
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Poly(styrene-ran-cinnamic acid) (SCA), an approach to modified polystyrene with enhanced impact toughness, heat resistance and melt strength
Jie Wang,
Zixin Yu,
Peihua Li,
Dachuan Ding,
Xuan Zheng,
Chuanqun Hu,
Tao Hu,
Xinghou Gong,
Ying Chang and
Chonggang Wu*
and
Chonggang Wu*
Hubei Provincial Key Laboratory of Green Materials for Light Industry, Collaborative Innovation Centre of Green Light-weight Materials and Processing, School of Materials and Chemical Engineering, Hubei University of Technology, Wuhan, Hubei 430068, P. R. China. E-mail: cgwu@mail.hbut.edu.cn
First published on 2nd December 2019
Abstract
A poly(styrene-ran-cinnamic acid) (SCA) containing 6.8 mol% of CA, with a ![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) w (∼217
w (∼217![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) comparable to commercial polystyrene (PS), was successfully synthesised via emulsion free-radical copolymerisation as evidenced by 1744 and 1703 cm−1 infrared peak occurrences, respectively characteristic of free and dimeric carboxyl C
000) comparable to commercial polystyrene (PS), was successfully synthesised via emulsion free-radical copolymerisation as evidenced by 1744 and 1703 cm−1 infrared peak occurrences, respectively characteristic of free and dimeric carboxyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretches. Upon the interchain hydrogen bond cross-linking by CA, the impact toughness of the SCA was considerably improved by 47.2% against PS, the glass transition, heat deflection and Vicat softening temperatures were significantly enhanced until 117.0, 108.0 and 118.3 °C, respectively, compared with PS (95.2, 87.6 and 96.0 °C), while the extensional viscosities were near one order-of-magnitude higher than PS by which the temperature window required for appropriate melt-strengths would be greatly broadened. Meanwhile, the SCA displayed other properties basically analogous to PS. This work presents a modified PS, SCA, with enhanced toughness, heat resistance and melt strength that potentially extend its styrofoam and commodity applications.
O stretches. Upon the interchain hydrogen bond cross-linking by CA, the impact toughness of the SCA was considerably improved by 47.2% against PS, the glass transition, heat deflection and Vicat softening temperatures were significantly enhanced until 117.0, 108.0 and 118.3 °C, respectively, compared with PS (95.2, 87.6 and 96.0 °C), while the extensional viscosities were near one order-of-magnitude higher than PS by which the temperature window required for appropriate melt-strengths would be greatly broadened. Meanwhile, the SCA displayed other properties basically analogous to PS. This work presents a modified PS, SCA, with enhanced toughness, heat resistance and melt strength that potentially extend its styrofoam and commodity applications.
Introduction
Since its commercialisation, polystyrene (PS), a commodity resin second in consumption only to polyethylene and poly(vinyl chloride) (PVC), has widely been used in packaging materials, automotive interiors, electronic components, etc. because of its good transparency, high modulus, superior electrical insulation, satisfactory dyeability, low price and excellent processability. Nevertheless, owing to the presence of bulky, rigid pendant phenyl groups as well as weak, nonpolar chain-segmental interactions, PS exhibits poor (impact) toughness, i.e. high brittleness; PS does not have high enough heat resistance as well to extend itself to engineering applications, and more importantly, the low melt-strength of PS limits its foamability during styrofoam production. To overcome these disadvantages especially high brittleness, researchers have made a number of modifications to improve the toughness or heat resistance of PS. Some melt-blended elastomers, e.g. natural rubber,1 butadiene rubber (BR),2,3 ethylene–propylene–(diene monomer) rubber,4 etc., into PS matrices to enhance the toughness at the sacrifice of the heat resistance of PS, among which the BR-modified PS constituted commercialised high-impact polystyrene (HIPS); meanwhile, rigid polymers, e.g. poly(2,6-dimethyl-1,4-phenylene oxide),5 are blended with PS to enhance its heat resistance as well as mechanical strength, however, with its toughness reduced. Ceramic fillers, e.g. nano-CaCO3,6 nano-Ca3(PO4)2,7 functionalised graphene oxide,8 SiO2,9 expanded graphite,10 TiO2,11 etc., also fill PS to raise its heat resistance and/or mechanical strength, while its toughness is not elevated; even worse, most of the fillers cause serious wear and tear upon processing equipment such as screw extruders. (Covalent) cross-linking modification12,13 is used as well to increase the heat resistance and mechanical strength of PS, which, nevertheless, is only applicable to PS products instead of resins. Others modified PS by copolymerisation to increase its heat resistance and/or mechanical strength (e.g. commercial styrene-maleic anhydride (SMA)14–16 and styrene-acrylonitrile (SAN),17,18 styrene-N-phenyl maleimide,19 etc.) or to enhance its toughness (e.g. styrene–methyl methacrylate (SMMA),1,20–22 acrylonitrile–butadiene–styrene (ABS),23,24 etc.), whereas nearly all of the comonomers reported in the literature are hazardous or harmful to the environment. Obviously, although people's PS-modification endeavours have given rise to commercial styrenics like HIPS, SMA, SAN, SMMA and ABS, few modifications improve both the toughness and heat resistance of PS.Herein, an innovative modification of PS is made by copolymerising styrene with a fully green monomer, cinnamic acid (CA), recommended as a “generally recognised as safe” (GRAS) compound by the U.S. Food and Drug Administration,25 for improving both its toughness and heat resistance, as well as melt strength, by the introduction of reversible hydrogen bond cross-links.26 First, the molecular weight (w) of the product, supposedly a poly(styrene-ran-cinnamic acid) (SCA), is tailored to a commercial grade of 200–250k by adjustment of the process conditions of the emulsion free-radical copolymerisation. Then, the chemical structure and CA content, respectively, of the SCA are verified by infrared spectroscopy and acid–base titration. Subsequently, as a styrenic, the SCA is systematically characterised in terms of its mechanical, thermal, processing, dielectric, fire and other physical properties, in which an emphasis is placed on the examination of its toughness, heat resistance and melt strength.
