
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient microwave synthesis of rhodanine and 2-thiohydantoin derivatives and determination of relationships between their chemical structures and antibacterial activity†
Waldemar Tejchman a,
Bartosz Orwat*bc,
Izabela Korona-Głowniakd,
Anna Barbasza,
Ireneusz Kownackibc,
Gniewomir Latacze,
Jadwiga Handzlike,
Ewa Żesławskaa and
Anna Malmd
a,
Bartosz Orwat*bc,
Izabela Korona-Głowniakd,
Anna Barbasza,
Ireneusz Kownackibc,
Gniewomir Latacze,
Jadwiga Handzlike,
Ewa Żesławskaa and
Anna Malmd
aDepartment of Chemistry, Institute of Biology, Pedagogical University of Cracow, Podchorążych 2, 30-084 Kraków, Poland
bFaculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland. E-mail: b.orwat@amu.edu.pl
cCentre for Advanced Technology, Uniwersytetu Poznańskiego 10, 61-614 Poznań, Poland
dDepartment of Pharmaceutical Microbiology, Medical University of Lublin, Chodźki 1, 20-093 Lublin, Poland
eDepartment of Technology and Biotechnology of Drugs, Jagiellonian University Medical College, Medyczna 9, 30-688 Kraków, Poland
First published on 29th November 2019
Abstract
Here we report studies on the synthesis of 12 new heterocyclic derivatives that differ in three structural motifs and the simultaneous evaluation of the impact of these three variables on the biological properties. The examined compounds are based on rhodanine and 2-thiohydantoin cores equipped with hydrogen or carboxymethyl substituents at the N-3 position and linked to a triphenylamine moiety through 1,4-phenylene, 1,4-naphthalenylene and 1,9-anthracenylene spacers at the C-5 position of the heterocycles. All the compounds were synthesized very quickly, selectively and in high yields according to the developed microwave-assisted Knoevenagel condensation protocol, and they were characterized thoroughly with NMR, FT-IR and ESI-HRMS techniques. The derivatives were tested for their activity against selected strains of Gram-positive and Gram-negative bacteria and yeast. Two compounds showed good activity against Gram-positive bacteria, and all of them showed low cytotoxicity against three cell lines of the human immune system. Based on membrane permeability assays it was demonstrated that the active compounds do not penetrate the cell membrane, and thus they must act on the bacterial cell surface. Finally, we proved that the evaluated structure modifications had a synergistic effect and the simultaneous presence of a 1,4-phenylene spacer and carboxymethyl group at N-3 caused the highest boost in antimicrobial activity.
Introduction
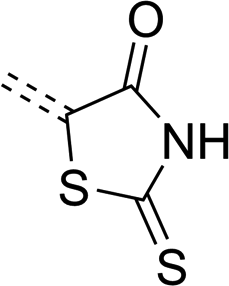
Throughout evolution, many pathogenic microorganisms have gained resistance to antibiotics, and this constitutes a threat to public health.1 This ongoing process is a serious challenge for modern medicinal chemistry, but it also stimulates development in the field. For this reason, many research groups seek new substances that can efficiently act against pathogenic microorganisms without posing a risk to patients. Among the pathogens, many dangerous ones belong to the kingdom of bacteria. These organisms can be classified into two groups based on the Gram staining result that indicates differences in their structure, physiology and drug resistance. Gram-negative bacteria have an outer membrane of their cell walls that makes it difficult for antibiotics to penetrate into the cell.2–4 In addition, in the periplasmic space located between the outer and the inner membrane, there are enzymes that deactivate antibiotics.5 Gram-positive bacteria do not have an outer membrane, but the structure of their peptidoglycan is more complicated than in Gram-negative bacteria.6 The lack of an outer membrane facilitates the penetration of antibiotics into the cells and increases the number of molecular targets on the cell wall surface that can be attacked by substances with antibacterial activity.There are well-known and particularly dangerous lethal diseases such as tuberculosis, leprosy and gas gangrene which are caused by Gram-positive bacteria; cholera, brucellosis, plague, syphilis and salmonellosis that are caused by Gram-negative bacteria. In the search for a cure against them, many scientists have synthesized various heterocyclic compounds and studied their bactericidal properties.7,8 Among others, derivatives of five-membered heterocyclic systems based on the core of 1,3-thiazolidine or imidazolidine have been studied due to their broad biological activity.9,10 Of particular interest are their derivatives containing exocyclic sulfur and oxygen atoms, such as rhodanine (2-sulfanylidene-1,3-thiazolidin-4-one)11 or 2-thiohydantoin (2-sulfanylidene-1,3-diazolidin-4-one).12 The presence of these double-bonded atoms with other functional groups results in particularly high density of binding sites for polar interactions and hydrogen bonds, which are responsible for their interesting biological properties.13 A spectrum of their biological activity includes antifungal,14,15 anticancer,16–18 antidiabetic,19,20 anti-inflammatory,21 antiviral,22,23 anticonvulsant24–26 and enzyme-inhibiting properties.27,28 Another noteworthy feature of such cyclic systems is their strong polar and electron-withdrawing character, making them popular anchoring and electron-accepting groups in construction of merocyanine dyes for organic dye-sensitized solar cells (DSSC).29–31 Additionally, the popularity of these heterocycles as building blocks is supported by the feasibility of their multi-functionalization in various positions (Scheme 1).
 | ||
| Scheme 1 General structure of rhodanine (left) and 2-thiohydantoin (right). R groups mark positions of their potential functionalization. | ||
In our previous research we proved that antibacterial activity of the examined rhodanine-3-carboxylic acid series were strengthened with the increase in hydrophobic character of the substituent in position 5.32 Moreover, the lowest minimal inhibitory concentration (MIC) and minimal bactericidal concentration (MBC) were observed for derivatives equipped with a triphenylamine moiety. This result indicated that a coexistence of aromatic rings and multi-interacting heterocycles within the same molecule might be a key requirement for enabling its activity. This conclusion prompted us to explore the topic for similar reports in the literature. In the search, we encountered some works describing activity of familiar derivatives. Tomašić et al. also reported that the increase of hydrophobicity of a benzylidene substituent in the rhodanine C-5 position led to an increase in antimicrobial activity.33 Among all examined compounds, 5-(2,3,4-trifluorobenzylidene)-rhodanine exhibited the highest activity, inhibiting Staphylococcus aureus and MRSA growth at concentrations of 0.5 and 32 mg L−1, respectively. A series of substituted rhodanines bearing benzylidene-derived fragments of various steric hindrance in position 5 showed also activity towards MRSA.34 Derivatives of hydantoin, an oxygen analogue of 2-thiohydantoin bearing a bulky 4-methyl-2-phenyl-furo[(3,2-d)pyrazole(3,2-d)imidazole] moiety in C-5, also showed antimicrobial activity against Escherichia coli and Staphylococcus aureus.35 Furthermore, it was proved that the steric and electronic properties of the C-5 substituent affect bactericidal activity in the series of various chlorinated 5-arylidene rhodanines.36 It is noteworthy that compounds containing lipophilic, condensed aromatic moieties like naphthalene might exhibit better antibacterial activity than those containing unfused aromatics in the C-5 position (e.g. 1,1′-biphenyl or p-methoxyphenylazosalicylidene moiety).37,38 The incorporation of such moieties might also affect other biological properties. Sing et al. proved that the incorporation of larger and more hydrophobic substituents in the C-5 position of rhodanine 3-acetic acid can increase HCV NS3 protease inhibition selectivity up to 25 times compared to a derivative bearing biphenyl moiety.39 It is therefore prerequisite that the size and lipophilicity of C-5 substituents can modulate biological activity of 5-substituted rhodanine and its analogues. We mentioned before that the C-5 substituent effect might not be the only parameter modulating activity. As Trotsko et al. showed, antibacterial activity of 2-thiohydantoin derivatives was slightly higher than the corresponding rhodanine derivatives.40 Consequently, we believe that the heterocycle is another binding point for interaction with enzymes or receptors, and that its core structure might alter the final compound's activity. Finally, the substituent in the N-3 position is also very important. As Sim et al. observed, rhodanine derivatives containing alkyl or aryl groups in this position do not show antibacterial activity. The highest antibacterial activity is exhibited in compounds equipped with hydrogen or carboxylic acid in the N-3 position.34 To sum up, we are convinced that in order to design a substance with high antibacterial activity, the total effect of three variable points, namely the C-5 substituent, the heterocycle core structure and the N-3 substituent, should be taken into account. These variables should be examined together within the same molecule since they might synergistically influence binding to active centers, but we did not find such an approach for rhodanine and 2-thiohydantoin derivatives in the literature.
Due to the lack of thorough evaluation of aromatic linker size effect evaluation on the biological properties of rhodanine and 2-thiohydantoin derivatives, we decided to fill in this gap. Therefore, we designed and synthesized a series of merocyanine dyes consisting of the mentioned heterocycles with a triphenylamine moiety bonded through a hydrocarbon aromatic linker in position C-5. For this purpose, 1,4-phenylene, 1,4-naphthalenylene and 1,10-anthracenylene were selected as the simplest homologue linkers. Guided by the literature, we have undertaken to find a cumulative effect of linker size, heterocycle core component and ancillary N-3 substituent (Scheme 2) on biological properties. Our goal was to determine how the variation of the three interacting points influence antibacterial activity and cytotoxicity of the tested compounds, and to shed some light on their mechanisms of activity.
