
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClickable poly-L-lysine for the formation of biorecognition surfaces†
Daniele Di Iorio ,
Almudena Marti,
Sander Koeman and
Jurriaan Huskens*
,
Almudena Marti,
Sander Koeman and
Jurriaan Huskens*
Molecular NanoFabrication Group, MESA+ Institute for Nanotechnology, University of Twente, Enschede, The Netherlands. E-mail: j.huskens@utwente.nl
First published on 4th November 2019
Abstract
Biomolecules are immobilized onto surfaces employing the fast and stable adsorption of poly-L-lysine (PLL) polymers and the versatile copper-free click chemistry reactions. This method provides the combined advantages of versatile surface adsorption with density control using polyelectrolytes and of the covalent and orthogonal immobilization of biomolecules with higher reaction rates and improved yields of click chemistry. Using DNA attachment as a proof of concept, control over the DNA probe density and applicability in electrochemical detection are presented.
Introduction
The immobilization of biomolecules onto surfaces is of fundamental importance in several fields such as analytical chemistry, medical diagnostics, tissue engineering and biomolecular chemistry.1–3 Monolayers have been prepared on surfaces by several methods in order to warrant the bio-activity of the biomolecules such as enzymes, antibodies or nucleic acids, and to exert control over their functionalization at the molecular scale.4–6 The monolayers are often used as an intermediate, thus to provide stable anchoring onto the material surface and, at the same time, present functional groups to allow the subsequent binding of (bio)molecules.Important routes for the ligation of molecules onto surfaces have been exploited by using click chemistry. The strain-promoted alkyne–azide cycloaddition (SPAAC) reactions between ring-strained cyclooalkynes and azides have been used for the catalyst-free modification of surfaces.7–10 More recently, the inverse electron-demand Diels–Alder (iEDDA) reaction of tetrazine with strained alkenes reported by Fox et al. introduced a broader range of compounds with even faster kinetics into the area of click chemistry.11 Tetrazine can react with trans-cyclooctyne (TCO) or norbornene groups at room temperature, atmospheric pressure and micromolar concentrations with faster kinetics and allowing up to 100% conversion, while avoiding byproducts.12–14
Alternative to the covalent modification of substrates, the physisorption of modified polyelectrolytes on surfaces for the immobilization of biomolecules has presented several advantages.15 In particular, poly-L-lysine (PLL) has been reported to present excellent properties such as biocompatibility and hydrophilicity. At physiological pH, the amino groups of the lysine sidechains are positively charged, allowing a fast adsorption of the PLL on a broad variety of negatively charged substrates through multiple electrostatic interactions.16,17 The modification of the polymer with additional functional groups and non-ionic, anti-fouling side-chains, such as oligo(ethylene glycol) (OEG), enables the engineering of surfaces with selective receptors as well as antifouling properties. A variety of different functional groups has been explored, such as biotin (for conjugation with streptavidin),18 RGD peptide (to promote cell binding),19 and maleimide (to bind thiol-PNA with control over the density at the surface).20 However, the modification of PLL with ‘clickable’ units to achieve bioconjugation at surfaces, has so far not been reported.
Here we report a surface functionalization method for fast and selective immobilization of molecules onto substrates by using PLL modified with click chemistry moieties. The employment of these polymers provides the combined advantages of providing fast and versatile surface adsorption with density control using polyelectrolytes and of the covalent and orthogonal immobilization of biomolecules with higher reaction rates and improved yields of click chemistry.
