.
- T. G. Zhang, Y. Zhao, I. Asselberghs, A. Persoons, K. Clays and M. J. Therien, J. Am. Chem. Soc., 2005, 127, 9710–9720 CrossRef CAS PubMed.
- Z.-J. Zhou, G.-T. Yu, F. Ma, X.-R. Huang, Z.-J. Wu and Z.-R. Li, J. Mater. Chem. C, 2014, 2, 306–311 RSC.
- J. Wu, W. Wang, C. Gong, Q. Li, Z. Li, G. Deng, X. Zhang, K. Chen, Y. Gong and K. s. Chiang, J. Mater. Chem. C, 2017, 5, 7472–7478 RSC.
- Q. Feng, Y. Li, G. Shi, L. Wang, W. Zhang, K. Li, H. Hou and Y. Song, J. Mater. Chem. C, 2016, 4, 8552–8558 RSC.
- S. Muhammad, H. Xu, M. R. Janjua, Z. Su and M. Nadeem, Phys. Chem. Chem. Phys., 2010, 12, 4791–4799 RSC.
- M. Schulze, M. Utecht, T. Moldt, D. Przyrembel, C. Gahl, M. Weinelt, P. Saalfrank and P. Tegeder, Phys. Chem. Chem. Phys., 2015, 17, 18079–18086 RSC.
- C. Tonnele, B. Champagne, L. Muccioli and F. Castet, Phys. Chem. Chem. Phys., 2018, 20, 27658–27667 RSC.
- A. Zhang, H. Xiao, C. Peng, S. Bo, H. Xu, M. Zhang, G. Deng, Z. Zhen and X. Liu, RSC Adv., 2014, 4, 65088–65097 RSC.
- J. Dalal, N. Sinha, H. Yadav and B. Kumar, RSC Adv., 2015, 5, 57735–57748 RSC.
- K. Nivetha, S. Kalainathan, M. Yamada, Y. Kondo and F. Hamada, RSC Adv., 2016, 6, 35977–35990 RSC.
- S. Muhammad, A. R. Chaudhry, A. Irfan and A. G. Al-Sehemi, RSC Adv., 2017, 7, 36632–36643 RSC.
- H.-L. Yu, W.-Y. Wang, B. Hong, Y.-L. Si, T.-L. Ma and R. Zheng, RSC Adv., 2017, 7, 41830–41837 RSC.
- Y. Y. Liang, B. Li, X. Xu, F. Long Gu and C. Zhu, J. Comput. Chem., 2019, 40, 971–979 CrossRef CAS PubMed.
- S. Muhammad, H. Xu, Y. Liao, Y. Kan and Z. Su, J. Am. Chem. Soc., 2009, 131, 11833–11840 CrossRef CAS PubMed.
- M. Nakano, H. Fujita, M. Takahata and K. Yamaguchi, J. Am. Chem. Soc., 2002, 124, 9648–9655 CrossRef CAS PubMed.
- H. L. Xu, Z. R. Li, D. Wu, B. Q. Wang, Y. Li, F. L. Gu and Y. Aoki, J. Am. Chem. Soc., 2007, 129, 2967–2970 CrossRef CAS PubMed.
- G. Yu, X. Zhao, M. Niu, X. Huang, H. Zhang and W. Chen, J. Mater. Chem. C, 2013, 1, 3833 RSC.
- R. L. Zhong, H. L. Xu, Z. M. Su, Z. R. Li, S. L. Sun and Y. Q. Qiu, ChemPhysChem, 2012, 13, 2349–2353 CrossRef CAS PubMed.
- Z. J. Zhou, X. P. Li, F. Ma, Z. B. Liu, Z. R. Li, X. R. Huang and C. C. Sun, Chem.–Eur. J., 2011, 17, 2414–2419 CrossRef CAS PubMed.
- C. Tu, G. Yu, G. Yang, X. Zhao, W. Chen, S. Li and X. Huang, Phys. Chem. Chem. Phys., 2014, 16, 1597–1606 RSC.