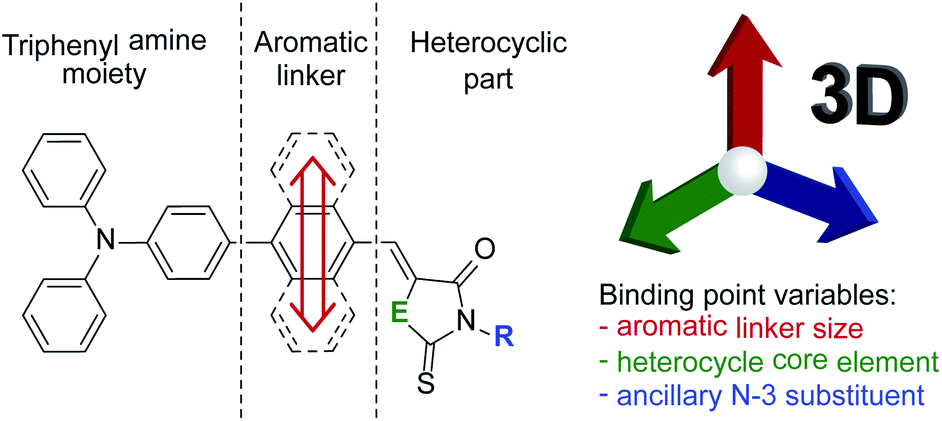 | ||
| Scheme 2 Illustration of the concept and the differences between the compounds investigated in this work. | ||
Results and discussion
Synthesis
The proposed synthetic protocol for the target molecules was based on Knoevenagel condensation of 4′-(diphenylamino)-[1,1′-biphenyl]-4-carbaldehyde (1a), 4-(4-(diphenylamino)phenyl)-1-naphthalenecarbaldehyde (1b) and 10-(4-(diphenylamino)phenyl)anthracene-9-carbaldehyde (1c) with rhodanines (2a, 2b) and thiohydantoins (3a, 3b). The former aldehydes were prepared in the course of a Suzuki–Miyaura cross-coupling reaction, the most commonly used of the palladium-mediated reactions in the synthesis of active pharmaceutical ingredients (APIs).41,42 Therefore, in the first step of synthetic work, 4-(diphenylamino)phenylboronic acid, 4-bromo-1-naphthalenecarbaldehyde and 10-bromo-9-anthracenecarbaldehyde were prepared with slightly modified literature methods.43–45 Then we started the synthesis of the intermediate aldehydes 1a–c. At the beginning we decided to examine the reliability of the reported synthetic protocol for 1a.46 To our surprise, we obtained the product in 70% yield, which differs significantly from the reported value of 98%. Moreover, this method proved to be completely ineffective in the coupling of the two other bromoarenecarbaldehydes. We suppose that this was due to their low solubility in the 2-propanol/water reaction medium used. Even after raising the reaction temperature to 100 °C, there were formed sticky clumps which probably hampered further progress of the reaction. In the face of this obstacle, we decided to use a different solvent system to facilitate the solubility of the reactants. Fortunately, changing the reaction medium to a mixture of toluene, ethanol and 2 M aqueous K2CO3 solution enabled us to efficiently perform all cross-coupling reactions with very good yields (Scheme 3). As a result, aldehydes 1a–c were isolated by the means of flash chromatography purification in the form of yellow or orange solids.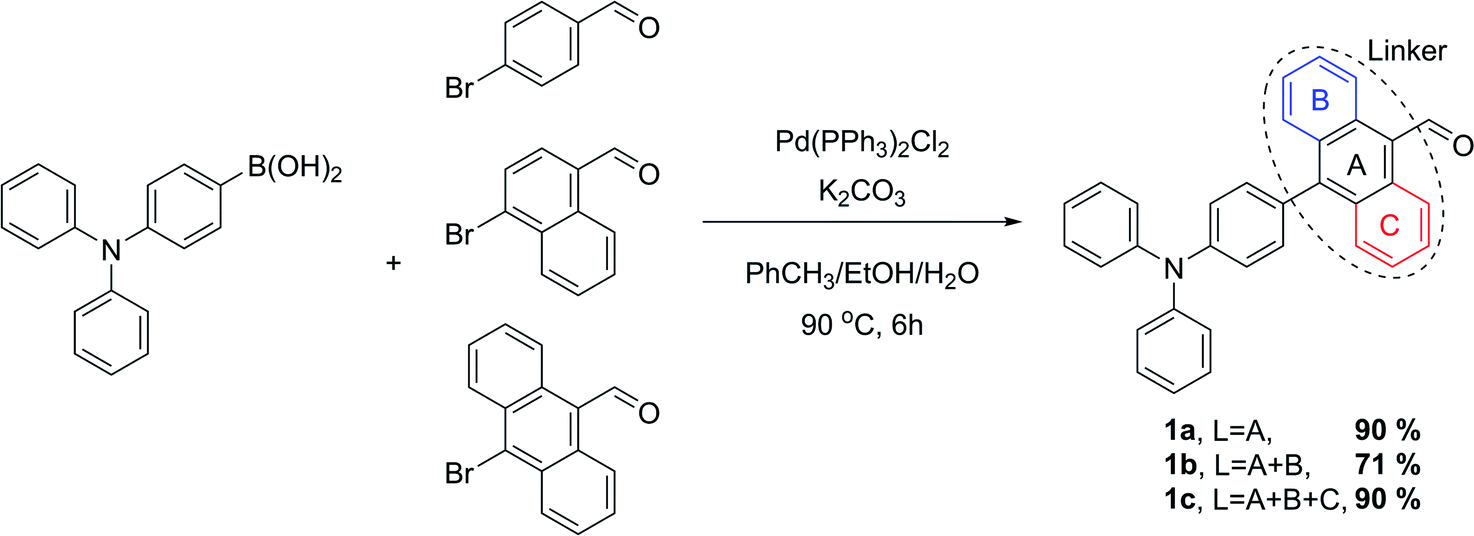 | ||
| Scheme 3 Synthesis of the aldehyde intermediates through Suzuki–Miyaura cross-coupling. | ||
After successful preparation of intermediates 1a–c, we focused on the Knoevenagel condensation of the aldehydes with selected heterocycles. Such transformation is a well-known method for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond formation; however, the results of this reaction are not always satisfactory. Additionally, in the most reported protocols, prolonged heating of the reaction mixture in reflux conditions followed by recrystallization of the product were required, resulting in medium yields.47–49 During our thorough literature review, we encountered the promising concept of microwave-accelerated Knoevenagel condensation.50 Because this unconventional heat source has recently been proved to increase the efficiency of various catalytic and stoichiometric reactions in our group,51 we decided to examine its effectiveness in our current work. Preliminary tests in which anthracene-9-carbaldehyde and rhodanine were involved as a model reagent system confirmed the superior efficiency of microwave irradiation compared to a classical oil bath. Thus, we decided to apply the former as a heat source in our synthetic protocol. The first reported reactions were conducted with rhodanines 2a and 2b according to Scheme 4.
C bond formation; however, the results of this reaction are not always satisfactory. Additionally, in the most reported protocols, prolonged heating of the reaction mixture in reflux conditions followed by recrystallization of the product were required, resulting in medium yields.47–49 During our thorough literature review, we encountered the promising concept of microwave-accelerated Knoevenagel condensation.50 Because this unconventional heat source has recently been proved to increase the efficiency of various catalytic and stoichiometric reactions in our group,51 we decided to examine its effectiveness in our current work. Preliminary tests in which anthracene-9-carbaldehyde and rhodanine were involved as a model reagent system confirmed the superior efficiency of microwave irradiation compared to a classical oil bath. Thus, we decided to apply the former as a heat source in our synthetic protocol. The first reported reactions were conducted with rhodanines 2a and 2b according to Scheme 4.
 | ||
| Scheme 4 Knoevenagel condensation of rhodanines (2a, 2b) with aldehydes (1a, 1b, 1c). | ||
During the synthesis of the desired compounds, we observed that the reactions started almost immediately at elevated temperature, evidenced by the change of reaction mixture color mostly from yellow to intense red or orange. At this point, we noticed that the progress of the condensation correlated with the size of the aromatic linker. The reason for this might be the increase in steric hindrance in the surrounding of the formyl group from 1a to 1c, resulting in a decrease in yield from 4a to 4c and 5a to 5c. Because of this, reaction time was extended to 6 and 7 minutes for aldehydes bearing 1,4-naphthalenylene and 9,10-anthracenylene linkers in their structures, respectively. Nevertheless, a slight trend in decreasing yield was still observed.
The same reaction conditions were applied in the condensation processes involving 2-thiohydantoins 3a and 3b. It is worth emphasizing that in the condensations involving 3a, the substrate was equipped with an acetyl group in position N-1 due to superior feasibility of its synthesis compared to related unprotected 2-thiohydantoin.52 However, the acetyl group was cleaved in situ in an acidic environment during the Knoevenagel reaction, yielding N-1 deprotected derivatives of 2-thiohydantoin (Scheme 5).
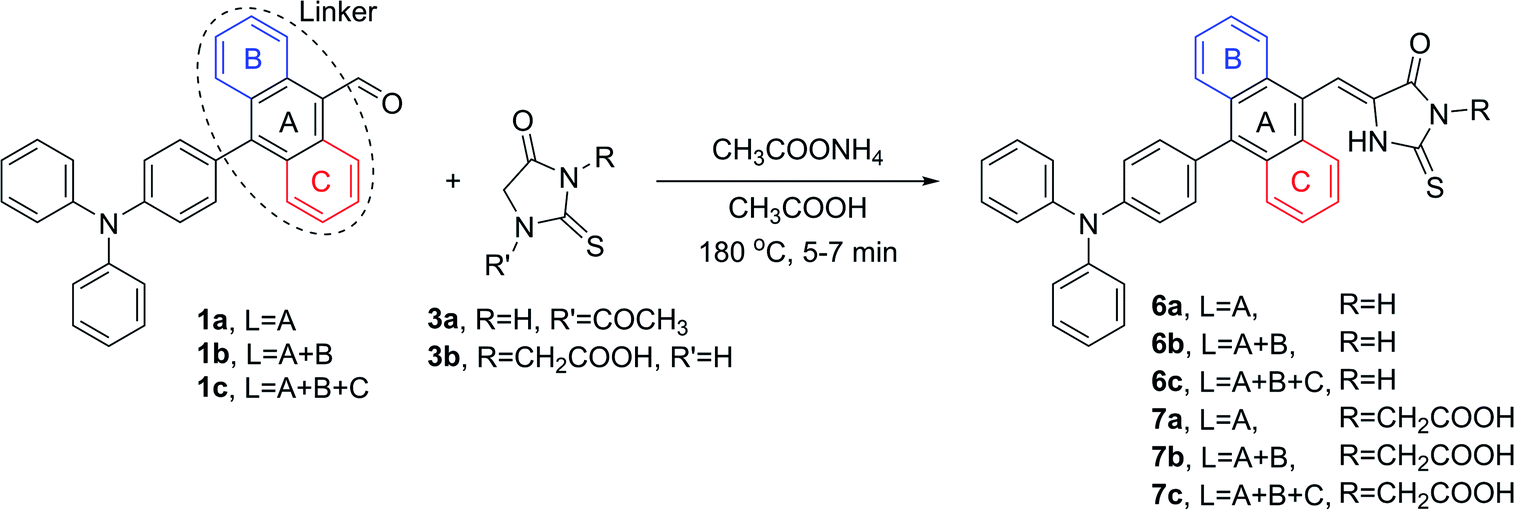 | ||
| Scheme 5 Knoevenagel condensation of 2-thiohydantoins (3a, 3b) with aldehydes (1a, 1b, 1c). | ||
Table 1 summarizes the yields of all isolated target compounds. It is evident that 2-thiohydantoin derivatives are characterized by lower yields than rhodanines. This difference is most likely due to the replacement of the less electronegative sulfur atom in position 1 for an –NH– moiety. Such a trend is more pronounced for the compounds bearing a 1,4-phenylene and 9,10-anthracenylene linker in their structures than those comprising a 1,4-naphthalenylene moiety. It should be emphasized that all the compounds were obtained with good to excellent yields, and most were isolated by simple filtration and rinsing. In the case of compounds 6c and 7c, conversion of the starting aldehyde was insufficiently low, causing contamination of the crude product with an observable amount of unreacted 1c that could not be separated by simple rinsing of the obtained solid. This was probably caused by π–π stacking of condensed aromatic linker that is more favored with the increase in aromatic moiety size. It may also explain the lack of contamination problems in the case of products bearing smaller aromatic linkers in their structures. Nonetheless, this problem was solved by simple recrystallization. In this way we have finally developed a successful protocol for the synthesis of the desired compounds, providing ourselves materials for further studies of their biological activity. Additionally, aldehydes 1a–c and heterocyclic derivatives 4a–7c were fully characterized by nuclear magnetic resonance spectroscopy (NMR), high resolution mass spectrometry (HRMS) and Fourier-transform infrared spectroscopy (FTIR), providing high quality identification of the new compounds. All spectra were included in the ESI.†
| Molecule's fragment |
|
|
|
|---|---|---|---|
 |
96% (4a) | 94% (4b) | 90% (4c) |
 |
94% (5a) | 90% (5b) | 89% (5c) |
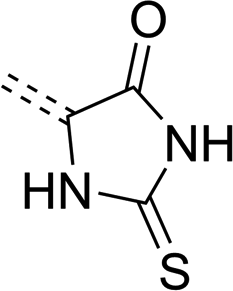 |
72% (6a) | 86% (6b) | 74% (6c) |
 |
84% (7a) | 85% (7b) | 74% (7c) |


Antibacterial activity
In order to examine biological activity of the synthesized compounds, MIC and MBC values were determined to quantify their influence on selected strains of Gram-positive and Gram-negative bacteria, as well as yeast. Escherichia coli ATCC25922, Salmonella typhimurium ATCC14028, Klebsiella pneumoniae ATCC13883, Pseudomonas aeruginosa ATCC9027 and Proteus mirabilis ATCC12453 were selected as model strain of Gram-negative bacteria. Staphylococcus aureus ATCC25923, Staphylococcus aureus ATCC6538, Staphylococcus aureus ATCC43300, Staphylococcus epidermidis ATCC12228, Micrococcus luteus ATCC10240, Bacillus subtilis ATCC6633, Bacillus cereus ATCC10876, Streptococcus pyogenes ATCC19615, Streptococcus pneumoniae ATCC49619 and Streptococcus mutans ATCC25175 were selected as Gram-positive bacteria. Finally, the yeast Candida albicans ATCC10231, Candida albicans ATCC2091, Candida parapsilosis ATCC22019, Candida glabrata ATCC 90030 and Candida crusei ATCC14243 were selected. Ciprofloxacin and vancomycin were used as reference positive controls (Table 3S†). None of the tested compounds demonstrated activity against Gram-negative bacteria and yeast (MIC > 1000 mg L−1, data not shown). Tables 1S and 2S,† containing a complete set of measured data against Gram-positive bacteria, are included in the ESI.†Comparison of the data compiled in Tables 1S and 2S† prompted us to conclude that rhodanine derivatives are more biologically active than 2-thiohydantoin derivatives in general. Among all tested strains, M. luteus ATCC 10240 appeared to be the most sensitive to the examined compounds, while S. mutans ATCC 25175 was the most resistant (sensitive only to 7a however, in the high concentration of 62.5 mg L−1). As one can see, compounds 5a and 7a are characterized by significantly outstanding values of the determined parameters. Activity of these compounds is discussed in detail below.