Results and discussion
Fig. 1 shows a schematic representation of the stepwise approach used in this work to selectively functionalize surfaces with DNA. Hereto, PLL was grafted with OEG units and either methyl-tetrazine (Tz) or dibenzocyclooctyl (DBCO) functional groups and adsorbed onto surfaces for the subsequent immobilization of, respectively, TCO or azido-modified biomolecules by fast and catalyst-free bioorthogonal click reactions.21 The different PLLs were then adsorbed onto activated substrates, thus displaying the respective functional groups at the interface.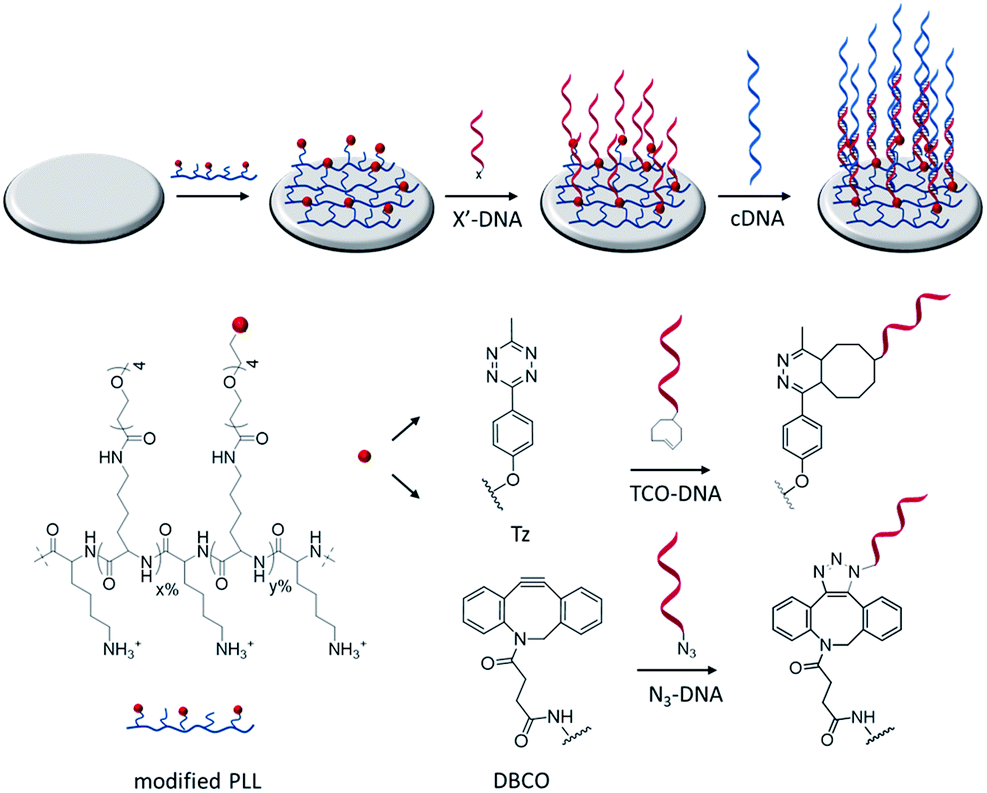 | ||
| Fig. 1 Schematic representation of the functionalization of activated surfaces with clickable PLL to bind complementarily functionalized DNA probes. After activation, silicon dioxide and gold surfaces are functionalized with either Tz or DBCO-functionalized PLL (PLL–OEG–X). Hereafter, TCO or azide-DNA (X′-DNA) sequences are selectively anchored on the PLL-modified surface by click chemistry reactions. The proper presentation of DNA probes on the surface is monitored by the hybridization with cDNA sequences. | ||
Modified PLLs were synthesized by adapting a previously reported procedure.22 PLL (MW 15–30 kDa) was functionalized in a one-step reaction, by mixing different molar ratios of NHS-(OEG)4-methyl (without a functional group) and either NHS-(OEG)4-Tz or NHS-(OEG)4-DBCO. The grafting of the polymer with short OEG ensures a better control over the PLL modification and improves the antifouling properties of PLL, thus preventing non-specific adsorption of molecules onto the PLL monolayer. By varying the molar ratios of the components in the mixture it is possible to tune the degree of functionalization of the PLL. Therefore the abbreviation PLL–OEG(x)–X(y) is used for a PLLs modified with x% of OEG and y% of functional group X (here: Tz or DBCO) grafted to the PLL. 1H-NMR was used to quantify the total degree of functionalization (x + y) of the polymer. Specifically, three different types of polymers have been synthesized: one displaying only OEG as functionalization (PLL–OEG), one presenting both OEG and DBCO (PLL–OEG–DBCO) and one with OEG and Tz (PLL–OEG–Tz). Table S1 (see ESI†) shows the PLLs synthesized and the degrees of functionalization calculated by NMR.
After the synthesis, the adsorption of PLL polymers was tested on both silicon dioxide and gold substrates. The modification of substrates with modified PLL was monitored in situ and in real time by quartz crystal microbalance with dissipation monitoring (QCM-D). Fig. 2 shows a typical QCM measurement where SiO2 surfaces were modified with PLL–OEG–Tz and PLL–OEG–DBCO to test the immobilization of TCO-DNA or azido-DNA respectively, and the subsequent recognition of cDNA. After the activation of the substrates by UV/ozone, the surfaces were flushed with a solution of PLL–OEG–Tz or PLL–OEG–DBCO (0.1 mg mL−1, in PBS at pH 7.4). Right after injection of the PLL solution, a clear and rapid frequency change (Δf5) was observed, confirming a fast self-assembly of the modified polymer onto the surface. An only moderate desorption of the PLL after rinsing with buffer indicated the stability of the remaining polymer, and the small dissipation signals confirmed the formation of a thin and rather rigid PLL film. No relevant differences between the two substrate materials were observed (Fig. S1†).
