
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-tuned magnetic exchange interactions in Dy2 systems ligated by a μ-phenolato heptadentate Schiff base†
Zhijie Jiangad,
Lin Sunac,
Min Lia,
Haipeng Wua,
Zhengqiang Xia *a,
Hongshan Kea,
Yiquan Zhang*b,
Gang Xiea and
Sanping Chen*a
*a,
Hongshan Kea,
Yiquan Zhang*b,
Gang Xiea and
Sanping Chen*a
aKey Laboratory of Synthetic and Natural Functional Molecule Chemistry of Ministry of Education, College of Chemistry and Materials Science, Northwest University, Xi'an, Shaanxi 710069, China. E-mail: northwindy@126.com; sanpingchen@126.com
bJiangsu Key Laboratory for NSLSCS, School of Physical Science and Technology, Nanjing Normal University, Nanjing 210023, China. E-mail: zhangyiquan@njnu.edu.cn
cCollege of Chemistry and Chemical Engineering, Henan University, Kaifeng 475001, China
dSchool of Chemistry & Chemical Engineering, Shaanxi Xueqian Normal University, Xi'an 710100, China
First published on 2nd December 2019
Abstract
A series of binuclear dysprosium compounds, namely, [Dy(api)]2 (1), [Dy(api)]2·2CH2Cl2 (2), [Dy(Clapi)]2·2C4H8O (3), and [Dy(Clapi)]2·2C3H6O (4) (H3api = 2-(2-hydroxyphenyl)-1,3-bis[4-(2-hydroxyphenyl)-3-azabut-3-enyl]-1,3-imidazoline; H3Clapi = 2-(2′-hydroxy-5′-chlorophenyl)-1,3-bis[3′-aza-4′-(2′′-hydroxy-5′′-chlorophenyl)prop-4′-en-1′-yl]-1,3-imidazolidine), have been isolated by the reactions of salen-type ligands H3api/H3Clapi with DyCl3·6H2O in different solvent systems. Structural analysis reveals that each salen-type ligand provides a heptadentate coordination pocket (N4O3) to encapsulate a DyIII ion and all of the DyIII centers in 1–4 adopt a distorted square antiprism geometry with D4d symmetry. Magnetic studies showed that compound 1 did not exhibit single-molecule magnetic (SMMs) behavior. With the introduction of different lattice solvents, compounds 2–4 showed filed-induced slow magnetic relaxation with barriers Ueff of 18.2 K (2), 28.0 K (3) and 16.4 K (4), respectively. Ab initio calculations were employed to interpret the magnetization behavior of 1–4. The combination of experimental and theoretical data reveal the importance of the weak exchange interaction between the DyIII ions in the observation of slow magnetic relaxation, and a relaxation mechanism has been developed to rationalize the observed difference in the Ueff values. The different lattice solvents influence Dy–O–Dy bond angles and thus alter the torsion of the square antiprism geometry, consequently resulting in distinct magnetic interactions and the magnetic behavior.
Introduction
Thanks the tremendous optical1 and magnetic properties of lanthanide ions,2 the design and study of original lanthanide-based coordination compounds have drawn wide attention in materials chemistry. The slow relaxation of the magnetization in lanthanide-based single-molecule magnets (SMMs) makes them promising and fascinating candidates for molecular magnetism researchers due to their strong spin-orbital coupling effect and large magnetic anisotropy for constructing SMMs with a higher relaxation energy barrier and blocking temperature.3–5 Such unique magnetic properties connecting the conventional and quantum world make SMMs good candidates for high-density information storage, quantum information processing and molecular spintronics.6 Recent rapid advances in this field have led to the design of lanthanide compounds exhibiting very high anisotropic barriers that can reach several thousand wavenumbers,7–11 as well as magnetic hysteresis up to 80 K,10,12 making such compounds potential applicants for integration into magnetic memory devices.Certainly, these forefront advances are concentrated on a single spin center. There are still little investigated on another important factors of molecular magnetism, namely, magnetic exchange interactions and magneto-structural correlations.13–18 There are mainly the following reasons for the lack of research in this area: the first reason is the deep-seated nature of the 4f orbitals and the associated very weak exchange interactions. The second reason is that it is difficult to understand even the nature of exchange interactions, because the profile of the product of susceptibility and temperature versus temperature is dominated by the variation in the population of the Stark levels. The last reason is that ligand changes have no significant effect on the expected magnetic exchange interaction of closely analogues. For the above reasons, little research has been done on the understanding of exchange interactions in closely related lanthanide compounds.
Compared to mononuclear lanthanide compounds known as single-ion magnets (SIMs), dilanthanide species are viewed as the simplest molecular units to conveniently study the nature and strength of magnetic exchange interactions between spin carriers and then elucidate magnetic relaxation mechanisms.19–21 In recent years, different kinds of ligands including phthalocyanine,22 Schiff base,23 β-diketone,24 carboxylic acid25 and their derivatives have been successfully utilized to construct binuclear lanthanide compounds. Among them, Schiff base ligands with versatile O, N-based multi-chelating sites to impart different ligand fields and bridges have invoked increasing interest in the study of the magnetic anisotropy and exchange interactions of binuclear systems. Based on above all, two heptadentate Schiff base ligands has been elected to prepared four closely related Dy2 compounds, with the formula [Dy(Xapi)]2·2Y, (X = H (1, 2), Cl (3, 4); Y = no solvent (1), CH2Cl2 (2), C4H8O (3), C3H6O (4); H3api = 2-(2-hydroxyphenyl)-1,3-bis[4-(2-hydroxyphenyl)-3-azabut-3-enyl]-1,3-imidazoline; H3Clapi = 2-(2′-hydroxy-5′-chlorophenyl)-1,3-bis[3′-aza-4′-(2′′-hydroxy-5′′-chlorophenyl)prop-4′-en-1′-yl]-1,3-imidazolidine). Magnetic properties measurements indicate that the slight changes of coordination geometry around the DyIII induced by the guest solvent molecules produce different coupling interactions and distinct magnetic behaviour. Ab initio calculations are employed to interpret the magnetic anisotropy and exchange interactions of the compounds and the results give a more thorough understanding of the effects of structural factors on the relaxation dynamics in Dy2III SMMs.