Fig. 1 presents the activity of compound 5a. MIC values for 5a range from 0.98 mg L−1 for S. epidermidis ATCC12228 and M. luteus ATCC10240, to a level of 1000 mg L−1 against M. mutans ATCC25175 (not presented in the chart to maintain clarity). MBC values were also determined in order to establish whether the examined substance has bacteriostatic or bactericidal effect. For most strains, MBC was significantly higher than MIC, allowing us to assume less bactericidal activity. For this quantification the MBC/MIC ratio was calculated, since if it is ≤4, the examined compound is recognized as bactericidal.53 5a showed the satisfactory MBC/MIC ratios against B. subtilis ATCC 6633, S. aureus ATCC 6538, S. aureus ATCC 43300 and B. cereus ATCC 10876, while for the other strains the determined ratios were in the range of 8–32.
 | ||
| Fig. 1 Activity of 5a against selected strains of Gram-positive bacteria. | ||
Fig. 2 presents activity of 7a with exclusion of data for S. mutans ATCC 25175 (MBC > 1000 mg L−1). Measured MIC values range from 1.95 mg L−1 against S. aureus ATCC6538, S. aureus ATCC43300, S. epidermidis ATCC12228 and M. luteus ATCC10240 to 125 mg L−1 for B. cereus ATCC10876. The corresponding MBC values range from 3.9 mg L−1 to 250 mg L−1, which indicates a less varied response of 7a compared to 5a. For the nine presented strains, MIC/MBC ratios were not satisfactory only in the case of M. luteus ATCC 10240, which had a value of 16.
 | ||
| Fig. 2 Activity of 7a against selected strains of Gram-positive bacteria. | ||
Comparison of the MBC/MIC ratios, determined for the two most active compounds, leads to the conclusion that 7a has better bactericidal characteristics than 5a. In the case of 7a, values of MBC/MIC ratio usually range from 1 to 4, while for 5a the typical range is from 1 to 32. However, compound 5a is generally characterized by lower MIC values, proving its higher inhibitory activity. It should be emphasized that the mentioned differences are caused only by the replacement of the sulfur atom with the –NH– moiety in position 1 of the heterocycle. Another noted relationship is the influence of a carboxymethyl substituent on MIC and MBC values. If we compare 4a vs. 5a and 6a vs. 7a, it is seen that the presence of the mentioned substituent in position 3 of the heterocycle has an enormous boosting effect on biological activity. This very significant observation might serve as confirmation of the role of the carboxylic group in the activity of the examined compounds. Our findings might be supported by the results of Talele et al., who investigated the antiviral activity of 5-(3-phenoxybenzylidene) rhodanine-3-acetic acid derivatives. Their target was HC5 hepatitis C virus (HCV) polymerase.54 They observed that a carboxylic group in the N-3 position of the rhodanine ring formed a hydrogen bond with the guanidine group of arginine. The exocyclic oxygen atom in the C-4 position of the rhodanine ring also took part in the formation of hydrogen bonds. Based on the former, we suppose that rhodanine and 2-thiohydantoin derivatives without a carboxyl group in the N-3 position have fewer binding centers and thus cannot produce sufficiently strong hydrogen bonds with amino acid side chains present in the enzymes/receptors. In consequence, their antibacterial activity is much lower than the derivatives that can form hydrogen bonds.
A previously examined 5-(4′-N,N-diphenylaminobenzylidene)-rhodanine-3-acetic acid demonstrated biological activity (MIC 1.95–500 mg L−1, MBC/MIC 2–4) similar to the activity of 5a.32 The chemical structures of both compounds differed by the presence of an additional 1,4-phenylene linker between the triphenylamine and rhodanine moieties. Introducing the phenylene linker to the structure of 5-(4′-N,N-diphenylaminobenzylidene)-rhodanine-3-acetic acid resulted in a slight increase in the biological activity, especially against S. pyogenes ATCC19615 and S. pneumoniae ATCC49619. However, bactericidal properties of the obtained derivative became lower at the same time. Further increase in the aromatic linker size (1,4-naphthalenylene or 9,10-anthracenylene) resulted in a decrease or even total block of biological activity. Unfortunately, we could not perform a similar comparison for 7a since analogous 5-(4′-N,N-diphenylaminobenzylidene)-2-thiohydantoin-3-acetic acid has not been reported in literature.
Based on these results, we concluded that the presence of both a carboxymethyl group in the N-3 position and a triphenylamine moiety connected through a 1,4-phenylene linker in the C-5 position are crucial for the desirable biological activity of rhodanine and 2-thiohydantoin derivatives. Moreover, the activity can be modulated to some extent with introduction of an aromatic linker.
Cytotoxicity toward human immune cell lines
After evaluation of the antibacterial properties of 4a–7c, we focused our research on their influence on three human immune system cell lines. The immune cells present in the body are responsible for protection against infections caused by microorganisms or xenobiotics; thus, potential antimicrobial compounds should be characterized by low cytotoxicity towards immune cells in order to support its efficacy. The group of three chosen cell lines consisted of U-937 monocytic cells, a non-adherent cells differentiable to monocytes and macrophages;55,56 T-cell lymphoma HUT-78 line, exhibiting the features of a mature T-cell line with inducer/helper phenotype57 and the B lymphoblastoid COLO-720L cell line, growing in the form of characteristic clumps.58 The results are presented below in the graphical form of charts, while the raw data are grouped in Table 4S.†Fig. 3 (top) presents the LD50 of rhodanine derivatives against the examined cell lines. As can be clearly seen, the cytotoxicity for series 4a–c is inversely proportional to the increase of the aromatic linker size, for all cell lines. In contrast, introduction of an acidic moiety in position N-3 results in higher cytotoxicity in general. In the series 5a–c, LD50 is less diversified with only one anomalously high value of 5a for the HUT-78 line. However, the origin of this deviation remains unknown. In the case of rhodanine derivatives, the highest cytotoxicity was observed for compounds bearing a 1,4-phenylene linker, for all cell lines (except for the mentioned example of 5a against the HUT-78 cell line).
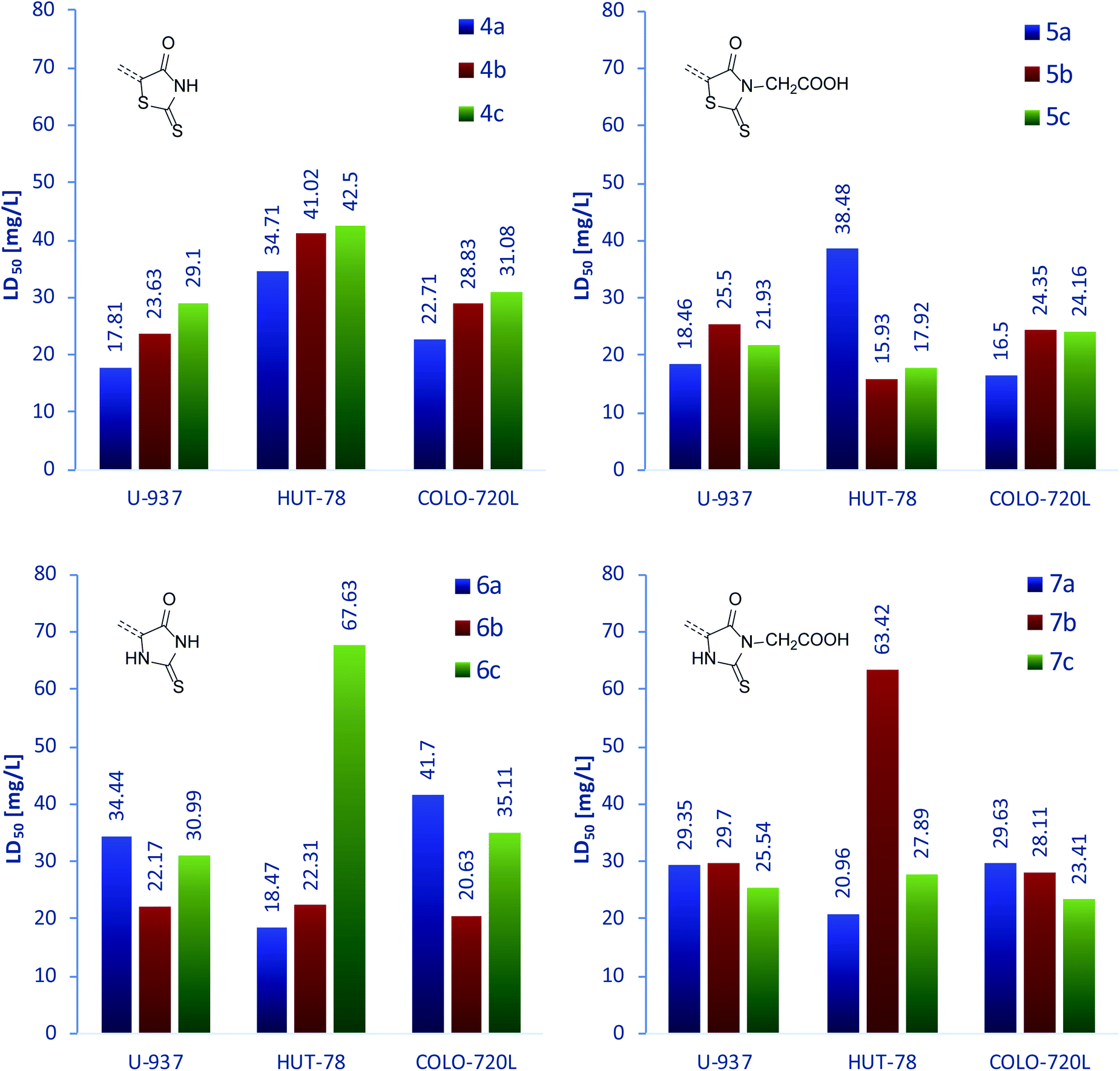 | ||
| Fig. 3 LD50 of rhodanine (4a–c, top left), rhodanine-3-acetic acid (5a–c, top right), 2-thiohydantoin (6a–c, bottom left), and 2-thiohydantoin-3-acetic acid (7a–c, bottom right) derivatives, determined for three human immune system cell lines. | ||
A similar set of data for the derivatives of 2-thiohydantoin is presented in the Fig. 3 (bottom). The demonstrated compounds show more varied cytotoxicity with respect to the rhodanine analogs. Compounds 6a–c are characterized by the same relative values of LD50 against U-937 and COLO-720L lines; however, their activity towards HUT-78 line does not follow these trends. Interestingly, compounds 7a–c exhibited similar behavior. Moreover, the highest LD50 values from all the compounds were recorded for 6c and 7b against the HUT-78 line, representing the highest anomalies in the 6a–c and 7a–c series. Leaving behind these exceptions, 2-thiohydantoin derivatives 6b, 6c, 7b and 7c showed almost unchanged cytotoxicity against the three examined cell lines.