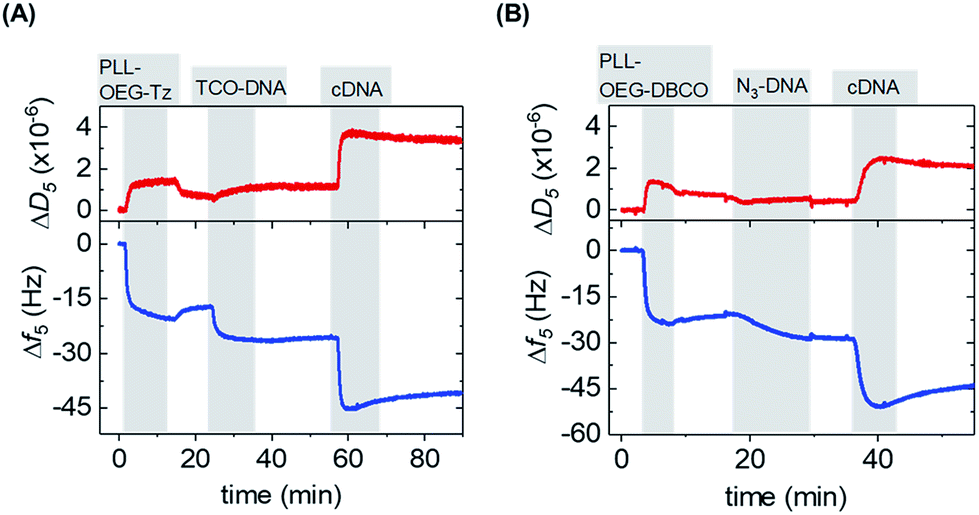 | ||
| Fig. 2 QCM-D sensograms of the assembly of (A) PLL–OEG(26.0)–Tz(4.9) (0.1 mg mL−1 in PBS) and (B). PLL–OEG(31.2)–DBCO(6.7) (0.1 mg mL−1 in PBS) on silicon dioxide surfaces, followed by (A) TCO-DNA (1 μM in PBS) and cDNA (1 μM in PBS) or (B) N3-DNA (1 μM in PBS) and either cDNA (1 μM in PBS). The frequency shift (Δf5) is represented by the blue lines and the dissipation signal (ΔD5) by the red lines. All adsorption steps are shown in grey vertical bars, while the washing steps with PBS are in white. | ||
After surface functionalization with PLL–OEG–Tz and PLL–OEG–DBCO, TCO or azide-modified DNA (15 nts) was added for the catalyst-free coupling with Tz and DBCO, respectively. X′-DNA (1 μM) was flushed over the PLL-modified substrates. A clear frequency shift, observed upon DNA addition, demonstrated a fast attachment of the DNA probes at the surface, thus confirming the successful Tz–TCO and DBCO-azide coupling. Interestingly, the observed reaction time differed for the two types of reactions. As expected, the reaction rate of the coupling of DBCO with azido-DNA appeared to be somewhat slower than the rate observed for the reaction between Tz and TCO-DNA.7 The half-time of the reaction of DBCO with azido-DNA measured by QCM resulted to be approx. 5 min, while, under the same conditions the Tz–TCO coupling showed a half-time of approx. 1 min.
Subsequently, the correct formation of a DNA probe layer onto the surface and the possibility of forming DNA duplexes was tested by the addition of fully complementary DNA (cDNA) sequences (36 nts, see ESI†). The clear frequency change, and concomitant dissipation change, upon binding of cDNA and the stability of the signal upon rinsing with buffer demonstrate the effective hybridization of the cDNA strands. The formation of DNA duplexes at the interface, stretching out from the surface, caused a change of the viscoelastic properties of the layers which is represented by an increase of the dissipation signal.23 In contrast, the absence of both frequency and dissipation changes upon addition of ncDNA confirmed that DNA binds exclusively in the presence of the complementary sequence (Fig. S1†).