- L. Chen, G. Yu, W. Chen, C. Tu, X. Zhao and X. Huang, Phys. Chem. Chem. Phys., 2014, 16, 10933–10942 RSC.
- B. Li, C. Xu, X. Xu, C. Zhu and F. L. Gu, Phys. Chem. Chem. Phys., 2017, 19, 23951–23959 RSC.
- F. Liu, Y. Yang, S. Cong, H. Wang, M. Zhang, S. Bo, J. Liu, Z. Zhen, X. Liu and L. Qiu, RSC Adv., 2014, 4, 52991–52999 RSC.
- W. Chen, Z. R. Li, D. Wu, R. Y. Li and C. C. Sun, J. Phys. Chem. B, 2005, 109, 601–608 CrossRef CAS PubMed.
- J. L. Dye, Inorg. Chem., 1997, 36, 3816–3826 CrossRef CAS.
- J. L. Dye, Acc. Chem. Res., 2009, 42, 1564–1572 CrossRef CAS PubMed.
- N. Ma, J. Gong, S. Li, J. Zhang, Y. Qiu and G. Zhang, Phys. Chem. Chem. Phys., 2017, 19, 2557–2566 RSC.
- M. Miyakawa, S. W. Kim, M. Hirano, Y. Kohama, H. Kawaji, T. Atake, H. Ikegami, K. Kono and H. Hosono, J. Am. Chem. Soc., 2007, 129, 7270–7271 CrossRef CAS PubMed.
- K. Shimizu, H. Ishikawa, D. Takao, T. Yagi and K. Amaya, Nature, 2002, 419, 597 CrossRef CAS PubMed.
- S. Watanabe, T. Watanabe, K. Ito, N. Miyakawa, S. Ito, H. Hosono and S. Mikoshiba, Sci. Technol. Adv. Mater., 2016, 12, 034410 CrossRef PubMed.
- H. Yanagi, K.-B. Kim, I. Koizumi, M. Kikuchi, H. Hiramatsu, M. Miyakawa, T. Kamiya, M. Hirano and H. Hosono, J. Phys. Chem. C, 2009, 113, 18379–18384 CrossRef CAS.
- X. Zhang, R. Guo, L. Jin, X. Dai and G. Liu, J. Mater. Chem. C, 2018, 6, 575–581 RSC.
- M. Kitano, S. Kanbara, Y. Inoue, N. Kuganathan, P. V. Sushko, T. Yokoyama, M. Hara and H. Hosono, Nat. Commun., 2015, 6, 6731 CrossRef CAS PubMed.
- S. Zhao, E. Kan and Z. Li, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 430–440 CAS.
- S. G. Dale and E. R. Johnson, Phys. Chem. Chem. Phys., 2016, 18, 27326–27335 RSC.
- Y. Li, Z.-R. Li, D. Wu, R.-Y. Li, X.-Y. Hao and C.-C. Sun, J. Phys. Chem. B, 2004, 108, 3145–3148 CrossRef CAS.
- Z. Li, J. Yang, J. G. Hou and Q. Zhu, J. Am. Chem. Soc., 2003, 125, 6050–6051 CrossRef CAS PubMed.
- Z.-J. Li, Z.-R. Li, F.-F. Wang, C. Luo, F. Ma, D. Wu, Q. Wang and X.-R. Huang, J. Phys. Chem. A, 2009, 113, 2961–2966 CrossRef CAS PubMed.
- Z. R. Li, F. F. Wang, D. Wu, Y. Li, W. Chen, X. Y. Sun, F. L. Gu and Y. Aoki, J. Comput. Chem., 2006, 27, 986–993 CrossRef CAS PubMed.
- R. L. Zhong, H. L. Xu, Z. R. Li and Z. M. Su, J. Phys. Chem. Lett., 2015, 6, 612–619 CrossRef CAS PubMed.
- J.-J. Wang, Z.-J. Zhou, Y. Bai, Z.-B. Liu, Y. Li, D. Wu, W. Chen, Z.-R. Li and C.-C. Sun, J. Mater. Chem., 2012, 22, 9652 RSC.