Experimental
Materials and methods
All commercial reagents and solvents were purchased from Aldrich, Adamas and TCI and were used without further purification. 1H-NMR spectra were recorded on a Bruker AV-400 or AV-100 spectrometer. Chemical shifts (δ) reported in parts per million (ppm) are referenced relative to the residual solvent peak in the NMR solvent (CDCl3: δ 7.26 (CHCl3)). Data are represented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), integration, and coupling constants in Hertz (Hz). The phase purity of the bulk samples was confirmed by powder X-ray diffraction (PXRD) measurements executed on a Rigaku RU200 diffractometer at 60 kV, 300 mA, and Cu Kα radiation (l = 1.5406 Å), with a scan speed of 51 min−1 and a step size of 0.02° in 2θ. Magnetic measurements were performed in the temperature range of 2.0–300 K under 0 Oe, using a Quantum Design MPMS-XL-7 SQUID magnetometer on polycrystalline samples. The diamagnetic corrections for the complexes were estimated using Pascal's constants. Alternating current (ac) susceptibility experiments were performed using an oscillating ac field of 0 Oe at ac frequencies ranging from 1.0 to 1000 Hz. The magnetization was measured in the field range of 0–70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Oe.
000 Oe.
Synthetic procedures
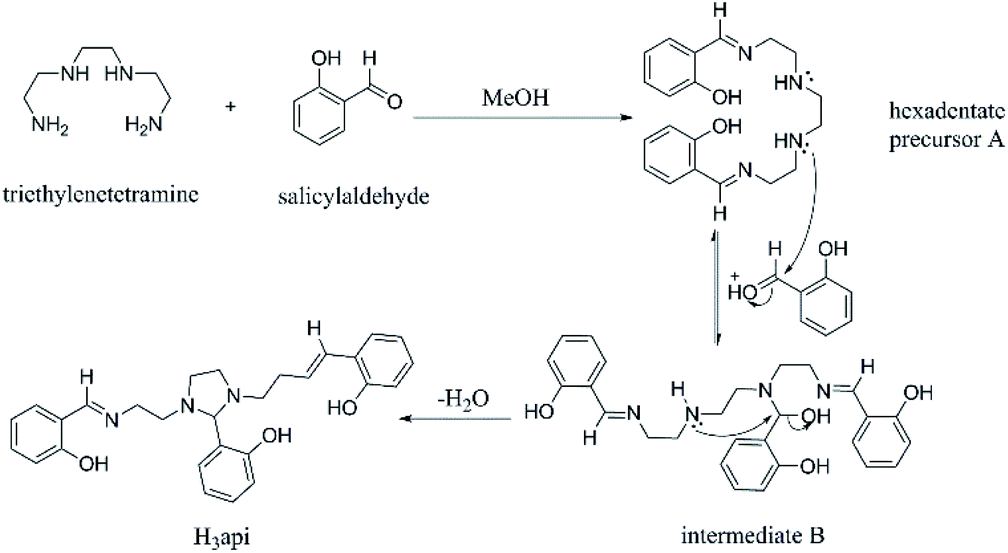 | ||
| Scheme 1 Probable mechanism for the formation of the ligand H3api. | ||
1H NMR (400 MHz, CDCl3) δ 13.21 (br s, 2H, OH18), 10.66 (br s, 1H, OH19), 8.25 (s, 2H, H5), 7.31–7.29 (m, 2H, H9), 7.25 (d, J = 6.9 Hz, 2H, H7), 7.22 (m, 2H, H13 & H15), 7.01 (m, 1H, H14), 6.93 (d, J = 8.3 Hz, 2H, H10), 6.86 (t, J = 7.4 Hz, 2H, H8), 6.82–6.79 (m, 1H, H16), 3.84 (s, 1H, H4), 3.60 (t, J = 6.5 Hz, 4H, H3), 3.43 (q, J = 4.7 Hz, 2H, Heq.1), 2.97 (dt, J = 12.7, 6.4 Hz, 2H, Heq.2), 2.73–2.63 (m, 4H, Hax1 & Hax2)(Fig. S1†).
13C {1H} NMR (101 MHz, CDCl3) δ 166.2 (C5), 161.2 (C11), 158.4 (C17), 132.4 (C9), 131.5 (C7), 131.0 (C13), 130.4 (C15), 121.0 (C6), 118.86 (C14), 118.85 (C12), 118.7 (C8), 117.13 (C10), 117.09 (C16), 89.8 (C4), 58.6 (C3), 53.0 (C2), 51.3 (C1) (Fig. S2†).
1H NMR (400 MHz, CDCl3) δ 13.07 (br s, 2H, OH18), 10.43 (br s, 1H, OH19), 8.17 (s, 2H, H5), 7.23 (dd, J = 8.8, 2.4 Hz, 2H, H9), 7.18 (d, J = 2.4 Hz, 2H, H7), 7.15 (d, J = 2.4 Hz, 1H, H15), 6.96 (d, J = 2.4 Hz, 1H, H13), 6.87 (d, J = 8.8 Hz, 2H, H10), 6.68 (d, J = 8.7 Hz, 1H, H16), 3.79 (s, 1H, H4), 3.59 (t, J = 6.1 Hz, 4H, H3), 3.44–3.40 (m, 2H, Heq.1), 2.99–2.92 (m, 2H, Heq.2), 2.73–2.64 (m, 4H, Hax1 & Hax2) (Fig. S3†).
13C {1H} NMR (101 MHz, CDCl3) δ 165.2 (C5), 159.8 (C11), 156.9 (C17), 132.3 (C9), 130.7 (C7), 130.34 (C13), 130.28 (C15), 123.4 (C14), 123.3 (C8), 122.5 (C6), 119.6 (C12), 118.7 (C10), 118.6 (C16), 89.0 (C4), 58.3 (C3), 52.8 (C2), 51.2 (C1) (Fig. S4†).