After proving the fast DNA coupling, the effect of the reactive Tz content in the PLL backbone on the adsorption of DNA was assessed by QCM. Different amounts of Tz anchored to the PLL are expected to lead to varying surface densities of DNA probes and therefore different responses for the cDNA adsorption. Hereto, PLLs containing different degrees of functionalization with Tz groups (0%, 0.5%, 4.9% and 14.6%) were tested, and the adsorption steps were plotted as a function of the percentage of Tz in the PLL backbone, for both gold and SiO2 surfaces. Fig. S2† shows the full QCM time traces obtained upon adsorption of PLL, TCO-DNA and cDNA for different PLLs on both types of surfaces. In the case of PLL–OEG (Tz = 0%), the absence of Tz groups on the surface prevented the binding of TCO-DNA, thus confirming the specificity of the Tz-TCO conjugation on the surface (Fig. S2†). The low adsorption of cDNA (≤2 Hz) confirmed also the antifouling properties of the designed PLL. For low Tz contents (i.e., 0.5 and 4.9), the data shows a clear correlation between the Tz density at the interface and TCO-DNA adsorption (Fig. 3). At higher Tz contents, instead, no further increase of the amount of TCO-DNA on the surface was observed. These results indicate that a saturation of the surface with TCO-DNA probes is obtained at approx. 5% of Tz in the PLL backbone. This is attributed to electrostatic repulsion and steric hindrance between the DNA probes. Very similar trends were observed for the adsorption steps of cDNA, indicating that the hybridization efficiency was not affected by variations of the degree of Tz functionalization.
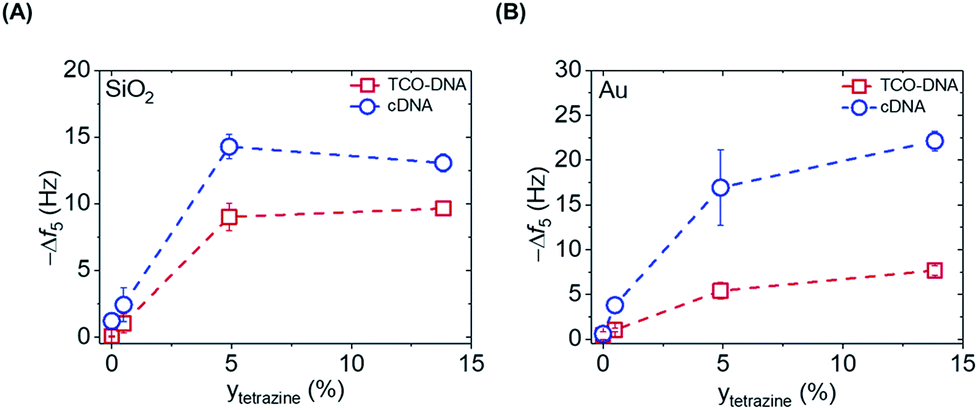 | ||
| Fig. 3 Correlation between the Δf5 obtained upon addition of TCO-DNA (red) and cDNA (blue) for increasing degrees (y) of functionalization of PLL with Tz, measured for both SiO2 (A) and gold (B) substrates. Dashed lines are guides to the eye. | ||
The apparent hybridization efficiency, defined as the ratio of the frequency shifts induced by cDNA and TCO-DNA, was higher for gold surfaces compared to silicon dioxide. By taking into account the different lengths of the TCO-DNA (15 nts) and cDNA (36 nts), hybridization efficiencies (uncorrected for possible differences and changes in hydration) of 130% and 65% were obtained for gold and silicon dioxide surfaces, respectively (see also ESI, Fig. S3†). Different DNA lengths as well as the formation of a DNA duplex can cause a change in the degree of hydration, which is detected by QCM.23,24
Control over the probe density is important for an optimal interaction of the immobilized probes with the target molecule.25 For example, when considering the design of sensors for DNA detection, steric hindrance and electrostatic repulsion can cause a loss of hybridization efficiency. When deriving probe densities from QCM measurements, the hydration of the molecules needs to be taken into account. Here, we assume that the 80% of the mass adsorbed in both the DNA step is due to water.23 This leads, in case of TCO-DNA, to a calculated DNA probe at the SiO2 substrates ranging from 0.43 to 4.8 × 1012 molecules per cm2, whereas for gold substrates a density between 0.44 and 3.3 × 1012 molecules per cm2 was obtained. The hybridization step leads to values ranging respectively between 0.47 and 2.8 × 1012 molecules per cm2, and between 0.73 and 4.3 × 1012 molecules per cm2 for silicon dioxide and gold surfaces, respectively. A density of 3.5 × 1012 molecules per cm2 was obtained instead for the N3-DNA, and 3.0 × 1012 molecules per cm2 for the cDNA. Notably, the density obtained from the measurements appear to be below the reported limit for electrostatic repulsion, where values of above 5 × 1012 molecules per cm2 are known to affect the hybridization efficiency.25
Recently, our group demonstrated that using PLL functionalized with maleimide (Mal), the density of PNA probes on the surface can be controlled in the preceding synthetic step of the customized PLL.20 Based on the empirical prediction of PNA probes on the surface by using PLL–OEG–(Mal), in which each percent of grafted Mal in the PLL corresponds to 1.24 ± 0.02 × 1012 probes per cm2, it was possible to predict the density of DNA also with the current system. By adopting the same relationship for PLL–OEG–Tz and PLL–OEG–DBCO, we calculated probe densities ranging between 0.66 and 6.1 × 1012 DNA probes per cm2 for the former, and 8.3 × 1012 molecules per cm2 for the latter. These values are in good agreement with the quantification of the DNA probes obtained from the QCM measurements described above. The differences observed between the predicted and the calculated values can be attributed to a different water content that cannot be precisely estimated by QCM. Another reason may be found in differences between the Mal and tetrazine or DBCO moieties which may induce a different adsorption behavior due to differences in size and hydrophobicity. At the same time, the predicted values do not take into account the steric hindrance and the electrostatic repulsion that might occur at such DNA probe densities.