The measured LD50 for line U-937 was between 17.81 and 34.44 mg L−1; for cell line HUT-78 between 15.93 and 67.63 mg L−1; and for cell line COLO-720L it was between 21.34 and 44.58 mg L−1. As demonstrated, the examined compounds were shown to be the most cytotoxic against U-937 line and the least toxic against HUT-78 line. Moreover, the latter cell line showed significantly variable response to treatment with 4a–7c, thus proving its sensitivity towards the chemical structure of the examined compounds. However, no regularity was observed. In contrast to that, cytotoxicity towards U-937 and COLO-720L was much less variable. Notwithstanding, we observed very similar trends of activity towards these two cell lines for compounds bearing 1,4-phenylene (compounds a) and 9,10-anthracenylene (compounds c) linkers. It is also worth noting that among compounds 4–7c, derivatives containing a carboxymethyl group in the N-3 position always exhibited higher cytotoxicity toward all examined cell lines, compared to derivatives with a hydrogen substituent in the same position.
To sum up this section, we conclude that all examined compounds showed very low cytotoxicity towards the examined U-937, HUT-78 and COLO-720L cell lines. For all the compounds, significantly higher LD50 values than MIC values were measured for compounds 5a and 7a, thus confirming their selective toxicity against bacteria and non-toxicity towards selected human immune system cells. Based on the results obtained so far, we decided to briefly explore their probable mechanisms of action.
Membrane permeability
Our attention was drawn to the differences in general activity of 4a–7c against various microorganisms. The fact that the compounds affected only Gram-positive bacteria, and not Gram-negative bacteria or yeast, allowed us to speculate about the mechanism of interaction. According to the literature, potential antibiotics can interfere with various molecular targets, causing malfunction of pathogens' systems.59 These interactions can be classified into five main categories: (1) inhibition of specific enzymes (especially gene expression enzymes e.g., topoisomerases); (2) inhibition of RNA synthesis; (3) inhibition of protein synthesis; (4) inhibition of cytoplasmic membrane precursor synthesis; and (5) blocking the functions of the cytoplasmic membrane. The first four actions can occur only if the antibiotic is successfully transferred through the cell membrane and into the cytoplasm. In contrast, the last process requires antibiotic molecules to bind to receptors or enzymes present on the cell surface, as penicillin and other β-lactam antibiotics do. In the search for potential targets for rhodanine and 2-thiohydantoin derivatives, we referred to the literature.In almost all bacteria, the main component of the cell wall is peptidoglycan. This polymer protects the cell from changes in osmotic pressure as well as physical, chemical and mechanical agents. Therefore, disturbance of its synthesis or rendering it dysfunctional might induce the cell's death.60,61 Mendgen et al. examined the inhibitory effect of 5-arylideno rhodanines and 2-thiohydantoins towards MurA, one of the enzymes responsible for peptidoglycan synthesis.62 They discovered that their activity might be caused by the special electronic properties conferred by the exocyclic double-bonded sulfur atom, empowering molecule-to-enzyme binding. Another important enzyme, MurB reductase, was also examined regarding its interaction with 5-substituted rhodanines. In the reported theoretical studies, it was shown that molecular docking might be particularly preferred in the case of naphthyl-derived rhodanines due to their interaction with the lipophilic area of the enzyme's active site. On the other hand, the disturbance of peptidoglycan synthesis might also occur on the cell's surface as a result of interaction with penicillin-binding proteins (PBPs). These proteins catalyze the final steps of peptidoglycan synthesis (maturing), thus affecting its proper construction.63 Disturbance of such a process was examined for 5-arylalkylidenerhodanines and 5-arylalkylidene-2-iminothiazolidin-4-ones, and it proved to inhibit cell wall growth in Gram-positive bacteria in the case of some tested compounds.64 As one can see, it is possible that the compounds examined in our work can hamper the peptidoglycan synthetic pathway due to structural similarity.
In order to test whether our compounds inhibit the activity of enzymes operating in the cytoplasm, or rather act only on the cell's surface, we performed a parallel artificial membrane permeability assay (PAMPA) test for several of them. We chose the most active compound 5a along with 5b and 5c in order to verify any possible relationship between linker size and membrane permeability. The results of the studies clearly showed that none of the tested compounds had the ability to penetrate the cell membrane. During PAMPA tests we observed that the examined compounds underwent isomerization process which might be attributed to the sunlight irradiation65,66 or the interaction with the membrane. Due to the structural similarities of all studied compounds, we suppose that none of 4a–7c will be able to reach the interior of the cells. Despite the inability to penetrate the cell wall, compounds 5a and 7a showed high antibacterial activity. Based on the PAMPA test results, their Gram-positive antibacterial activity must be due to outer-cell interaction. We suspect that the compounds exhibiting antibacterial activity may bind to the active centers of the enzymes responsible for the cross-linking of the peptidoglycan structure, causing them to inactivate and consequently impair the protective function of the cell wall. This may particularly occur for derivatives containing a carboxymethyl group in the N-3 position, a group capable of forming hydrogen bonds with nucleophilic sites of the enzymes. However, a necessary condition also appears to be the presence of a 1,4-phenylene linker in combination with a triphenylamine moiety, which results in a good fit of the entire C-5 substituent to the hydrophobic pocket of the enzyme. If both conditions are met, the change of –S– to –NH– group in the C-1 position of the heterocycle shows a little effect on MIC reduction and a significant improvement in bactericidal properties by reducing the MIC/MBC ratio. The significantly lower antibacterial activity of the other derivatives, or even its lack, confirms the key role of the abovementioned structural motifs. In addition, significantly lower activity of compounds subjected to a single structure modification in reference to 5a and 7a indicates a synergistic effect of the three structural variables. Due to inactivity of 4a–7c towards Gram-negative bacteria, we believe that these compounds are neutralized within the periplasmic space.
Conclusions
In summary, we obtained two series of new rhodanine and 2-thiohydantoin derivatives containing a triphenylamine moiety, differing in the heterocyclic core as well as the number of condensed aromatic rings in the linker and the N-3 substituent of the heterocycles. The target compounds were obtained in a two-step process involving Suzuki–Miyaura cross-coupling and highly efficient microwave-assisted Knoevenagel condensation. In the course of further research, we determined antibacterial properties, cytotoxicity against human immune system cells and permeability through cell membranes. The most active turned out to be derivatives containing a 1,4-phenylene moiety, and as the size of the aromatic linker increased, their activity decreased noticeably. In addition, the antibacterial properties of the compounds bearing a 1,4-phenylene linker were significantly enhanced by introducing the carboxymethyl substituent in the N-3 position, proving the synergy of these structural modifications. Among the tested compounds, 5a and 7a showed the highest activity, while rhodanine derivative 5a was characterized by a higher activity in the inhibition of the bacterial growth, whereas 2-thiohydantoin derivative 7a was more bactericidal. In addition, the cytotoxicity of the examined compounds against three cell lines of the human immune system: U-937, HUT-78 and COLO-720L was found to be very low. PAMPA tests excluded the ability of the compounds to penetrate the cells which suggested that the activity of 4a–7c is associated only with their interaction with molecular targets located on the surface of the cell membrane. A more detailed explanation of the antimicrobial mechanism of activity of compounds 5a and 7a will constitute the next stage of our work.Experimental section
General information
Acetic acid (AA), acetic anhydride, acetonitrile (ACN), aminoethanoic acid, ammonium thiocyanate, carbon disulfide, chloroacetic acid, ethanol, dichloromethane (DCM), N,N-dimethylformamide (DMF), methanol, pentane, tetrahydrofuran (THF), toluene, 4-bromo-N,N-diphenylaniline, n-butyllithium (1.6 M solution in hexanes), trimethyl borate, triphenylphosphine, naphthalene, bromine, potassium carbonate, potassium hydroxide, sodium hydroxide and 9-anthracenecarbaldehyde were obtained from Sigma-Aldrich; 2-propanol (IPA), hexanes, ethyl acetate, 4-bromobenzaldehyde and palladium(II) chloride from POCH, while deuterated solvents were purchased from Deutero GmbH and Sigma-Aldrich. Rhodanine,67 rhodanine-3-acetic acid,68 1-acetyl-2-thiohydantoin,50,69 2-thiohydantoin-3-acetic acid70 and dichlorobis (triphenylophosphine)palladium(II)71 were prepared following the cited papers. 4-(diphenylamino)phenylboronic acid, 1,4-dibromonaphthalene, 4-bromo-1-naphthalenecarbaldehyde and 10-bromo-9-anthracenecarbaldehyde,72,73 were synthesized according to the published procedures with slight modifications.Lithiation reactions and Suzuki–Miyaura couplings were performed under an inert atmosphere, using standard vacuum-gas manifold (99.999% argon). For these transformations, all reagents and solvents were deoxygenated and moreover, DMF and THF were dehydrated. DMF was dried over calcium hydride, while THF over sodium/benzophenone, and both distilled prior to use. Other reactions were conducted under air atmosphere without special precautions, using reagents and solvents as received from the suppliers. Reactions accelerated with microwave irradiation were conducted in CEM microwave 10 mL vials with CEM Discover, a pressurized microwave reactor.