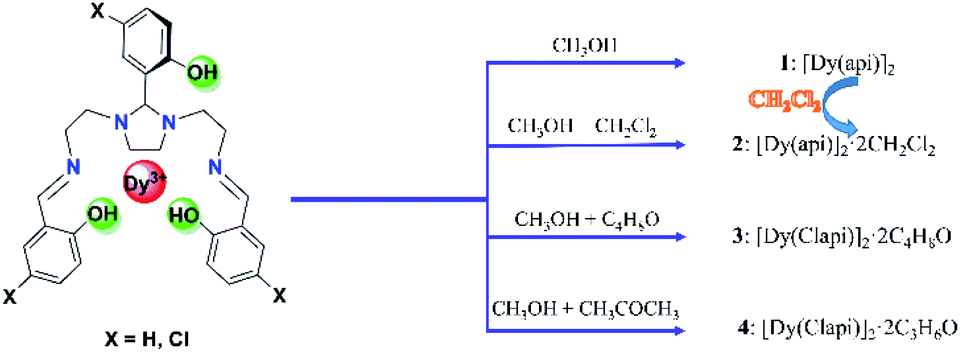 | ||
| Scheme 2 Syntheses of Dy2 compounds 1–4. | ||
(i) The synthesis method of compound 2 is similar to that of compound 1, except that it is replaced with the mixed methanol (5 ml) and dichloromethane (10 ml). Pale-yellow blocky crystals were gathered after three days in a yield of 28% (based on DyIII salts). Elem. anal. calcd: C, 47.84; H, 4.16; N, 7.97%. Found: C 47.79; H 4.15; N 7.95%.
(ii) Single crystals of compound 1 (1.0 mmol, 0.277 g) were soaked into dichloromethane solution (30 ml), which also yielded pale-yellow blocky crystals of compound 2 within one week at room temperature (Scheme 2).
X-ray crystallography
The crystal data for all compounds in our system have been collected on a Bruker SMART APEX-CCD-based diffractometer (Mo Kα radiation, λ = 0.710 73 Å) at low temperature. Raw area detector data integration and reduction were performed with SAINT+ program. Absorption correction based on multiscan was processed using the SADABS software.26 All structures were solved by the direct methods and refined by a full-matrix least-squares method on F2 using the SHELXL-2014 software.27 Non-hydrogen atoms were refined with anisotropic thermal parameters. The experimental details of crystal data, data collection parameters, and refinement statistics are presented in Table S1,† and the selected bond lengths and angles are summarized in Table S2.†Computational methodology
Theoretical studies using CASSCF calculations on the DyIII nodes in four compounds based on the single-crystal diffraction measured geometries were performed by MOLCAS 8.0 software package.28,29 For the calculations, the basis sets are atomic natural orbitals from the MOLCAS ANO-RCC library: ANO-RCCVTZP for DyIII cation; VTZ for close N atom and O atom; VDZ for distant atoms. The calculations used the second-order Douglas–Kroll–Hess Hamiltonian, in which scalar relativistic contractions were adopted in the basis sets. The spin–orbit coupling was treated individually in the RASSI process. In the CASSCF calculations, the active electrons in seven active spaces contain all f electrons CAS (9 in 7) for all compounds. All the roots were computed in the active space to eliminate possible doubts. The maximal value of spin-free state has been mixed, which might be related with the computer hardware (all from 21 sextets, 128 from 224 quadruplets and 130 from 490 doublets for DyIII nodes).Results and discussion
Synthesis and characterization of ligand H3api
The ligand H3api is prepared from the condensation of triethylenetetramine and salicylaldehyde in 1:3 molar ratio in methanol and characterized by 1H and 13C NMR spectroscopic techniques. It is noted that in these Schiff bases ligand, five-membered imidazolidine rings were formed at the backbone after the condensation and that the middle arm was, therefore, unique from the other two arms (clearly shown in the 1H NMR spectra). Two phenol hydroxyl resonances were observed at about 10 and 13 ppm in a 1:2 ratio. These two downfield signals can be explained by the intramolecular hydrogen bonding of the phenol hydroxyl with unsaturated azomethine nitrogen or tertiary nitrogen which caused decrease in the shielding at the hydroxy hydrogen. However, OH resonances on the terminal outside arms are further downfield than that of the middle arm. The explanation is that, for the terminal arm, the O–H⋯N group lies coplanar with the aromatic ring; hence, the proton is further deshielded by the induced aromatic ring current.
A plausible mechanism was proposed for generating ligand H3api (Scheme 1). First, the reaction between triethylenetetramine and salicylaldehyde, thus producing the precursor A. The nucleophilic addition of A to another salicylaldehyde gives the intermediate B, which can be converted into the imidazolidine ring compound H3api through the internal SN2 reaction.
Preparation and structural description of 1–4