As an additional method to study the probe density and to show the applicability of the click chemistry/PLL method for electrochemical biosensing schemes, we employed chronocoulometry (CC) as a convenient electrochemical method for the detection of the adsorbed analytes on an electrode surface.26,27 In this method, the surface-bound DNA probe moieties are exposed to a cationic redox probe in solution, such as ruthenium(II/III)hexamine (RuHex), which electrostatically interacts with the phosphate groups of the DNA probe. The CC signal is proportional to the density of phosphate groups of the nucleic acids present at the surface, and a follow-up hybridization step can therefore be observed by an increase of the adsorbed RuHex probe molecules.27 Accordingly, we used here the interaction of RuHex to DNA to obtain information about the coupling of DNA probes at the surface, and to detect the surface coverage before and after the hybridization step. PLL–OEG–Tz bearing 0.5 and 4.9% of Tz were used in these measurements. The gold electrodes were modified with PLL–OEG–Tz (0.25 mg mL−1) in PBS solution. After rinsing the surface, a solution of TCO-DNA (1 μM in PBS) was added onto the electrode for 1 h, and subsequently cDNA (1 μM in PBS) was adsorbed during an additional 1 h. Typical CC curves for the gold electrodes were obtained in the presence of 50 μM RuHex in 20 mM Tris buffer (Fig. 4), while the type and density of PLL were varied. The surface densities for TCO-DNA, resulting from the CC measurements (see ESI, Fig. S4†), were 1.1 × 1012 molecules per cm2 and 2.3 × 1012 molecules per cm2 for PLL–OEG–Tz(y) with y = 0.5 and 4.9% respectively. The densities of cDNA were found to be 2.0 and 3.4 × 1012 molecules per cm,2 respectively. These values are in reasonable agreement with the previously reported results in which densities between 1 and 10 × 1012 molecules per cm2 were measured.28 Most importantly, both in the case of the DNA probe and the cDNA, these values are very comparable to the ones calculated from the QCM measurements. The relatively small observed differences can be, once more, attributed to the error in estimating the water content in the QCM measurements. At the same time, the hybridization efficiency calculated for this system appears to be higher than 100%. Interestingly, a different behavior was observed, instead, in case of N3-DNA. As shown in Fig. 4, the measured DNA probe density appeared to be lower than the measured cDNA, thus resulting in a lower hybridization efficiency. At the same time, no clear difference was observed between values of cDNA densities obtained with PLL–OEG(26.0)–Tz(4.9) and PLL–OEG(31.2)–DBCO(6.7). This observation can find an explanation in the different degrees of functionalization of the PLL. In particular, a higher amount of DBCO can lead to the (observed) surface functionalization with DNA probes. However, due to steric hindrance and electrostatic repulsion, not all the probes can hybridize with cDNA, resulting in a lower hybridization efficiency. Therefore, these results support the previous observations, where at a PLL functionalization above 5%, no further increase of cDNA was observed.