Experimental
Materials
Styrene (≥98.0%), NaOH (≥96.0%), ethanol, anhydrous (≥99.7%), Al2(SO4)3·18H2O (≥99.0%), toluene (≥99.5%), KOH (≥85.0%), all of analytical reagent, and phenolphthalein (indicator grade) were purchased from Sinopharm (Shanghai) Chemical Reagents Co., Ltd., China. Na2SO4, anhydrous (≥99.0%) and sodium dodecylsulfate (SDS), both of analytical purity, were supplied by Tianjin City Fuchen Chemical Reagents Plant, China. Potassium persulfate (K2S2O8) (analytical purity, ≥99.5%) was obtained from Younaide Initiators (Shanghai) Co., Ltd., China. (Trans-)CA (analytical reagent, 99.0%) was provided by Shanghai Macklin Biochemical Co., Ltd., China. A PS resin (injection grade, GPPS 123P) was received from Shanghai Secco Petrochemical Co., Ltd., China. Tetrahydrofuran (THF) (standard for GC, >99.9%) and KBr (spectral reagent, 99.0%) were offered by Aladdin Industrial Corp., China. Potassium biphthalate (working chemical, ≥99.9%) was afforded by Tianjin City Chemical Reagents Institute Co., Ltd., China. Deionised water was home made in our laboratory using an ultra aqua pura machine (Ulupure, China, UPT-I-10T).Prior to its use, styrene was purified by extractions with a 5 wt% of NaOH aqueous solution to remove polymerisation inhibitor(s) and impurities and then with deionised water to eliminate the residual NaOH, followed by drying with anhydrous Na2SO4. K2S2O8 was also refined by recrystallisation in a 0 °C ice-water bath from 50 °C deionised water, followed by filtration, washing with ice water and then with anhydrous ethanol repeatedly, air-drying in a fume hood overnight and finally vacuum drying at 50 °C for at least 12 h. All the other chemicals were used as received without any further purification unless otherwise specified below.
Emulsion free-radical copolymerisation of styrene and cinnamic acid
Styrene was copolymerised with a small amount (10 wt%) of CA using an emulsion method. An overhead stirrer as well as a laboratory condenser was fitted to a 2000 mL three-necked round-bottom flask clamped and immersed in a 50 °C water bath, into which deionised water (600 mL) and SDS emulsifier (∼0.8080 g) were added. Upon 250 rpm stirred emulsification of the mixture for 0.5 h, the (purified) styrene monomer (60 mL) and CA comonomer (20.20 g) were added to the flask, after which the water-bath temperature was increased to 75 °C; meantime, the (refined) K2S2O8 (3.2320 g) was dissolved in deionised water (75 mL) in a 100 mL beaker to prepare an initiator solution. While the bath temperature was stabilised at 75 °C, the initiator solution (25 mL) was poured into the stirred reaction system. Once the reaction mixture appeared bluish or little liquid was observed to reflux, the bath temperature further was increased to 80 °C, when the styrene (70 mL) and initiator solution (25 mL) were sequentially decanted into the system. On their constant-temperature reaction for 1 h, the remaining styrene (70 mL) and initiator solution (25 mL) were successively dumped in, which were then reacted, again, at 80 °C for 1 h. At this juncture, the system was allowed to react for another 0.5 h at an enhanced temperature of 85 °C, contingent upon whether there was noticeably refluxed liquid (i.e. styrene) observed. After the copolymerisation reaction was completed, the emulsion was transferred to a 1000 mL beaker and cooled to room temperature (RT), into which Al2(SO4)3 demulsifier (∼1.6 g) was then poured to break the emulsion under vigorous stirring. The obtained white product was Büchner filtrated, then transferred to a 1000 mL beaker and subsequently soaked in hot (70–80 °C) deionised water (200–300 mL) for 5 min under stirring; such a process was repeated at least 3 times. Afterwards, a similar process was repeated at least 3 times with RT anhydrous ethanol as the soaking solvent. Finally, the product was Büchner filtrated, then air-dried in a fume hood for at least 3 days and subsequently vacuum dried at 90 °C for at least 1 day to supposedly obtain an SCA resin.Characterisations of the SCA
w of 200–250k to the (commercial) PS resin, the (potential SCA) product resin was measured at 35 °C against the PS using a gel permeation chromatograph (Waters, Waters 1525). For the runs, THF was used as the eluent at a constant flow rate of 1.0 mL min−1 and monodisperse molecular weights (up to 1200k) of PS resins as the standards.
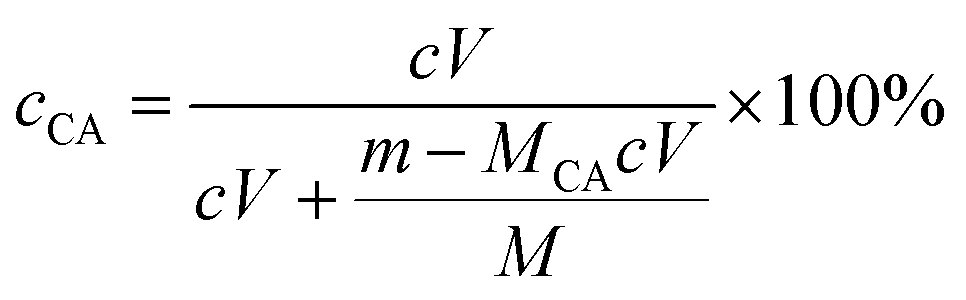 | (1) |
![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) ’s) (∼102–104 s−1), of the resins were measured at 200 °C by a twin-bore capillary rheometer (Malvern Panalytical, Rosand RH10) with one die aspect-ratio (L/D = 10/1) against the other (L/D = 0.17/1) for the Bagley correction. The acquired raw data were analysed by the Rabinowitch correction to obtain the flow-curve results.δ) of the SCA and PS resins according to the IEC 60250 testing standard. The resonance voltages and resonant capacitors were measured with and without the specimens at 60 Hz and 1 MHz to evaluate the ε's and tanδ's, using an LCR digital bridge (Shanghai Tongbei Testing Technologies Co., Ltd., China, TH2826).