- Z. B. Liu, Z. J. Zhou, Y. Li, Z. R. Li, R. Wang, Q. Z. Li, Y. Li, F. Y. Jia, Y. F. Wang, Z. J. Li, J. B. Cheng and C. C. Sun, Phys. Chem. Chem. Phys., 2010, 12, 10562–10568 RSC.
- W. Chen, Z. R. Li, D. Wu, Y. Li, C. C. Sun and F. L. Gu, J. Am. Chem. Soc., 2005, 127, 10977–10981 CrossRef CAS PubMed.
- V. Postils, M. Garcia-Borras, M. Sola, J. M. Luis and E. Matito, Chem. Commun., 2015, 51, 4865–4868 RSC.
- O. El Bakouri, V. Postils, M. Garcia-Borras, M. Duran, J. M. Luis, S. Calvello, A. Soncini, E. Matito, F. Feixas and M. Sola, Chem.–Eur. J., 2018, 24, 9853–9859 CrossRef CAS PubMed.
- L. A. Burton, F. Ricci, W. Chen, G.-M. Rignanese and G. Hautier, Chem. Mater., 2018, 30, 7521–7526 CrossRef CAS.
- W. M. Sun, D. Wu, Y. Li and Z. R. Li, ChemPhysChem, 2013, 14, 408–416 CrossRef CAS PubMed.
- P. A. Gale, J. L. Sessler and V. Král, Chem. Commun., 1998, 1–8 RSC.
- P. S. Liyanage, R. M. de Silva and K. M. N. de Silva, J. Mol. Struct.: THEOCHEM, 2003, 639, 195–201 CrossRef CAS.
- L. Yan, G. Yang, W. Guan, Z. Su and R. Wang, J. Phys. Chem. B, 2005, 109, 22332–22336 CrossRef CAS PubMed.
- Y. Tawada, T. Tsuneda, S. Yanagisawa, T. Yanai and K. Hirao, J. Chem. Phys., 2004, 120, 8425–8433 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- J. Zyss, J. Chem. Phys., 1979, 70, 3333–3340 CrossRef CAS.
- J. Zyss, J. Chem. Phys., 1979, 70, 3341–3349 CrossRef CAS.
- J. Zyss, J. Chem. Phys., 1979, 71, 909–916 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- K. Okuno, Y. Shigeta, R. Kishi, H. Miyasaka and M. Nakano, J. Photochem. Photobiol., A, 2012, 235, 29–34 CrossRef CAS.
- Y. Y. Hu, S. L. Sun, W. T. Tian, W. Q. Tian, H. L. Xu and Z. M. Su, ChemPhysChem, 2014, 15, 929–934 CrossRef CAS PubMed.
- H.-M. He, J. M. Luis, W.-H. Chen, D. Yu, Y. Li, D. Wu, W.-M. Sun and Z.-R. Li, J. Mater. Chem. C, 2019, 7, 645–653 RSC.
- X. Li, J. Mater. Chem. C, 2018, 6, 7576–7583 RSC.
- Y. Bai, Z. J. Zhou, J. J. Wang, Y. Li, D. Wu, W. Chen, Z. R. Li and C. C. Sun, J. Phys. Chem. A, 2013, 117, 2835–2843 CrossRef CAS PubMed.
- Q. K. Timerghazin, I. Rizvi and G. H. Peslherbe, J. Phys. Chem. A, 2011, 115, 13201–13209 CrossRef CAS PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, New York, 1990 Search PubMed.
- R. F. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- B. Silvi and A. Savin, Nature, 1994, 371, 683 CrossRef CAS.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev., 1994, 94, 195–242 CrossRef CAS.
- J. L. Oudar and D. S. Chemla, J. Chem. Phys., 1977, 66, 2664–2668 CrossRef CAS.
- E. Zurek, J. Am. Chem. Soc., 2011, 133, 4829–4839 CrossRef CAS PubMed.
- W.-M. Sun, D. Wu, J. Kang, C.-Y. Li, J.-H. Chen, Y. Li and Z.-R. Li, J. Alloys Compd., 2018, 740, 400–405 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08758c |
| This journal is © The Royal Society of Chemistry 2019 |