The reaction progress was monitored by means of a gas chromatograph (Bruker 450-GC) coupled to a mass detector (Bruker MS-320) and thin layer chromatography (TLC). Chemical structures of the obtained compounds were confirmed by nuclear magnetic resonance (NMR) spectroscopy using Bruker Ultrashield 300 MHz and Bruker Ascend 600 MHz spectrometers. CDCl3, CD2Cl2 and DMSO-d6 were used for NMR analysis with residual solvent peak as the internal standard (chloroform 1H δ = 7.26 ppm, 13C δ = 77.16 ppm; dichloromethane 1H δ = 5.32 ppm, 13C δ = 54.00 ppm; DMSO 1H δ = 2.50 ppm, 13C δ = 39.52 ppm). Infrared spectra were recorded using Thermo Fisher Nicolet iS50 FT-IR spectrometer with attenuated total reflectance (ATR) unit. Low- and high-resolution mass spectroscopy (LR- and HR-MS) analysis was conducted with mass spectrometer equipped with deep exposure probe (Bruker MS-320, GCMS or DEPMS mode) and electrospray ionization (ESI) time of flight (TOF) mass detector (ABSciex QTOF 5600), respectively. ESI-HRMS spectra were recorded in negative or positive ionization modes and the analytes were dissolved in HPLC grade ACN or 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DCM:methanol mixture. Elemental analysis was performed on a Thermo Scientific Flash 2000 analyzer. Chromatographic purifications were carried out with preparative flash chromatogram Isolera One from Biotage using self-packed columns with silica gel 60 Å 230–400 mesh (Merck). Celite 545 0.02–0.1 mm (Merck) was used as a Samplet® filler.
:1 DCM:methanol mixture. Elemental analysis was performed on a Thermo Scientific Flash 2000 analyzer. Chromatographic purifications were carried out with preparative flash chromatogram Isolera One from Biotage using self-packed columns with silica gel 60 Å 230–400 mesh (Merck). Celite 545 0.02–0.1 mm (Merck) was used as a Samplet® filler.
All target compounds were screened for antibacterial and antifungal activities by micro-dilution broth method using Mueller–Hinton broth and Mueller–Hinton broth with 5% lysed sheep blood for growth of non-fastidious and fastidious bacteria or Mueller–Hinton broth with 2% glucose for growth of fungi. Minimal inhibitory concentration (MIC) and minimal bactericidal concentration (MBC) of the tested compounds were evaluated for the panel of the reference microorganism from American Type Culture Collection (ATCC), including Escherichia coli ATCC25922, Salmonella typhimurium ATCC14028, Klebsiella pneumoniae ATCC13883, Pseudomonas aeruginosa ATCC9027 and Proteus mirabilis ATCC12453 were selected as exemplary Gram-negative bacteria; Staphylococcus aureus ATCC25923, Staphylococcus aureus ATCC6538, Staphylococcus aureus ATCC43300, Staphylococcus epidermidis ATCC12228, Micrococcus luteus ATCC10240, Bacillus subtilis ATCC6633, Bacillus cereus ATCC10876, Streptococcus pyogenes ATCC19615, Streptococcus pneumoniae ATCC49619 and Streptococcus mutans ATCC25175 as Gram-positive bacteria; and Candida albicans ATCC10231, Candida albicans ATCC2091, Candida parapsilosis ATCC22019, Candida glabrata ATCC 90030 and Candida crusei ATCC14243 as yeast. A detailed procedure for the determination of the MIC and MBC value has been presented in the earlier publication.32
Human histiocytic lymphoma cell line U-937 (obtained from American Type Culture Collection), human T-cell lymphoma HUT-78 and human normal B lymphocytes COLO-720L (obtained from European Collection of Authenticated Cell Cultures) were cultured in suspension in RPMI 1640 containing 5% FBS and 0.01% penicillin–streptomycin. Stock solution of compounds 1–12 were diluted with DMSO to the required concentration.
The MTT tetrazolium salt colorimetric assay was used to detect cytotoxicity of compounds 1–12. Cells were cultured in 96-well plates in amount of 0.2 × 106 U-937 cells per well in a volume of 0.2 mL. After incubation 50 μL MTT solution (5 mg mL−1) in sterile water was applied to each well and left for 2 h incubation at 37 °C. Then 0.4 mL of dimethylsulfoxide (DMSO) was added to each Eppendorf tube and left for 5 min. After centrifugation the optical density of the supernatant at 570 nm was read. Data were expressed as mean ± SD of at least 5 independent experiments.
Based on the results of the cell survival presented in this paper, the values of the median lethal dose (LD50 median lethal dose) for cells were calculated using Behrens' method74 with the following assumptions: the number of cells per measurement point is constant, and the absorbance determined in the MTT method is correlated with the number of live cells. Besides, if the cell is experiencing a higher dose, it has survived all lower doses; if the cell dies at the lower dose, it would die at all higher doses. Then, the percentage of mortality was calculated for each dose of the compound and LD50 values were obtained from appropriate graphs.
Pre-coated PAMPA Plate System Gentest™ was purchased from Corning, Tewksbury, MA, USA. The stock solutions of 5a, 5b, 5c and caffeine (10 mM) were prepared in DMSO. Before the assay the examined compounds were diluted in PBS buffer (pH 7.4) to 200 μM and added to the donor wells (300 μL per well). The PBS buffer was added to the acceptor wells (200 μL per well). Each compound was tested in triplicate. The plate was incubated next at room temperature without shaking for five hours. The solutions from donor and acceptor wells were analyzed by LC/MS Waters ACQUITY™ TQD system with the TQ Detector (Waters, Milford, USA). The analyte/internal standard (IS) peak height ratios were used to determine the concentrations of caffeine (CFN) in donor and acceptor wells. The permeability coefficient (Pe, cm s−1) was calculated using the following formula: Pe = {−ln[1 − CA/Ceq]}/[A × t × (1/VD + 1/VA)] where: A = filter area (0.3 cm2), VD = donor well volume (0.3 mL), VA = acceptor well volume (0.2 mL), t = incubation time (s), CA = compound concentration in the acceptor well after incubation, CD = compound concentration in the donor well after incubation, Ceq = [CD × VD + CA × VA]/(VD + VA).
General procedure for the synthesis of 1a, 1b and 1c
A 100 mL Schlenk flask was loaded with 4-(diphenylamino)phenylboronic acid, aryl bromoaldehyde and dichlorobis(triphenylophosphine)palladium(II). Subsequently, the flask was evacuated and backfilled with argon three times. Next, toluene, ethanol and deoxygenated aqueous potassium carbonate solution was added. The reaction mixture was vigorously stirred at 90 °C for 6 h. After complete consumption of bromoaldehyde confirmed by GC-MS, the volume of the organic phase was almost completely reduced under reduced pressure. Then, 20 mL of DCM was added and water phase was removed. The organic phase was washed three times with 50 mL of water and evaporated to dryness under reduced pressure. The residue was adsorbed on Celite and loaded into a Biotage Samplet®. The product was purified by column chromatography using gradient elution from 100% hexanes to 60% DCM:40% hexanes. The fractions containing desired product were collected and concentrated under reduced pressure. The residue was recrystallized by slow evaporation from DCM:hexanes and subsequently washed twice with pentane.
4′-(Diphenylamino)-[1,1′-biphenyl]-4-carbaldehyde (1a)
The product was synthesized according to the general procedure, using the following substances: 4-(diphenylamino)phenylboronic acid (1.272 g, 4.40 mmol), 4-bromobenzaldehyde (0.74 g, 4.0 mmol), dichlorobis(triphenylophosphine)palladium(II) (28.0 mg, 0.04 mmol), 2 M potassium carbonate aqueous solution (11.99 mL, 23.96 mmol), 23 mL of toluene 7.6 mL of ethanol. The product was obtained as yellow needles (1.36 g, 3.90 mmol). Yield: 90%. 1H NMR (300 MHz, CD2Cl2) δ 10.02 (s, 1H), 7.92 (d, J = 8.4 Hz, 2H), 7.75 (d, J = 8.4 Hz, 2H), 7.55 (d, J = 8.8 Hz, 2H), 7.36–7.23 (m, 4H), 7.18–7.02 (m, 8H). 13C NMR (75 MHz, CD2Cl2) δ 192.20, 149.00, 147.93, 146.97, 135.40, 133.30, 130.72, 129.95, 128.54, 127.39, 125.45, 124.05, 123.57. IR (ATR, νmax, cm−1): 1690 (CO). MS (DEP-LRMS): 350.3 (28.0%), 349.2 (100.0%). MS (ESI-HRMS): calculated for [C25H20NO]+ 350.1539, measured 350.1565 (error 7.4 ppm).
4-(4-(Diphenylamino)phenyl)-1-naphthalenecarbaldehyde (1b)
The product was synthesized according to the general procedure, using the following substances: 4-(diphenylamino)phenylboronic acid (1.178 g, 4.08 mmol), 4-bromo-1-naphthalenecarbaldehyde (0.871 g, 3.71 mmol), dichlorobis(triphenylophosphine)palladium(II) (26.0 mg, 0.037 mmol), 2 M potassium carbonate aqueous solution (11.12 mL, 23.96 mmol), 22 mL of toluene 7 mL of ethanol. The product was obtained as yellow powder (1.05 g, 2.63 mmol). Yield: 71%. 1H NMR (300 MHz, CD2Cl2) δ 10.40 (s, 1H), 9.34 (d, J = 7.6 Hz, 1H), 8.13 (d, J = 8.4 Hz, 1H), 8.03 (d, J = 7.4 Hz, 1H), 7.71 (t, J = 6.9 Hz, 1H), 7.65–7.53 (m, 2H), 7.42–7.26 (m, 6H), 7.24–7.14 (m, 6H), 7.09 (t, J = 7.3 Hz, 2H). 13C NMR (75 MHz, CD2Cl2) δ 193.74, 148.51, 148.11, 147.70, 136.88, 133.82, 132.56, 131.73, 131.30, 130.85, 129.95, 129.25, 127.40, 127.36, 126.60, 125.48, 125.37, 123.92, 123.28. IR (ATR, νmax, cm−1): 1680 (CO). MS (DEP-LRMS): 400.3 (31.4%), 399.3 (100%). MS (ESI-HRMS): calculated for [C29H22NO]+ 400.1696, measured 400.1728 (error 8.0 ppm).