’s) (∼102–104 s−1), of the resins were measured at 200 °C by a twin-bore capillary rheometer (Malvern Panalytical, Rosand RH10) with one die aspect-ratio (L/D = 10/1) against the other (L/D = 0.17/1) for the Bagley correction. The acquired raw data were analysed by the Rabinowitch correction to obtain the flow-curve results.δ) of the SCA and PS resins according to the IEC 60250 testing standard. The resonance voltages and resonant capacitors were measured with and without the specimens at 60 Hz and 1 MHz to evaluate the ε's and tanδ's, using an LCR digital bridge (Shanghai Tongbei Testing Technologies Co., Ltd., China, TH2826).To ensure statistical significance, the medians from five parallel results were taken as the data for analysis for the titration, dielectric-property and specific-gravity measurements, from at least eight results for the mechanical-property and MFR tests and from three results for the HDT, VST, αm, thermal-conductivity, smoke-density and water-absorption measurements.
Results and discussion
Reproducibility of the emulsion free-radical copolymerisation
In Table 1, the (potential SCA) product resin (Table 1(A)) synthesised had molecular weights (n, w, Mp, z and z+1) and their distributions (D1, D2 and D3) all comparable to those of the (commercial) PS. More synthetic batches (Table 1(B–F)) of the product resin were all not changed considerably in molecular weights and their distributions compared with Table 1(A), indicating that the polymerisation process was controlled at high reproducibility.
| Resin | Polymerisation no. | n |
w |
Mp | z |
z+1 |
w/n(D1) |
z/w(D2) |
z+1/w(D3) |
|---|---|---|---|---|---|---|---|---|---|
| a Not applicable. | |||||||||
| PS | —a | 60000 |
198000 |
151000 |
414000 |
611000 |
3.3 | 2.1 | 3.1 |
| SCA | A | 60000 |
217000 |
157000 |
467000 |
685000 |
3.6 | 2.2 | 3.2 |
| B | 64000 |
209000 |
155000 |
439000 |
656000 |
3.3 | 2.1 | 3.1 | |
| C | 30000 |
224000 |
188000 |
483000 |
698000 |
7.2 | 2.2 | 3.1 | |
| D | 30000 |
210000 |
174000 |
463000 |
681000 |
6.9 | 2.2 | 3.3 | |
| E | 60000 |
227000 |
176000 |
474000 |
690000 |
3.8 | 2.1 | 3.0 | |
| F | 38000 |
227000 |
188000 |
486000 |
704000 |
6.0 | 2.1 | 3.1 | |
Evidence for the successful synthesis of SCA
Shown in Fig. 1 is the FTIR absorption spectrum (Trace 2) of the product resin as opposed to that (Trace 1) of the PS. In Trace 1 for the PS, the absorption bands at 3080, 3059 and 3025 cm−1 were attributed to the stretches of the phenyl C–H bonds,27 the 2923 and 2850 cm−1 bands, respectively, to the antisymmetric and symmetric stretches of the –CH2– groups,28 the 1943, 1872, 1803, 1745 and 1664 cm−1 bands to the C–C and C–H overtone and combination stretches of the phenyl groups,29 the 1600, 1492 and 1448 cm−1 absorption peaks to the stretches of the phenyl C–C bonds (i.e. phenyl skeleton vibrations),27,30 the 1378 cm−1 peak to the in-plane rocking of the aliphatic CH groups in the main chain,29 the 1069 and 1026 cm−1 and 754 and 697 cm−1 peaks, respectively, to the in-plane and out-of-plane bending of the phenyl C–H bonds.31 Compared with Trace 1, Trace 2 for the product resin exhibited, in addition to all the above featured absorptions, new peaks at 1744 and 1703 cm−1 characteristic of the stretches of free and dimeric carboxyl CO bonds, respectively.13,32 Right around there, part of the phenyl overtone and combination stretches (at 1745 and 1664 cm−1) were found to be overshadowed due to their weaker intensities, which does not prevent one inferring that the 1744 and 1703 cm−1 bands were indeed new from the introduced carboxyls.
 | ||
| Fig. 1 Fourier transform infrared (FTIR) absorption spectra of (1) a commercial polystyrene (PS) and (2) a resin, supposedly poly(styrene-ran-cinnamic acid), laboratory synthesised by emulsion free-radical copolymerisation of styrene with a small amount (10 wt%) of cinnamic acid. | ||
Contingent upon the reactivity ratios of styrene and CA monomers, the product resin might primarily comprise one of three possible species, PS-poly(cinnamic acid) (PCA) blend, PS and SCA. From the resin's FTIR sample reprecipitated from toluene into ethanol (cf. Par. 2, Section 2.3), free CA and/or PCA molecules, if any, were effectively eliminated by the dissolving ethanol. Therefore, if it had been a PS–PCA blend or PS, the resin would have demonstrated an FTIR spectrum basically of neat PS (Trace 1, Fig. 1), which was impossible. In other words, upon the removal of any free (i.e. unpolymerised) CA from it, the resin actually displayed an FTIR spectrum (Trace 2, Fig. 1) bearing the carboxyls' characteristic bands, which proves success in the copolymerisation of CA into PS to form an SCA. More quantitatively, the CA content of the SCA was evaluated from eqn (1) by acid–base titration to be 6.8 mol%, which was indeed significant considering the CA-comonomer feed ratio of 10 wt% (∼7.2 mol%). This value, along with the SCA's typical w and w/n against those of the PS (Table 1), is listed in Table 2.