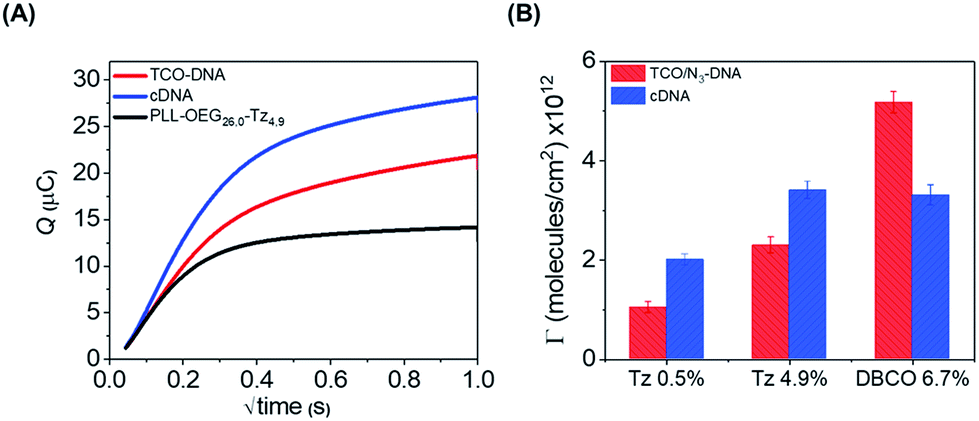 | ||
| Fig. 4 (A) Example of CC curves measured for the gold electrodes modified with 0.25 mg mL−1 of PLL–OEG(26.0)–Tz(4.9), 1 μM TCO-DNA and cDNA in the presence of 50 μM RuHex in 20 mM Tris buffer. (B) Surface coverage of Au electrode surfaces modified with PLL–OEG–Tz(0.5), PLL–OEG–Tz(4.9) and PLL–OEG–DBCO(6.7) and subsequently with TCO-DNA and N3-DNA, before and after hybridization with cDNA. | ||
Conclusions
In conclusion, a surface modification method has been developed, based on PLL polymers functionalized with catalyst-free click chemistry moieties, for a fast and selective immobilization of biomolecules. Gold and silicon dioxide surfaces have been functionalized selectively with DNA probe molecules in less than 20 min using two different coupling chemistries, and the modified surfaces showed hybridization with fully complementary DNA sequences while not responding to non-complementary DNA. The correlation between PLL functionalization and DNA density was assessed, resulting in a surface saturation with DNA for PLL functionalization above 5%. This surface modification method appears to be suitable for the modification of substrates for biomedical applications, in which biosensors can be developed with a precise control of the probe density. Furthermore, we believe that this method can be applied for fast surface immobilization of a several types of biomolecules, such as proteins and antibodies, thus allowing the recognition of a broad range of analytes in solution.Experimental
Materials
Phosphate-buffered saline tablets (PBS, pH 7.4), and poly-L-lysine–HBr (PLL–HBr) (15–30 kDa), DMSO ≥99.9% anhydrous, sodium dodecyl sulfate (SDS) ≥98.5%, and DBCO-OEG4-NHS were purchased from Sigma Aldrich and used without further purification. Methyl-OEG4-NHS was purchased from ThermoFischer Scientific, while methyltetrazine-OEG4-NHS was purchased from Click chemistry tools. TCO-DNA (15 nt, 5′-TCO-PEG4-C5-TCGTACCATCTATCC-3′) and N3-DNA (15 nt, 5′-Azido-PEG4-C5-TCGTACCATCTATCC-3′) were obtained from Biomers, cDNA (36 nt, 5′-CGCGGTCTCAGGATACCCCCCGGATAGATGGTACGA-3′) and ncDNA (36 nt, 5′AATGCTTCTCGCGCTTTTTTTTAGACTTCGCGCGTT-3′) sequences were purchased from Eurofins Genomic. Au and SiO2 QCM chips (AT cut, 5 MHz, 14 mm diameter) were purchased from Biolin Scientific. Milli-Q water with a resistivity >18 MΩ cm was used in all experiments.Synthesis of PLL–OEG–Tz
PLL–OEG(x)–Tz(y) with varying percentages of functionalization where synthesized based on a procedure reported by Duan et al. (see Scheme S1†).22 PLL was dissolved in PBS (pH = 7.2) at a concentration of 10 mg mL−1. NHS-OEG4-methyl and NHS-OEG4-methyltetrazine, both dissolved in DMSO at a concentration of 250 mM, were added to the PLL solution simultaneously under nitrogen atmosphere in desired ratios. The total volume of the mixture was adjusted with PBS in order to obey the maximum solubility of 1.8 mM of NHS-OEG4-methyltetrazine. The reaction was conducted for 4 h at room temperature under vigorous stirring. Afterwards, the reaction mixture was dialyzed with a dialysis membrane (cut-off 6–8 kDa) for 3 days against PBS and water. The solution was then freeze-dried overnight. The final product was stored at −20 °C as a stock solution of 10 mg mL−1 in Milli-Q water.1H NMR of PLL–OEG–Tz (400 MHz D2O) δ [ppm] = 1.26–1.56 (lysine γ-CH2), 1.61–1.82 (lysine β, δ-CH2), 2.49 (ethylene glycol CH2 from both OEG and Tz coupled, –CH2–C(=O)–NH), 2.99 (free lysine, H2N–CH2), 3.16 (ethylene glycol CH2 of coupled lysine from both OEG and Tz, C(=O)–NH–CH2–), 3.35 (OEG methoxy, –O–CH3), 3.58–3.79 (ethylene glycol from both OEG and Tz, CH2–O–), 4.29 (lysine backbone, NH–CH–C(O)–), 8.4 and 7.2 (tetrazine from coupled Tz, HCAr = CArH).