10-(4-(Diphenylamino)phenyl)anthracene-9-carbaldehyde (1c)
The product was synthesized according to the general procedure, using the following substances: 4-(diphenylamino)phenylboronic acid (0.829 g, 2.87 mmol), 10-bromo-9-anthracenecarbaldehyde (0.743 g, 2.61 mmol), dichlorobis(triphenylophosphine)palladium(II) (18.0 mg, 0.026 mmol), 2 M potassium carbonate aqueous solution(7.82 mL, 15.63 mmol), 15 mL of toluene 5 mL of ethanol. The product was obtained as orange powder (1.06 g, 2.36 mmol). Yield: 90%. 1H NMR (300 MHz, CD2Cl2) δ 11.55 (s, 1H), 9.00 (d, J = 9.0 Hz, 2H), 7.90 (d, J = 8.8 Hz, 2H), 7.73–7.62 (m, 2H), 7.53–7.42 (m, 2H), 7.41–7.31 (m, 4H), 7.30–7.22 (m, 8H), 7.10 (t, J = 7.2 Hz, 2H). 13C NMR (75 MHz, CD2Cl2) δ 193.79, 148.32, 148.20, 146.06, 132.20, 132.09, 132.01, 130.68, 129.98, 129.08, 128.67, 125.98, 125.53, 125.36, 124.00, 123.90, 123.29. IR (ATR, νmax, cm−1): 1664 (CO). MS (DEP-LRMS): 450.3 (36.2%), 449.3 (100.0%). MS (ESI-HRMS): calculated for [C33H24NO]+ 450.1852, measured 400.1876 (error 5.3 ppm).
Rhodanine (2a)
A solution of chloroacetic acid (23.62 g, 0.25 mol) and ammonium thiocyanate (38.06 g, 0.50 mol) in 150 mL of water was heated for 20 minutes under reflux. After cooling down, a precipitate was formed. The crude product was filtered and recrystallized from water. The product was obtained as a yellowish powder (14.96 g, 0.113 mol), yield: 45%. 1H NMR (300 MHz, DMSO-d6) δ 13.14 (s, 1H), 4.26 (s, 2H). 13C NMR (75 MHz, DMSO-d6) δ 205.43, 176.81, 39.42. MS (ESI-HRMS): calculated for [C3H2NOS2]− 131.9583, measured 131.9572 (error 8.3 ppm).Rhodanine-3-acetic acid (2b)
A solution of potassium hydroxide (28.00 g, 0.5 mol) in 100 mL of water was added to the suspension of aminoethanoic acid (18.75 g, 0.25 mol). The resulting solution was cooled to 5 °C and carbon disulfide (19.03 g, 0.25 mol) was added. The content of the flask was mixed at 5 °C for 6 h. The cooling bath was removed and mixing was continued at room temperature for 20 h. Then, a solution of chloroacetic acid (23.62 g, 0.25 mol) in 100 mL of water was added. The reaction mixture was stirred for 8 h at the temperature below 15 °C. Next, a mixture of 150 mL concentrated hydrochloric acid and 200 mL of water was added slowly. The resulting mixture was heated at 90 °C for 25 min. After cooling down, a precipitate was formed, which was next filtered and recrystallized from water. The product was obtained as a white crystalline solid (21.49 g, 0.275 mol), yield: 55%. 1H NMR (300 MHz, DMSO-d6) δ 4.55 (s, 2H), 4.40 (s, 2H). 13C NMR (75 MHz, DMSO-d6) δ 202.96, 173.88, 167.47, 44.90, 36.08. MS (ESI-HRMS): calculated for [C5H4NO3S2]− 189.9638, measured 189.9625 (error 6.8 ppm).1-Acetyl-2-thiohydantoin (3a)
A flask containing a mixture of aminoethanoic acid (37.5 g, 0.5 mol), ammonium thiocyanate (38.06 g, 0.5 mol), 150 mL of acetic anhydride and 15 mL of acetic acid was heated in a water bath at 110 °C for 30 minutes under reflux. Next, the flask was cooled to 60 °C and heated again at 100 °C for 15 minutes. Then, the reaction mixture was poured into a beaker containing 1000 mL of cold water. The resulting precipitate was filtered and recrystallized from acetic acid. The product was obtained as a brown crystalline solid (55.3 g, 0.35 mol), yield: 70%. 1H NMR (300 MHz, DMSO-d6) δ 12.58 (s, 1H), 4.39 (s, 2H), 2.67 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 182.57, 170.46, 169.44, 52.26, 26.70, 26.67. MS (ESI-HRMS): calculated for [C5H5N2O2S]− 157.0077, measured 157.0052 (error 15.9 ppm).2-Thiohydantoin-3-acetic acid (3b)
A solution of sodium hydroxide (12.0 g, 0.3 mol) was added to a well stirred suspension of aminoethanoic acid (18.75 g, 0.25 mol) in 20 mL of water. To the resulting solution carbon disulfide (11.42 g, 0.15 mol) was added under nitrogen atmosphere and the reaction mixture was refluxed for 8 h. After the heating was finished, the excess of carbon disulfide was evaporated on a rotary evaporator. Next, a mixture of 100 mL of concentrated hydrochloric acid and 200 mL of water was added. The resulting mixture was refluxed for 2 h. After cooling, a precipitate was formed, which was filtered and subsequently recrystallized from water. The product was obtained as a white crystalline solid (39.15 g, 0.225 mol), yield: 75%. 1H NMR (300 MHz, DMSO-d6) δ 10.34 (s, 1H), 4.34 (s, 2H), 4.23 (d, J = 1.3 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 182.87, 172.27, 168.44, 48.64, 41.47. MS (ESI-HRMS): calculated for [C5H5N2O3S]− 173.0026, measured 173.0008 (error 10.4 ppm).General procedure for the synthesis of 4a–7c
1a (0.1 g, 0.286 mmol), 1b (0.114 g, 0.286 mmol) or 1c (0.129 g, 0.286 mmol), respective rhodanine (0.046 g, 0.343 mmol), rhodanine-3-acetic acid (0.066 g, 0.343 mmol), 1-acetyl-2-thiohydantoin (0.054 g, 0.343 mmol) or 2-thiohydantoin-3-acetic acid (0.06 g, 0.343 mmol) and ammonium acetate (44 mg, 0.57 mmol) were added to a CEM microwave vial. Subsequently, 2 mL of AA was added and the vessel was closed with a cap. The mixture was heated with maximum power 200 W at 180 °C for 5–7 min (5 min with 1a, 6 min with 1b and 7 min with 1c). During heating the color of the mixtures turned red very quickly and became homogenous. After cooling back to room temperature the precipitate was formed (some products tended to form supercooled solutions, in that case precipitation was initiated by ultrasonification). The solid was centrifuged and the supernatant was sucked off with a syringe. The solid was washed with 1 mL of AA and 4 times with 4 mL portions of water in the same manner. The obtained product was dried in an oven at 110 °C overnight. In some cases the product was purified by subsequent recrystallization from 2-propanol.(Z)-5-((4′-(Diphenylamino)-(1,1′-biphenyl)-4-yl)methylene)-2-thioxothiazolidin-4-one (4a)
Brick-red crystalline solid (128 mg, 0.276 mmol). Yield: 96%. 1H NMR (300 MHz, DMSO-d6) δ 7.79 (d, J = 8.2 Hz, 2H), 7.71–7.55 (m, 5H), 7.33 (t, J = 7.8 Hz, 4H), 7.14–6.95 (m, 8H). 13C NMR (75 MHz, DMSO-d6) δ 195.64, 169.62, 147.75, 146.89, 141.66, 132.02, 131.50, 131.42, 129.84, 127.94, 126.90, 124.86, 124.72, 123.82, 122.64. IR (ATR, νmax, cm−1): 1689 (CO), 1068 (CS). MS (ESI-HRMS): calculated for [C28H19N2OS2]− 463.0944, measured 463.0947 (error 0.6 ppm).
(Z)-5-((4-(4-(Diphenylamino)phenyl)naphthalen-1-yl)methylene)-2-thioxothiazolidin-4-one (4b)
Dark-brown crystalline solid (139 mg, 0.270 mmol). Yield: 94%. 1H NMR (300 MHz, DMSO-d6) δ 8.31 (s, 1H), 8.23 (d, J = 8.4 Hz, 1H), 8.01 (d, J = 8.1 Hz, 1H), 7.75–7.53 (m, 4H), 7.45–7.30 (m, 6H), 7.18–7.04 (m, 8H). 13C NMR (151 MHz, DMSO-d6) δ 196.17, 168.97, 147.16, 146.97, 142.33, 132.77, 131.66, 131.19, 130.85, 129.75, 129.34, 128.60, 128.17, 127.52, 127.17, 126.68, 126.63, 126.57, 124.58, 123.93, 123.60, 122.26. IR (ATR, νmax, cm−1): 1683 (CO), 1073 (CS). MS (ESI-HRMS): calculated for [C32H21N2OS2]− 513.1101, measured 513.1103 (error 0.4 ppm).
(Z)-5-((10-(4-(Diphenylamino)phenyl)anthracen-9-yl)methylene)-2-thioxothiazolidin-4-one (4c)
Orange powder (145 mg, 0.257 mmol). Yield: 90%. 1H NMR (300 MHz, DMSO-d6) δ 8.52 (s, 1H), 8.10 (d, J = 8.6 Hz, 2H), 7.75 (d, J = 8.7 Hz, 2H), 7.68–7.58 (m, 2H), 7.58–7.47 (m, 2H), 7.39 (t, J = 7.9 Hz, 4H), 7.30 (d, J = 8.5 Hz, 2H), 7.24–7.08 (m, 8H). 13C NMR (151 MHz, DMSO-d6) δ 147.24, 132.02, 129.97, 129.56, 127.88, 127.42, 127.04, 126.27, 125.58, 124.76, 124.70, 123.78, 122.36. IR (ATR, νmax, cm−1): 1734 (CO), 1068 (CS). MS (ESI-HRMS): calculated for [C36H23N2OS2]− 563.1257, measured 563.1259 (error 0.4 pm).
(Z)-2-(5-((4′-(Diphenylamino)-(1,1′-biphenyl)-4-yl)methylene)-4-oxo-2-thioxothiazolidin-3-yl)acetic acid (5a)
Red powder (140 mg, 0.268 mmol). Yield: 94%. 1H NMR (300 MHz, DMSO-d6) δ 7.90 (s, 1H), 7.84 (d, J = 8.3 Hz, 2H), 7.70 (dd, J = 8.5, 6.3 Hz, 4H), 7.34 (t, J = 7.8 Hz, 4H), 7.15–7.04 (m, 6H), 7.02 (d, J = 8.6 Hz, 2H), 4.74 (s, 2H). 13C NMR (151 MHz, DMSO) δ 193.21, 167.49, 166.56, 147.89, 146.88, 142.17, 133.79, 131.89, 131.76, 131.28, 129.87, 128.01, 127.01, 124.80, 123.90, 122.56, 121.12, 45.19. IR (ATR, νmax, cm−1): 1722 (CO), 1701 (CO), 1056 (CS). MS (ESI-HRMS): calculated for [C30H21N2O3S2]− 521.0999, measured 521.0994 (error 1.0 ppm).
(Z)-2-(5-((4-(4-(Diphenylamino)phenyl)naphthalen-1-yl)methylene)-4-oxo-2-thioxothiazolidin-3-yl)acetic acid (5b)
Orange crystalline solid (148 mg, 0.258 mmol). Yield: 90%. 1H NMR (300 MHz, DMSO-d6) δ 8.55 (s, 1H), 8.26 (d, J = 8.3 Hz, 1H), 8.02 (d, J = 8.3 Hz, 1H), 7.79 (d, J = 7.6 Hz, 1H), 7.75–7.61 (m, 2H), 7.59 (d, J = 7.6 Hz, 1H), 7.45–7.31 (m, 6H), 7.17–7.04 (m, 8H), 4.78 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 193.84, 167.51, 166.03, 147.34, 147.06, 142.95, 132.76, 131.79, 131.32, 130.97, 130.73, 129.89, 129.21, 127.82, 127.41, 127.18, 126.79, 126.73, 124.95, 124.73, 124.09, 123.78, 122.29, 45.22. IR (ATR, νmax, cm−1): 1725 (CO), 1708 (CO), 1062 (CS). MS (ESI-HRMS): calculated for [C34H23N2O3S2]− 571.1156, measured 571.1137 (error 3.3 ppm).