Incorporating the ligand H3api/H3Clapi with DyCl3·6H2O in different solvents containing an excess of triethylamine brings about four binuclear DyIII species share the formula [DyXapi]2·solvent (Scheme 2): [Dyapi]2 (1), [Dy(api)]2·2CH2Cl2 (2), [Dy(Clapi)]2·2C4H8O (3) and [Dy(Clapi)]2·2C3H6O (4), respectively. Because each ligand carries a triple negative charge, the positive charge of the two trivalent DyIII is counterbalanced and the binuclear compounds are neutral; hence no counterions are necessary and these symmetric Dy2 compounds are structurally similar. Instead of forcing N4O3 donor atoms exclusively from one ligand onto one metal ion, here the homodinuclear compounds contain two DyIII ions where each of the metal ions is coordinated by two N2O donor sets, each coming from one ligand. Two phenolate oxygen atoms, from the middle arm of each ligand, act as μ-bridges between the two metal centres and complete the eight-membered coordination sphere, overall furnishing a sandwich type dimeric structure. This no doubt arises from the rigid five-membered imidazolidine ring in the ligand backbone; this forces the ligand into a more open configuration rendering impossible coordination of all the donor atoms of one ligand to one metal ion. Instead, these two ligands cooperate with each other to form two compartments to accommodate two DyIII (Fig. 1a).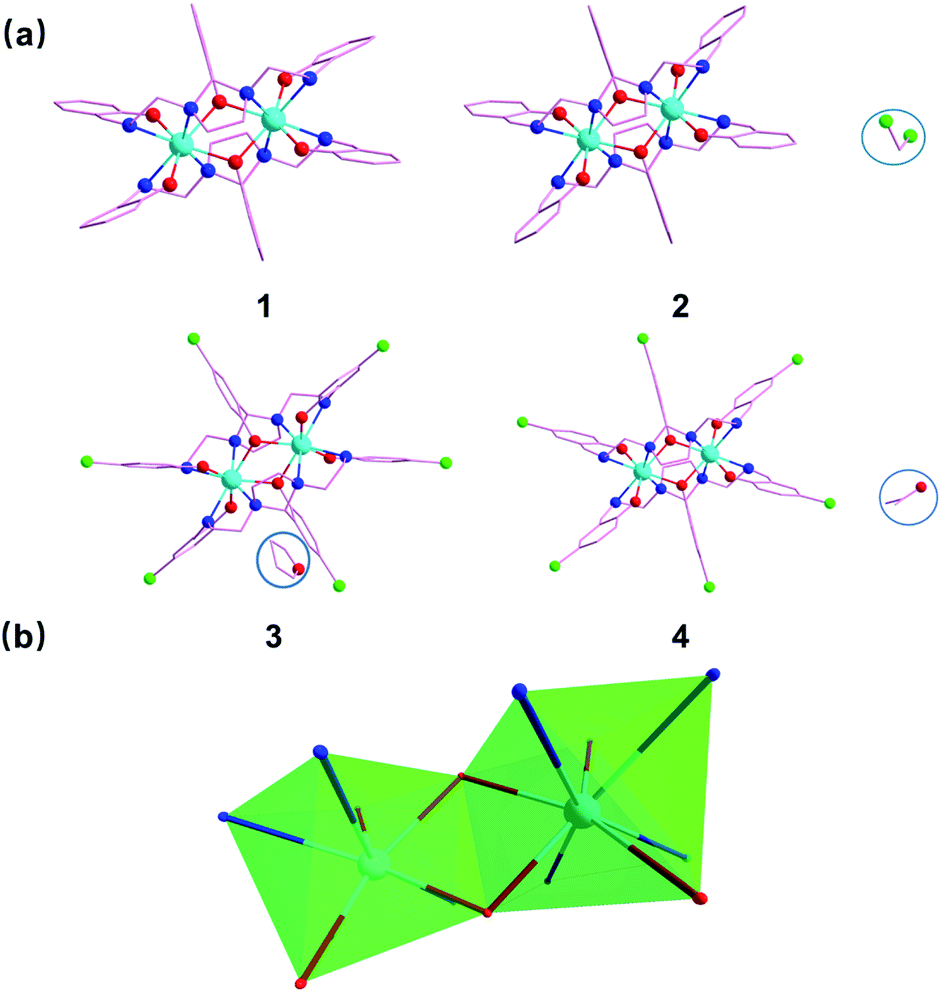 | ||
| Fig. 1 (a) X-ray crystal structure of compounds of 1–4. (b) Local coordination polyhedron of the DyIII ions in 1–4. Color code: cyan = DyIII; green = Cl; red = O; blue = N. Hydrogen atoms and carbon atom are omitted for clarity. | ||
The distortions from the ideal eight-coordinate geometries for the {DyN4O4} core is computed using SHAPE 2.1 software30 and the least deviation has been found for the distorted square antiprism geometry, suggesting the presence of pseudo-D4d symmetry (Fig. 1b) for the core geometry but with different distortions from the ideal geometry (Table S3†). The Dy–N (imidazolidine) bond lengths are approximately 0.3 Å larger than the Dy–N (imine) bond lengths in all the cases (Table S2†).
The unit cell of compound 2 contains binuclear units with two dichloromethane molecules in the crystal lattice for each dimer, and each compound 3 and 4 has two solvent molecules tetrahydrofuran and acetone. But no free solvent molecules in compound 1. Because of the uncoordinated solvent molecules and, most importantly, the different coordination geometry, the supramolecular interactions are different in the structures. The 3D Hirshfeld surface analysis and 2D fingerprint plot of 1–4 were applied to analyze the intermolecular interactions. As illustrated in Fig. S7–S10,† the major contribution (73.6% for 1, 58.5% for 2, 50.1% for 3 and 41.7% for 4) is provided by weak van der Waals H⋯H interactions, while C–H⋯Cl hydrogen-bond interactions provide partial contributions of 18.5%, 24.2% and 28.8% for 2–4, respectively.
Magnetic studies
For all samples, the variable-temperature dc susceptibility measurements were performed at an applied field of 1000 Oe from 2 to 300 K (Fig. 2); the variable-field magnetization measurements were performed at 2 K, 3 K and 5 K from 0 to 7 T (Fig. S11†). In summary, the general behaviour of all compounds is consistent with other DyIII compounds reported in the literature.31–33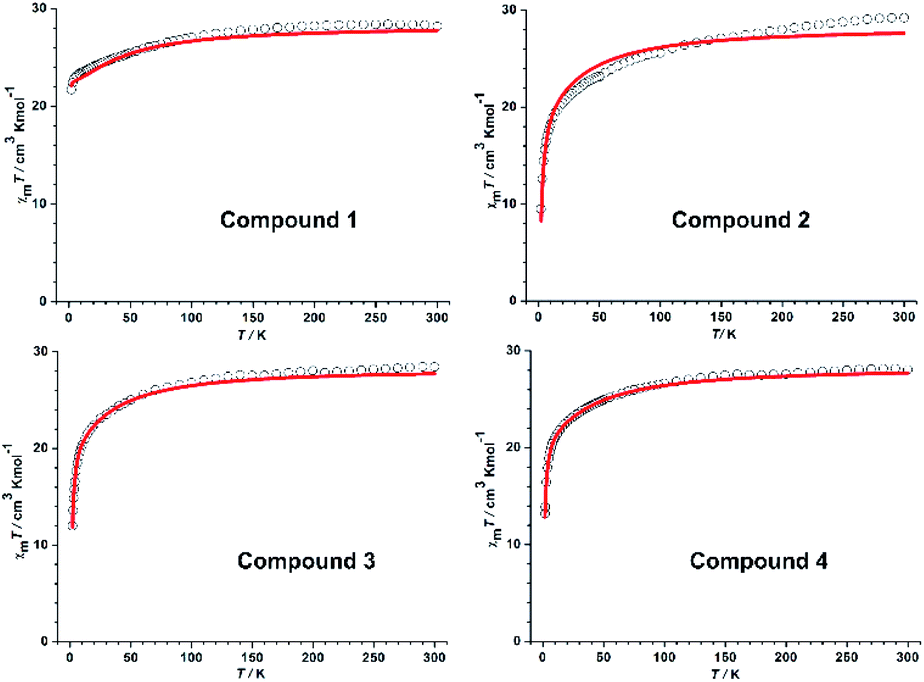 | ||
| Fig. 2 χmT vs. T plots for 1–4 (red solid lines represent the simulation from ab initio calculation). | ||
Direct-current (dc) magnetic susceptibilities
The room temperature χMT value for all compounds (see Fig. 2) is in good agreement with the expected value for two noninteracting DyIII ions (7H15/2, S = 5/2, L = 5, g = 4/3, C = 14.17 cm3 K mol−1).34 As the temperature decreases, χMT is almost constant down to ∼50 K and then slightly decreases to reach a minimum at 2 K, which could be ascribed to an intramolecular antiferromagnetic exchange coupling between the two DyIII ions. The M = f(μ0H) curve increases with the applied magnetic field and reaches saturation at 7 T. It is possible to fit the susceptibility and the magnetization data (see Fig. S11†) using a model based on the spin Hamiltonian H = −JSDy1 × SDy2 + gβHzSz, where g is the Lande factor and J the interaction parameter between the local S = 15/2 spins of the DyIII ions.Ac susceptibility measurements