Synthesis of PLL–OEG–DBCO
PLL–OEG–DBCO was synthesized in a similar way as PLL–OEG–Tz (see Scheme S1†). NHS-OEG4-DBCO was dissolved in DMSO at a concentration of 50 mM, and this solution was added together with a solution of NHS-OEG4-methyl to the PLL solution simultaneously under nitrogen atmosphere in the desired ratio. The reaction was conducted for 4 h at room temperature under vigorous stirring. Afterwards, the reaction mixture was dialyzed with a dialysis membrane (cut-off 6–8 kDa) for 3 days against PBS and water. The solution was freeze-dried overnight. The final product was stored at −20 °C as a stock solution of 10 mg mL−1 in Milli-Q water.1H NMR of PLL–OEG–Tz (400 MHz D2O) δ [ppm] = 1.26–1.56 (lysine γ-CH2), 1.61–1.82 (lysine β, δ-CH2), 2.49 (ethylene glycol CH2 from both OEG and DBCO coupled, –CH2–C(=O)–NH), 2.99 (free lysine, H2N–CH2), 3.16 (ethylene glycol CH2 of coupled lysine from both OEG and DBCO, C(=O)–NH–CH2–), 3.35 (OEG methoxy, –O–CH3), 3.58–3.79 (ethylene glycol from both OEG and DBCO, CH2–O–), 4.29 (lysine backbone, NH–CH–C(O)–), 7.42 (DBCO from coupled DBCO, CArH).
Methods
Quartz crystal microbalance with dissipation monitoring (QCM-D)
QCM-D measurements were performed with a Q-Sense E4 4-channel quartz crystal microbalance with a peristaltic pump (Biolin Scientific). All experiments were performed in PBS buffer (0.01 M phosphate and 0.138 M NaCl and 0.0027 M KCl, pH 7.4) using a flow rate of 100 μL min−1 at 22 °C and operated with four parallel flow chambers.Gold surfaces were cleaned using basic Piranha solution (Milli-Q water: H2O2 (30%): NH4OH (25%), in ratio 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) for 5 min, then rinsed with Milli-Q water, dried in a N2 stream and treated with UV-ozone for 10 min. Silicon dioxide surfaces were submerged in 2% SDS in water and sonicated for 10 min. Afterwards, the surfaces were rinsed with Milli-Q water and activated for 20 min in UV-ozone.
:1:1) for 5 min, then rinsed with Milli-Q water, dried in a N2 stream and treated with UV-ozone for 10 min. Silicon dioxide surfaces were submerged in 2% SDS in water and sonicated for 10 min. Afterwards, the surfaces were rinsed with Milli-Q water and activated for 20 min in UV-ozone.
The relationship between the measured frequency shift (Δf) and the adsorbed mass per unit area (Δm) was established using the Sauerbrey equation (eqn (1)):
| Δf = −CΔm | (1) |
Electrochemical measurements
All electrochemical measurements were performed with a CHI 760D potentiostat (CH Instruments, Austin, Texas), connected to an electrochemical cell containing the modified gold chip as a working electrode, an Ag/AgCl reference electrode, and a platinum wire as a counter electrode. Chronocoulometry (CC) for the quantification of the DNA (TCO-DNA or N3-DNA) surface density and monitoring of the hybridization with the cDNA was carried out in 50 μM of RuHex in 20 mM Tris buffer (pH = 7.4). The buffer was purged with nitrogen for 15 min before the experiments. To obtain a stable signal, the solution was left to equilibrate for 5 min before each measurement. All measurements were taken under the same conditions where a two-step potential was employed with the following parameters: initial potential = −0.5 V; final potential = 0.2 V; pulse width = 1 s; pulse period = 1000 ms; sample interval = 0.002 s; quiet time = 2 s.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Marie Curie Innovative Training Network MULTI-APP (No. 642793) and by the MESA+ Institute for Nanotechnology (Early Diagnostics program).Notes and references
- H. Lee, J. Rho and P. B. Messersmith, Adv. Mater., 2009, 21, 431–434 CrossRef CAS PubMed.
- A. Kuzmin, A. Poloukhtine, M. A. Wolfert and V. V. Popik, Bioconjugate Chem., 2010, 21, 2076–2085 CrossRef CAS PubMed.
- L. Nebhani and C. Barner-Kowollik, Adv. Mater., 2009, 21, 3442–3468 CrossRef CAS.