(Z)-2-(5-((10-(4-(Diphenylamino)phenyl)anthracen-9-yl)methylene)-4-oxo-2-thioxothiazolidin-3-yl)acetic acid (5c)
Red crystalline solid (159 mg, 0.255 mmol). Yield: 89%. 1H NMR (300 MHz, DMSO-d6) δ 8.83 (s, 1H), 8.06 (d, J = 8.7 Hz, 2H), 7.76 (d, J = 8.7 Hz, 2H), 7.69–7.58 (m, 2H), 7.59–7.48 (m, 2H), 7.38 (t, J = 7.8 Hz, 4H), 7.28 (d, J = 8.5 Hz, 2H), 7.23–7.07 (m, 8H), 4.79 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 193.63, 167.57, 165.05, 147.18, 139.73, 132.57, 131.94, 131.10, 130.85, 129.92, 129.53, 127.89, 127.51, 127.27, 126.99, 126.31, 125.30, 124.76, 123.75, 122.25, 45.29. IR (ATR, νmax, cm−1): 1727 (CO), 1713 (CO), 1056 (CS). MS (ESI-HRMS): calculated for [C38H25N2O3S2]− 621.1312, measured 621.1289 (error 3.7 ppm).
(Z)-5-((4′-(Diphenylamino)-(1,1′-biphenyl)-4-yl)methylene)-2-thioxoimidazolidin-4-one (6a)
Orange crystalline solid (92 mg, 0.206 mmol). Yield: 72%. 1H NMR (300 MHz, DMSO-d6) δ 12.38 (s, 1H), 12.20 (s, 1H), 7.80 (d, J = 8.2 Hz, 2H), 7.67 (t, J = 8.2 Hz, 4H), 7.33 (t, J = 7.8 Hz, 4H), 7.13–6.97 (m, 8H), 6.51 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ 179.17, 166.05, 147.45, 147.05, 140.32, 132.81, 131.06, 129.88, 127.83, 127.61, 126.46, 124.59, 123.73, 123.05, 111.63. IR (ATR, νmax, cm−1): 1720 (CO), 1080 (CS). MS (ESI-HRMS): calculated for [C28H20N3OS]− 446.1333, measured 446.1321 (error 2.7 ppm).
(Z)-5-((4-(4-(Diphenylamino)phenyl)naphthalen-1-yl)methylene)-2-thioxoimidazolidin-4-one (6b)
Yellow crystalline solid (122 mg, 0.245 mmol). Yield: 86%. 1H NMR (300 MHz, DMSO-d6) δ 12.34 (s, 2H), 8.15 (d, J = 8.2 Hz, 1H), 7.98 (d, J = 7.9 Hz, 1H), 7.85 (d, J = 7.5 Hz, 1H), 7.70–7.53 (m, 2H), 7.49 (d, J = 7.5 Hz, 1H), 7.45–7.31 (m, 6H), 7.17–7.05 (m, 9H). 13C NMR (151 MHz, DMSO-d6) δ 130.99, 129.93, 124.64, 123.72. IR (ATR, νmax, cm−1): 1714 (CO), 1087 (CS). MS (ESI-HRMS): calculated for [C32H22N3OS]− 496.1489, measured 496.1465 (error 4.8 ppm).
(Z)-5-((10-(4-(Diphenylamino)phenyl)anthracen-9-yl)methylene)-2-thioxoimidazolidin-4-one (6c)
The pure product was obtained after recrystallization from 2-propanol. Red crystalline solid (116 mg, 0.212 mmol). Yield: 74%. 1H NMR (300 MHz, DMSO-d6) δ 12.37 (s, 1H), 11.67 (s, 1H), 8.06 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 8.6 Hz, 2H), 7.62–7.46 (m, 4H), 7.46–7.35 (m, 4H), 7.34–7.25 (m, 3H), 7.22 (d, J = 7.1 Hz, 6H), 7.12 (t, J = 7.3 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 179.13, 164.64, 147.15, 146.88, 137.76, 133.63, 131.86, 131.51, 129.79, 129.64, 129.08, 127.09, 126.77, 126.18, 125.85, 125.68, 124.57, 123.54, 122.38, 108.17. IR (ATR, νmax, cm−1): 1742 (CO), 1099 (CS). MS (ESI-HRMS): calculated for [C36H24N3OS]− 546.1646, measured 546.1629 (error 3.1 ppm).
(Z)-2-(5-((4′-(Diphenylamino)-(1,1′-biphenyl)-4-yl)methylene)-4-oxo-2-thioxoimidazolidin-3-yl)acetic acid (7a)
Orange crystalline solid (122 mg, 0.241 mmol). Yield: 84%. 1H NMR (300 MHz, DMSO-d6) δ 12.58 (s, 1H), 7.85 (d, J = 8.2 Hz, 2H), 7.72 (d, J = 8.2 Hz, 2H), 7.68 (d, J = 8.4 Hz, 2H), 7.34 (t, J = 7.7 Hz, 4H), 7.15–6.97 (m, 8H), 6.72 (s, 1H), 4.51 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 178.26, 168.37, 163.78, 147.49, 147.00, 140.67, 132.65, 131.27, 130.76, 129.84, 127.84, 126.47, 125.72, 124.60, 124.58, 123.73, 122.93, 113.61, 41.97. IR (ATR, νmax, cm−1): 1735 (CO), 1647 (CO), 1076 (CS). MS (ESI-HRMS): calculated for [C30H22N3O3S]− 504.1387, measured 504.1383 (error 0.8 ppm).
(Z)-2-(5-((4-(4-(Diphenylamino)phenyl)naphthalen-1-yl)methylene)-4-oxo-2-thioxoimidazolidin-3-yl)acetic acid (7b)
Yellow powder (135 mg, 0.243 mmol). Yield: 85%. 1H NMR (300 MHz, DMSO-d6) δ 8.17 (d, J = 8.2 Hz, 1H), 7.99 (d, J = 8.3 Hz, 1H), 7.88 (d, J = 7.5 Hz, 1H), 7.70–7.55 (m, 2H), 7.52 (d, J = 7.5 Hz, 1H), 7.44–7.29 (m, 7H), 7.17–7.05 (m, 8H), 4.54 (s, 2H). 13C NMR (151 MHz, DMSO) δ 178.48, 168.44, 163.38, 147.15, 147.09, 141.08, 133.36, 131.75, 131.19, 130.94, 129.88, 128.57, 128.37, 128.14, 127.25, 126.86, 126.73, 126.48, 124.63, 124.58, 124.38, 123.69, 123.64, 122.59, 122.53, 110.33, 42.00. IR (ATR, νmax, cm−1): 1707 (CO), 1649 (CO), 1073 (CS). MS (ESI-HRMS): calculated for [C34H24N3O3S]− 554.1544, measured 554.1527 (error 3.1 ppm).
(Z)-2-(5-((10-(4-(Diphenylamino)phenyl)anthracen-9-yl)methylene)-4-oxo-2-thioxoimidazolidin-3-yl)acetic acid (7c)
The pure product was obtained after recrystallization from 2-propanol. Orange powder (128 mg, 0.211 mmol). Yield: 74%. 1H NMR (300 MHz, DMSO-d6) δ 12.09 (s, 1H), 8.05 (d, J = 8.6 Hz, 2H), 7.76 (d, J = 8.6 Hz, 2H), 7.64–7.48 (m, 5H), 7.40 (t, J = 7.7 Hz, 4H), 7.33–7.26 (m, 2H), 7.25–7.17 (m, 6H), 7.12 (t, J = 7.3 Hz, 2H), 4.55 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 178.29, 172.10, 168.35, 162.43, 147.16, 146.93, 138.11, 131.85, 131.81, 131.42, 129.81, 129.66, 129.09, 127.15, 126.36, 125.76, 125.69, 124.61, 123.58, 122.36, 110.38, 41.85. IR (ATR, νmax, cm−1): 1718 (CO), 1643 (CO), 1075 (CS). MS (ESI-HRMS): calculated for [C38H26N3O3S]− 604.1700, measured 604.1692 (error 1.3 ppm).
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The research was partially financed from the Polish Statutory Research Projects K/ZDS/007886 and BS-465/G/2018.Notes and references
- S. N. Farouk, J. Adv. Microbiol., 2017, 7, 1 Search PubMed.
- M. Exner, S. Bhattacharya, B. Christiansen, J. Gebel, P. Goroncy-Bermes, P. Hartemann, P. Heeg, C. Ilschner, A. Kramer, E. Larson, W. Merkens, M. Mielke, P. Oltmanns, B. Ross, M. Rotter, R. M. Schmithausen, H. G. Sonntag and M. Trautmann, GMS Hyg. Infect. Control, 2017, 12, Doc05, DOI:10.3205/dgkh000290.
- K. C. Huanga, R. Mukhopadhyay, B. Wena, Z. Gitaia and N. S. Wingreena, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19282 CrossRef PubMed.
- N. Kashef, Y. Y. Huang and M. R. Hamblin, Nanophotonics, 2017, 6, 853 CAS.
- S. I. Miller and N. R. Salama, PLoS Biol., 2018, 16, e2004935 CrossRef PubMed.
- K. Li, X. X. Yuan, H. M. Sun, L. S. Zhao, R. Tang, Z. H. Chen, Q. L. Qin, X. L. Chen, Y. Z. Zhang and H. N. Su, Microbiology, 2018, 9, 620, DOI:10.3389/fmicb.2018.00620.
- A. Al-Mulla, Der Pharma Chem., 2017, 9, 141 CAS.
- R. Kumar and S. Patil, Hygeia J. Drugs Med., 2017, 9, 80 CAS.
- S. K. Manjal, R. Kaur, R. Bhatia, K. Kumar, V. Singh, R. Shankar, R. Kaur and R. K. Rawal, Bioorg. Chem., 2017, 75, 406 CrossRef PubMed.
- S. Vengurlekar, R. Sharma and T. Trivedi, Lett. Drug Des. Discov., 2012, 9, 549 CrossRef CAS.
- D. Kaminskyy, A. Kryshchyshyn and R. Lesyk, Eur. J. Med. Chem., 2017, 140, 542 CrossRef CAS PubMed.
- M. A. Metwally and E. Abdel-Latif, J. Sulfur Chem., 2012, 33, 229 CrossRef CAS.
- T. Mendgen, C. Steuer and C. D. Klein, J. Med. Chem., 2012, 55, 743 CrossRef CAS PubMed.
- H. A. Elhady, H. S. Al-Nathali and R. El-Sayed, Int. J. Adv. Res., 2017, 5, 1716 CrossRef.
- C. Mathew, B. Saraswati, N. Lal and J. Varkey, Int. J. Pharm. Chem., 2018, 8, 25 CAS.