In order to probe the magnetic dynamic behaviour of these compounds, the ac susceptibilities at various frequencies and temperatures measurements were performed. Under zero applied dc field, both the in-phase (χ′) and out-of-phase (χ′′) susceptibilities did not display a frequency and temperature dependence for all samples in our system. Lanthanide ions are well known to exhibit significant quantum tunnelling of the magnetization (QTM), but in the case of DyIII ions, the QTM is reduced as a result of a spin-parity effect. The spin-parity effect predicts that the QTM should be suppressed at zero field when the total spin of the magnetic system is a half-integer but allowed in integer-spin systems. However, QTM can be induced experimentally even in half-integer spin systems through effects such as environmental degrees of freedom as well as hyperfine and dipolar coupling via transverse field components.35 In order to suppress or minimize the QTM, optimum dc field measurements were carried out at optimum fields of 1500 Oe for 2, 700 Oe for 3 and 500 Oe for 4, respectively (Fig. 3). Subsequent ac measurements were carried out at these optimum fields, both χ′ and χ′′ susceptibilities show significant frequency dependence peaks at a relatively high temperature range, which clearly indicates that the SMMs behaviour of compounds 2, 3 and 4 (Fig. S12–S14†). In contrast, compound 1 display no out-of-phase component of the ac susceptibilities in the applied field for frequencies of 1 and 1000 Hz down to 2 K (Fig. S12 and S13†). Really, in such measurements the degeneracy of the ±MS energy levels can be removed, consequently preventing the tunnelling of electrons from the +MS state to the −MS state through the spin-reversal barrier. The frequency magnitude of the first maximum peaks for all compounds reflect the impact of QTM on the SMMs, where the lower one mostly indicates the existence of a slower QTM process where the thermally activated Orbach relaxation process occurs prominently.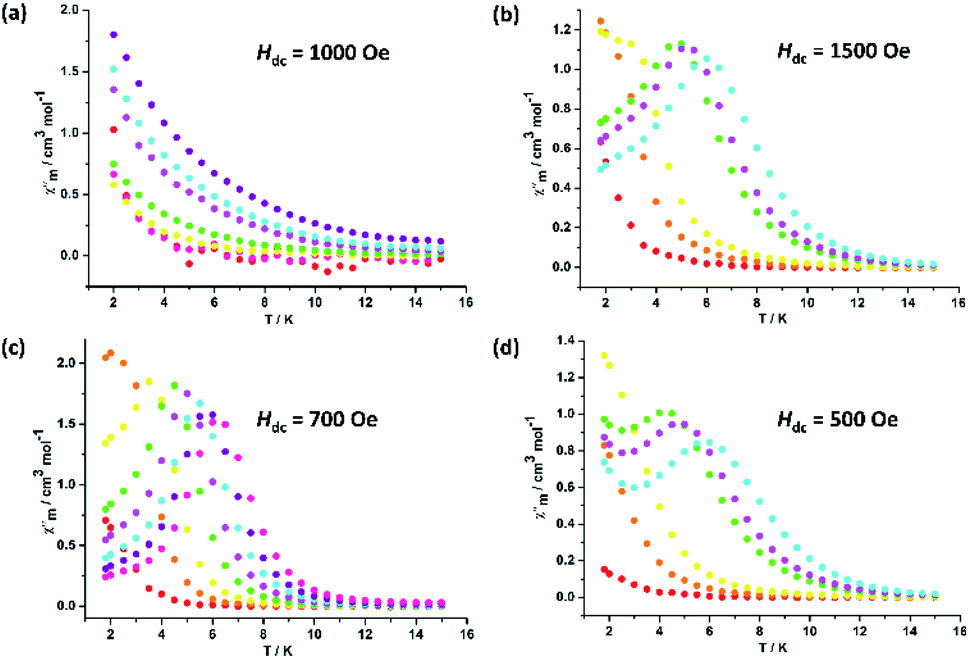 | ||
| Fig. 3 Temperature dependence of the out-of-phase ac susceptibility signals under an applied dc field for 1 (a), 2 (b), 3 (c) and 4 (d). | ||
The Cole–Cole plots36 for all samples are shown in Fig. 4 and exhibit semicircular shape which can be fitted to the generalized Debye model. The fitting parameters τ (relaxation time) and α (α determines the width of the distribution of relaxation times) are given in Table S4.† The α values are <0.25 in the higher temperature regions for 2 and 4, indicating the presence of a relatively narrow width of relaxation processes most likely due to a combination of QTM and thermally assisted relaxation pathways. The presence of multiple relaxation processes is possible, as reported in other literatures.37–39 For compound 3, the α values are <0.05 which indicates a single relaxation mechanism. The thermally induced relaxation can be fit using the Arrhenius law (τ = τ0exp(Ueff/kT)) yielding effective energy barriers and extrapolated relaxation times were Ueff = 18.19 K, τ0 = 10.9 × 10−6 (2) under 1500 dc field, 27.99 K, τ0 = 2.92 × 10−6 (3) under 700 dc field and 16.35 K, τ0 = 12.6 × 10−6 (4) under 500 dc field (Fig. 4). These relaxation barriers are comparable to those of other reported similar Dy2 SMMs,40 suggesting that the slow relaxation behavior of 2–4 originates primarily from the presence of anisotropic DyIII ions. The magnetic coupling between lanthanide ions as a secondary consideration may also mediate the magnetic relaxation of Dy2 compounds.