- D. Samanta and A. Sarkar, Chem. Soc. Rev., 2011, 40, 2567–2592 RSC.
- L. A. Ruiz-Taylor, T. L. Martin, F. G. Zaugg, K. Witte, P. Indermuhle, S. Nock and P. Wagner, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 852–857 CrossRef CAS.
- Y. Liu and J. Yu, Microchim. Acta, 2016, 183, 1–19 CrossRef CAS.
- J. Escorihuela, A. T. M. Marcelis and H. Zuilhof, Adv. Mater. Interfaces, 2015, 2, 1500135 CrossRef.
- R. Manova, T. A. vanBeek and H. Zuilhof, Angew. Chem., Int. Ed., 2011, 50, 5428–5430 CrossRef CAS PubMed.
- M. A. Wijdeven, C. Nicosia, A. Borrmann, J. Huskens and F. L. van Delft, RSC Adv., 2014, 4, 10549–10552 RSC.
- D. Prim, F. Rebeaud, V. Cosandey, R. Marti, P. Passeraub and M. E. Pfeifer, Molecules, 2013, 18, 9833–9849 CrossRef CAS.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS.
- F. Ehret, H. Wu, S. C. Alexander and N. K. Devaraj, J. Am. Chem. Soc., 2015, 137, 8876–8879 CrossRef CAS.
- C.-J. Zhang, C. Y. J. Tan, J. Ge, Z. Na, G. Y. J. Chen, M. Uttamchandani, H. Sun and S. Q. Yao, Angew. Chem., Int. Ed., 2013, 52, 14060–14064 CrossRef CAS.
- O. Roling, A. Mardyukov, S. Lamping, B. Vonhören, S. Rinnen, H. F. Arlinghaus, A. Studer and B. J. Ravoo, Org. Biomol. Chem., 2014, 12, 7828–7835 RSC.
- M. Schena, D. Shalon, R. W. Davis and P. O. Brown, Science, 1995, 270, 467–470 CrossRef CAS.
- G. L. Kenausis, J. Vörös, D. L. Elbert, N. Huang, R. Hofer, L. Ruiz-Taylor, M. Textor, J. A. Hubbell and N. D. Spencer, J. Phys. Chem. B, 2000, 104, 3298–3309 CrossRef CAS.
- N.-P. Huang, R. Michel, J. Voros, M. Textor, R. Hofer, A. Rossi, D. L. Elbert, J. A. Hubbell and N. D. Spencer, Langmuir, 2001, 17, 489–498 CrossRef CAS.
- G. Zhen, V. Eggli, J. Vörös, P. Zammaretti, M. Textor, R. Glockshuber and E. Kuennemann, Langmuir, 2004, 20, 10464–10473 CrossRef CAS.
- S. VandeVondele, J. Vörös and J. A. Hubbell, Biotechnol. Bioeng., 2003, 82, 784–790 CrossRef CAS.
- J. Movilli, A. Rozzi, R. Ricciardi, R. Corradini and J. Huskens, Bioconjugate Chem., 2018, 29, 4110–4118 CrossRef CAS.
- Z. Han, Y. Wang and X. Duan, Anal. Chim. Acta, 2017, 964, 170–177 CrossRef CAS.
- X. Duan, L. Mu, S. D. Sawtelle, N. K. Rajan, Z. Han, Y. Wang, H. Qu and M. A. Reed, Adv. Funct. Mater., 2015, 25, 2279–2286 CrossRef CAS.
- C. Larsson, M. Rodahl and F. Höök, Anal. Chem., 2003, 75, 5080–5087 CrossRef CAS PubMed.
- X. Su, Y.-J. Wu and W. Knoll, Biosens. Bioelectron., 2005, 21, 719–726 CrossRef CAS PubMed.
- A. W. Peterson, R. J. Heaton and R. M. Georgiadis, Nucleic Acids Res., 2001, 29, 5163–5168 CrossRef CAS.
- J. Ji, Y. Liu, W. Wei, Y. Zhang and S. Liu, Biosens. Bioelectron., 2016, 85, 25–31 CrossRef CAS.
- R. Lao, S. Song, H. Wu, L. Wang, Z. Zhang, L. He and C. Fan, Anal. Chem., 2005, 77, 6475–6480 CrossRef CAS.
- B. Yao, Y. Liu, M. Tabata, H. Zhu and Y. Miyahara, Chem. Commun., 2014, 50, 9704–9706 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthetic procedures, QCM-D sensograms. See DOI: 10.1039/c9ra08714a |
| This journal is © The Royal Society of Chemistry 2019 |