| Properties | Parameter | Testing method | Testing condition(s) | Value | |
|---|---|---|---|---|---|
| PS | SCA | ||||
a Measured by titration of a toluene solution of the purified SCA against a KOH ethanol solution titrated by potassium-biphthalate standard, with a phenolphthalein solution in an ethanol/water (19/1 v/v) mixed solvent as the indicator.b Measured at 35 °C by gel permeation chromatography (GPC), with tetrahydrofuran as the eluent at a constant flow rate of 1.0 mL min−1 and monodisperse PS resins up to 1200k as the standards.c Not applicable.d Measured by thermogravimetric analysis (TGA) at a heating rate of 20 °C min−1 under a N2 atmosphere, with the temperature at 5% weight loss defined as the Td.e Characterised by the extensional viscosity (ηe) at a practical extensional strain rate (![[small epsi, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a1.gif) ) of 0.5 s−1 in the ηe vs. flow curves.f Measured at 200 °C (PS)/220 °C (SCA) by an extensional rheometer, with a die-exit melt extrusion rate of 11.3 mm s−1, a nip-rollers’ linear acceleration of 12 mm s−2, and a gauge length (i.e. distance between the die exit and the nip rollers) of 100 mm.g Measured at 200 °C by a twin-bore capillary rheometer with one die aspect-ratio (L/D = 10/1) against the other (L/D = 0.17/1) for the Bagley correction. ) of 0.5 s−1 in the ηe vs. flow curves.f Measured at 200 °C (PS)/220 °C (SCA) by an extensional rheometer, with a die-exit melt extrusion rate of 11.3 mm s−1, a nip-rollers’ linear acceleration of 12 mm s−2, and a gauge length (i.e. distance between the die exit and the nip rollers) of 100 mm.g Measured at 200 °C by a twin-bore capillary rheometer with one die aspect-ratio (L/D = 10/1) against the other (L/D = 0.17/1) for the Bagley correction. |
|||||
| Structural | Cinnamic-acid (CA) content (mol%) | —a | —a | —c | 6.8 |
| Weight-average molecular weight (w) |
—b | 35 °C | 198000 |
217000 |
|
| Polydispersity index (w/n) |
—b | 35 °C | 3.3 | 3.6 | |
| Mechanical | Charpy notched impact strength (kJ m−2) | ISO 179-1 | 23 ± 2 °C, 2 mm min−1 | 5.3 | 7.8 |
| Young's modulus (MPa) | ISO 527 | 23 ± 2 °C, 2 mm min−1 | 2.8 × 102 | 3.7 × 102 | |
| Tensile strength (MPa) | ISO 527 | 23 ± 2 °C, 2 mm min−1 | 52.3 | 57.1 | |
| Elongation at break (%) | ISO 527 | 23 ± 2 °C, 2 mm min−1 | 10.5 | 9.7 | |
| Flexural modulus (MPa) | ISO 178 | 23 ± 2 °C, 2 mm min−1 | 2.1 × 104 | 2.4 × 104 | |
| Flexural strength (MPa) | ISO 178 | 23 ± 2 °C, 2 mm min−1 | 138.4 | 167.6 | |
| Thermal | Glass transition temperature (°C) | ISO 11357 | 20 °C min−1 | 95.2 | 117.0 |
| Heat deflection temperature (°C) | ISO 75 | 0.45 MPa, 120 °C h−1 | 87.6 | 108.0 | |
| Vicat softening temperature (°C) | ASTM D1525 | 10 N, 50 °C h−1 | 96.0 | 118.3 | |
| Thermal decomposition temperature (°C) | ASTM E2402 | 20 °C min−1 | 391.9d | 360.1d | |
| Linear thermal expansion coefficient (μm m−1 °C−1) | ASTM E831 | 25–75 °C, 5 °C min−1 | 78.4 | 79.5 | |
| Thermal conductivity (W m−1 K−1) | ASTM D5930 | 23 ± 0.1 °C | 0.160 | 0.165 | |
| Processing | Melt strengthe (Pa s) | —f | 0.5 s−1, 220 °C (SCA)/200 °C (PS) | 4.2 × 104 | 3.3 × 105 |
| Melt flow rate (g per 10 min) | ISO 01133 | 200 °C, 5.0 kg | 6.9 | 0.6 | |
| Shear viscosity (Pa s) | —g | 200 °C, 103 s−1 | 101.7 | 278.9 | |
| Dielectric | Dielectric constant | IEC 60250 | 60 Hz | 2.8 | 2.8 |
| 1 MHz | 2.8 | 2.7 | |||
| Dielectric loss | IEC 60250 | 60 Hz | 3.7 × 10−3 | 3.3 × 10−3 | |
| 1 MHz | 0.6 × 10−3 | 1.6 × 10−3 | |||
| Fire | Oxygen index (vol%) | ISO 4589 | 23 ± 2 °C | 19.7 | 19.9 |
| Density of smoke (%) | ASTM D2843 | 23 ± 2 °C | 88.8 | 92.5 | |
| Other physical | Intrinsic viscosity (dL g−1) | ISO 1628-1 | 25 ± 0.1 °C, toluene | 8.4 × 10−2 | 2.6 × 10−2 |
| Specific gravity | ASTM D792 | 23 ± 2 °C | 1.05 | 1.07 | |
| Water absorption (wt%) | ISO 62 | 23 ± 2 °C, 24 h | 0.06 | 0.47 | |
Mechanical properties of the SCA