- X. Xu, R. Ge, L. Li, J. Wang, X. Lu, S. Xue, X. Chen, Z. Li and J. Bian, Eur. J. Med. Chem., 2018, 143, 1325 CrossRef CAS PubMed.
- J. Handzlik, G. Spengler, B. Mastek, A. Dela, J. Molnar, L. Amaral and K. Kieć-Konowicz, Acta Pol. Pharm., 2012, 61, 149 Search PubMed.
- A. Y. Hassan, M. M. Said, M. T. Sarg, H. S. Al-Zahabi and E. M. Hussein, Life Sci., 2013, 10, 1993 Search PubMed.
- S. Prachand, A. K. Gupta and R. Gilhotra, Int. J. Pharm. Sci. Nanotechnol., 2018, 11, 3977 CAS.
- N. Daş-Evcimen, N. O. Bozdağ-Dündar, M. Sarikaya and R. Ertan, J. Enzyme Inhib. Med. Chem., 2008, 23, 297 CrossRef PubMed.
- S. Rekha, U. Shantharam and C. Vineet, Int. Res. J. Pharm., 2011, 2, 81 CAS.
- Y. Huang, Z. Guo, H. Song, Y. Liu, L. Wang and Q. Wang, J. Agric. Food Chem., 2018, 66, 8253 CrossRef CAS PubMed.
- C. Tintori, G. Iovenitti, E. R. Ceresola, R. Ferrarese, C. Zamperini, A. Brai, G. Poli, E. Dreassi, V. Cagno, D. Lembo, F. Canducci and M. Botta, PLoS One, 2018, 13, e0198478 CrossRef PubMed.
- J. Gagoria, A. Khatkar, S. Khatkar, R. Rao, K. Singh and A. Nanda, Orient. J. Chem., 2008, 24, 673 CAS.
- N. Lopez, T. C. Jose, G. Krisnan and W. Sam, Int. J. Pharm. Sci. Rev. Res., 2016, 41, 242 CAS.
- D. K. Mahapatra, D. Das, R. S. Shivhare and S. S. Borkar, MOJ Bioorganic & Organic Chemistry, 2018, 2, 46 Search PubMed.
- C. J. R. Bataille, M. B. Brennan, S. Byrne, S. G. Davies, M. Durbin, O. Fedorov, K. V. M. Huber, A. M. Jones, S. Knapp, G. Liu, A. Nadali, C. E. Quevedo, A. J. Russell, R. G. Walker, R. Westwood and G. M. Wynne, Bioorg. Med. Chem., 2017, 25, 2657 CrossRef CAS PubMed.
- Y. Huang, Z. Guo, H. Song, Y. Liu, L. Wang and Q. Wang, J. Agric. Food Chem., 2018, 66, 8253 CrossRef CAS PubMed.
- J. Sivanadanam, R. Mukkamala, S. Mandal, R. Vedarajan, N. Matsumi, I. S. Aidhen and K. Ramanujam, Int. J. Hydrogen Energy, 2018, 43, 4691 CrossRef CAS.
- D. M. Chang, D. Y. Kwon and Y. S. Kim, Mol. Cryst. Liq. Cryst., 2013, 585, 100 CrossRef CAS.
- K. Narayanaswamy, T. Swetha, G. Kapil, S. S. Pandey, S. Hayase and S. P. Singh, Electrochim. Acta, 2015, 169, 256 CrossRef CAS.
- W. Tejchman, I. Korona-Głowniak, A. Malm, M. Żylewski and P. Suder, Med. Chem. Res., 2017, 26, 1316 CrossRef CAS PubMed.
- T. Tomasić, N. Zidar, M. Mueller-Premru, D. Kikelj and L. P. Masic, Eur. J. Med. Chem., 2010, 45, 1667 CrossRef PubMed.
- M. M. Sim, S. B. Ng, A. D. Buss, S. C. Crasta, K. L. Goh and S. K. Lee, Bioorg. Med. Chem. Lett., 2002, 12, 697 CrossRef CAS PubMed.
- H. A. Shindy, A. K. Khalafalla, M. M. Goma and A. H. Eed, Chem. Int., 2016, 2, 114 CAS.
- A. Abdel Khalek, C. R. Asby, B. A. Patel, T. T. Talele and M. N. Selem, PLoS One, 2016, 11, e0164227 CrossRef PubMed.
- R. Bajpai, International Journal of Engineering and Science Invention, 2013, 2, 23 Search PubMed.
- M. N. Song, X. Q. Deng, Y. R. Li, C. J. Zheng, L. Hong and H. R. Piao, J. Enzyme Inhib. Med. Chem., 2014, 29, 647 CrossRef CAS PubMed.
- W. T. Sing, C. L. Lee, S. L. Yeo, S. P. Lim and M. M. Sim, Bioorg. Med. Chem. Lett., 2001, 11, 91 CrossRef CAS PubMed.
- N. Trotsko, U. Kosikowska, A. Paneth, M. Wujec and A. Malm, Saudi Pharm. J., 2018, 26, 568 CrossRef PubMed.
- A. F. P. Biajoli, C. S. Schwalm, J. Limberger, T. S. Claudino and A. L. Monteiro, J. Braz. Chem. Soc., 2014, 25, 2186 CAS.
- A. J. Burke, C. S. Marques, N. J. Turner and G. J. Hermann, Active Pharmaceutical Ingredients in Synthesis: Catalytic Processes in Research and Development, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2018, ISBN: 978-3-527-80726-0 Search PubMed.
- W. Wu, C. Cheng, W. Wu, H. Guo, S. Ji, P. Song, K. Han, J. Zhao, X. Zhang, Y. Wu and G. Du, Eur. J. Inorg. Chem., 2010, 29, 4683 CrossRef.
- W. Guo, E. Faggi, R. M. Sebastián, A. Vallribera, R. Pleixats and A. Shafir, J. Org. Chem., 2013, 78, 8169 CrossRef CAS PubMed.
- S. Sasaki, Y. Niko, K. Igawa and G. Konishi, RSC Adv., 2014, 4, 33474 RSC.
- C. Liu, Q. Ni and J. Qiu, Eur. J. Org. Chem., 2011, 16, 3009 CrossRef.
- X. Jin, C. J. Zheng, M. X. Song, Y. Wu, L. P. Sun, Y. J. Li, L. J. Yu and H. R. Piao, Eur. J. Med. Chem., 2012, 56, 203 CrossRef CAS PubMed.
- M. A. El-Moneim, E. H. Elsayed and M. El-Ashry, J. Pharm. Pharm. Sci., 2017, 6, 20 CAS.
- C. Ozen, M. Ceylan-Unlusoy, N. Aliary, M. Ozturk and O. Bozdag-Dundar, J. Pharm. Sci., 2017, 20, 415 CAS.
- C. Nitsche, V. N. Schreier, M. A. M. Behnam, K. Kumar, R. Bartenschlager and C. D. Klein, J. Med. Chem., 2013, 56, 8389 CrossRef CAS PubMed.
- B. Orwat, M. Oh, M. Kubicki and I. Kownacki, Adv. Synth. Catal., 2018, 360, 3331 CrossRef CAS.
- S. Reyes and K. Burgess, J. Org. Chem., 2006, 71, 2507 CrossRef CAS PubMed.
- G. L. French, J. Antimicrob. Chemother., 2006, 58, 1107 CrossRef CAS PubMed.
- T. T. Talele, P. Arora, S. S. Kulkarni, M. R. Patel, S. Singh, M. Chudayeu and N. Kaushik-Basu, Bioorg. Med. Chem., 2010, 18, 4630 CrossRef CAS PubMed.
- P. Ralph, M. A. Moore and K. Nilsson, J. Exp. Med., 1976, 143, 1528 CrossRef CAS PubMed.
- P. Harris and P. Ralph, J. Leukocyte Biol., 1985, 37, 407 CrossRef CAS PubMed.
- P. A. Bunn Jr and F. M. Foss, J. Cell. Biochem. Suppl., 1996, 63, 123 Search PubMed.
- M. S. Anglesio, K. C. Wiegand, N. Melnyk, C. Chow, C. Salamanca, L. M. Prentice and M. K. Shumansky, PLoS One, 2013, 8, e72162 CrossRef CAS PubMed.
- A. Coates, Y. Hu, R. Bax and C. Page, Nat. Rev. Drug Discovery, 2002, 1, 895 CrossRef CAS PubMed.
- W. Vollmer, D. Blanot and M. A. de Pedro, FEMS Microbiol. Rev., 2008, 32, 149 CrossRef CAS PubMed.
- V. N. Tra and D. H. Dube, Chem. Commun., 2014, 50, 4659 RSC.
- T. Mendgen, C. Steuer and C. D. Klein, J. Med. Chem., 2012, 55, 743 CrossRef CAS PubMed.
- M. I. Konaklieva, Antibiotics, 2014, 3, 128 CrossRef PubMed.
- A. Zervosen, W. P. Lu, Z. Chen, R. E. White, T. P. Demuth Jr and J. M. Frere, Antimicrob. Agents Chemother., 2004, 48, 961 CrossRef CAS PubMed.
- T. Ishida, Y. In, M. Inoue, Y. Ueno, C. Tanaka and N. Hamanaka, Tetrahedron Lett., 1989, 30, 959 CrossRef CAS.
- O. P. Dimitriev, K. P. Grytsenko, O. I. Tolmachev, Y. L. Slominskii, M. A. Kudinova and S. Schrade, Effect of Concentration on Isomerization of Rhodanine Derivatives of Merocyanine Dyes in Polar Solvents, Adv. Phys. Chem., 2014, 468910 Search PubMed.
- M. Nencki, J. Prakt. Chem., 1877, 16, 1 CrossRef.
- W. Tejchman, A. Skórska-Stania and E. Żesławska, J. Chem. Crystallogr., 2016, 46, 181 CrossRef CAS.
- T. B. Johnson and B. H. Nicolet, J. Am. Chem. Soc., 1911, 33, 1973 CrossRef CAS.
- E. J. Tartlon and A. F. McKay, Can. J. Chem., 1958, 36, 496 CrossRef.
- F. E. Hahn, T. Lügger and M. Beinhoff, Z. Naturforsch., B: J. Chem. Sci., 2004, 59, 196 CAS.
- S. Boudiba, A. Růžička, C. Ulbricht, S. Enengl, C. Enengl, J. Gasiorowski, C. Yumusak, V. Pokorná, D. Výprachtický, K. Hingerl, D. R. T. Zahn, F. Tinti, N. Camaioni, S. Bouguessa, A. Gouasmia, V. Cimrová and D. A. M. Egbe, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 129 CrossRef CAS.
- F. de Montigny, G. Argouarch and C. Lapinte, Synthesis, 2006, 2, 293 Search PubMed.
- G. Zbinden and M. Flury-Roversi, Arch. Toxicol., 1981, 47, 77 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR, IR, LR-MS and ESI-HRMS spectra, MIC and MBC values for selected Gram-positive bacteria, LD50 values for human immune cell lines, and PAMPA experiments HPLC chromatograms. See DOI: 10.1039/c9ra08690k |
| This journal is © The Royal Society of Chemistry 2019 |