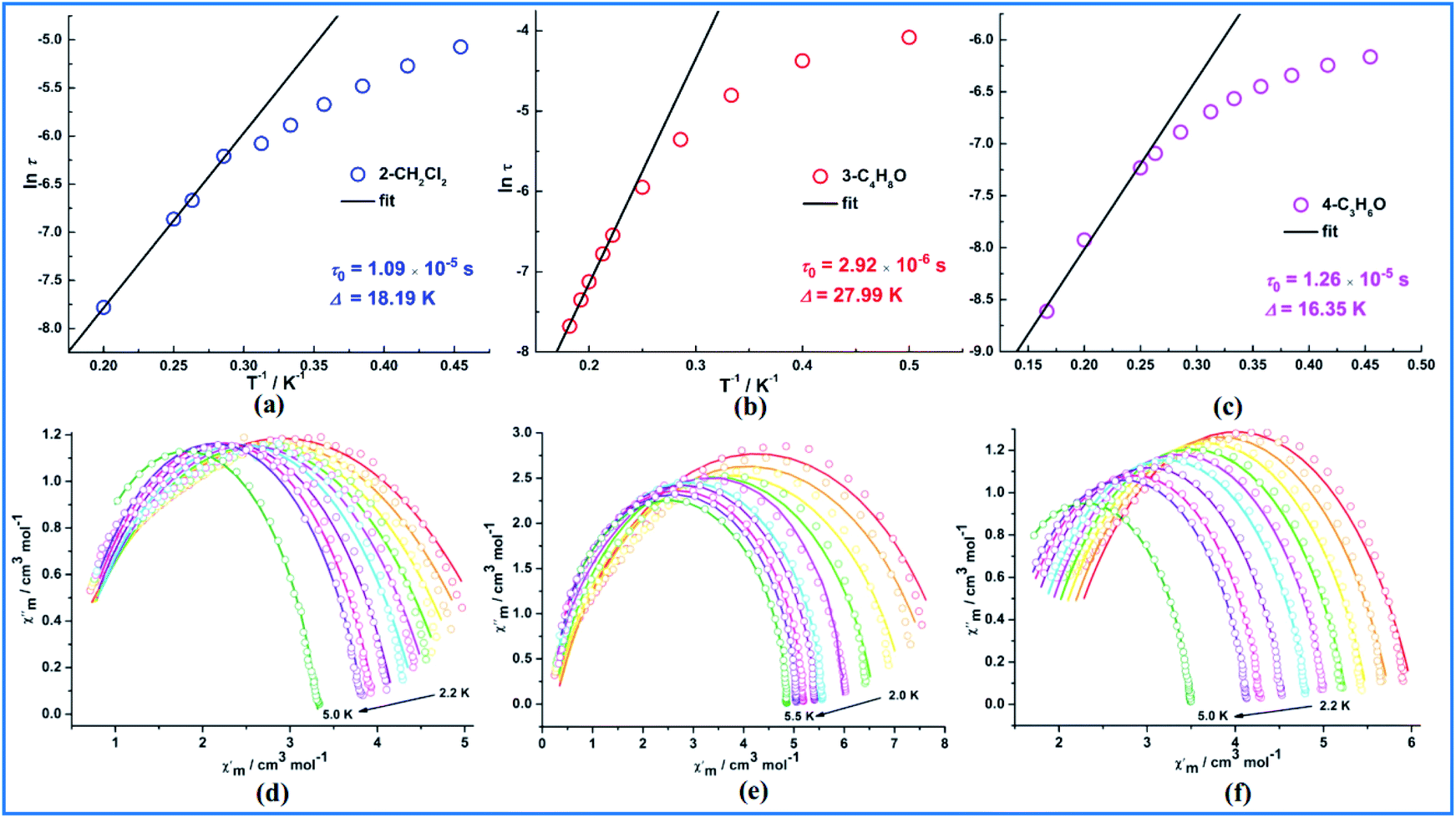 | ||
| Fig. 4 The Arrhenius plots constructed from the ac magnetization relaxation dynamics under an indicated dc field for 2 (a), 3 (b) and 4 (c). The solid lines represent the fitting with the Arrhenius law. Cole–Cole diagram under 1500 Oe dc field for 2 (d), under 700 Oe dc field for 3 (e) and under 500 Oe dc field for 4 (f). The solid lines represent the best fit to the measured results. | ||
Structure–property relationship
Although compounds 1–4 show a very similar crystal structure, they exhibit different magnetic relaxation behaviours. To probe the structure–property relationship in all samples, some crucial parameters of structure and property, including Dy–O–Dy angles in Dy2O2 core, the CShM of D4d, and Ueff have been presented in Fig. S15.† These distinctive magnetic behaviours must be caused by subtle but crucial differences among the respective structures. According to the single crystal X-ray diffraction study and the semiquantitative method of polytopal analysis, for compounds 1–4, the most reasonable geometry around the Dy atom is D4d. However, on a closer comparison of the four Dy2 structures, the Dy–O–Dy angles are found to be slightly different. The Dy–O–Dy angle of compound 3 is 110.509°, which is a bit smaller than other three (1, 111.027; 2, 110.596; 4, 110.626, respectively). Whereas 1 shows the largest Dy–O–Dy angle. These slight differences of important bond angles may have an influence on the distorted degree of the coordination geometry of the DyIII ion, which can lead to the fast quantum tunnelling arising introduced by a geometrical distortion of the coordinated sphere. These differences can also have a significant influence on the magnetic interaction between the DyIII ions. Additionally, considering that the single-ion anisotropy contributions from the two DyIII centers in the bimetallic units are symmetry related, the CSM method was employed to evaluate the deviation from an ideal square antiprism geometry geometry. Herein, the larger the calculated CSAPR-8 parameter, the greater the deviation from an ideal D4d symmetry. The calculated CSAPR-8 parameters are 1.423, 1.318, 1.268 and 1.389 for compounds 1–4, respectively, indicating the largest deviation from the ideal symmetry for 1. It is evident that the introduction of guest molecules in 2, 3 and 4 induces decreased Dy–O–Dy angles and enhanced symmetry, and thus triggering the SMM behaviour for the compounds. The improvement of SMM property of Dy2 complex by introducing free molecule has be observed in some reported systems.19b,41 However, there are no clear electronic effects on the magnetic relaxation behaviors between 1–2 and 3–4 induced by the different substituents (–H or –Cl) of terminal ligands.Further insights into the electronic structure and magnetic blocking in the present dysprosium complexes were obtained via ab initio calculations. Ab initio CASSCF/RASSI/SINGLE_ANISO calculations have been carried out with MOLCAS 8.0 (ref. 28 and 29) on individual DyIII fragments of compounds 1–4 on the basis of X-ray determined structures (see the ESI† for details). Each Dy centre was calculated, keeping the experimentally determined structure of the corresponding complex while replacing the neighbouring DyIII ion with diamagnetic Lu. The lowest Kramers doublets and the g tensors corresponding to the pseudospin S = 1/2 of DyIII ions for all compounds are shown in Table S5.† Herein, the calculated gz values of the DyIII fragments of 1–4 are close to 20, which shows that the DyIII–DyIII exchange interactions for each of them can be approximately regarded as the Ising type.