As shown in Table 2, the Charpy notched impact strength, tensile and flexural properties (primarily modulus and strength) of the SCA were all enhanced compared with the (commercial) PS, which may arise from, besides the volume fraction (6.8 mol%) of rigid CA comonomer, essentially the reversible hydrogen bond cross-links (i.e. dimeric carboxyls) introduced by the copolymerisation of CA. Particularly worth noting is that the impact toughness of the SCA was significantly improved by 47.2% against the PS. According to the craze theory,33,34 this might be associated with balanced initiation of new crazes by unzipping (i.e. de-hydrogen-bonding) of SCA segments and stabilisation of preexisting crazes propagation by segmental zipping (i.e. re-hydrogen-bonding), both of which contributed to absorption of impact energy.Thermal properties of the SCA
Fig. 2 gives the DSC thermograms of the SCA and PS, from which their Tg's were analysed as summarised in Table 2; it is seen that the Tg (117.0 °C) of the SCA was noticeably (∼20 °C) higher than that (95.2 °C) of the PS. Also observed from Table 2 is that the HDT and VST of the SCA were both significantly enhanced until 108.0 and 118.3 °C, respectively, compared with those (87.6 and 96.0 °C) of the PS. The large increases in the Tg, HDT and VST indicate that the heat resistance of the SCA was greatly improved as opposed to the PS. Again, this may be basically due to the 6.8 mol% of carboxyl groups incorporated into the SCA by copolymerisation: the steric hindrance of the pendant carboxyls limited the internal rotation of the SCA backbone chains; more remarkably, the hydrogen bond cross-linking applied by dimeric carboxyls strengthened the SCA chain-segmental interactions. Both of these led to rigidification of the SCA chains that constituted pronounced enhancement of the SCA heat resistance. | ||
| Fig. 2 Differential scanning calorimetry (DSC) thermograms in the second heating cycle at a rate of 20 °C min−1 for (1) a commercial polystyrene (PS) and (2) a poly(styrene-ran-cinnamic acid) (SCA), i.e. PS copolymerised with 6.8 mol% of cinnamic acid. | ||
It can be seen from Fig. 3 that the thermal decomposition temperature (360.1 °C) of the SCA was ∼30 °C lower than the PS (391.9 °C), the results of which are listed in Table 2. This may originate from higher chemical activity of the SCA bearing carboxyl groups, which were subject to thermal decomposition into CO2. Finally, as shown in Table 2, the linear thermal expansion coefficient (αm) and thermal conductivity (λ) of the SCA, 79.5 μm m−1 °C−1 and 0.165 W m−1 K−1 respectively, were comparable to those (78.4 μm m−1 °C−1 and 0.160 W m−1 K−1) of the PS, suggesting that the αm and λ of the PS were both little changed upon the carboxylation modification.
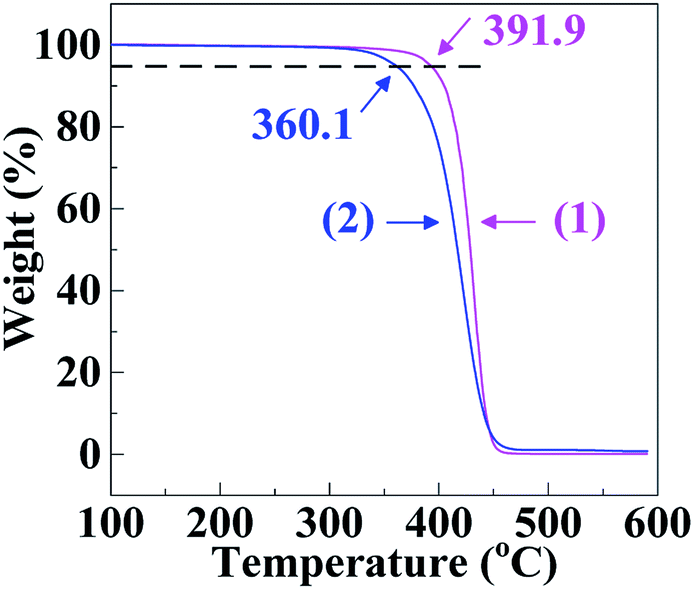 | ||
| Fig. 3 Thermogravimetric analysis (TGA) thermograms at a rate of 20 °C min−1 for (1) a commercial polystyrene (PS) and (2) a poly(styrene-ran-cinnamic acid) (SCA), i.e. PS copolymerised with 6.8 mol% of cinnamic acid. | ||
Melt strength of the SCA
In the molten states, with melt flow temperatures (Tf) defined by HDT + 112 °C, of the PS (Tf = 200 °C) and SCA (Tf = 220 °C), the drawdown force (F) vs. draw ratio (V) (Fig. 4a), extensional stress (σe) vs. V (Fig. 4b) and extensional viscosity (ηe) vs. extensional strain rate () (Fig. 4c) curves demonstrate that the F, σe and ηe of the SCA were all significantly larger than those of the PS, suggesting a markedly enhanced melt strength of the SCA compared with the PS. This is presumably owing to the reversible hydrogen bond cross-links as well as rigid CA inclusion; the former significantly strengthened the chain-segmental interactions, which in turn would cause a much milder decrease in the SCA melt strength with increasing temperature. In other words, compared with the PS, the SCA may exhibit a considerably broadened temperature window required for the appropriate melt strengths for stabilisation of its foaming (i.e. styrofoam production) operations; this constitutes a more essential sense of the SCA melt-strength improvement. As a typical measure of melt strength, the ηe (3.3 × 105 Pa s) at a practical melt-processing (e.g. foaming) of 0.5 s−1 for the SCA is given against the PS (4.2 × 104 Pa s) in Table 2.
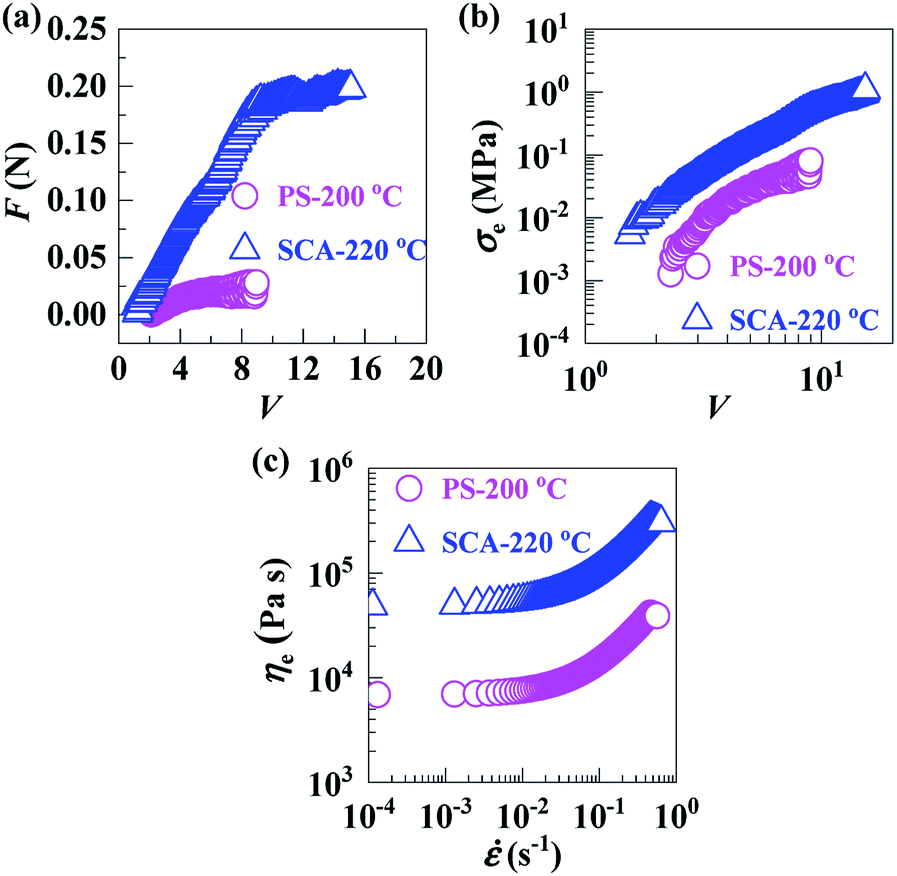 | ||
Fig. 4 (a) Drawdown forces (F's) and (b) extensional stresses (σe's) as functions of draw ratio (V) and (c) extensional viscosities (ηe's) as functions of extensional strain rate (), for ( ) a commercial polystyrene (PS) at 200 °C and ( ) a commercial polystyrene (PS) at 200 °C and ( ) a poly(styrene-ran-cinnamic acid) (SCA), i.e. PS copolymerised with 6.8 mol% of cinnamic acid, at 220 °C. ) a poly(styrene-ran-cinnamic acid) (SCA), i.e. PS copolymerised with 6.8 mol% of cinnamic acid, at 220 °C. | ||
Processability of the SCA
Compared with the PS, the apparently sharply-reduced MFR of the SCA (0.6 vs. 6.9 g 10−1 min−1) shown in Table 2 does not necessarily indicate that the processing fluidity of the SCA was dramatically deteriorated at the same typical melt-processing shear rate. This is because the greatly increased viscosity of the SCA originated from not only the hydrogen bond cross-linking as well as rigid volume-fraction of CA, but, preponderantly, its much smaller shear rate (and thus shear thinning) under the same load applied during the MFR testing.As can be seen from Fig. 5a, the SCA and PS were both in the linear viscoelastic region below ε's of ∼5%; their |η*| vs. ω behaviours were therefore compared at a small ε of 0.1% ensuring linear viscoelasticity (Fig. 5b), which were then reduced to η vs. (10−1–102 s−1) relationships (Fig. 6) according to the Cox–Merz rule. The capillary rheology at higher ’s (102–104 s−1) was subsequently measured as well, and finally added to Fig. 6 to form flow mastercurves of the SCA and PS. It is seen that, over the entire range of ’s investigated, the η's of the SCA at 200 °C were higher than the PS presumably from its hydrogen bond cross-linking and rigid CA copolymerisation combined effects. Further, at low ’s, the SCA displayed a stronger shear-thinning behaviour than the PS since its hydrogen bond cross-links were inclined to dissociate upon shearing, while, at the higher ’s, the SCA and PS exhibited similar shear-thinning behaviours. Under these circumstances, the η (e.g. 278.9 Pa s) of the SCA at a typical melt-processing (e.g. 103 s−1) was just a little higher relative to the PS (e.g. 101.7 Pa s) (Table 2), which constitutes an inheritance of the PS's excellent processability by the SCA without significant compromisation.