The magnetic susceptibilities of compounds 1–4 were simulated with the program POLY_ANISO42 using the exchange parameters from Table 1. All parameters were calculated with respect to the pseudospin S = 1/2 of the Dy ions. From Table 1, the slight differences of the Jdipolar values are found in complexes 1–4, and the Jexch is ferromagnetic for 1 whereas antiferromagnetic for 2–4, the total interaction J in 2–4 being around 10 times larger than that in 1. The DyIII–DyIII interactions J of all compounds within the Lines model43,44 are antiferromagnetic. The distinct magnetic interactions observed for complexes 1 and 2–4 are most likely induced by different Dy–O–Dy angles. And the total coupling parameters J (dipolar and exchange) were included to fit the magnetic susceptibilities. As shown in Fig. 2, a great agreement was reached between the calculated and experimental χMT versus T plots of 1–4. The main magnetic axes on the DyIII ions for 1–4 are indicated in Fig. S17,† where the magnetic axes on DyIII for each compound are antiparallel to each other because of the DyIII–DyIII antiferromagnetic interaction.
| Compound | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Jdipolar | −2.56 | −2.51 | −2.47 | −2.69 |
| Jexch | 2.25 | −1.25 | −0.50 | −0.50 |
| J | −0.31 | −3.76 | −2.97 | −3.19 |
The foregoing results clearly reveal the origin of the magnetization dynamics: (a) the similar energy barriers are mainly from the single-ion anisotropy of the DyIII centres because of the similar coordination environments; (b) the subtle differences caused by either the guest molecules or no guest molecule lead to different magnetic interactions and symmetry which tune the relaxation rate to give different effective relaxation barriers. The weaker magnetic interactions and lower symmetry of the coordination geometry for 1 most likely associate with the poorer SMMs behavior than 2–4. This is a typical example in which guest molecules are employed to adjust the molecule symmetry as well as the magnetic interactions between lanthanides and thereby tune the relaxation rates arising from incoherent quantum tunnelling of the magnetization.
Conclusions
A series of Dy2 SMMs were prepared as well as characterized structurally and magnetically. The structural analyses demonstrated that we successfully prevented the coordination of solvent molecules to DyIII ions utilizing the larger steric hindrance of Schiff base ligand api3−, and gave rise to the D4d analogues with enhanced local symmetry and enhanced Ueff. The different anisotropy of individual DyIII ions and intramolecular couplings are most likely derived from the different local symmetry, which ultimately leads to different SMM behaviors. The work verifies the sensitivity of magnetic relaxation to the remote alteration on individual spin centers, which provides an opportunity to shed light on tuning of the magnetic properties of SMMs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (Grant No. 21727805, 21673180, 21703135, 21803042, 21973074 and 21973046), the Natural Science Basic Research Program of Shaanxi (Grant No. 2017JZ002, 2018JM5180, 2019JQ-067, 2019JM-079) and the 64th China Postdoctoral Science Foundation Funded Project (2018M643706).Notes and references
- K. Binnemans, Coord. Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS PubMed
.
- J. Luzon and R. Sessoli, Dalton Trans., 2012, 41, 13556–13567 RSC
-
(a) N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed
-
(a) Y. C. Chen, J. L. Liu, L. Ungur, J. Liu, Q. W. Li, L. F. Wang, Z. P. Ni, L. F. Chibotaru, X. M. Chen and M. L. Tong, J. Am. Chem. Soc., 2016, 138, 2829–2837 CrossRef CAS PubMed
-
(a) L. Norel, L. E. Darago, B. L. Guennic, K. Chakarawet, M. I. Gonzalez, J. H. Olshansky, S. Rigaut and J. R. Long, Angew. Chem., Int. Ed., 2018, 57, 1933–1938 CrossRef CAS PubMed
-
(a) R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS
- J. Liu, Y.-C. Chen, J.-L. Liu, V. Vieru, L. Ungur, J.-H. Jia, L. F. Chibotaru, Y. Lan, W. Wernsdorfer, S. Gao, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2016, 138, 5441–5450 CrossRef CAS PubMed
- S. K. Gupta, T. Rajeshkumar, G. Rajaraman and R. Murugavel, Chem. Sci., 2016, 7, 5181–5191 RSC
- L. Ungur and L. F. Chibotaru, Inorg. Chem., 2016, 55, 10043–10056 CrossRef CAS PubMed
- F. S. Guo, B. M. Day, Y. C. Chen, M. L. Tong, A. Mansikkamaki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445–11449 CrossRef CAS PubMed
- Y.-S. Ding, N. F. Chilton, R. E. P. Winpenny and Y.-Z. Zheng, Angew. Chem., Int. Ed., 2016, 55, 16071–16074 CrossRef CAS PubMed
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400–1403 CrossRef CAS PubMed
- O. Kahn, Molecular Magnetism, VCH Publications, New York, 1993 Search PubMed
-
(a) S. Hazra, S. Bhattacharya, M. K. Singh, L. Carrella, E. Rentschler, T. Weyhermueller, G. Rajaraman and S. Mohanta, Inorg. Chem., 2013, 52, 12881–12892 CrossRef CAS PubMed
- E. Ruiz, J. Cano, S. Alvarez and P. Alemany, J. Am. Chem. Soc., 1998, 120, 11122–11129 CrossRef CAS
- J.-P. Costes, F. Dahan and A. Dupuis, Inorg. Chem., 2000, 39, 165–168 CrossRef CAS PubMed
-
(a) L. E. Roy and T. Hughbanks, J. Am. Chem. Soc., 2006, 128, 568–575 CrossRef CAS PubMed
- K. Zhang, D. Liu, V. Vieru, L. Hou, B. Cui, F.-S. Guo, L. F. Chibotaru and Y.-Y. Wang, Dalton Trans., 2017, 46, 638–642 RSC
-
(a) O. Sun, P. Chen, H.-F. Li, T. Gao, W.-B. Sun, G.-M. Li and P.-F. Yan, CrystEngComm, 2016, 18, 4627–4635 RSC
-
(a) K. Katoh, R. Asano, A. Miura, Y. Horii, T. Morita, B. K. Breedlove and M. Yamashita, Dalton Trans., 2014, 43, 7716–7725 RSC
-
(a) K. L. M. Harriman, J. J. Le Roy, L. Ungur, R. J. Holmberg, I. Korobkov and M. Murugesu, Chem. Sci., 2017, 8, 231–240 RSC
-
(a) N. Ishikawa, M. Sugita, N. Tanaka, T. Ishikawa, S. Koshihara and Y. Kaizu, Inorg. Chem., 2004, 43, 5498–5500 CrossRef CAS PubMed
-
(a) P. H. Lin, T. J. Burchell, R. Clerac and M. Murugesu, Angew. Chem., Int. Ed., 2008, 47, 8848–8851 CrossRef CAS PubMed
-
(a) G. J. Chen, Y. N. Guo, J. L. Tian, J. K. Tang, W. Gu, X. Liu, S. P. Yan, P. Cheng and D. Z. Liao, Chem.–Eur. J., 2012, 18, 2484–2487 CrossRef CAS PubMed
-
(a) Z. L. Wu, J. Dong, W. Y. Ni, B. W. Zhang, J. Z. Cui and B. Zhao, Dalton Trans., 2014, 43, 16838–16845 RSC
- G. M. Sheldrick, SADABS, Universität Göttingen, Germany, 2011 Search PubMed
-
(a) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
- F. Aquilante, L. De Vico, N. Ferre, G. Ghigo, P. A. Malmqvist, P. Neogrady, T. B. Pedersen, M. Pitonak, M. Reiher, B. O. Roos, L. Serrano-Andres, M. Urban, V. Veryazov and R. Lindh, J. Comput. Chem., 2010, 31, 224–247 CrossRef CAS PubMed
- F. Aquilante, J. Autschbach, R. K. Carlson, L. F. Chibotaru, M. G. Delcey, L. De Vico, I. Fdez Galvan, N. Ferre, L. M. Frutos, L. Gagliardi, M. Garavelli, A. Giussani, C. E. Hoyer, G. Li Manni, H. Lischka, D. Ma, P. A. Malmqvist, T. Muller, A. Nenov, M. Olivucci, T. B. Pedersen, D. Peng, F. Plasser, B. Pritchard, M. Reiher, I. Rivalta, I. Schapiro, J. Segarra-Marti, M. Stenrup, D. G. Truhlar, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, V. P. Vysotskiy, O. Weingart, F. Zapata and R. Lindh, J. Comput. Chem., 2016, 37, 506–541 CrossRef CAS PubMed
- B. O. Roos, R. Lindh, P. Malmqvist, V. Veryazov and P. Widmark, J. Phys. Chem. A, 2005, 109, 6575–6579 CrossRef CAS PubMed
- S. A. Sulway, R. A. Layfield, F. Tuna, W. Wernsdorfer and R. E. P. Winpenny, Chem. Commun., 2012, 48, 1508–1510 RSC
-
(a) J. Tang, I. Hewitt, N. T. Madhu, G. Chastanet, W. Wernsdorfer, C. E. Anson, C. Benelli, R. Sessoli and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 1729–1733 CrossRef CAS PubMed
- J. r. m. Long, F. Habib, P.-H. Lin, I. Korobkov, G. Enright, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 5319–5328 CrossRef CAS PubMed
-
(a) Y. Ma, G.-F. Xu, X. Yang, L.-C. Li, J. Tang, S.-P. Yan, P. Cheng and D.-Z. Liao, Chem. Commun., 2010, 46, 8264–8266 RSC
-
(a) W. Wernsdorfer, S. Bhaduri, C. Boskovic, G. Christou and D. N. Hendrickson, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 180403 CrossRef
-
(a) K. S. Cole and R. H. J. Cole, J. Chem. Phys., 1941, 9, 341–351 CrossRef CAS
- S. Titos-Padilla, J. Ruiz, J. M. Herrera, E. K. Brechin, W. Wersndorfer, F. Lloret and E. Colacio, Inorg. Chem., 2013, 52, 9620–9626 CrossRef CAS PubMed
- J. L. Liu, K. Yuan, J. D. Leng, L. Ungur, W. Wernsdorfer, F. S. Guo, L. F. Chibotaru and M. L. Tong, Inorg. Chem., 2012, 51, 8538–8544 CrossRef CAS PubMed
- E. Colacio, J. Ruiz, E. Ruiz, E. Cremades, J. Krzystek, S. Carretta, J. Cano, T. Guidi, W. Wernsdorfer and E. K. Brechin, Angew. Chem., Int. Ed., 2013, 52, 9130–9134 CrossRef CAS PubMed
-
(a) M. Nematirad, W. J. Gee, S. K. Langley, N. F. Chilton, B. Moubaraki, K. S. Murray and S. R. Batten, Dalton Trans., 2012, 41, 13711–13715 RSC
-
(a) W. B. Sun, B. Yan, L. H. Jia, B. W. Wang, Q. Zhang, X. Cheng, H. F. Li, P. Chen, Z. M. Wang and S. Gao, Dalton Trans., 2016, 45, 8790–8794 RSC
-
(a) L. F. Chibotaru, L. Ungur and A. Soncini, Angew. Chem., Int. Ed., 2008, 47, 4126–4129 CrossRef CAS PubMed
- M. E. Lines, J. Chem. Phys., 1971, 55, 2977–2984 CrossRef CAS
- K. C. Mondal, A. Sundt, Y. H. Lan, G. E. Kostakis, O. Waldmann, L. Ungur, L. F. Chibotaru, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2012, 51, 7550–7554 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic files in CIF format, experimental and computational details, crystallographic data, and additional structural and magnetic figures and tables. CCDC 1543462–1543464 and 1543466. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra08754k |
| This journal is © The Royal Society of Chemistry 2019 |