 | ||
Fig. 5 (a) Complex viscosity magnitudes (|η*|‘s) as functions of strain amplitude (ε) at 200 °C and 60 Hz, and (b) |η*|‘s as functions of angular frequency (ω) at 200 °C and an ε of 0.1%, for ( ) a commercial polystyrene (PS) and ( ) a commercial polystyrene (PS) and ( ) a poly(styrene-ran-cinnamic acid) (SCA), i.e. PS copolymerised with 6.8 mol% of cinnamic acid. ) a poly(styrene-ran-cinnamic acid) (SCA), i.e. PS copolymerised with 6.8 mol% of cinnamic acid. | ||
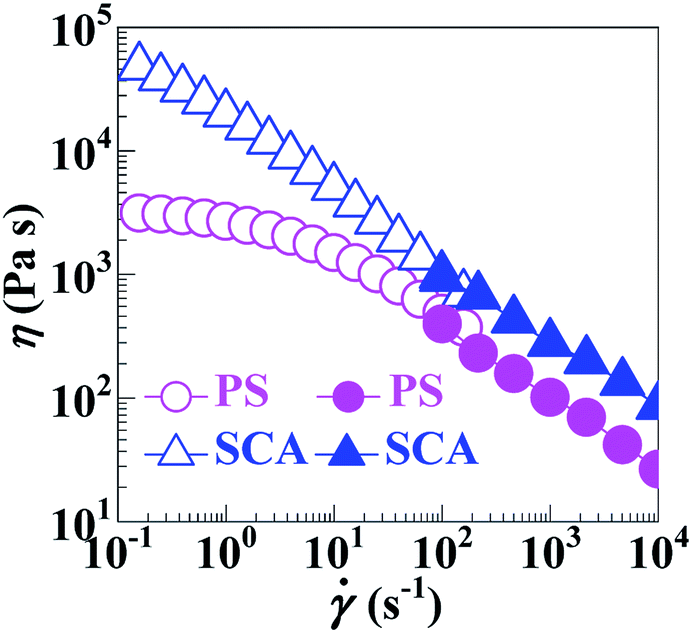 | ||
Fig. 6 Shear-viscosity (η) vs. shear-rate () flow mastercurves at 200 °C for ( ) a commercial polystyrene (PS) and ( ) a commercial polystyrene (PS) and ( ) a poly(styrene-ran-cinnamic acid) (SCA), i.e. PS copolymerised with 6.8 mol% of cinnamic acid. Wherein, the data points ( ) a poly(styrene-ran-cinnamic acid) (SCA), i.e. PS copolymerised with 6.8 mol% of cinnamic acid. Wherein, the data points ( ) are derived from oscillatory-shear rotational rheometry and the other data points ( ) are derived from oscillatory-shear rotational rheometry and the other data points ( ) are obtained from capillary rheometry. ) are obtained from capillary rheometry. | ||
Dielectric properties of the SCA
At a low frequency (e.g. 60 Hz), the electrical insulation (ε of 2.8 and tanδ of 3.3 × 10−3) of the SCA was comparable to the PS (ε of 2.8 and tan δ of 3.7 × 10−3); at a higher frequency (e.g. 1 MHz), the ε (2.7) of the SCA was still approximate to the PS (2.8) whereas the tanδ (1.6 × 10−3) of the SCA larger than the PS (0.6 × 10−3), indicating a deterioration of the SCA's electrical insulation compared with the PS (Table 2). In other words, the ε's of the SCA were equivalent to the PS at both the frequencies (Table 2), which may result from the hydrogen bond cross-links of the former that essentially repressed the polarisation orientation of its carboxyls.35,36 However, the tanδ of the SCA remained close to the PS at the low frequency while became larger than the PS at the higher frequency (Table 2), the latter of which should also be associated with the weaker mobility of the SCA chain segments constrained by their hydrogen bond cross-links.
Fire properties of the SCA
The oxygen index (19.9) of the SCA was comparable to the PS (19.7), and the density of smoke (92.5) of the SCA a bit higher than the PS (88.8) (Table 2). These results seem to discover that the fire properties of the SCA were basically in the proximity of the PS.Other physical properties of the SCA
The intrinsic viscosity (2.6 × 10−2 dL g−1) of the SCA was considerably lower than the PS (8.4 × 10−2 dL g−1) (Table 2), which probably arises from the hydrogen bond cross-links of the former shrinking its chain segments in radial size. The specific gravity (1.07) of the SCA was almost equal to the PS (1.05) (Table 2). And the water absorption (0.47) of the SCA was significantly increased relative to the PS (0.06) (Table 2), obviously due to its introduction of 6.8 mol% of polar, hydrophilic carboxy groups.Conclusions
Styrene is copolymerised via emulsion radical polymerisation with cinnamic acid (CA), a green compound to produce a modified polystyrene (PS), poly(styrene-ran-cinnamic acid) (SCA), of comparablew of ∼200000 to commercial PS bearing 6.8 mol% of CA. Compared with commercial PS, the SCA displays significantly improved impact toughness (by 47.2%), heat resistance (glass-transition, heat deflection and Vicat softening temperatures all by ∼20 °C) and melt strength (extensional viscosity by one order of magnitude, and foaming temperature window). Regarding other properties, despite decreases in its thermal decomposition stability, electrical insulation and moisture resistance, the SCA exhibits mechanical strengths, linear thermal expansion coefficient, thermal conductivity, processability, fire properties, specific gravity, etc. all basically approximate to commercial PS. These overall properties of the SCA relative to commercial PS, from a chain topological viewpoint, are all attributed to its rigid volume-fraction of CA and/or hydrogen bond cross-linking from dimerisation of CA carboxyls. As a modified version of PS, the SCA demonstrates both-improved, balanced impact toughness and heat resistance superior to commercial HIPS, SMA, SAN, SMMA and ABS, as well as enhanced melt strength, which potentially extend its styrofoam and commodity applications.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge with gratitude that this work was financially supported by the Overseas High-level Talents Scientific-research Starting Fund of Hubei University of Technology, China (Contract no. HBUT-science-[2005]2), as well as by the Select Overseas Chinese Scholars Science and Technology Activities Foundation of the Ministry of Human Resources and Social Security of China (Grant no. [2013]277) and the Natural Science Foundation of the Hubei Province of China (Contract nos. 2014CFA094 and 2016CFB205).References
- S. B. Neoh and A. S. Hashim, J. Appl. Polym. Sci., 2004, 93, 1660 CrossRef CAS.
- J. L. Amos, J. L. McCurdy and O. R. Mclntire, US Pat., US18157650A, 1954.
- G. H. Gao, C. Zhou, H. D. Yang and H. X. Zhang, J. Appl. Polym. Sci., 2007, 103, 738 CrossRef CAS.
- S. Shaw and R. P. Singh, J. Appl. Polym. Sci., 2010, 40, 701 CrossRef.
- A. Z. Aroğuz and B. M. Baysal, J. Appl. Polym. Sci., 2015, 75, 225 Search PubMed.
- Y. Gao, L. Liu and Z. Zhang, Acta Mech. Solida Sin., 2009, 22, 555 CrossRef.
- S. P. Thomas and S. Thomas, J. Phys. Chem. C, 2009, 113, 97 CrossRef CAS.
- S. L. Qiu, W. Z. Hu, B. Yu, B. H. Yuan, Y. L. Zhu, S. H. Jiang, B. B. Wang, L. Song and Y. Hu, Ind. Eng. Chem. Res., 2015, 54, 3309 CrossRef CAS.
- O. Bera, B. Pilić, J. Pavličević, M. Jovičić, B. Holló, K. M. Szécsényi and M. Špirkova, Thermochim. Acta, 2011, 515, 1 CrossRef CAS.
- G. H. Chen, D. J. Wu, W. G. Weng, B. He and W. L. Yan, Polym. Int., 2001, 50, 980 CrossRef CAS.
- T. P. Selvin, J. Kuruvilla and T. Sabu, Mater. Lett., 2004, 58, 281 CrossRef CAS.
- A. P. Mathew, S. Packirisamy and S. Thomas, Polym. Degrad. Stab., 2001, 72, 423 CrossRef CAS.
- M. P. Tsyurupa and V. A. Davankov, React. Funct. Polym., 2002, 53, 193 CrossRef CAS.
- E. R. Moore, Ind. Eng. Chem. Prod. Res. Dev., 1986, 25, 315 CrossRef CAS.
- T. Alfrey and E. Lavin, J. Am. Chem. Soc., 1945, 67, 2044 CrossRef CAS.
- M. Narayana, H. Keskkula and J. E. Mason, US Pat., US3641212DA, 1972.
- N. V. Tsarevsky, T. Sarbu, B. Göbelt and K. Matyjaszewski, Macromolecules, 2002, 35, 6142 CrossRef CAS.
- M. E. Fowler, H. Keskkula and D. R. Paul, Polymer, 1987, 28, 1703 CrossRef CAS.
- G. D. Liu, X. C. Li, L. C. Zhang, X. W. Qu, P. G. Liu, L. T. Yang and J. G. Gao, J. Appl. Polym. Sci., 2002, 83, 417 CrossRef CAS.
- F. R. Mayo and F. M. Lewis, J. Am. Chem. Soc., 1994, 66, 1594 CrossRef.
- P. Bataille and F. Granger, Polym. Eng. Sci., 1985, 25, 1164 CrossRef CAS.
- B. J. P. Jansen, S. Rastogi, H. E. H. Meijer and P. J. Lemstra, Macromolecules, 2001, 34, 3998 CrossRef CAS.
- L. E. Dlay, US Pat., US54612555A, 1961.
- W. Brostow, H. E. H. Lobland and S. Khoja, Mater. Lett., 2015, 159, 478 CrossRef CAS.
- Food and Drug Administration, Food additives permitted for direct addition to food for human consumption, Food and Drug Administration, Washington, DC, 2018 Search PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601 CrossRef CAS PubMed.
- S. H. Kim, W. K. Son, Y. J. Kim, E. G. Kang, D. W. Kim, C. W. Park, W. G. Kim and H. J. Kim, J. Appl. Polym. Sci., 2003, 88, 595 CrossRef CAS.
- G. M. Chen, S. H. Liu, S. J. Chen and Z. N. Qi, Macromol. Chem. Phys., 2001, 202, 1189 CrossRef CAS.
- R. A. Nyquist, Appl. Spectrosc., 1984, 38, 264 CrossRef CAS.
- V. K. Thakur, E. J. Tan, M. F. Lin and P. S. Lee, Polym. Chem., 2011, 2, 2000 RSC.
- A. A. Bhutto, D. Vesely and B. J. Gabrys, Polymer, 2003, 44, 6627 CrossRef CAS.
- J. Dong, Y. Ozaki and K. Zakashima, Macromolecules, 1997, 30, 1111 CrossRef CAS.
- C. B. Bucknall, Polymer, 2007, 48, 1030 CrossRef CAS.
- W. J. Si, W. Q. Yuan, Y. D. Li, Y. K. Chen and J. B. Zeng, Polym. Test., 2018, 65, 249 CrossRef CAS.
- I. M. El-Anwar, O. M. El-Nabawy, S. A. El-Hennwii and A. H. Salama, Chaos, Solitons Fractals, 2000, 11, 1303 CrossRef CAS.
- P. S. Alegaonkar, V. N. Bhoraskar, P. Balaya and P. S. Goyal, Appl. Phys. Lett., 2002, 80, 640 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |